

Abstract
Direct interaction of RAS with the PI3K p110α subunit mediates RAS-driven tumor development: however, it is not clear how p110α/RAS-dependant signaling mediates interactions between tumors and host tissues. Here, using a murine tumor cell transfer model, we demonstrated that disruption of the interaction between RAS and p110α within host tissue reduced tumor growth and tumor-induced angiogenesis, leading to improved survival of tumor-bearing mice, even when this interaction was intact in the transferred tumor. Furthermore, functional interaction of RAS with p110α in host tissue was required for efficient establishment and growth of metastatic tumors. Inhibition of RAS and p110α interaction prevented proper VEGF-A and FGF-2 signaling, which are required for efficient angiogenesis. Additionally, disruption of the RAS and p110α interaction altered the nature of tumor-associated macrophages, inducing expression of markers typical for macrophage populations with reduced tumor-promoting capacity. Together, these results indicate that a functional RAS interaction with PI3K p110α in host tissue is required for the establishment of a growth-permissive environment for the tumor, particularly for tumor-induced angiogenesis. Targeting the interaction of RAS with PI3K has the potential to impair tumor formation by altering the tumor-host relationship, in addition to previously described tumor cell–autonomous effects.
Introduction
Class I PI3Ks are expressed in all mammalian cell types and consist of 4 different isoforms: p110α, p110β, p110γ, and p110δ. Class I PI3Ks share some common structural characteristics, such as a p85-binding domain (except p110γ), a C2 domain, a helical domain, and a RAS-binding domain (RBD). Previous results from our laboratory have shown that the PI3K p110α subunit (encoded by Pik3ca) is able to bind directly to RAS through the RBD (1, 2). In vitro work has shown that this interaction is required for proper signaling propagation through some receptor tyrosine kinases (RTKs), like EGFR or FGFR, and that its loss reduces cell growth (3, 4). In vivo, the interaction of RAS with PI3K is also needed for proper development of embryonic lymphangiogenesis. Moreover, when the RBD of Pik3ca is mutated in such a way that RAS cannot bind to and activate PI3K, RAS-driven tumor development in the lung and skin is abrogated (5), and preexisting RAS-driven lung tumors undergo partial regression and long-term stasis (6).
PI3Ks control a wide variety of signaling networks that modulate many processes, including cellular survival and proliferation, and constitute an important convergence signaling point for many other signaling molecules, from RTKs and G protein–coupled receptors to intracellular proto-oncogenic proteins such as RAS. Many of these are mutated in cancer, promoting an unregulated activation of the PI3K signaling pathway that in turn leads to uncontrolled cellular proliferation and survival. Thus, the importance of the interaction of RAS with PI3K for the signaling of all these molecules, under both physiological and tumor conditions, acquires special relevance. Drugs targeting different components and members of the PI3K signaling pathway are currently being evaluated in clinical trials and show some promise, although it is still too early to predict whether they will become effective anticancer tools. Among the main problems of such inhibitors are the lack of information regarding the amount of PI3K inhibition needed to achieve a clinical effect and, linked to this, the appearance of dose-dependent side effects (7).
Drugs available against other RAS-regulated signaling proteins, such as BRAF and MEK, have also shown moderate efficacy, although this is limited due to rapid development of acquired resistance, in many cases due to aberrant activation of parallel signaling pathways that in turn stimulate the PI3K pathway (8–10). Moreover, RAS itself has proved impossible to target effectively to date (11, 12).
Our knowledge of tumor interactions with host tissues has evolved very quickly in the last decade and has proven to be extremely important for understanding all the steps of tumor natural history, from initial tumor formation to metastasis. It is well established that the tumor microenvironment is needed to support the growth and metastasis of tumors, and that tumor cells induce changes in the microenvironment to facilitate those processes (13–15). The stromal tissue supports the growth of tumors, orchestrating a completely new structural and molecular microenvironment. In building the new niche, 2 of the main steps involve the generation of new blood vessels and the suppression of an effective immune response. While tumor-induced angiogenesis is a process that has been considered for many years to be an important clinical target to halt tumor growth (16), new strategies to modulate the tumor-induced immune response have only recently been developed (17).
In the present work, we aimed to extend our studies of the role of the interaction of RAS with PI3K beyond tumor cell–intrinsic effects to the relationship of tumors with host tissue. We developed a mouse model in which we could tightly control the generation of WT and RBD mutant Pik3ca backgrounds to study how the disruption of the interaction of RAS with PI3K in the host tissue affects the growth of tumors, and to what extent this affects tumor metastasis. We found that tumors grew more slowly in mice with RBD mutant versus WT Pik3ca, and overall survival of tumor-bearing mice was increased. Furthermore, experimental metastasis assays showed that tumor cells had a highly reduced ability to anchor and grow in the RBD mutant Pik3ca environment. Disruption of the interaction of RAS with PI3K in the host had a major effect on tumor-induced angiogenesis, without any obvious deleterious effect in the normal homeostasis of the mice. Moreover, we observed a clear difference in the morphology and distribution of the tumor-infiltrating macrophage population, which additionally showed a shift to an M1-like, tumor suppressor phenotype. Disruption of the interaction of RAS with PI3K may therefore represent an outstanding molecular target for inhibiting tumors, not only directly in a cell-autonomous manner, but also indirectly via their interplay with the tumor microenvironment.
Results
Generation of a conditional RBD mutant Pik3ca mouse model.
In order to study the role of interaction of RAS with PI3K in tumor processes, our laboratory developed a mouse model in which 2 point mutations in the RBD of Pik3ca prevent physical interaction with RAS proteins (5). Animals homozygous for this mutation (Pik3caMUT/MUT mice) constitutively express only mutant p110α and show profound resistance to RAS-driven tumor onset. However, the relatively low percentage of Pik3caMUT/MUT mice obtained (due to developmental defects in lymphangiogenesis), the difficulty in generating murine tumor cell lines from naturally arising tumors, and the lack of control over the generation of mutant or WT genetic backgrounds prompted us to generate a conditional mouse model to study the effect of blocking PI3K interaction with RAS. We crossed mice in which WT Pik3ca can be conditionally deleted via floxed Pik3ca (18) onto a line ubiquitously expressing a tamoxifen-inducible, estrogen receptor (ER) hormone binding domain–fused Cre recombinase, Gt(ROSA)26Sortm9(cre/ESR1)Arte (referred to herein as Cre-ERT2 mice). These Pik3cafl/fl Cre-ERT2 mice were crossed with mice heterozygous for the RAS mutant Pik3ca allele. All resulting mice (referred to herein as Pik3caWT/fl [being totally WT] and Pik3caMUT/fl [being heterozygous for the RBD mutation]) harbored a copy of the ubiquitously expressed Cre-ERT2, allowing us to manipulate the timing of floxed Pik3ca deletion by tamoxifen administration, thus avoiding the developmental problems observed in Pik3caMUT/MUT homozygotes (Supplemental Figure 1A; supplemental material available online with this article; doi:10.1172/JCI74134DS1).
We monitored the percentage of mice obtained for each genotype and their growth progression to determine whether having a Pik3ca RBD mutant allele shows any effect in terms of development. In the absence of tamoxifen treatment, we did not find any difference in either birth percentage or weight gain between genotypes (Supplemental Figure 1B), which suggests that 1 WT Pik3ca allele is enough for maintaining homeostasis. Moreover, mice did not show any differential phenotype or adverse effect during their whole lifespan.
We next fed mice with tamoxifen-containing food pellets to activate the ubiquitously expressed Cre-ERT2 in order to obtain pure WT or mutant Pik3ca genetic backgrounds (referred to herein as Pik3caWT/– and Pik3caMUT/– mice, respectively). After 2 weeks, we sacrificed a group of animals and analyzed the degree of recombination in several tissues (Figure 1A), observing complete recombination of the floxed Pik3ca allele. A group of mice was maintained after recombination to monitor for any adverse effect or differential phenotype. These mice did not show any deleterious effects, and their lifespan was similar to that of mice containing 2 alleles. These data suggest that 1 Pik3ca allele — either WT or RBD mutant — is sufficient to maintain homeostasis, as previously reported (19).
Figure 1. Generation of a conditional RBD mutant Pik3ca mouse model.

(A) In order to assess the degree of recombination of the floxed Pik3ca allele, different tissues from a group of mice were collected no earlier than 21 days after tamoxifen food was withdrawn. DNA extracts were prepared and subjected to PCR analysis for detection of the floxed Pik3ca allele (upper band, 500 bp; lower band, 400 bp). (B–E) Primary MEFs were obtained from tamoxifen-untreated mice containing either the RBD mutant or the WT Pik3ca allele. Cells were treated with vehicle or tamoxifen (1 μM) for 16 hours followed by 24 hours in complete, tamoxifen-free media. Subsequently, cells were starved for 16 hours and treated with EGF (20 ng/ml) (B and C), PDGF (20 ng/ml) (D and E), or vehicle for 10 minutes. Protein extracts were prepared and subjected to Western blot analysis (B and D). Fluorescent secondary antibodies were used, and subsequent quantification in a LI-COR system was performed (C and E). A representative experiment is shown.
We previously reported that mouse embryonic fibroblasts (MEFs) obtained from homozygous Pik3caMUT/MUT mice are not able to activate the PI3K/AKT pathway upon stimulation by some growth factors, such as EGF (5). To verify that the conditional model retained the molecular characteristics observed in the constitutive mouse model, we generated Pik3caWT/fl, Pik3caMUT/fl, Pik3caWT/–, and Pik3caMUT/– MEFs and analyzed their response to growth factors. In response to EGF, Pik3caWT/fl MEFs activated AKT normally, whereas AKT activation was reduced in Pik3caMUT/fl and Pik3caWT/– MEFs and totally abrogated in Pik3caMUT/– MEFs (Figure 1, B and C). No differences in ERK activation were observed, in line with our results in Pik3caMUT/MUT MEFs, or for PDGF-induced AKT signaling, as previously reported (Figure 1, D and E, and ref. 5).
These data confirmed that the inducible mouse model does not show any developmental defect, that the generation of pure genetic backgrounds can be tightly controlled in time without any adverse effect, and that at the molecular level, the Pik3caMUT/– inducible model retains the characteristics of the Pik3caMUT/MUT constitutive model.
Disruption of the interaction of RAS with PI3K exclusively in host tissue reduces tumor growth.
We used lung carcinoma–derived LLC1 (Nras mutant) and melanoma-derived B16F10 (no Ras or RAS pathway mutations) murine cell lines to analyze the growth of tumor cells with normal PI3K signaling in Pik3caWT/– and Pik3caMUT/– syngeneic hosts. Cells were injected subcutaneously, and tumor growth was measured over time. Growth of B16F10 tumors was continuous regardless of genetic background, but was slower in Pik3caMUT/– versus Pik3caWT/– mice (Figure 2A). LLC1 tumors also grew more slowly in Pik3caMUT/– mice (Figure 2D). After 21 days of engraftment, tumors had grown much less in Pik3caMUT/– than in Pik3caWT/– mice for both lines (B16F10, 56% less; LLC1, 63% less; Figure 2, B and E). We also analyzed survival rates and found that for both cell lines, Pik3caMUT/– mice had a longer survival rate than Pik3caWT/– mice (Figure 2, C and F). 2 Pik3caMUT/– mice harboring LLC1 tumors did not reach the endpoint, and the tumors seemed to remain stable beyond the end of the analysis. Samples from the host mouse tissues were collected at the end of the experiment and subjected to genotyping to ensure that recombination was complete (Supplemental Figure 2A).
Figure 2. Disruption of the interaction of RAS with PI3K in the host reduces tumor growth.

B16F10 (A–C) and LLC1 (D–F) cells were injected into the indicated mice. (A and D) Tumor volumes, measured by caliper over time. n = 11 (A); 6 (D). (B and E) Tumor volume 18 (B) or 21 (E) days after cell injection. Horizontal bars represent the median. Mann-Whitney nonparametric statistical analysis was performed. (C and F) Overall survival. The endpoint of the analysis was set in 2 cm of length in any direction, as stated in the approved Animal Project License. Kaplan-Meier survival curves are shown. Mantel-Cox (Log-rank) statistical analysis was performed. n = 6 (C); 6 (F, WT/– and MUT/–); 10 (F, WT/fl and MUT/fl).
Together, these data indicated that disrupting the interaction of RAS with PI3K in host tissues reduces the rate of tumor growth in transplanted tumors with normal PI3K signaling.
Disruption of the interaction of RAS with PI3K in the host reduces metastasis.
We also analyzed the effect of disrupting interaction of RAS with PI3K in the host in an experimental metastasis assay. B16F10 cells harboring a luciferase expression gene were injected through the tail vein into syngeneic host mice with a WT or mutant Pik3ca background. Bioluminescence in vivo was measured after cell injection to ensure equal cell number in all mice (Supplemental Figure 3A). After 26 days, mice were sacrificed, and lungs were imaged ex vivo to avoid any interference with fur or skin (Supplemental Figure 3B). While no statistical differences in bioluminescence emission were found between Pik3caWT/fl and Pik3caMUT/fl mice, an 87% decrease was detected in Pik3caMUT/– compared with Pik3caWT/– mice (1.7 × 107 vs. 2.9 × 106; Figure 3A). We also observed reduced tumor burden using LLC1 cells (Supplemental Figure 3C). To further explore whether these differences were due to reduced number and/or growth of metastatic foci, lungs were subjected to histopathology analysis. Lungs from Pik3caWT/– mice showed multiple solid nodules located both in the periphery and inside the lung structure, whereas in Pik3caMUT/– lungs, only small, diffuse nodules were found, mostly located on the surface (Figure 3B). Using Nikon NIS-Elements software, the number of foci and overall tumor burden per whole lung was measured. Lungs from Pik3caMUT/– mice showed a much lower number of metastatic foci (75% less) and a drastic reduction in tumor burden (93% less) compared with Pik3caWT/– lungs (Figure 3, C and D). While the presence of fewer foci in the lungs could be explained by a lower degree of extravasation or tissue anchoring, foci were also 76% smaller in Pik3caMUT/– mice (Figure 3E), which indicated that the growth of established foci was also reduced.
Figure 3. Disruption of the interaction of RAS with PI3K in the host reduces metastasis.

(A) Luciferase activity of B16F10 cells injected in the tail vein was measured ex vivo 21 days after injection. Bars represent the median. Mann-Whitney nonparametric statistical analysis was performed. (B) H&E staining of lung sections. Arrows indicate diffuse nodules in the periphery of the lung. Scale bars: 1,000 μm (left); 100 μm (right). (C) Number of tumor foci per whole lung. (D) Tumor area, expressed as a percentage of total lung area. (E) Average foci area, expressed as a percentage of lung area. (C–E) Mann-Whitney statistical analysis was performed. n = 6 per group.
These results show that intact interaction of RAS with PI3K is required for efficient establishment of tumor foci and growth of tumor cells in an experimental metastasis model.
Disruption of the interaction of RAS with PI3K reduces tumor-induced angiogenesis.
We next sought to explore why tumors grew less in the mutant Pik3ca genetic background. We subjected transplanted B16F10 subcutaneous tumors and experimental metastasis samples to histopathology analysis. There were no differences in the number of apoptotic cells in necrotic interfaces (we did not detect apoptotic cells in non-necrotic tissue) or in the rate of proliferation of subcutaneous tumor samples (Supplemental Figure 4, A–C). However, we clearly observed a marked reduction in the amount of blood vessels and an increase in necrotic areas in Pik3caMUT/– versus Pik3caWT/– samples (Figure 4A). Specific staining for SMA also showed that general vessel architecture was somehow altered in Pik3caMUT/– samples, presenting a rather tortuous pattern (Figure 4A).
Figure 4. Disruption of the interaction of RAS with PI3K reduces tumor-induced angiogenesis.

B16F10 subcutaneous tumors were collected after 14 days of injection. (A) Paraffin sections were stained with endomucin and SMA specific antibodies. (B and C) Endomucin-stained sections were subjected to quantitative analysis using Leica’s Digital Pathology capture and analysis platform to analyze vessel density (B) and total vessel area (C). Mann-Whitney statistical analysis was performed (n = 5 per group). (D) Lung sections from experimental metastasis assays were also collected, and paraffin sections were stained with endomucin and nestin specific antibodies. Representative single nodules for each genotype are shown. Arrows denote preexisting, nesting-negative vessels in the periphery of the nodules. Scale bars: 100 μm (A, right, and D); 500 μm (A, middle); 1,000 μm (A, left).
To get further insight into these differences, we performed automated quantification of the number, distribution, and size of vessels inside the subcutaneous tumors using the Angiosight module from Leica’s Digital Pathology capture and analysis platform. We found that vessel density and total vessel area were highly reduced in the Pik3caMUT/– mice (about 50% less in both cases; Figure 4, B and C). We also analyzed the necrotic tissue inside the tumor and found that Pik3caMUT/– samples contained a much larger percentage of it than did Pik3caWT/– samples (30% more; Supplemental Figure 4D). A similar reduction in vessel density and increase in necrotic tissue was observed in LLC1 subcutaneous tumors (Supplemental Figure 4H). Analysis of vessel density restricted to healthy tissue (non-necrotic) also showed a marked decrease in Pik3caMUT/– samples (43.75% less; Supplemental Figure 4E). Interestingly, we did not find a statistical difference in average vessel area, although there was a small tendency to be smaller in Pik3caWT/– samples (Supplemental Figure 4F). We also analyzed the distribution of vessels in metastatic nodules from the experimental metastasis assay. While we clearly found vessels both inside and in the periphery of the Pik3caWT/– samples, we were not able to find vessels included in Pik3caMUT/– nodules, regardless of the their size (Supplemental Figure 4G). Only vessels on the periphery of these nodules were found. To further demonstrate that metastatic foci in the Pik3caMUT/– mice have reduced vascularization, we stained samples with endomucin and nestin, a marker for newly formed vessels. While nodules in the Pik3caWT/– mice showed a rich vasculature and clear nestin staining, the Pik3caMUT/– samples were virtually absent of both endomucin and nestin staining, exhibiting marginal staining clearly restricted to the periphery of the nodules (Figure 4D and Supplemental Figure 4I).
All these data demonstrate that disrupted interaction of RAS with PI3K in the host tissue impairs tumor-induced angiogenesis, even if the interaction of RAS with PI3K is intact in the tumor cells.
Functional interaction of RAS with PI3K is needed for efficient angiogenesis.
To further demonstrate that the defects in angiogenesis are not due to the reduced capability of tumor cells to induce angiogenesis, and that this event can be attributed entirely to the host tissue, we performed a Matrigel plug assay. VEGF-A was diluted in Matrigel and injected subcutaneously. Mice were sacrificed after 2 weeks, and Matrigel plugs were extracted and subjected to histopathology analysis. Plugs from Pik3caWT/– mice showed a moderate presence of blood vessels, whereas Pik3caMUT/– plugs were almost devoid of blood vessels (71% less; Figure 5, A and C). In addition, we performed the plug assay again using FGF-2, which is considered a more powerful angiogenesis inducer than VEGF-A (20). Using FGF-2, we observed an even greater difference in vessel formation; Pik3caWT/– plugs were massively filled with new vessels, whereas Pik3caMUT/– plugs were almost empty (81% less; Figure 5, B and C).
Figure 5. Functional interaction of RAS with PI3K is needed for efficient angiogenesis.

(A and B) Angiogenesis induced by Matrigel plugs containing VEGF-A (A) or FGF-2 (B). Paraffin-embedded sections were stained for endomucin. Dashed outlines denote the Matrigel plug. (C) Matrigel plugs were quantified from paraffin-embedded sections stained for endomucin. n = 8. (D) The ability of endothelial cells to form capillary-like structures was analyzed in a tubulogenesis assay. Cells were treated with VEGF-A (40 ng/ml) or FGF-2 (10 ng/ml). (E) Matrigel plug sections stained for Ki67. (F) Quantification of E using NIS-Elements program. n = 5 per group. Scale bars: 100 μm.
To investigate whether these observations were due to defective capability of endothelial cells to form new vessels in response to angiogenic factors, we isolated endothelial cells for a functional tubulogenesis assay. Pik3caWT/fl endothelial cells formed tube-like structures on Matrigel when stimulated with VEGF-A or FGF-2, with the latter being clearly more effective (Figure 5D and Supplemental Figure 5, A and B). Strikingly, both Pik3caMUT/fl and Pik3caMUT/– endothelial cells grew in colonies without forming any kind of connection between them and not showing any obvious deleterious effect. Pik3caWT/– cells exhibited cell protrusions that were not sufficient to establish the typical tube network. In the case of FGF-2, Pik3caMUT/fl endothelial cells formed networks to a lesser degree than Pik3caWT/fl cells, while Pik3caWT/– and Pik3caWT/fl cells behaved similarly. Pik3caMUT/– endothelial cells grew in colonies and did not form connections between them.
Analysis of the cellular signaling in these cells revealed that the PI3K/AKT pathway was reduced in response to VEGF-A, whereas ERK activation was only slightly impaired (Supplemental Figure 5C). p-AKT and p-ERK activation were lower in both Pik3caMUT/fl and Pik3caMUT/– endothelial cells compared with Pik3caWT/fl and Pik3caWT/– cells. Signaling induced by FGF-2 showed that efficient activation of AKT only occurred in Pik3caWT/fl endothelial cells, whereas ERK activation was intact in all cell types (Supplemental Figure 5D). We also analyzed levels of Ki67+ and TUNEL+ cells in the Matrigel plug samples to determine whether there was any indication of increased proliferation or apoptosis. As expected based on our results regarding the PI3K and ERK pathways, Pik3caMUT/– samples had a highly reduced number of Ki67+ cells compared with Pik3caWT/– samples (Figure 5, E and F, and Supplemental Figure 5F), indicative of a defect in proliferation. We did not find TUNEL+ cells in these samples. To further analyze the possibility of increased apoptosis, we analyzed the levels of PARP and cleaved PARP and did not observe any difference between isolated Pik3caWT/– and Pik3caMUT/– endothelial cells (Supplemental Figure 5G).
These results indicate that an intact interaction of RAS with PI3K is needed for new blood vessel formation induced by either VEGF-A or FGF-2, and that its disruption in endothelial cells results in deficient cellular signaling that translates into a reduced capability to form new vessels.
Interaction of RAS with PI3K and the immune system.
We finally investigated whether there is any difference in immune cell tumor infiltration or whether any immune cell population is differentially represented in B16F10 subcutaneous tumors. First, we subjected tumor samples to histopathology analysis using specific markers for different populations. We clearly detected drastic differences in the morphologies of cells expressing the F4/80 antigen: Pik3caWT/– samples showed cells with a stellate-like phenotype forming prominent cellular networks, whereas Pik3caMUT/– samples showed cells with a round phenotype that were rather dispersed (Figure 6A).
Figure 6. Interaction of RAS with PI3K and the immune system.

B16F10 subcutaneous tumors were collected after 14 days of injection. (A and B) Paraffin sections stained with F4/80 antibody. Arrows in A indicate typical macrophages. (C) Quantification of F4/80+ cells in the viable tumor area. n = 5 per group. (D) Total mRNA from whole tumor samples was isolated, and Vegfa expression was analyzed (n = 4 per genotype, pooled in groups of 2). (E) Unsorted tumor cells were plated overnight in a petri dish, and supernatant was collected for ELISA analysis of secreted VEGF-A. n = 3. Scale bars: 100 μm (A, top, and B); 50 μm (A, bottom).
Analysis of the histopathology samples revealed striking differences in the distribution of the macrophages among the different areas in the tumors. Macrophages from Pik3caMUT/– samples accumulated in the periphery, with little infiltration into the cortical area, whereas Pik3caWT/– samples showed less accumulation in the periphery and larger infiltration (Figure 6B, top). In the interface between the cortical, viable areas, and the necrotic areas, much higher immunoreactivity was observed in Pik3caWT/– samples (Figure 6B, middle). Interestingly, the networking observed in the Pik3caWT/– samples was even more prominent in areas with infolding vasculature, a feature totally absent in Pik3caMUT/– samples (Figure 6B, bottom). Independent quantification of the total number of F4/80+ cells in the viable areas (excluding tumor periphery and necrotic areas) revealed an increased number in the Pik3caWT/– samples (63% more; Figure 6C). We observed a similar defect in F4/80+ cell infiltration in LLC1 subcutaneous tumors growing in Pik3caMUT/– mice (Supplemental Figure 6K).
We also analyzed the presence of different immune populations by FACS. We did not find any difference in T cell or neutrophil infiltration (Supplemental Figure 6, A–C). It was previously suggested that FOXP3+ Tregs may play an important role in tumor development by suppressing local antitumor immune response, and that the migration of these cells into the tumor microenvironment is promoted by VEGF-A (21). Analysis of this specific population also showed no differences (Supplemental Figure 6D). Moreover, analysis of F4/80+ cell numbers did not show any statistical difference (Supplemental Figure 6E), which suggests that the differences observed in the histopathology analysis may reflect a difference in the ability of cells to migrate from the periphery into the tumor.
To gain further insight into the possible relationship between the macrophage population and the defects observed in angiogenesis, we analyzed the expression of different genes in the tumors and observed highly reduced Vegfa expression in Pik3caMUT/– compared with Pik3caWT/– samples (Figure 6D). Moreover, we also analyzed the amount of VEGF-A and FGF-2 protein secreted by cells within the tumor; levels of both were slightly lower in cells from Pik3caMUT/– versus Pik3caWT/– tumors (Figure 6E and Supplemental Figure 6H). We also verified that the cell lines used expressed detectable levels of angiogenic factors like VEGF-A and FGF-2 in vitro (Supplemental Figures 6, I and J). To clarify whether this difference may be, at least in part, due to reduced expression of VEGF-A by the macrophages, we isolated F4/80+ cells from the tumors and extracted mRNA for expression profile analysis. We found that the F4/80+ population from the Pik3caMUT/– mice had an expression profile closer to an M1-like, tumor-resistant phenotype, expressing higher levels of Ccl5, Cxcl11, Fas, Ikbkb, Il6, and Il12a compared with F4/80+ cells from Pik3caWT/– mice (Table 1). Interestingly, this population also expressed lower levels of Vegfa and Tgfb1 compared with Pik3caWT/– mouse F4/80+ cells. We finally analyzed whether these populations express different surface markers typical for M1 (e.g., major histocompatibility complex II [MHCII]) and M2 (e.g., CD206; also known as mannose receptor) populations, and did not find any statistical difference (Supplemental Figure 6, F and G). These data indicate that these populations do not have a canonical M1 or M2 phenotype, but rather show a mixed one, with Pik3caMUT/– being closer to the M1 phenotype and Pik3caWT/– being closer to the M2 phenotype.
Table 1.
Gene expression analysis in Pik3caWT/– and Pik3caMUT/– macrophage populations
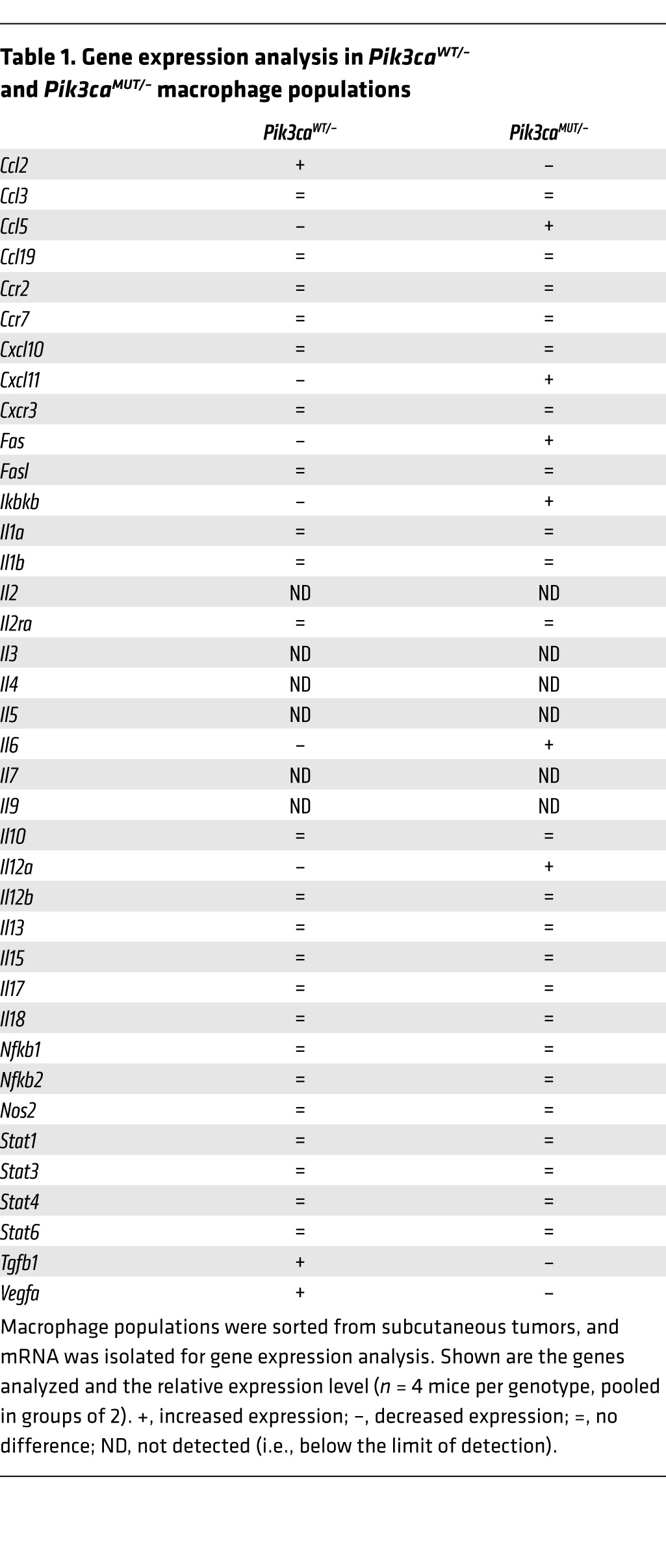
Together, these results show that disrupting the interaction of RAS with PI3K has an effect on the tumor macrophage population, consistent with a defect in migration and/or invasion and with the generation of a less tumor-permissive environment.
Discussion
In this work, we generated a conditional Pik3ca mouse model to block the direct binding of RAS with PI3K p110α in host tissue and examine its significance to host-tumor interaction. Disrupting the interaction of RAS with PI3K in the host reduced tumor growth, even when this interaction was intact in the tumor cells. Interestingly, tumors growing in the Pik3caMUT/fl background grew less than those in the Pik3caWT/– background, which suggests that the mutant allele may possess some dominant-negative activity. Importantly, our mouse model also showed that permanent disruption of the interaction of RAS with PI3K in all tissues did not promote the appearance of any obvious toxicities, even in the long term. Inhibitors of PI3K block its catalytic activity, virtually blocking all PI3K cellular activity and altering cellular homeostasis. While desirable in tumor cells, this inhibition may give rise to damaging adverse effects, due to the wide variety of cellular mechanisms affected in other cell types. Disrupting the interaction of PI3K with RAS did not block its intrinsic catalytic activity, so cells may be able to readjust signaling networks to compensate in part, while being relatively independent of extracellular signaling through several RTKs or through direct intracellular input from RAS. Interestingly, the results shown seem to indicate that the RBD mutant form of Pik3ca may posses some dominant-negative capabilities: AKT activation seemed to be higher in Pik3caWT/– versus Pik3caMUT/fl MEFs (Figure 1B), and tumors grew slower in the Pik3caMUT/fl background compared with the Pik3caWT/– background. Although we do not have further data that may explain this observation, a possible explanation may be a degree of competition by adaptor proteins or docking sites by the WT and mutant forms of Pik3ca.
Intervening in the interaction of RAS with PI3K p110α might represent a good opportunity to manipulate the host-tumor niche, as tumor cells induce changes in the stromal compartment mainly through the alteration of extracellular signals. One such affected extracellular signaling network controls angiogenesis. Antiangiogenic drugs targeting key components of VEGF signaling pathways have proven relatively disappointing so far in the clinic for various tumor types (22). While not active in melanoma (23), pancreatic cancer (24, 25), and prostate cancer (26), they have shown some effectiveness when combined with cytotoxic therapy in non–small cell lung cancer (27–29) and colorectal cancer (30), while the benefit in breast cancer remains controversial (31–34). Among the main problems with these drugs are toxicity and rapid acquired resistance (35, 36). In the present study, we showed that disruption of the interaction of RAS with PI3K p110α in host tissue impaired tumor-induced angiogenesis. Tumors growing in the RBD mutant Pik3ca genetic background showed a reduced ability to promote efficient vascularization (Figure 4, A and B, and Supplemental Figure 4, D–F). Interestingly, the ability of tumor cells to establish metastatic colonies was also reduced: in experimental metastasis assays, we showed that highly metastatic B16F10 cells gave rise to a much lower number of metastatic foci in the Pik3caMUT/– background (Figure 3, A–D). These differences may account for the impaired capability of metastatic cells to extravasate and/or anchor in the target tissue. However, the observation that already-stabilized metastatic foci were much smaller in Pik3caMUT/– mice (Figure 3E) can only be explained by a reduced growth capability. In this regard, the finding that Pik3caMUT/– foci were not able to induce normal angiogenesis, and that this was restricted to the periphery of the nodules (Figure 4C and Supplemental Figure 4G), is fully in line with the phenotype observed. Interestingly, even though the lung is a well-vascularized organ, necrosis is a common event in lung cancer, even in early stages (37). Further work is needed to unveil the relevance of the interaction between RAS and PI3K in all steps of cancer metastasis.
PI3K p110α has been previously implicated in physiological angiogenesis. A mouse model with a constitutive kinase-dead Pik3ca showed primary angiogenic remodeling defects that abrogated embryo development. Deletion of Pik3ca alleles in endothelial cells resulted in a similar outcome (38). Moreover, intact RAS-PI3K signaling is needed for proper hematopoiesis and angiogenesis in zebrafish (39). Recently, Soler et al. described similar growth impairment of transplanted tumors in a mouse model heterozygous for expression of a kinase-dead Pik3ca in all host tissues (40). Presumably, these animals have partial attenuation of all PI3K p110α activity, whereas in ours, the defect in PI3K activity is selective to its regulation by interaction with RAS. There are some key differences compared with our results; for example, in our model we observed a reduced number of blood vessels in the mutant compared with the WT background, but with similar vessel size, whereas Soler et al. observed an increased number of vessels, but with reduced lumen size in the mutant (40).
Interestingly, either a heterozygous kinase-dead Pik3ca allele or a sole RBD mutant Pik3ca did not appear deleterious for endothelial cells, and we did not observe any obvious apoptotic events in the tubulogenesis assay or TUNEL staining of Matrigel samples (Figure 5D, Supplemental Figure 5, F and G, and ref. 40). Moreover, Pik3caWT/– cells were able to form small tube structures, in contrast to those formed by Pik3caWT/fl cells (Figure 2), suggestive of a dose-dependent effect and of inhibitory behavior of the mutant Pik3ca allele similar to that observed on subcutaneous tumor growth.
We found that Pik3caMUT/– mice did not respond to VEGF-A or FGF-2 in terms of new vessel formation, and that this effect might be mediated by the inability of endothelial cells to respond to these growth factors at the molecular level (Figure 5, A–D, and Supplemental Figure 5, A–D), with possible effects on migration, proliferation, and formation of new vessels. In this regard, Ki67+ cell number in Pik3caMUT/– samples from the Matrigel plug assay showed a clear defect in proliferation (Figure 5, E and F).
Our finding that Pik3caMUT/– mice, and more specifically their endothelial cells, were not able to form vessels upon FGF-2 stimulation (Figure 5, B–D, and Supplemental Figure 5B) is also relevant, because one of the main mechanisms for acquired resistance in anti-VEGF antiangiogenic therapy is upregulation of the FGF signaling pathway (41). A synergistic effect between FGF and members of the VEGF family in promoting angiogenesis (42, 43) and lymphangiogenesis (44) has been documented, and new drugs that target both VEGF and FGF pathways are currently being tested in the clinic. Examples of such drugs are brivanib, a VEGFR/FGFR inhibitor currently being tested for hepatocellular carcinoma (45), and nintedanib, a broad VEGFR/FGFR/PDGFR inhibitor currently being tested for non–small cell lung cancer (46). The disruption of the interaction of RAS with PI3K targets both VEGF and FGF pathways at the same time, which may slow the appearance of resistance to some extent.
Finally, we analyzed whether the immune system was somehow differentially modulated depending on RAS interaction with PI3K in the tumor niche. We found a striking difference in the phenotype of F4/80+ cells: while they had a star-shaped morphology in tumors growing in a Pik3caWT/– genetic background, they were round-shaped in the tumors growing in Pik3caMUT/– mice (Figure 6A). This result suggested a possible defect in the migration capabilities of the Pik3caMUT/– macrophages, with elongated macrophage morphologies recently being associated with a tumor-promoting M2 phenotype (47). Our finding that these macrophages accumulated in the periphery and that the number of infiltrating cells in the tumor was clearly lower further supports the concept of a migration defect. We were unable to find any quantitative difference in cell infiltration or in the percentage of different populations of T cells, including FOXP3+ Tregs (Supplemental Figure 6, A–D). Analysis of the expression profile in the infiltrating macrophages showed that in Pik3caMUT/– mice, numerous markers typical of an M1-like, tumor-resistant population were upregulated compared with the Pik3caWT/– population, in addition to the increased expression of Ccl5, Cxcl11, and Il12a; conversely, markers typical of an M2-like, tumor-permissive population were downregulated, including Ccl2, Tgfb, and Vegfa (Table 1). However, we failed to detect differences in typical surface markers of M1 or M2 populations (Supplemental Figure 6, F and G), possibly reflecting the additional complexity of regulation that takes place in vivo compared with the differentiation of these populations in vitro, where consistent and clear differentiation can be achieved. Among the markers with differential expression, Il12a and Vegfa are of special relevance, as they are directly implicated in angiogenesis. In the case of Il12a, there is compelling evidence for its antiangiogenic properties (48, 49).
The nature and role of the different macrophage populations is currently the subject of much debate (50, 51). Although the differences found in the immune population are in accordance with the general phenotype observed, further work is needed to more fully understand their nature and to what extent they contribute to tumor-induced angiogenesis and growth. However, our findings in Matrigel plug assays in the absence of tumor cells showed that Pik3caMUT/– mice exhibited a fundamental defect in their ability to induce angiogenesis, and that their endothelial cells showed defects in intracellular signaling (Figure 5, A–C, and Supplemental Figure 5, C and D), which indicates that the changes observed in the macrophage population may contribute only partially to the general phenotype.
Overall, we showed that the direct binding of RAS to PI3K plays a fundamental role in the tumor-host interaction, enhancing tumor-induced angiogenesis and facilitating the establishment of metastatic colonies. We previously established that the disruption of the interaction of RAS with PI3K represents a significant opportunity for inducing tumor cell–autonomous inhibition of tumor growth (6), but the current work extends this to show that this interaction may also be important in host tissue for the building of a favorable niche for tumor growth. The observed effects on tumor-induced angiogenesis were achieved by defective signaling of the VEGF and FGF pathways, anticipating acquired resistance and without any obvious deleterious effect in the long term. The differences observed in the macrophage populations are suggestive of a more hostile environment for tumor growth in Pik3caMUT/– mice and provide an interesting opportunity for further research.
Methods
Further information can be found in Supplemental Methods.
Mouse strains and in vivo recombination.
The RBD mutant Pik3ca mouse strain was previously generated in our lab (5). The floxed Pik3ca mouse strain was provided by J.J. Zhao and T.M. Roberts (Dana-Farber Cancer Institute, Boston, Massachusetts, USA; ref. 18). The Cre-ERT2 [Gt(ROSA)26Sortm9(cre/ESR1)Arte] mouse strain was from TaconicArtemis GmbH. In vivo recombination of the floxed Pik3ca allele was achieved by feeding mice for 14 days with tamoxifen-containing food pellets (400 ppm; Harlan Teklad) ad libitum.
Matrigel plug assay.
500 ml Matrigel (BD Biosciences) containing either 1 mg/ml VEGF-A (R&D Systems) or 1 mg/ml FGF-2 (Sigma-Aldrich) was injected subcutaneously into mice. Samples were collected and subjected to histopathological analysis.
Tumor burden and angiogenesis analysis.
H&E-stained tissue sections were used for tumor burden and necrosis analysis, and caspase-3 and Ki67 were used to analyze apoptosis and proliferation, respectively. Quantification was performed using NIKON NIS-Elements software according to the manufacturer’s instructions. Angiogenesis analysis was performed using the Angiosight module from Leica’s Digital Pathology capture and analysis platform according to the manufacturer’s instructions.
Bioluminescence quantification.
B16F10 cells expressing a luciferase vector were purchased from PerkinElmer-Caliper. Bioluminescence was measured using an IVIS Spectrum Pre-Clinical In Vivo Imagine System according to the manufacturer’s instructions.
Tubulogenesis assay.
Endothelial cells were serum starved and seeded on top of pre-polymerized Matrigel (BD Biosciences). After attachment, cells were incubated with media containing either 40 ng/ml VEGF-A (R&D Systems) or 40 ng/ml FGF-2 (Sigma-Aldrich) for 8 hours, and images were taken with a NIKON AZ100 microscope.
ELISA.
For analysis of secreted VEGF-A and FGF-A from tumors harvested from mice, tumors were digested, and cell suspensions were plated overnight. For analysis of secreted factors from B16F10 and LLC1 cell lines, the supernatant of these cells growing in a petri dish was collected. All samples were analyzed using Abcam ELISA kits (catalog nos. ab100751 and ab100670).
Statistics.
2-tailed Student’s t test was used when the samples compared had a normal distribution. When samples were not normally distributed, the Mann-Whitney U nonparametric test was used. Prior to selecting one test or the other, a Shapiro-Wilk test was performed in each sample population to determine whether the samples had a normal distribution. Data are presented as mean ± SEM. For survival analysis, a Kaplan-Meier estimator curve followed by a log-rank test analysis was performed. In all cases, a P value less than 0.05 was considered statistically significant.
Study approval.
Studies in animals were reviewed and approved by the Animal Ethics Committee of the London Research Institute and were compliant with UK Home Office Project Licence provisions.
Supplementary Material
Acknowledgments
M.M. Murillo was supported by a Cancer Research UK postdoctoral fellowship and an EMBO Long Term fellowship. This work was funded by Cancer Research UK, by the European Union Framework Programme 7 grant “LUNGTARGET,” and by the European Research Council grant “RASTARGET.”
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(8):3601–3611. doi:10.1172/JCI74134.
References
- 1.Kodaki T, Woscholski R, Hallberg B, Rodriguez-Viciana P, Downward J, Parker PJ. The activation of phosphatidylinositol 3-kinase by Ras. Curr Biol. 1994;4(9):798–806. doi: 10.1016/S0960-9822(00)00177-9. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Viciana P, et al. Phosphatidylinositol-3-OH kinase as a direct target of Ras. Nature. 1994;370(6490):527–532. doi: 10.1038/370527a0. [DOI] [PubMed] [Google Scholar]
- 3.Rodriguez-Viciana P, et al. Role of phosphoinositide 3-OH kinase in cell transformation and control of the actin cytoskeleton by Ras. Cell. 1997;89(3):457–467. doi: 10.1016/S0092-8674(00)80226-3. [DOI] [PubMed] [Google Scholar]
- 4.Rodriguez-Viciana P, Warne PH, Vanhaesebroeck B, Waterfield MD, Downward J. Activation of phosphoinositide 3-kinase by interaction with Ras and by point mutation. EMBO J. 1996;15(10):2442–2451. [PMC free article] [PubMed] [Google Scholar]
- 5.Gupta S, et al. Binding of ras to phosphoinositide 3-kinase p110alpha is required for ras-driven tumorigenesis in mice. Cell. 2007;129(5):957–968. doi: 10.1016/j.cell.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 6.Castellano E, et al. Requirement for interaction of PI 3-kinase p110α with RAS in lung tumor maintenance. Cancer Cell. 2013;24(5):617–630. doi: 10.1016/j.ccr.2013.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–153. doi: 10.1038/nrclinonc.2013.10. [DOI] [PubMed] [Google Scholar]
- 8.Mao M, et al. Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res. 2013;19(3):657–667. doi: 10.1158/1078-0432.CCR-11-1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roberts PJ, et al. Combined PI3K/mTOR and MEK inhibition provides broad antitumor activity in faithful murine cancer models. Clin Cancer Res. 2012;18(19):5290–5303. doi: 10.1158/1078-0432.CCR-12-0563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villanueva J, et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell. 2010;18(6):683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3(1):11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 12.Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11(11):761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Engels B, Rowley DA, Schreiber H. Targeting stroma to treat cancers. Semin Cancer Biol. 2012;22(1):41–49. doi: 10.1016/j.semcancer.2011.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Polyak K, Haviv I, Campbell IG. Co-evolution of tumor cells and their microenvironment. Trends Genet. 2009;25(1):30–38. doi: 10.1016/j.tig.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 15.Tlsty TD, Coussens LM. Tumor stroma and regulation of cancer development. Annu Rev Pathol. 2006;1:119–150. doi: 10.1146/annurev.pathol.1.110304.100224. [DOI] [PubMed] [Google Scholar]
- 16.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285(21):1182–1186. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 17.Dougan M, Dranoff G. Immune therapy for cancer. Annu Rev Immunol. 2009;27:83–117. doi: 10.1146/annurev.immunol.021908.132544. [DOI] [PubMed] [Google Scholar]
- 18.Zhao JJ, et al. The p110α isoform of PI3K is essential for proper growth factor signaling and oncogenic transformation. Proc Natl Acad Sci U S A. 2006;103(44):16296–16300. doi: 10.1073/pnas.0607899103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110α subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274(16):10963–10968. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 20.Cao R, et al. Angiogenic synergism, vascular stability and improvement of hind-limb ischemia by a combination of PDGF-BB and FGF-2. Nat Med. 2003;9(5):604–613. doi: 10.1038/nm848. [DOI] [PubMed] [Google Scholar]
- 21.Hansen W, et al. Neuropilin 1 deficiency on CD4+Foxp3+ regulatory T cells impairs mouse melanoma growth. J Exp Med. 2012;209(11):2001–2016. doi: 10.1084/jem.20111497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jayson GC, Hicklin DJ, Ellis LM. Antiangiogenic therapy — evolving view based on clinical trial results. Nat Rev Clin Oncol. 2012;9(5):297–303. doi: 10.1038/nrclinonc.2012.8. [DOI] [PubMed] [Google Scholar]
- 23.Hauschild A, et al. Results of a phase III, randomized, placebo-controlled study of sorafenib in combination with carboplatin and paclitaxel as second-line treatment in patients with unresectable stage III or stage IV melanoma. J Clin Oncol. 2009;27(17):2823–2830. doi: 10.1200/JCO.2007.15.7636. [DOI] [PubMed] [Google Scholar]
- 24.Kindler HL, et al. Gemcitabine plus bevacizumab compared with gemcitabine plus placebo in patients with advanced pancreatic cancer: phase III trial of the Cancer and Leukemia Group B (CALGB 80303). J Clin Oncol. 2010;28(22):3617–3622. doi: 10.1200/JCO.2010.28.1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Cutsem E, et al. Phase III trial of bevacizumab in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. J Clin Oncol. 2009;27(13):2231–2237. doi: 10.1200/JCO.2008.20.0238. [DOI] [PubMed] [Google Scholar]
- 26.Kelly WK, et al. Randomized, double-blind, placebo-controlled phase III trial comparing docetaxel and prednisone with or without bevacizumab in men with metastatic castration-resistant prostate cancer: CALGB 90401. J Clin Oncol. 2012;30(13):1534–1540. doi: 10.1200/JCO.2011.39.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandler A, et al. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355(24):2542–2550. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
- 28.Scagliotti G, et al. Phase III study of carboplatin and paclitaxel alone or with sorafenib in advanced non-small-cell lung cancer. J Clin Oncol. 2010;28(11):1835–1842. doi: 10.1200/JCO.2009.26.1321. [DOI] [PubMed] [Google Scholar]
- 29.Scagliotti GV, et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol. 2012;30(17):2070–2078. doi: 10.1200/JCO.2011.39.2993. [DOI] [PubMed] [Google Scholar]
- 30.Hurwitz H, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350(23):2335–2342. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 31.Bear HD, et al. Bevacizumab added to neoadjuvant chemotherapy for breast cancer. N Engl J Med. 2012;366(4):310–320. doi: 10.1056/NEJMoa1111097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller K, et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357(26):2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- 33.Robert NJ, et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J Clin Oncol. 2011;29(10):1252–1260. doi: 10.1200/JCO.2010.28.0982. [DOI] [PubMed] [Google Scholar]
- 34.von Minckwitz G, et al. Neoadjuvant chemotherapy and bevacizumab for HER2-negative breast cancer. N Engl J Med. 2012;366(4):299–309. doi: 10.1056/NEJMoa1111065. [DOI] [PubMed] [Google Scholar]
- 35.Cortes J, et al. Adverse events risk associated with bevacizumab addition to breast cancer chemotherapy: a meta-analysis. Ann OncolAnn Oncol. 2012;23(5):1130–1137. doi: 10.1093/annonc/mdr432. [DOI] [PubMed] [Google Scholar]
- 36.Ranpura V, Hapani S, Wu S. Treatment-related mortality with bevacizumab in cancer patients: a meta-analysis. J Am Med Assoc. 2011;305(5):487–494. doi: 10.1001/jama.2011.51. [DOI] [PubMed] [Google Scholar]
- 37.Park SY, Lee H-S, Jang H-J, Lee GK, Chung KY, Zo JI. Tumor necrosis as a prognostic factor for stage IA non-small cell lung cancer. Ann Thoracic Surg. 2011;91(6):1668–1673. doi: 10.1016/j.athoracsur.2010.12.028. [DOI] [PubMed] [Google Scholar]
- 38.Graupera M, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453(7195):662–666. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 39.Liu L, Zhu S, Gong Z, Low BC. K-ras/PI3K-Akt signaling is essential for zebrafish hematopoiesis and angiogenesis. PLoS One. 2008;3(8):e2850. doi: 10.1371/journal.pone.0002850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Soler A, et al. Inhibition of the p110α isoform of PI 3-kinase stimulates nonfunctional tumor angiogenesis. J Exp Med. 2013;210(10):1937–1945. doi: 10.1084/jem.20121571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lieu C, Heymach J, Overman M, Tran H, Kopetz S. Beyond VEGF: inhibition of the fibroblast growth factor pathway and antiangiogenesis. Clin Cancer Res. 2011;17(19):6130–6139. doi: 10.1158/1078-0432.CCR-11-0659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Giavazzi R, et al. Distinct role of fibroblast growth factor-2 and vascular endothelial growth factor on tumor growth and angiogenesis. Am J Pathol. 2003;162(6):1913–1926. doi: 10.1016/S0002-9440(10)64325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pepper MS, Ferrara N, Orci L, Montesano R. Potent synergism between vascular endothelial growth factor and basic fibroblast growth factor in the induction of angiogenesis in vitro. Biochem Biophys Res Commun. 1992;189(2):824–831. doi: 10.1016/0006-291X(92)92277-5. [DOI] [PubMed] [Google Scholar]
- 44.Cao R, et al. Collaborative interplay between FGF-2 and VEGF-C promotes lymphangiogenesis and metastasis. Proc Natl Acad Sci U S A. 2012;109(39):15894–15899. doi: 10.1073/pnas.1208324109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn RS, et al. Phase II, open-label study of brivanib as second-line therapy in patients with advanced hepatocellular carcinoma. Clin Cancer Res. 2012;18(7):2090–2098. doi: 10.1158/1078-0432.CCR-11-1991. [DOI] [PubMed] [Google Scholar]
- 46.Doebele RC, et al. A phase I, open-label dose-escalation study of continuous treatment with BIBF 1120 in combination with paclitaxel and carboplatin as first-line treatment in patients with advanced non-small-cell lung cancer. Ann Oncol. 2012;23(8):2094–2102. doi: 10.1093/annonc/mdr596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McWhorter FY, Wang T, Nguyen P, Chung T, Liu WF. Modulation of macrophage phenotype by cell shape. Proc Natl Acad Sci U S A. 2013;110(43):17253–17258. doi: 10.1073/pnas.1308887110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Airoldi I, et al. IL-12 can target human lung adenocarcinoma cells and normal bronchial epithelial cells surrounding tumor lesions. PLoS One. 2009;4(7):e6119. doi: 10.1371/journal.pone.0006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Strasly M, et al. IL-12 inhibition of endothelial cell functions and angiogenesis depends on lymphocyte-endothelial cell cross-talk. J Immunol. 2001;166(6):3890–3899. doi: 10.4049/jimmunol.166.6.3890. [DOI] [PubMed] [Google Scholar]
- 50.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11(11):723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sica A, Bronte V. Altered macrophage differentiation and immune dysfunction in tumor development. J Clin Invest. 2007;117(5):1155–1166. doi: 10.1172/JCI31422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.