

Abstract
Metabolic disorders, including obesity, diabetes, and cardiovascular disease, are widespread in Westernized nations. Gut microbiota composition is a contributing factor to the susceptibility of an individual to the development of these disorders; therefore, altering a person’s microbiota may ameliorate disease. One potential microbiome-altering strategy is the incorporation of modified bacteria that express therapeutic factors into the gut microbiota. For example, N-acylphosphatidylethanolamines (NAPEs) are precursors to the N-acylethanolamide (NAE) family of lipids, which are synthesized in the small intestine in response to feeding and reduce food intake and obesity. Here, we demonstrated that administration of engineered NAPE-expressing E. coli Nissle 1917 bacteria in drinking water for 8 weeks reduced the levels of obesity in mice fed a high-fat diet. Mice that received modified bacteria had dramatically lower food intake, adiposity, insulin resistance, and hepatosteatosis compared with mice receiving standard water or control bacteria. The protective effects conferred by NAPE-expressing bacteria persisted for at least 4 weeks after their removal from the drinking water. Moreover, administration of NAPE-expressing bacteria to TallyHo mice, a polygenic mouse model of obesity, inhibited weight gain. Our results demonstrate that incorporation of appropriately modified bacteria into the gut microbiota has potential as an effective strategy to inhibit the development of metabolic disorders.
Introduction
Obesity dramatically increases the risks for cardiovascular disease and diabetes as well as other diseases, and rates of obesity have increased dramatically in the past 25 years (1, 2). Unfortunately, current medical and lifestyle treatments for obesity have largely failed to achieve long-term success (3–6). A major challenge is the need for lifelong adherence to maintain modest effects in the face of evolutionarily driven compensatory responses to weight loss induced by voluntary caloric restriction. Thus, alternative and novel strategies are urgently needed for long-term prevention and treatment of obesity. One such alternative is suggested by studies showing a reciprocal relationship between the composition of the gut microbiota and metabolic disorders (7–13). Transfer of gut microbiota from obese mice into germ-free mice demonstrates that this microbiota directly contributes to metabolic phenotypes (9, 11). Thus, appropriate alteration of the gut microbiota might provide long-term protection against obesity. One potential strategy to beneficially alter the gut microbiota is to incorporate bacteria that have been genetically modified to express therapeutic factors that increase satiety and sensitivity to adipose-derived negative feedback signals such as leptin. This strategy could also be used to sustainably deliver other therapeutic molecules beneficial in the treatment of obesity-related diseases or which could not be readily delivered by other therapeutic routes.
Although a number of therapeutic molecules could be potentially biosynthesized by bacteria for the treatment of obesity, we focused on N-acyl-phosphatidylethanolamines (NAPEs), the immediate precursors of N-acylethanolamides (NAEs), a family of the potent anorexigenic lipids. NAPEs are synthesized in the proximal small intestine in response to feeding (14). A high-fat diet impairs feeding-induced NAPE synthesis (15) so that bacterially-synthesized NAPEs could help compensate for this reduced NAPE synthesis. After their synthesis, NAPEs are rapidly converted to the active NAEs through hydrolysis by NAPE-hydrolyzing phospholipase D (NAPE-PLD). Intraperitoneal administration of NAEs, where the N-acyl chains are saturated or monounsaturated (e.g., N-oleoyl-ethanolamide [OEA]), or of their precursors (e.g., C16:0NAPE) markedly reduces food intake and obesity in mice fed a high-fat diet (15–24). Chronic NAE administration appears to modulate adiposity through multiple mechanisms including inhibition of fat absorption (25), delayed gastric emptying (26, 27), reduced food intake (19, 20, 28, 29), and increased fatty acid oxidation (23, 25). NAEs act as ligands for at least 3 receptors: PPARα, TRPV1, and GPR119 (23, 28, 30), and genetic ablation of PPARα or TRPV1 significantly blunts the anorectic effects of NAEs (28, 31). The goal of this study was to determine whether incorporating genetically modified bacteria that biosynthesize NAPEs into the gut of mice would result in lasting attenuation of obesity induced by a high-fat diet.
Results
We previously transformed the C41-DE3 laboratory strain of E. coli (Ec) with At1g78690, an N-acyltransferase from Arabidopsis thaliana that catalyzes the synthesis of NAPEs. This markedly increased NAPE levels in these bacteria (pNAPE-Ec) compared with those transformed with empty vector (pEc) (32, 33). To determine whether NAPEs synthesized by intestinal E. coli could markedly alter food intake, we administered a daily bolus of 1011 CFU pNAPE-Ec or pEc bacteria by gavage to lean male C57BL/6J mice for 7 days. Additional groups of mice were administered either vehicle or pNAPE-Ec bacteria that had been killed by treatment with kanamycin prior to gavage. Cumulative food intake did not differ in mice receiving pEc compared with those receiving vehicle, but was reduced by 15% in mice receiving living pNAPE-Ec (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI72517DS1). pNAPE-Ec are sensitive to kanamycin, and pNAPE-Ec pretreated with kanamycin prior to gavage did not reduce food intake to the same extent as untreated pNAPE-Ec, indicating that viable bacteria expressing NAPEs are needed for maximum effectiveness. Our follow-up studies revealed that administration of 1011 pNAPE-Ec bacteria once a day for 7 consecutive days resulted in an approximately 2-fold increase in NAPE levels in the colon (Supplemental Figure 2A). NAPE levels were not as markedly increased in other gastrointestinal tissues such as stomach, small intestine, and cecum, nor were they increased in plasma (Supplemental Figure 2, B–E).
To examine the effect of persistent NAPE expression by gut bacteria on food intake and obesity, we transformed the probiotic wild-type strain of E. coli, Nissle 1917 (EcN), with At1g78690 (pNAPE-EcN) (Supplemental Figure 3). To facilitate monitoring of colonization by transformed EcN, we also inserted the P. luminescens luciferase operon (Lux) into the recA gene of the EcN chromosome. We analyzed phospholipid extracts from both pNAPE-EcN and pEcN by negative ion mass spectrometry (Supplemental Figure 4), and the identified NAPE species were consistent with N-acylation of the major bacterial PE species (Supplemental Table 1). We observed that pNAPE-EcN had markedly higher levels of saturated and monounsaturated NAPEs expected to exert anorexigenic effects (Supplemental Figure 5). In addition to NAPEs, several species with m/z ions consistent with O-acyl-phosphatidylglycerols (acyl-PG) species (34) were also enriched in pNAPE-EcN (Supplemental Table 2).
We then tested whether incorporating pNAPE-EcN into the gut microbiota would protect against the development of obesity, insulin resistance, and liver steatosis that occurs in C57BL6 mice fed an ad libitum high-fat diet (60% of calories from fat). For these experiments, we administered pNAPE-EcN via the drinking water at 5 × 109 CFU bacteria/ml with 0.125% gelatin added to the water to keep the bacteria suspended and viable for at least 48 hours (Supplemental Figure 6). Supplementation of drinking water with pNAPE-EcN resulted in bioluminescence that could be readily detected in the intestinal tract (Supplemental Figure 7). Bacteria were administered for a total of 8 weeks (Supplemental Figure 8), and food intake, weight gain, and body composition were monitored. We also measured consumption of kaolin (pica), a measure of gastrointestinal distress in rodents (35), to assess whether bacterial administration had an adverse effect on the mice. At the end of the bacterial treatment period, we performed an oral glucose tolerance test. Mice were continued on the high-fat diet for an additional 4 weeks to determine the persistence of the altered gut microbiota and the resulting changes in food intake and obesity.
During the 8-week treatment period, pNAPE-EcN–treated mice gained less body weight (Figure 1A) and accumulated less fat mass (Figure 1B) compared with control mice. Consistent with their reduced adiposity, pNAPE-EcN mice also had lower plasma leptin and insulin levels (Table 1). In contrast to the striking differences in adiposity, we observed no significant differences in lean body mass between the groups (Figure 1C). One of the major bioactivities of the NAE metabolites of NAPEs is the reduction of food intake, and pNAPE-EcN–treated mice had markedly lower cumulative food intake than those treated with standard water, vehicle, or pEcN (Figure 1D).
Figure 1. Treatment with pNAPE-EcN, but not pEcN, inhibits gain in body weight and adiposity.

All values are the mean ± SEM (n = 10 mice per group). Solid bars indicate time points with significant differences between pNAPE-EcN and other groups (P < 0.05 by Bonferroni’s multiple comparison test). (A) Effect of treatments on gain in body weight from start of treatment (2-way RM ANOVA, for treatment P = 0.0073, for time P < 0.0001). (B) Effect of treatments on fat mass (2-way RM ANOVA, for treatment P = 0.0127, for time P < 0.0001). (C) Effect of treatments on lean body mass (2-way RM ANOVA, for treatment P = 0.8113, for time P < 0.001). (D) Effect of treatments on cumulative food intake from start of treatment (2-way RM ANOVA, for treatment P = 0.0035, for time P < 0.0001).
Table 1.
Effects of NAPE-secreting bacteria on metabolic biomarkers during an 8-week treatment and a 4-week follow-up period
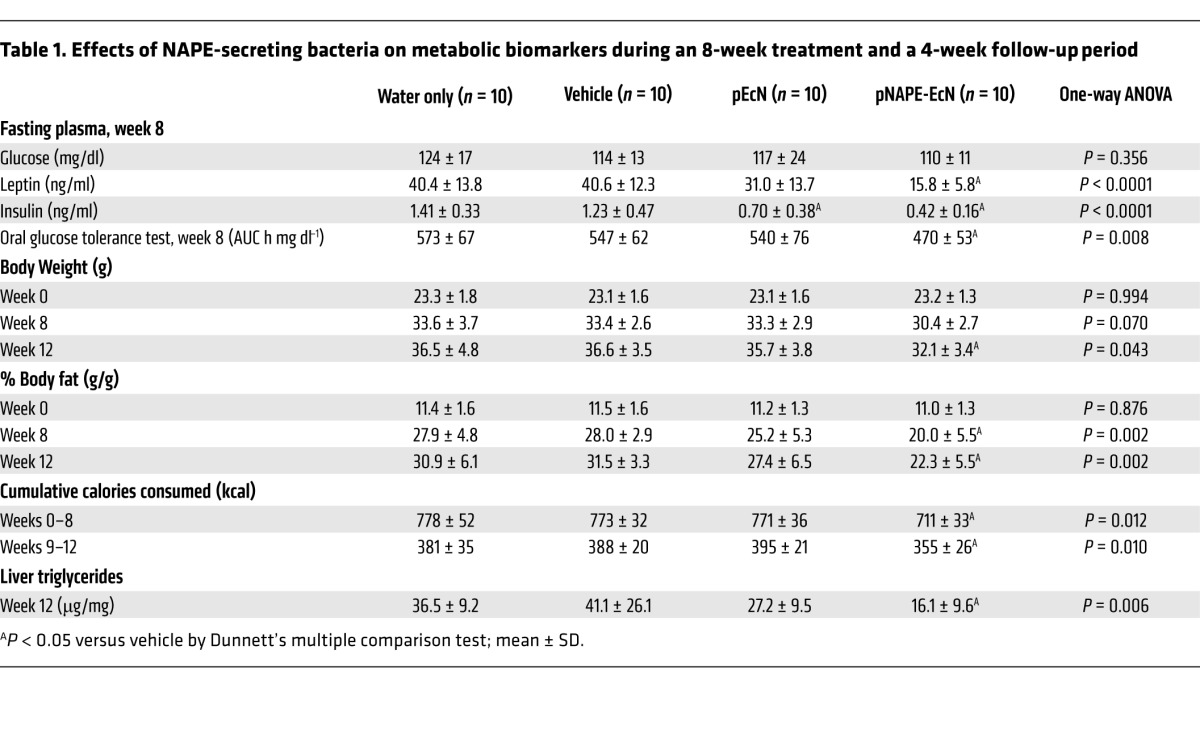
Importantly, we found no evidence that changes in body weight and adiposity were the result of an adverse health effect of treatment with pNAPE-EcN. For instance, there was no difference between treatment groups in the consumption of kaolin (Supplemental Figure 9), so bacterial administration did not produce gastrointestinal distress. Nor did bacterial overgrowth appear to be the cause of reduced food intake and adiposity in pNAPE-EcN–treated mice, as levels of EcN retained in the intestinal tract did not significantly differ between pNAPE-EcN– and pEcN-treated mice (Supplemental Figure 10). Scores on simple tests of muscle strength, coordination, and speed either did not differ or were improved with pNAPE-EcN treatment (Supplemental Table 3). Perhaps most importantly, since it is well established that endotoxemia impairs glucose tolerance and insulin sensitivity (12, 13), mice administered pNAPE-EcN had significantly improved glucose tolerance compared with that observed in the other groups (Table 1). Overall, these findings support the conclusion that the reduced adiposity found in high-fat diet–fed mice receiving pNAPE-EcN results from the action of NAPEs rather than from an adverse effect of bacterial administration.
Our larger goal was to incorporate NAPE-secreting bacteria into the gut microbiota in order to endow their host with persistent resistance to obesity. Mice initially treated with either pNAPE-EcN or pEcN excreted luminescent bacteria in their feces for at least 4 weeks after the cessation of bacterial administration (Supplemental Figure 11), consistent with persistence of pNAPE-EcN in the gut. We therefore investigated whether the beneficial effects of pNAPE-EcN on adiposity would persist after stopping its administration. Food intake, body weight, and fat mass during the follow-up period (experimental weeks 9–12) remained significantly lower in mice initially treated with pNAPE-EcN than was the case in the other groups (Table 1), consistent with the notion that incorporation of pNAPE-EcN in the gut lumen leads to long-lasting delivery of NAPEs and concomitant inhibition of obesity. Subsequent studies in a separate cohort of mice found that body weight was still significantly lower than that of vehicle- or pEcN-treated mice even 12 weeks after ending administration of pNAPE-EcN (Figure 2). The difference in body weight between vehicle- and pNAPE-EcN–treated mice continued to increase in magnitude up until 6 weeks after cessation of administration. Bioluminescence was no longer detectable in feces at this time, suggesting that the loss of inhibited weight gain at this time was the result of loss of pNAPE-EcN from the gut.
Figure 2. Effects of pNAPE-EcN persist for more than 6 weeks after ending administration.

Groups of mice were fed a high-fat diet for a total of 20 weeks (n = 10 mice per group). For the first 8 weeks, mice were administered either vehicle (0.125% gelatin), 5 × 109 CFU pEcN/ml, or 5 × 109 CFU pNAPE-EcN/ml. For the remaining 12 weeks, all mice received standard drinking water. (A) Effect of treatment on total body weight. (B) Body weight of group treated with pNAPE-EcN normalized to the vehicle-treated group. (C) Percentage of body fat for all 3 groups. (D) Percentage of body fat of pNAPE-EcN mice normalized to the vehicle-treated mice. (E) Daily average food intake for each group as a 2-week rolling average. A rolling average was used to minimize day-to-day variations to better determine the overall trends. The dip in food intake that occurred in all groups after day 70 is likely the result of changing the housing location of all the mice due to institutional requirements. (F) Daily average food intake of pNAPE-EcN–treated mice normalized to the vehicle-treated mice.
Molecules absorbed through the colon enter the portal circulation, so that bacterially secreted NAPEs that had been subsequently converted to the biologically active NAEs by NAPE-PLD might exert effects in the liver in addition to their effects within the intestinal tract. When we examined liver NAE levels in mice that were euthanized 2 days after ending bacterial administration, we found that pNAPE-EcN treatment increased the levels of total NAE in liver by 44% (Figure 3). Treatment with pNAPE-EcN did not significantly alter either total plasma levels of NAE or NAPE or their N-acyl chain profiles (Supplemental Figure 12). Previous studies showed that orally administered NAEs induce a number of effects relevant to protection against obesity including the activation of fatty acid oxidation, inhibition of lipogenesis, and inhibition of lipid absorption (23, 25). We found no difference in lipid absorption between mice given standard drinking water and those treated with pNAPE-EcN (Supplemental Figure 13.) When we examined gene expression in livers collected from mice euthanized 4 weeks after ending bacterial administration, we found that mice treated with pNAPE-EcN had increased liver mRNA expression for genes linked to fatty acid oxidation such as peroxisome proliferator activator receptor α (Ppara) and delta (Ppard), carnitine palmitoyl transferase-1 (Cpt1), and acyl CoA oxidase (AOX), but not for genes linked to fatty acid synthesis such as fatty acid synthase (FAS) and sterol regulatory element–binding protein-1c (SREBP-1c) or to gluconeogenesis such as phosphoenolpyruvate carboxykinase (PEPCK) (Figure 4). They also had reduced expression of the stearoyl CoA desaturase-1 (Scd1) and lipid transporter Cd36 as well as reduced expression of inflammatory genes including Cd68 and monocyte chemotactic protein-1/chemokine C-C motif ligand 2 (Ccl2). To further examine the effect of pNAPE-EcN treatment on hepatic inflammation, we immunostained for infiltrating monocytes/macrophages using antibodies against F4/80 and CD11b. The livers of control mice had large numbers of immunoreactive cells (Figure 5). In contrast, pNAPE-EcN mice had markedly fewer immunoreactive cells. We also examined triglyceride levels in these livers and found significantly lower levels in pNAPE-EcN–treated mice than those in the other 3 groups (Table 1). Histological examination with both H&E and Oil Red O staining revealed markedly less hepatosteatosis in the livers of mice treated with pNAPE-EcN (Supplemental Figure 14).
Figure 3. Treatment with pNAPE-EcN increases hepatic NAE levels.

NAEs were measured by LC/MS in liver collected from mice 2 days after ending a 9-week treatment with pNAPE-EcN or from untreated mice receiving standard drinking water (Water) during this same period (n = 10 mice per group; mean ± SEM). NAE levels were significantly different (2-way ANOVA, for treatment P < 0.0001, for NAE species P < 0.0001). Summed NAE levels were 1.70 ± 0.18 versus 2.45 ± 0.21 nmol/g liver for untreated versus pNAPE-EcN–treated mice, respectively.
Figure 4. Treatment with pNAPE-EcN induces expression of genes encoding for fatty acid oxidation, but not fatty acid synthesis, and reduces expression of inflammatory genes in the liver.

Liver mRNA was measured by qRT-PCR using primers specific for each gene. β-actin (Actb) was used as a control, and all values were normalized to the vehicle group (mean ± SEM, n = 10 mice per group). *P < 0.05 by 1-way ANOVA for individual gene expression and by Dunnett’s multiple comparison test versus vehicle for pNAPE-EcN, but not for pEcN.
Figure 5. Treatment with pNAPE-EcN reduces infiltration of F4/80 and CD11b immunopositive leukocytes into liver.
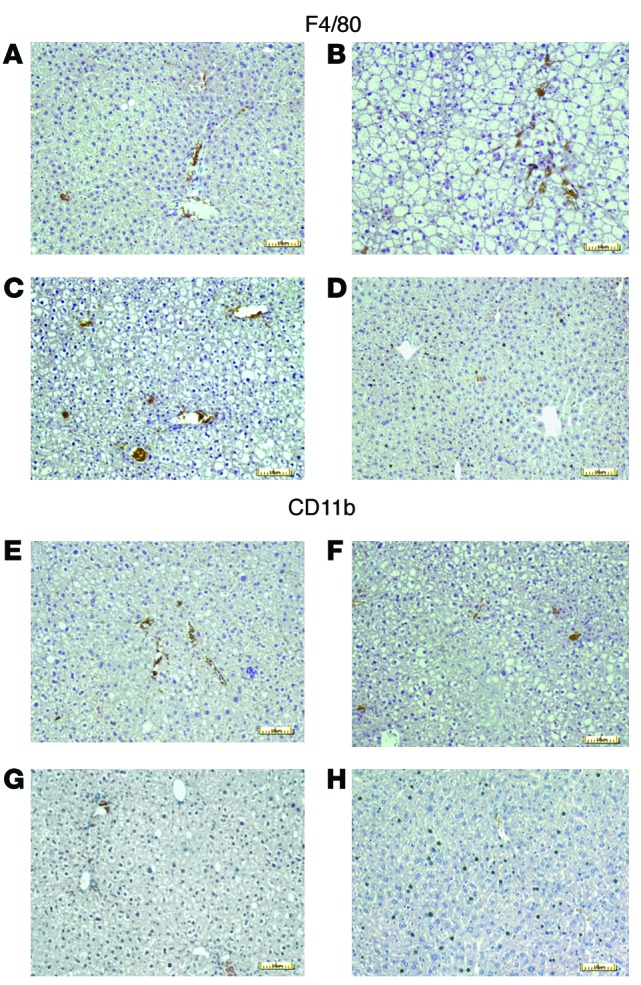
Slides were immunostained with either anti-F4/80 (A–D) or anti-CD11b (E–H) antibodies (DAB brown stain) and counterstained with hematoxylin. Representative photomicrographs are shown. Scale bars: 10 μm. (A and E) Standard drinking water only. (B and F) Vehicle-treated (0.125% gelatin) mice. (C and G) pEcN-treated mice. (D and H) pNAPE-EcN–treated mice.
To determine whether the effect of pNAPE-EcN was mediated solely by its ability to reduce food intake, we pair-fed a group of mice treated with pEcN with the same amount of food consumed each day by pNAPE-EcN–treated mice for a period of 4 weeks. The body weight of mice that were pair-fed for 4 weeks was significantly lower than that of mice treated with pEcN or standard water, but was still significantly higher than that of mice treated with pNAPE-EcN (Figure 6A). Pair-fed mice also had significantly less body fat than did pEcN- or water-only–treated mice, but they trended toward higher body fat levels than were seen in pNAPE-EcN–treated mice (Figure 6B). When we calculated feeding efficiency over the 4-week treatment period, both pEcN- and water-treated mice gained significantly more weight per kcal of food consumed than did pNAPE-EcN–treated mice, while the pair-fed mice showed intermediate feeding efficiency (Figure 6C). We then examined whether the changes in body composition induced by food restriction were sufficient to induce the expression of the fatty acid oxidation genes seen in pNAPE-EcN–treated mice. Despite having similarly low levels of triglyceride levels in their livers (Figure 7A), pair-fed mice failed to induce increased expression of hepatic fatty acid oxidation in a manner similar to that of pNAPE-EcN–treated mice (Figure 7B). We interpreted these results as indicating that this gene induction was the direct result of the delivery of NAEs to the liver afforded by pNAPE-EcN treatment rather than an indirect response to lower food intake or body fat.
Figure 6. Pair-feeding fails to fully recapitulate the effect of pNAPE-EcN treatment on body weight and body fat gain.

A group of mice treated with pEcN (E) were were pair-fed (PF) by restricting them to the same number of calories of food as the pNAPE-EcN mice (N). An ad libitum–fed control group received standard drinking water (W). All values are the mean ± SEM; n = 9–10 mice per group. (A) Body weight gain during 4 weeks of treatment and pair-feeding. Two-way RM ANOVA, P < 0.0001 for time, P = 0.0003 for treatment. P < 0.05 by Bonferroni’s multiple comparison test for pair-fed versus pNAPE-EcN for days 16 to 25. P < 0.05 for pair-fed versus pEcN and versus standard water for day 28. (B) Body fat at week 0 (before beginning treatment) and week 4 (28 days after beginning treatment). No group differences were observed at week 0. Week 4 1-way ANOVA, P < 0.0001 by 1-way ANOVA; P < 0.05 by Dunnett’s multiple comparison test for pair-fed versus water or versus pEcN. Pair-fed versus pNAPE-EcN, not significant. (C) Calculated feeding efficiency, P < 0.0001 by 1-way ANOVA; P < 0.05 BY Dunnett’s multiple comparison test for pair-fed versus pEcN; pair-fed versus standard water or versus pNAPE-EcN, not significant.
Figure 7. Pair-feeding does not induce hepatic expression of fatty acid oxidation genes but does reduce lipid accumulation.

All values are the mean ± SEM; n = 9–10 mice per group. (A) Liver triglyceride (TG) levels in mice given standard drinking water (W), or drinking water with pEcN (E), pNAPE-EcN (N), or pEcN with pair-feeding to pNAPE-EcN mice (PF). P = 0.0012 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. (B) Expression of mRNA encoding for proteins related to fatty acid oxidation. Ppara, P < 0.0001 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test; Cpt1a, P < 0.0233 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test; AOX, P = 0.1326 by 1-way ANOVA.
Although these findings were consistent with pNAPE-EcN treatment directly altering food intake and energy expenditure by the action of NAEs, we considered an alternative hypothesis for pNAPE-EcN action, which is that treatment with pNAPE-EcN indirectly leads to changes in adiposity by altering global gut microbiota. Changes in global gut microbiota, including an increased ratio of Firmicutes to Bacteroidetes, have been associated with obesity (9, 36–38). We anticipated that our treatment strategy would have very little long-term effect on the global microbial composition of the gut if our genetically modified EcN primarily colonized the gut by occupying niches previously held by native E. coli or other Proteobacteria. This is because Proteobacteria typically account for only a very small percentage of gut bacteria (~1% of adherent bacteria in the distal small intestine, ~3% of adherent bacteria in the descending colon, and <0.1% of bacteria in feces; R.L. Walzem, unpublished observations). To assess the effect of pNAPE-EcN treatment on global gut microbial composition, we performed 454 pyrosequencing of bacterial 16S rRNA obtained from the feces of treated mice. Fecal microbiota composition is a noninvasive, surrogate measure of intestinal microbiota composition with contributions from both bacteria shed by established intestinal colonies and ingested bacteria, which pass through the gut without adhering. Treatment with either pNAPE-EcN or pEcN markedly increased fecal levels of Proteobacteria compared with the levels found in vehicle-treated mice at the 8-week time point (Figure 8 and Supplemental Table 4). This increase in fecal Proteobacteria levels is consistent with large numbers of ingested EcN passing through the gut without adhering. Ingestion of either form of EcN also temporarily reduced the proportion of Firmicutes in the feces by week 8. Because pEcN reduced fecal Firmicutes levels but failed to alter body weight and body fat, this result does not support the notion that the acute change in fecal global microbial composition is responsible for the beneficial effects of pNAPE-EcN treatment. This interpretation is further supported by the microbial composition of feces collected at week 12, 4 weeks after EcN bacteria were no longer being ingested. At this time point, the global microbial composition of feces was very similar in the 3 groups, suggesting that treatment with pEcN or pNAPE-EcN only led to colonization of the relatively low-abundance niches available for E. coli in the gut and did not result in long-term alterations of the global composition of the gut microbiota. Because the mice colonized with pNAPE-EcN continued to show reduced food intake and body weight (Figure 2), these results support the notion that global changes in microbiota composition are not required for the beneficial effects of pNAPE-EcN treatment and that these effects are instead the result of the direct action of NAEs on target host tissues.
Figure 8. Relative abundance in feces of major bacterial phylla.

Bacterial composition of feces collected during experimental week 8 (final week of treatment) and week 12 (fourth week of post-treatment follow-up) was determined by 16S rRNA sequencing (n = 9–10 mice per group). Treatment with either pNAPE-EcN (N) or pEcN (E) significantly decreased the abundance of Firmicutes compared with vehicle (V) and increased the abundance of Proteobacteria in excreted feces (week 8), but microbial composition reverted to that of the vehicle-treated animals by 4 weeks after ending bacterial administration (week 12).
To further characterize alterations in feeding behavior and energy expenditure induced by pNAPE-EcN, we performed metabolic monitoring after 4 weeks of treatment with pNAPE-EcN, pEcN, or standard drinking water. Mice were placed for 7 days in cages equipped with the Promethion system for monitoring indirect calorimetry, physical activity, and food and water intake. The mice were maintained on a high-fat diet and their designated treatment throughout these metabolic monitoring experiments. All groups had some body weight and fat loss during their time in the Promethion system, likely due to the novel environment (Supplemental Figure 15). Analysis was performed using data from the final 3 days of monitoring when mice had acclimated to the novel environment. Examination of the pattern of food intake over the 24 hours of the light-dark cycle showed that food intake primarily diverged during the dark phase, when mice typically are most active and consume the majority of their food (Figure 9A and Supplemental Figure 16). During the dark phase, we observed that meal duration was significantly shorter for mice treated with pNAPE-EcN (Figure 9B). We found no significant differences in meal duration between groups during the light phase. Intermeal intervals did not significantly differ between the groups in either the light or dark phase (Figure 9C). When meal size was categorized by food weight consumed during each 5-minute time block of monitoring, the pNAPE-EcN–treated mice consumed significantly fewer large meals (Figure 9D). These results suggest that treatment with pNAPE-EcN lowered the threshold of the high-fat diet consumption required to achieve satiation. The pattern of water intake was similar to that of food intake: pNAPE-EcN–treated mice consumed less water overall, and the greatest divergence in water intake occurred during the dark phase (Supplemental Figure 17).
Figure 9. Treatment with pNAPE-EcN reduces food intake and meal duration during the dark phase of the light cycle.

Mice were given standard drinking water only, water with pEcN, or water with pNAPE-EcN for 4 weeks prior to metabolic monitoring and continued to receive treated water throughout the monitoring period (n = 9–10 mice per group). (A) Average cumulative food intake during the 24-hour light cycle. For each mouse, the cumulative food intake through each 5-minute block of a 24-hour light cycle (6 am–6 am) for each of the 3 days of monitoring was averaged. The mean cumulative food intake for each treatment group is shown. The 72-hour cumulative food traces for individual mice are shown in Supplemental Figure 16. (B) Effect of treatment on meal duration in light and dark phases (mean ± SEM). P = 0.8972 by 1-way ANOVA for light phase; P = 0.0554 by 1-way ANOVA for dark phase; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. (C) Effect of treatment on intermeal interval (mean ± SEM). P = 0.0743 by 1-way ANOVA for light phase; P = 0.1085 by 1-way ANOVA for dark phase. (D) Effect of treatment on meal size (mean ± SEM). Each 5-minute block during which food hopper weight decreased by greater than 0.05 mg was scored as a meal and categorized from 1 (smallest) to 5 (largest), as described in Methods. Category 4, P = 0.0256 by 1-way ANOVA; all other meal categories, P = NS by 1-way ANOVA.
In addition to changes in energy intake, pNAPE-EcN treatment induced subtle changes in energy expenditure. While the lower body fat of pNAPE-EcN mice complicated direct comparisons of energy expenditure between groups, during the light phase of the 24-hour light-dark cycle (when mice are least active), the linear slope of the relationship between energy expenditure versus body weight increased more steeply for pNAPE-EcN–treated mice than for the other 2 groups (Figure 10A and Supplemental Figure 18A). The slopes during the dark phase were similar (Figure 10B and Supplemental Figure 18B). The increase in energy expenditure we observed for pNAPE-EcN–treated mice during the light phase was not due to increased physical activity, because pNAPE-EcN–treated mice did not show an increase in distance traveled within the cage, as calculated from beam breaks (Supplemental Figure 19). These results suggest that pNAPE-EcN might slightly increase resting metabolic rate.
Figure 10. Effect of pNAPE-EcN treatment on energy expenditure.

(A) Energy expenditure during 12-hour light phase of the 24-hour light-dark cycle. Slopes for each group differed: pNAPE-EcN, y = 0.01016x + 0.02295; pEcN, y = 0.006475x + 0.1201; standard water, y = 0.002726 + 0.001779; differences in slope, P = 0.04038, F = 3.702 (n = 9–10 mice per group). (B) Energy expenditure during the dark phase of the 24-hour light cycle. Slopes for each group did not differ: pNAPE-EcN, y = 0.002704 + 0.3448; pEcN, y = 0.003438+0.3304; standard water, y = 0.004455 + 0.2857; differences in slope, P = 0.8734, F = 0.1362.
To examine whether the increased energy expenditure during the light phase resulted from increased fatty acid oxidation while resting, we analyzed the effect of pNAPE-EcN treatment on the respiratory quotient (RQ). Because all mice ate the same high-fat diet, no large-scale changes in the RQ would be expected, but animals with an increased reliance on their fat reserves might have a slightly lower RQ (i.e., favoring fatty acid oxidation over carbohydrate and protein oxidation) during periods of rest. Indeed, the pNAPE-EcN–treated mice consistently showed a slightly lower RQ than did the other 2 groups, although this apparent difference failed to reach statistical significance (Supplemental Figure 20).
To further characterize the effects of pNAPE-EcN treatment on glucose intolerance and insulin resistance, we performed several follow-up studies. To estimate the extent of protection from glucose intolerance afforded by pNAPE-EcN treatment, we treated mice with a high-fat diet, a high-fat diet and pNAPE-EcN, or a low-fat chow diet for 8 weeks. While administering pNAPE-EcN to mice fed a high-fat diet significantly improved homeostatic model assessment–estimated insulin resistance (HOMA-IR) index scores and the insulin response to glucose when compared with untreated mice receiving this diet, impairment in glucose tolerance was still evident in the pNAPE-EcN–treated mice when compared with mice fed a low-fat chow diet (Figure 11).
Figure 11. Treatment with pNAPE-EcN reduces the insulin resistance index and increases insulin response in mice fed a high-fat diet.

All values are the mean ± SEM; n = 10 mice per group. (A) Treatment with pNAPE-EcN (HF + N) improved basal glucose levels and glucose tolerance compared with mice treated with standard drinking water (HF + W), but not a low-fat chow diet (LF + W). Fasting glucose levels differed significantly between groups (P < 0.001 by 1-way ANOVA; P < 0.05 by Bonferroni’s post-hoc multiple comparison for HF + W versus HF + N or LF + W, and HF + N versus LF + W. Glucose AUC differed significantly between each treatment group (P < 0.0009 by 1-way ANOVA); P <0.05 by Bonferroni’s post-hoc multiple comparison for HF + W (50,841 ± 2,191 mg/dl/min) versus either HF + N (39,463 ± 1,800) or LF + W (24,573 ± 517), and for HF + N versus LF + W. (B) Treatment with pNAPE-EcN increased insulin responsiveness. Fasting levels of insulin were significantly higher in HF + W versus HF + N or LF + W mice (P < 0.0001 by 1-way ANOVA); P < 0.05 by Bonferroni’s post-hoc multiple comparison for HF + W versus HF + N or LF + W, but not for HF + N versus LF + W. The insulin AUC for each treatment differed significantly: P < 0.0001 by 1-way ANOVA; P < 0.05 by Bonferroni’s post-hoc multiple comparison for LF + W (69.5 ± 5.7 ng/ml/min, mean ± SEM) versus HF + W (193.9 ± 17.0) or HF + N (158.7 ± 8.4, P < 0.05). (C) Treatment with pNAPE-EcN improved the HOMA-IR score compared with a high-fat diet–only treatment. P < 0.0001 by 1-way ANOVA; P < 0.05 by Bonferroni’s post-hoc multiple comparison for HF + W versus HF + N or LF + W, but not for HF + N versus LF + W.
The improvement in glucose tolerance raised the possibility that pNAPE-EcN treatment increased insulin sensitivity. Insulin sensitivity could be improved if pNAPE-EcN treatment inhibited high-fat diet–induced phosphorylation of insulin receptor substrates that reduce activation of downstream signals in response to insulin. We therefore examined this signaling in response to both endogenous and exogenous insulin. As expected, the 4-hour fasting plasma insulin levels were lower in mice treated with pNAPE-EcN (1.14 ± 0.12 ng/ml) compared with levels in mice treated with pEcN (1.78 ± 0.17) or standard drinking water (1.96 ± 0.24; mean ± SEM, 1-way ANOVA, P = 0.0102), which is consistent with increased insulin sensitivity. Phosphorylation of the serine-threonine protein kinase AKT at serine 473 (p-S473-AKT) is a sensitive indicator of downstream activation of insulin targets. Consistent with pNAPE-EcN preserving sensitivity to insulin action during high-fat feeding, mice treated with pNAPE-EcN had higher levels of p-S473-AKT in their liver per unit of circulating insulin (p-AKT-INS) than did control mice (Figure 12A). When stimulated with a bolus of 0.75 IU/kg insulin, all groups of mice responded by increasing the level of p-AKT to a similar extent in liver and muscle (Supplemental Figure 21A), consistent with the notion that insulin resistance can be overcome with a sufficiently high concentration of insulin. Stimulation with this bolus of insulin also reduced glucose levels to a similar extent in all groups (Supplemental Figure 21B). Downstream activation of signaling cascades in response to insulin is reduced by inhibitory phosphorylation of the insulin receptor substrate-1 on serine 307 (p-IRS1) by inflammatory kinases like JNK. With saline-only stimulation, pNAPE-EcN–treated mice had lower liver p-IRS1 levels compared with those in control mice (Figure 12B). These mice also had lower levels of activated JNKs (determined by the extent of phosphorylation of the p46 and p54 isoforms) in liver tissue compared with levels in the control mice (Figure 12C). Insulin-stimulated mice did not have appreciable differences in activated JNKs compared with the saline-only–stimulated mice (not shown). Together, these data suggest that pNAPE-EcN treatment leads to lower levels of inhibitory phosphorylation of IRS1 by JNKs, thereby maintaining greater sensitivity to insulin.
Figure 12. Treatment with pNAPE-EcN preserves insulin sensitivity in liver by protecting against inhibitory phosphorylation of IRS1 by JNKs.

Mice treated with standard water only (W), with pEcN (E), or with pNAPE-EcN (N) for 6 weeks were fasted for 4 hours and then injected intraperitoneally with either saline (to determine response to endogenous levels of insulin; n = 5 mice per group) or 0.75 IU/kg insulin (to determine response to exogenous, pharmacological levels of insulin; n = 4–5 mice per group). 15 minutes after injection, mice were euthanized and tissue collected. Data are the mean ± SEM. (A) Extent of activating phosphorylation of Ser473 of AKT (p-AKT) in liver of saline-injected mice. Values were normalized to plasma insulin (INS) and expressed relative to the average for water-only–treated mice. P = 0.0225 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. (B) Extent of inhibitory Ser307 phosphorylation of IRS1. Values were normalized to heat shock protein (HSP) and expressed relative to the average for standard water-only–treated mice. P = 0.0173 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. (C) Extent of activating phosphorylation of JNK isoforms p46 and p54. Values were normalized to HSP and expressed relative to the average for standard water-only–treated mice. For p46-JNK, P = 0.0084 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. For p54-JNK, P = 0.0134 by 1-way ANOVA; *P < 0.05 versus standard water by Dunnett’s multiple comparison test. There were no significant differences between groups stimulated with the exogenous insulin.
Although obesity in mice induced by a high-fat diet models many aspects of human obesity, consumption of a high-fat diet is not required for development of obesity in humans, particularly in individuals with a genetic predisposition toward overeating. Rodents carrying monogenic mutations in the gene for leptin or its receptor (e.g., ob/ob mice or Zucker rats) have been very useful models for understanding feeding and body weight regulation. However, mutations in these genes are rare in humans and cause extreme forms of obesity (39–41). The need to model the polygenic obesity more typical of human disease has led to the development of mice with polygenic obesity such as TallyHo/Jng (TallyHo) mice (42–45). Female TallyHo mice have been reported to rapidly gain excess weight and body fat (relative to C57BL6J mice) in the first 12 weeks of life, even while consuming a standard chow diet and without displaying the hyperglycemia seen in male TallyHo mice (45). We therefore tested whether pNAPE-EcN treatment could inhibit weight gain in female TallyHo mice fed a standard chow diet. TallyHo mice treated with pNAPE-EcN beginning at 5 weeks of age gained significantly less weight than did mice treated with vehicle or pEcN (Figure 13). This reduced body weight persisted for at least 4 weeks following the end of pNAPE-EcN treatment. The obesity induced by the low-fat chow diet in the vehicle-treated TallyHo mice was relatively modest in this study. For this reason, along with the difficulty of obtaining a large number of young TallyHo mice of similar age and body weight, our study was underpowered to detect statistically significant improvements in many of the metabolic parameters previously examined for C57BL6J mice fed a high-fat diet. Nevertheless, treatment of TallyHo mice with pNAPE-EcN tended to improve the same parameters such as lowering body fat, liver triglyceride levels, plasma leptin levels, expression of inflammatory genes in the liver, and infiltration of macrophages, while increasing expression of fatty acid oxidation genes in the liver (Supplemental Table 5 and Supplemental Figure 22). These data demonstrate that pNAPE-EcN can prevent obesity arising from a variety of causes and not simply obesity induced by high-fat diets.
Figure 13. Effect of pNAPE-EcN treatment on body weight of TallyHo mice fed a standard chow diet.

P < 0.0001 by 2-way ANOVA for time and P = 0.0357 for treatment. All values are the mean ± SEM; n = 5 mice per group.
Discussion
Our results demonstrate the feasibility of incorporating genetically modified bacteria that secrete NAPEs into the intestinal microbiota to provide sustained treatment for a chronic condition, obesity, and thus relieve the need for continuing daily administration of the therapeutic compound. The long-term effectiveness of current medical and lifestyle treatment regimens for obesity is poor because of the difficulty in sustaining compliance over long periods of time and compensatory responses to weight loss (3–6). Our findings that incorporation of pNAPE-EcN into the gut microbiota provided sustained attenuation of weight gain in mice consuming a high-fat diet for up to 6 weeks after ending administration of the bacteria and maintained total body weight and adiposity lower than that of control-treated animals even 12 weeks after ending administration demonstrates that this could be an effective strategy for ensuring sustained treatment. Additional studies are needed to determine the optimum interval and duration of booster administrations of pNAPE-EcN in order to prevent excess weight gain throughout the lifetime of the animals. However, our studies suggest that the interval between booster administrations would likely be at least 4 weeks. Thus, even without further optimization, pNAPE-EcN could represent a significantly less onerous treatment regimen than daily medication or voluntary caloric restriction.
Our studies also show that therapeutic compounds produced by colonic bacteria can be delivered to tissues beyond the intestinal tract. Within the intestinal tract, we found that pNAPE-EcN administration primarily increased NAPE levels in the colon, consistent with the predominant localization of E. coli in the colon. We also found significant elevation of liver NAE (but not NAPE) after sustained pNAPE-EcN administration, suggesting that NAPE absorbed from the colon is converted to NAE by NAPE-PLD and then enters the portal circulation. We did not find increased plasma levels of NAE (or NAPE), consistent with previous findings that elevations in liver NAE levels do not result in significant elevations in plasma NAE (14). The rapid metabolism of NAE by FAAH and NAAA likely accounts for this observation (46, 47). That the modest elevations in hepatic levels of NAE generated by pNAPE-EcN treatment were sufficient to induce beneficial effects was evident by the increased hepatic expression of genes involved in fatty acid oxidation (Ppara, Cpt1, and AOX) and the decreased infiltration of leukocytes and expression of genes involved in inflammation (Cd36, Ccl2, and Cd68). These changes in hepatic gene expression were the result of NAE action rather than of a downstream effect of altered body composition, because fatty acid oxidation gene expression was not increased in pair-fed mice. Thus, our studies demonstrate that genetically modified bacteria are capable of producing sufficient amounts of secreted small molecules to induce significant therapeutic effects, even when the target organ is not the intestinal tract. In theory, by choosing appropriate therapeutic molecules that are similarly well absorbed, but less rapidly metabolized, it should be possible to target even more distal organs such as the brain.
Treatment with pNAPE-EcN appeared to invoke a number of subtle effects on the patterns of energy intake and expenditure that led to reduced weight gain on the high-fat diet. For instance, the lower overall food intake appeared to be particularly driven by reductions in food intake during the dark phase (the active period), which was accompanied by shorter meal bouts and smaller meal size without an effect on intermeal intervals. This effect of pNAPE-EcN treatment on meal patterning for high-fat feeding differs somewhat from previous reports in chow-fed rats, in which increasing NAE levels decreased food intake primarily by increasing post-meal intervals (48–50). Our results suggest that increasing NAE levels via pNAPE-EcN–mediated biosynthesis increases sensitivity to short-term satiation signals induced by food intake that terminate feeding as well as long-term feeding drive. The effects on short-term satiation and long-term feeding drive by bacterially derived NAEs are consistent with activation of at least 3 established NAE receptors: GPR119, PPARα, and TRPV1. NAE activation of GPR119 induces GLP-1 secretion (51, 52), and GLP-1 receptor agonists reduce food intake and fat accumulation on a high-fat diet (53, 54). NAEs are also potent PPARα agonists, and PPARα agonists reduce long-term food intake and fat accumulation, while also increasing fatty acid oxidation (55–57). Importantly, NAEs fail to reduce food intake and body fat when PPARα is genetically ablated (18, 28, 58). NAEs activate TRPV1 (59, 60), and genetic ablation of TRPV1 also inhibits the effectiveness of NAEs at reducing food intake (31). The extent to which each of these receptors contributes to the reduction in food intake induced by pNAPE-EcN treatment requires further elucidation.
Because pair-feeding did not completely recapitulate the effect of pNAPE-EcN treatment on body weight and body fat gain, at least some of the effects must be the result of its increasing energy expenditure. Indeed, we found that NAPE-EcN–treated mice had increased energy expenditure relative to body weight during the light phase when mice are mostly inactive, but not during the dark phase when they are highly active. Because overall physical activity also did not differ between the groups, the increased energy expenditure is most likely the result of an increased metabolic rate during rest. We also found an increased expression of fatty acid oxidation genes and a small but consistent tendency toward a lower RQ in pNAPE-EcN–treated mice. Even small changes in resting metabolic rate and fatty acid oxidation may result in marked long-term differences in adiposity. A reduced RQ when switching to high-fat diets predicts lower long-term weight gain and body fat in humans (61). Conversely, individuals prone to obesity have blunted lowering of their RQ in response to deliberate short-term overeating of a balanced diet (62). Therefore, the ability of pNAPE-EcN treatment to increase expression of fatty acid oxidation genes and resting energy expenditure may be important mechanisms contributing to its long-term efficacy in lowering adiposity.
Of note, our finding that elevations of NAEs in the liver corresponded with slightly increased resting energy expenditure contrasts with a recent study that found decreased resting energy expenditure when NAE levels were elevated systematically by genetic ablation of fatty acid amide hydrolase or by subcutaneous infusion of synthetic NAE (63). In that study, NAE levels were significantly elevated both in plasma and in the brain. The elevated brain NAE levels corresponded with reduced mRNA expression of thyrotropin-releasing hormone in the hypothalamus and thyroid-stimulating hormone in the pituitary gland and with increased mRNA expression of deiodinase 2 in the hypothalamus. These changes resulted in lower circulating levels of triodothyronine (T3) and thyroxine (T4) and therefore decreased energy expenditure (63). Thus, the failure of the bacterially synthesized NAPE metabolites to reach the systemic circulation and the CNS may be somewhat beneficial, as this minimizes the likelihood that they will induce counterregulatory effects on energy expenditure in the CNS.
Our choice of NAPEs as the anorexigenic compound for testing our strategy was based on studies showing that NAEs, the primary metabolites of NAPEs, are endogenous regulators of food intake and adiposity (15–24). Biosynthesis of small-molecule therapeutic compounds like NAPEs may have several potential advantages over biosynthesis of anorexigenic peptides or the use of nonmodified probiotic bacteria. For instance, because NAPE biosynthesis requires heterologous expression of only a single enzyme that uses substrates already in high abundance in the bacteria, relatively high levels of NAPEs (>10 nmol/1011 CFU) can be produced. This is in contrast to bacterial expression and secretion of heterologous peptide, where the yield is typically quite low (64–67). The ability of EcN to tolerate high NAPE levels may in part result from NAPEs being endogenous phospholipids of E. coli (68), rather than a foreign peptide. Because NAPEs are also endogenous mammalian phospholipids that exert antiinflammatory effects after conversion to NAEs, NAPEs seem unlikely to invoke an immune response even when biosynthesized chronically by gut bacteria.
In addition to our genetically modified EcN, a number of unmodified probiotic bacteria also show antiobesity effects in mice (69–73). One clear advantage of using genetically modified gut microbiota rather than wild-type probiotics is the ability to choose both an appropriate carrier bacteria that can colonize the gut of the diseased individual and an appropriate therapeutic compound, whereas most probiotic bacteria are poor colonizers and the actual bioactive metabolites that confer their benefit poorly characterized. Without appropriate characterization of the bioactive metabolites, quality control during production and continuous culture of these probiotics may be very difficult. Another final possible advantage of genetically modified gut bacteria is that they could be made responsive to temporal cues such as food intake by use of appropriate promoters. Such food-dependent biosynthesis would mimic physiological regulation of many metabolic responses, potentially improving efficacy. Thus, if appropriate regulatory guidelines for use of genetically modified gut bacteria can be established, this approach may allow highly customized long-term treatments for individuals with different chronic medical conditions including obesity. Our finding that biosynthesis of NAPEs by EcN inhibits obesity induced by a high-fat diet and does so for an extended period after ending administration of bacteria illustrates the potential efficacy of this approach.
Methods
Bacterial strains and preparation.
The pDEST-At1g78690 expression plasmid and transformation into Ec have been previously described (32). EcN was obtained from ArdeyPharm, GmbH and the P. luminescens luciferase operon cloned from the pXen5 plasmid (Xenogen) inserted into the RecA gene. For expression of At1g78690 in EcN, pQE-80L (QIAGEN) was modified by removing one lac operator to enable basal expression of inserted genes without IPTG induction. This was accomplished by digesting pQE-80L with XhoI and EcoR1 and then annealing 2 pairs of oligonucleotides that had previously been annealed and then digested with the same enzymes. The sequences of these oligonucleotides are as follows: Pair 1, sense: TCGTCTTCAC CTCGAGAAAT CATAAAAAAT TTATTTGCTT TGTGAGCGGA TAACAATTAT AATAGATTCA ATCACACAGA ATTCATTAAA; antisense: TTTAATGAAT TCTGTGTGAT TGAATCTATT ATAATTGTTA TCCGCTCACA AAGCAAATAA ATTTTTTATG ATTTCTCGAG GTGAAGACGA. Pair 2, sense: TCGTCTTCAC CTCGAGAAAT CATAAAAAAT TTATTTGCTT TCAGGAAAAT TTTTCTGTAT AATAGATTCA ATCACACAGA ATTCATTAAA; antisense: TTTAATGAAT TCTGTGTGAT TGAATCTATT ATACAGAAAA ATTTTCCTGA AAGCAAATAA ATTTTTTATG ATTTCTCGAG GTGAAGACGA. The At1g78690 gene was obtained by high-fidelity PCR using pDEST-At1g78690 (32) as a template, and the following primers were used: sense: CGCGGATCC A TGGCTATGGG GAAGATAATGG; antisense: GAGAGAGCTC TCACAACCGC TTGGCTAAGA GTC and were subcloned in frame into pQE-80L1 digested with BamHI and SacI. Bioluminescent EcN were then transformed either with pQE-80L1 empty vector (pEcN) or pQE-80L1 with At1g78690 inserted (pNAPE-EcN).
NAPE measurement.
NAPE concentration was quantified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) after methylamine hydrolysis as we have previously described (33). Because Bulat and Garrett (34) reported that they failed to find evidence for significant synthesis of NAPEs in E. coli expressing At1g76890p, we performed additional experiments to confirm the synthesis of NAPEs in our engineered EcN. To determine whether the analyzed methylamine-hydrolyzed species did indeed arise from NAPEs and not from lysoNAPEs or acyl-PG, an aliquot was taken prior to methylamine hydrolysis and analyzed by limited mass scanning in negative ion mode after HPLC to separate phospholipid species. To confirm that the putative NAPE species did indeed contain N-acyl head groups and were not simply acyl-PG species, as proposed by Bulat and Garrett, we performed base hydrolysis with 0.35 M methanolic sodium hydroxide for 2 hours, removed salts from the neutralized reaction mixture via C18 solid-phase extraction and then analyzed the products on LC/MS in full scanning mode using the same HPLC gradient as for the methylamine-hydrolyzed products.
Animal studies.
Four- or 12-week-old male C57BL/6J mice and 3- to 5-week-old female TallyHo mice were purchased from The Jackson Laboratory. Mice were individually housed in the Vanderbilt University animal facility in a 12-hour light/12-hour dark cycle.
For preliminary studies to determine NAPE absorption after gavage and the effect on food intake, 12-week-old male C57BL/6J mice were treated with 500 mg/l ampicillin in drinking water for 7 days and then administered 1011 CFU of pEc or pNAPE-Ec by oral gastric gavage using a ball-point metal syringe. For studies on NAPE absorption and food intake, mice were given a single daily dose of bacteria for 7 days. For studies examining the effect on food intake, mice were given a single daily dose of 1011 CFU bacteria for 7 days. Daily food intake was measured by adding preweighed food pellets to each cage and then reweighing these pellets after 24 hours. Four hours after the last gavage, mice were sacrificed and blood and tissue collected.
For the main diet-induced obesity study, 40 male C57BL/6J mice were obtained at approximately 4 weeks of age and initially fed a standard chow diet (LabDiet 5001: 13.5% kcal from fat, 60% kcal from carbohydrate, 28.5% kcal from protein). After 1 week of adaptation to the animal facility (experimental day –7), the mice were separated into individual cages and were treated with 500 mg/l ampicillin in drinking water for 7 days. During this time, food intake, body weight, and body composition were determined for each mouse, and then the mice were divided into 4 groups of 10 mice each so that the mean and variation in body weight and body fat for each group were as similar as possible (Table 1). Each group was then randomly assigned to 1 of the 4 treatment groups. On experimental day 0, ampicillin treatment was stopped, mice began receiving their assigned treatment for 8 weeks and began the high-fat diet (TestDiet 58Y1, containing 60% fat by kcal), which they received for the remainder of the study. The 4 treatment groups received standard drinking water either with no additives (water), with 0.125% gelatin (vehicle), with 0.125% gelatin and 5 × 109 CFU/ml pNAPE-EcN (pNAPE-EcN), or with 0.125% gelatin and 5 × 109 CFU/ml pEcN (pEcN). Additionally, all mice were given preweighed kaolin pellets (Research Diets), and the change in kaolin pellet weight was measured once a week as an indicator of intestinal distress. On the final day of the study (day 87), mice were euthanized and blood and tissue collected.
Body weight was measured using a portable electronic scale. For body composition, mice were scanned by MRI using a Bruker Minispec MQ10 NMR Analyzer to determine fat mass, lean mass, and free fluid. All bioluminescence imaging (BLI) was performed using a Xenogen IVIS 200 CCD camera (Caliper Life Sciences). Equal areas for each region of interest (ROI) were centered over the bioluminescent region. Photon counting measurements summed bioluminescent intensity for all pixels within the ROI over the integration time.
For glucose tolerance testing after 8 weeks of bacterial treatment, all mice were returned to standard drinking water, fasted overnight, weighed, and then given a bolus of glucose (2 g glucose/kg body weight) by oral gastric gavage. Blood samples were taken 0, 15, 30, 60, and 120 minutes after gavage.
The 4 follow-up mouse studies were performed as described for the main study except that for the first follow-up study, no untreated (water-only) group of mice was used, and the follow-up period after ending bacterial administration was extended to 12 weeks rather than only 4 weeks as in the main study. Feces were collected for 16S rRNA analysis at the end of the eighth week of treatment and the fourth week of follow-up (week 12). Mice were euthanized on experimental day 140.
For the second follow-up study, the groups of mice were ad libitum fed a chow diet, a high-fat diet and standard drinking water, or a high-fat diet and drinking water supplemented with 5 × 109 CFU/ml pNAPE-EcN. After 9 weeks of treatment, the pNAPE-EcN group was returned to standard drinking water for 1 day, the mice were fasted for 6 hours, and an oral glucose tolerance test was performed. Mice were euthanized the following day (experimental day 65), and livers were collected for measurement of NAPE and NAE levels.
For the third follow-up study, 3 groups were a fed high-fat diet ad libitum, and a fourth group was pair-fed using the average weekly food intake of the pNAPE-EcN treatment group. The 4 treatment groups were: untreated mice (receiving standard drinking water only); mice treated with 5 × 109 CFU/ml pNAPE-EcN; mice treated with water supplemented with 5 × 109 CFU/ml pEcN; and mice that were pair-fed while treated with 5 × 109 CFU/ml pEcN. These 4 treatments were carried out in 3 cohorts staged 1 week apart, with the first 2 cohorts consisting of 5 animals from each of the first 3 treatment groups and the third cohort consisting of all 10 of the pair-fed animals. For pair-fed mice, the daily allowance of food was divided into 2 aliquots, with two-thirds of total calories provided in the evening prior to lights out and one-third of total calories provided in the morning after the lights were turned on. Any uneaten food at the end of the day was recorded and removed. No uneaten food was found in any cages after the first 2 weeks of pair-feeding. After 4 weeks of treatment, the first 3 groups were placed in cages equipped with the Promethion monitoring system (Sable Systems) for 7 days. After acclimation to the Promethion cages for 4 days, the water bottles were changed and mouse behavior and metabolism continuously monitored for 3 days. The Promethion system monitors food and water intake by changes in weight of the food hopper and water bottle, estimates physical activity as distance traveled in the cage by beam breaks, and measures energy expenditure by indirect calorimetry. For 4- or 12-hour time blocks used to analyze the effect of circadian time on meal patterning, each data point represented the average of data for that time block for 3 separate days. A malfunction of the water bottle led to 1 mouse receiving treatment with standard drinking water to be severely dehydrated and removed from the study. Pair-fed animals were not placed in the Promethion system because it was not possible to continue pair-feeding using the Promethion system.
For analysis of the effect of treatment on meal size, the change in food hopper weight for each 5-minute block during the 72 hours of monitoring was calculated, and any block during which food hopper weight decreased by greater than 0.05 mg was scored as a meal and categorized based on size relative to the combined group mean size for all meals (39.7 mg). Category 1: meals ≤19.84 mg (1/2 mean); category 2: meals >19.84 mg and ≤39.7 mg; category 3: meals >39.7 mg and ≤103.3 mg (mean + 1 SD); category 4: meals >103.3 mg and ≤166.8 mg (mean + 2 SD); category 5: meal events >166.8 mg and ≤230.4 mg (mean + 3 SD). Category X represents meal events >230.4 mg. This extreme change in food hopper weight may be the result of a mouse removing an entire food pellet from the hopper, rather than actually eating the pellet while in the hopper.
After completion of monitoring in the Promethion system, mice were returned to standard cages for 1 week while continuing treatment. On the first day after being returned to their standard cages, mice were fasted for 4 hours and blood samples drawn. One week after their return to standard cages, the mice were fasted for 4 hours starting at lights on prior to receiving either saline or 0.75 IU/kg body weight insulin and then euthanized 15 minutes later.
For the fourth follow-up study, TallyHo mice were used instead of C57BL6 mice. Because of the limited number of TallyHo mice available for purchase from the commercial vendor (The Jackson Laboratory), 2 different cohorts of mice were used. Only mice weighing between 21 and 24 g at 5 weeks of age were included in the study. The 3 treatment groups received: vehicle (0.125% gelatin in water), 5 × 109 CFU/ml pEcN, and 5 × 109 CFU/ml pEcN in drinking water. All TallyHo mice received a standard chow diet for the entire study. Ampicillin (500 mg/l) was given in the same drinking water as the EcN or the vehicle for 1 week at the beginning of the treatment. Treatment was continued for 11 weeks, and mice were euthanized and tissue harvested 4 weeks after stopping administration of EcN. Mice were fasted for 4 to 6 hours prior to being euthanized.
Biochemical measurements.
Blood glucose was measured using the Accu-Chek Diabetes monitoring kit (Roche). Insulin and leptin were measured by the Vanderbilt Hormone Assay and Analytical Services Core using radioimmunoassay. For triglyceride measurements in liver, lipids were extracted using chloroform/methanol (2:1), and individual lipid classes were separated by thin-layer chromatography. The triglycerides were scraped from the plate, and the fatty acids methylated using BF3/methanol. Fatty acid methyl esters were analyzed using an Agilent 7890A gas chromatograph equipped with flame ionization detectors and a capillary column (SP2380, 0.25 mm × 30 m, 0.25 μm film; Supelco). The esters were identified by comparing the retention times to those of known standards. Trieicosenoin (C20:1) was added to the total lipid extract, serving as an internal standard and permitting quantitation of the amount of triglycerides in the sample.
For measurement of gene expression in liver, total RNA was extracted from liver tissue using the RNeasy Micro Kit (QIAGEN), and the concentration was measured spectrophotometrically. The extracted RNA was reverse transcribed into cDNA using a cDNA reverse transcription kit (Applied Biosystems), and the RNA expression level was quantified by quantitative RT-PCR (qRT-PCR) using SYBR Green PCR Master Mix (QIAGEN) and the 7500 Real Time PCR system (Applied Biosystems) according to the manufacturer’s protocol. For Ppara, the primer pair used was GTACGGTGTGTATGAAGCCATCTT and GCCGTACGCGATCAGCAT. For Ppard, the primers were GCCATATTCCCAGGCTGTC and CAGCACAAGGGTCATCTGTG. For Cpt1a, the primers were GTGACTGGTGGGAGGAATAC and GAGCATCTCCATGGCGTAG. For AOX, the primers were GTGCAGCTCAGAGTCTG TCCAA and TACTGCTGCGTCTGAAAATCCA. For FAS, the primers were TGCTCCCAGCTGCAGGC and GCCCGGTAGCTCTGGGTGTA. For PEPCK, the primers were AAGCATTCAACGCCAGGTTC and GGGCGAGTCTGTCAGTTCAAT. For Cd36, the primers were CCTTAAAGGAATCCCCGTGT and TGCATTTGCCAATGTCTAGC. For Scd1, the primers were TTCTTACACGACCACCACCA and CCGAAGAGGCAGGTGTAGAG. For Ccl2, the primers were ACTGAAGCCAGCTCTCTCTTCCTC and TTCCTTCTTGGG GTCAGCACAGAC. For Cd68, the primers were CCATCCTTCACGATGACACCT and GGC AGGGTTATGAGTGACAGTT. For β-actin (Actb), the primers were GAGCGCAAGTACTCTGTGTG and CGGACTCATCGTACTCCTG. Relative quantification of gene expression with real-time PCR data was calculated relative to β-actin.
Fecal DNA extraction, pyrosequencing, and bacterial composition analysis.
Bacterial DNA was extracted by a bead-beating method using a commercial DNA extraction kit (Mobio Powersoil Kit) following the manufacturer’s instructions. The bead-beating step was performed on a homogenizer for 60 seconds at a speed of 4 m/s. Amplification of the 16S rRNA genes was carried out using universal bacterial primers (530F-1100R) to amplify DNA in a single-step, 30-cycle PCR using the HotStarTaq Plus Master Mix Kit (QIAGEN) under the following conditions: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds, 53°C for 40 seconds, and 72°C for 1 minute, after which a final elongation step at 72°C for 5 minutes was performed. Following PCR, all amplicon products from different samples were mixed in equal concentrations and purified using Agencourt Ampure beads. Samples were sequenced using Roche 454 FLX titanium instruments and reagents according to the manufacturer’s guidelines. The Q25 sequence data were processed using a proprietary analysis pipeline (www.mrdnalab.com) (74, 75). Briefly, sequences were depleted of barcodes and primers, then sequences less than 150 bp were removed, as were sequences with ambiguous base calls and homopolymer runs exceeding 6 bp. Operational taxonomic units (OTUs) were generated by clustering at 3% divergence (97% similarity) from de-noised sequences, and chimeras were removed. Final OTUs were taxonomically classified using BLASTn against a curated GreenGenes database (76) and compiled into each taxonomic level as the percentage of sequences within each sample that map to the designated taxonomic classification. A total of 423,513 sequences, 5,361 ± 262 per sample, were analyzed. Mean differences at each taxonomic level were evaluated by T-testing (Microsoft Excel) against vehicle controls. The Sequence Read Archive (SRA) accession number for our study is SRP042014.
Western blot analysis.
Frozen tissues were made into a powder using a tissue pulverizer, and then approximately 50 mg of powered tissue was sonicated with a Virsonic 100 Ultrasonic Cell Disrupter (at setting 8, for less than 10 seconds; Virtis) in cold T-Per Tissue Protein Extraction Buffer (10 μl/mg tissue; Thermoscientific) with added protease and phosphatase inhibitor cocktails (P8340, P5726, and P0044; Sigma-Aldrich). Diluted protein samples (10.5 μg total protein per lane) were subjected to denaturing electrophoresis on a 4%–12% Tris Acetate gel with XT MES Running Buffer using the Bio-Rad XT Criterion System. Proteins were transferred to 0.2-μm nitrocellulose membranes (Millipore), blocked in 5% BSA and 0.2% Tween-20, and incubated with primary antibodies diluted 1:1,000 in blocking buffer overnight at 4°C with gentle rocking. Primary antibodies used allowed the detection of AKT and its Ser473 and Thr308 phosphorylated forms (4691, 9271, and 9275, respectively; Cell Signaling Technology), the phosphorylated (Thr183/Tyr185) form of JNK (4668; Cell Signaling Technology), the phosphorylated (Ser307) form of IRS1 (2381; Cell Signaling Technology), and a gel loading control, HSC70/HSP73 (ADI-SPA-816; Enzo Life Sciences), was used. Blots were washed and incubated with a species-specific HRP-conjugated secondary antibody (goat anti-rabbit, W401B; Promega) in 50/50 blocking buffer and Starting Block T20 Blocking Buffer (37543; Thermo Scientific). After washing, membranes were developed using the PerkinElmer Western Lightning Plus-ECL Enhanced Chemiluminescence Substrate Kit (NEL105001; Amersham Biosciences) and GeneMate Blue Ultra Autorad Film (BioExpress). Films were scanned on an Epson 3200 scanner and band density quantified using ImageJ software (NIH). Densitometric values were reported after normalization for gel-loading differences and relative to the average value obtained from protein extracts of vehicle-treated control mice.
Statistics.
Statistical analysis was performed using GraphPad Prism 5.04 (GraphPad Software). A P value of less than 0.05 was considered statistically significant. For 2-way repeated-measures (RM) ANOVA, when overall significance for treatment effect was found, the Bonferroni’s multiple comparison post-hoc test was used to determine differences between treatment groups. For 1-way ANOVA, when overall significance for treatment effect was found, the Dunnett’s multiple comparison post-hoc test was used to determine groups that differed from the control group. A 2-tailed Student’s t test was used for studies with a fixed endpoint comparing pNAPE-EcN treatment to the control.
Study approval.
All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee and the Institutional Biosafety Committee of Vanderbilt University.
Supplementary Material
Acknowledgments
We thank Eric Skaar, Alyssa Hasty, Tim Cover, Italo Biaggioni, and Daniel Byrnes for helpful discussions. E. coli Nissle 1917 (Mutaflor) was obtained under a materials transfer agreement (MTA) from Ardeypharm GmbH, and pXen5 was purchased under an MTA from Xenogen. This work was funded by the NIH through the NIH New Innovator Award Program (DP2 OD003137). Information on the New Innovator Award Program, which is part of the NIH Roadmap for Medical Research, is available at https://commonfund.nih.gov/newinnovator/index. Additional support was provided by NIH grants AT07830 (to S.S. Davies); DK59637 (to the Vanderbilt Metabolic Mouse Phenotyping Center); DK20593 (to the Hormone Assay and Analytical Services Core, Vanderbilt Diabetes Research and Training Center); UL1 RR024975-01 (to the Vanderbilt Institute for Clinical and Translational Research); and U24 DK092993 (to the UC Davis Mouse Metabolic Phenotyping Center).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(8):3391–3406. doi:10.1172/JCI72517.
References
- 1.Lam DW, LeRoith D. The worldwide diabetes epidemic. Curr Opin Endocrinol Diabetes Obes. 2012;19(2):93–96. doi: 10.1097/MED.0b013e328350583a. [DOI] [PubMed] [Google Scholar]
- 2.Flegal KM, Carroll MD, Ogden CL, Curtin LR. PRevalence and trends in obesity among us adults, 1999–2008. JAMA. 2010;303(3):235–241. doi: 10.1001/jama.2009.2014. [DOI] [PubMed] [Google Scholar]
- 3.Wing RR, Hill JO. Successful weight loss maintenance. Annu Rev Nutr. 2001;21:323–341. doi: 10.1146/annurev.nutr.21.1.323. [DOI] [PubMed] [Google Scholar]
- 4.Stunkard A, Mc L-HM. The results of treatment for obesity: a review of the literature and report of a series. AMA Arch Intern Med. 1959;103(1):79–85. doi: 10.1001/archinte.1959.00270010085011. [DOI] [PubMed] [Google Scholar]
- 5.Kramer FM, Jeffery RW, Forster JL, Snell MK. Long-term follow-up of behavioral treatment for obesity: patterns of weight regain among men and women. Int J Obes. 1989;13(2):123–136. [PubMed] [Google Scholar]
- 6.Wadden TA, Sternberg JA, Letizia KA, Stunkard AJ, Foster GD. Treatment of obesity by very low calorie diet, behavior therapy, and their combination: a five-year perspective. Int J Obes. 1989;13(suppl 2):39–46. [PubMed] [Google Scholar]
- 7.Harris K, Kassis A, Major G, Chou CJ. Is the gut microbiota a new factor contributing to obesity and its metabolic disorders? J Obes. 2012;2012:879151. doi: 10.1155/2012/879151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472(7341):57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- 10.Turnbaugh PJ, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vijay-Kumar M, et al. Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science. 2010;328(5975):228–231. doi: 10.1126/science.1179721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cani PD, et al. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes. 2008;57(6):1470–1481. doi: 10.2337/db07-1403. [DOI] [PubMed] [Google Scholar]
- 13.Membrez M, et al. Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. FASEB J. 2008;22(7):2416–2426. doi: 10.1096/fj.07-102723. [DOI] [PubMed] [Google Scholar]
- 14.Fu J, et al. Food intake regulates oleoylethanolamide formation and degradation in the proximal small intestine. J Biol Chem. 2007;282(2):1518–1528. doi: 10.1074/jbc.M607809200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gillum MP, et al. N-acylphosphatidylethanolamine, a gut- derived circulating factor induced by fat ingestion, inhibits food intake. Cell. 2008;135(5):813–824. doi: 10.1016/j.cell.2008.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen HS, Diep TA. N-acylethanolamines, anandamide and food intake. Biochem Pharmacol. 2009;78(6):553–560. doi: 10.1016/j.bcp.2009.04.024. [DOI] [PubMed] [Google Scholar]
- 17.Petersen G, et al. Intestinal levels of anandamide and oleoylethanolamide in food-deprived rats are regulated through their precursors. Biochim Biophys Acta. 2006;1761(2):143–150. doi: 10.1016/j.bbalip.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz GJ, et al. The lipid messenger OEA links dietary fat intake to satiety. Cell Metab. 2008;8(4):281–288. doi: 10.1016/j.cmet.2008.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rodriguez de Fonseca F, et al. An anorexic lipid mediator regulated by feeding. Nature. 2001;414(6860):209–212. doi: 10.1038/35102582. [DOI] [PubMed] [Google Scholar]
- 20.Nielsen MJ, Petersen G, Astrup A, Hansen HS. Food intake is inhibited by oral oleoylethanolamide. J Lipid Res. 2004;45(6):1027–1029. doi: 10.1194/jlr.C300008-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Terrazzino S, et al. Stearoylethanolamide exerts anorexic effects in mice via down-regulation of liver stearoyl-coenzyme A desaturase-1 mRNA expression. FASEB J. 2004;18(13):1580–1582. doi: 10.1096/fj.03-1080fje. [DOI] [PubMed] [Google Scholar]
- 22.Srisai D, et al. Characterization of the hyperphagic response to dietary fat in the MC4R knockout mouse. Endocrinology. 2011;152(3):890–902. doi: 10.1210/en.2010-0716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzman M, Lo Verme J, Fu J, Oveisi F, Blazquez C, Piomelli D. Oleoylethanolamide stimulates lipolysis by activating the nuclear receptor peroxisome proliferator-activated receptor alpha (PPAR-α). J Biol Chem. 2004;279(27):27849–27854. doi: 10.1074/jbc.M404087200. [DOI] [PubMed] [Google Scholar]
- 24.Fu J, Oveisi F, Gaetani S, Lin E, Piomelli D. Oleoylethanolamide, an endogenous PPAR-α agonist, lowers body weight and hyperlipidemia in obese rats. Neuropharmacology. 2005;48(8):1147–1153. doi: 10.1016/j.neuropharm.2005.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Thabuis C, et al. Lipid transport function is the main target of oral oleoylethanolamide to reduce adiposity in high-fat-fed mice. J Lipid Res. 2011;52(7):1373–1382. doi: 10.1194/jlr.M013391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cluny NL, Keenan CM, Lutz B, Piomelli D, Sharkey KA. The identification of peroxisome proliferator-activated receptor α-independent effects of oleoylethanolamide on intestinal transit in mice. Neurogastroenterol Motil. 2009;21(4):420–429. doi: 10.1111/j.1365-2982.2008.01248.x. [DOI] [PubMed] [Google Scholar]
- 27.Aviello G, et al. Inhibitory effect of the anorexic compound oleoylethanolamide on gastric emptying in control and overweight mice. J Mol Med (Berl). 2008;86(4):413–422. doi: 10.1007/s00109-008-0305-7. [DOI] [PubMed] [Google Scholar]
- 28.Fu J, et al. Oleylethanolamide regulates feeding and body weight through activation of the nuclear receptor PPAR-α. Nature. 2003;425(6953):90–93. doi: 10.1038/nature01921. [DOI] [PubMed] [Google Scholar]
- 29.Thabuis C, Destaillats F, Landrier JF, Tissot-Favre D, Martin JC. Analysis of gene expression pattern reveals potential targets of dietary oleoylethanolamide in reducing body fat gain in C3H mice. J Nutr Biochem. 2010;21(10):922–928. doi: 10.1016/j.jnutbio.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Lo Verme J, et al. The nuclear receptor peroxisome proliferator-activated receptor-α mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67(1):15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- 31.Wang X, Miyares RL, Ahern GP. Oleoylethanolamide excites vagal sensory neurones, induces visceral pain and reduces short-term food intake in mice via capsaicin receptor TRPV1. J Physiol. 2005;564(Pt 2):541–547. doi: 10.1113/jphysiol.2004.081844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Faure L, et al. Discovery and characterization of an Arabidopsis thaliana N-acylphosphatidylethanolamine synthase. J Biol Chem. 2009;284(28):18734–18741. doi: 10.1074/jbc.M109.005744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo L, Amarnath V, Davies SS. A liquid chromatography-tandem mass spectrometry method for measurement of N-modified phosphatidylethanolamines. Anal Biochem. 2010;405(2):236–245. doi: 10.1016/j.ab.2010.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bulat E, Garrett TA. Putative N-acylphosphatidylethanolamine synthase from Arabidopsis thaliana is a lysoglycerophospholipid acyltransferase. J Biol Chem. 2011;286(39):33819–33831. doi: 10.1074/jbc.M111.269779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horn CC, De Jonghe BC, Matyas K, Norgren R. Chemotherapy-induced kaolin intake is increased by lesion of the lateral parabrachial nucleus of the rat. Am J Physiol Regul Integr Comp Physiol. 2009;297(5):R1375–R1382. doi: 10.1152/ajpregu.00284.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- 38.Turnbaugh PJ, Backhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe. 2008;3(4):213–223. doi: 10.1016/j.chom.2008.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montague CT, et al. Congenital leptin deficiency is associated with severe early-onset obesity in humans. Nature. 1997;387(6636):903–908. doi: 10.1038/43185. [DOI] [PubMed] [Google Scholar]
- 40.Rolland V, et al. Leptin receptor gene in a large cohort of massively obese subjects: no indication of the fa/fa rat mutation. Detection of an intronic variant with no association with obesity. Obes Res. 1998;6(2):122–127. doi: 10.1002/j.1550-8528.1998.tb00325.x. [DOI] [PubMed] [Google Scholar]
- 41.Dubern B, Clement K. Leptin and leptin receptor-related monogenic obesity. Biochimie. 2012;94(10):2111–2115. doi: 10.1016/j.biochi.2012.05.010. [DOI] [PubMed] [Google Scholar]
- 42.Leiter EH, Strobel M, O’Neill A, Schultz D, Schile A, Reifsnyder PC. Comparison of two new mouse models of polygenic type 2 diabetes at the Jackson Laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. J Diabetes Res. 2013;2013:165327. doi: 10.1155/2013/165327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim JH, et al. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics. 2001;74(3):273–286. doi: 10.1006/geno.2001.6569. [DOI] [PubMed] [Google Scholar]
- 44.Kim JH, Saxton AM. The TALLYHO mouse as a model of human type 2 diabetes. Methods Mol Biol. 2012;933:75–87. doi: 10.1007/978-1-62703-068-7_6. [DOI] [PubMed] [Google Scholar]
- 45.Kim JH, et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J Endocrinol. 2006;191(2):437–446. doi: 10.1677/joe.1.06647. [DOI] [PubMed] [Google Scholar]
- 46.Giang DK, Cravatt BF. Molecular characterization of human and mouse fatty acid amide hydrolases. Proc Natl Acad Sci U S A. 1997;94(6):2238–2242. doi: 10.1073/pnas.94.6.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ueda N, Tsuboi K, Uyama T. N-acylethanolamine metabolism with special reference to N-acylethanolamine-hydrolyzing acid amidase (NAAA). Prog Lipid Res. 2010;49(4):299–315. doi: 10.1016/j.plipres.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 48.Gaetani S, Oveisi F, Piomelli D. Modulation of meal pattern in the rat by the anorexic lipid mediator oleoylethanolamide. Neuropsychopharmacology. 2003;28(7):1311–1316. doi: 10.1038/sj.npp.1300166. [DOI] [PubMed] [Google Scholar]
- 49.Oveisi F, Gaetani S, Eng KT, Piomelli D. Oleoylethanolamide inhibits food intake in free-feeding rats after oral administration. Pharmacol Res. 2004;49(5):461–466. doi: 10.1016/j.phrs.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 50.Fu J, Kim J, Oveisi F, Astarita G, Piomelli D. Targeted enhancement of oleoylethanolamide production in proximal small intestine induces across-meal satiety in rats. Am J Physiol Regul Integr Comp Physiol. 2008;295(1):R45–R50. doi: 10.1152/ajpregu.00126.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Syed SK, et al. Regulation of GPR119 receptor activity with endocannabinoid-like lipids. Am J Physiol Endocrinol Metab. 2012;303(12):E1469–E1478. doi: 10.1152/ajpendo.00269.2012. [DOI] [PubMed] [Google Scholar]
- 52.Lauffer LM, Iakoubov R, Brubaker PL. GPR119 is essential for oleoylethanolamide-induced glucagon-like peptide-1 secretion from the intestinal enteroendocrine L-cell. Diabetes. 2009;58(5):1058–1066. doi: 10.2337/db08-1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mack CM, et al. Antiobesity action of peripheral exenatide (exendin-4) in rodents: effects on food intake, body weight, metabolic status and side-effect measures. Int J Obes (Lond). 2006;30(9):1332–1340. doi: 10.1038/sj.ijo.0803284. [DOI] [PubMed] [Google Scholar]
- 54.Tatarkiewicz K, Sablan EJ, Polizzi CJ, Villescaz C, Parkes DG. Long-term metabolic benefits of exenatide in mice are mediated solely via the known glucagon-like peptide 1 receptor. Am J Physiol Regul Integr Comp Physiol. 2014;306(7):R490–R498. doi: 10.1152/ajpregu.00495.2013. [DOI] [PubMed] [Google Scholar]
- 55.Perreault M, et al. Modulation of nutrient sensing nuclear hormone receptors promotes weight loss through appetite suppression in mice. Diabetes Obes Metab. 2010;12(3):234–245. doi: 10.1111/j.1463-1326.2009.01157.x. [DOI] [PubMed] [Google Scholar]
- 56.Harrington WW, et al. The effect of PPARα, PPARδ, PPARγ, and PPARpan agonists on body weight, body mass, and serum lipid profiles in diet-induced obese AKR/J mice. PPAR Res. 2007;2007:97125. doi: 10.1155/2007/97125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Larsen PJ, et al. Differential influences of peroxisome proliferator-activated receptors γ and -α on food intake and energy homeostasis. Diabetes. 2003;52(9):2249–2259. doi: 10.2337/diabetes.52.9.2249. [DOI] [PubMed] [Google Scholar]
- 58.Lo Verme J, Gaetani S, Fu J, Oveisi F, Burton K, Piomelli D. Regulation of food intake by oleoylethanolamide. Cell Mol Life Sci. 2005;62(6):708–716. doi: 10.1007/s00018-004-4494-0. [DOI] [PubMed] [Google Scholar]
- 59.Ahern GP. Activation of TRPV1 by the satiety factor oleoylethanolamide. J Biol Chem. 2003;278(33):30429–30434. doi: 10.1074/jbc.M305051200. [DOI] [PubMed] [Google Scholar]
- 60.Ho WS, Barrett DA, Randall MD. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol. 2008;155(6):837–846. doi: 10.1038/bjp.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Astrup A. The relevance of increased fat oxidation for body-weight management: metabolic inflexibility in the predisposition to weight gain. Obes Rev. 2011;12(10):859–865. doi: 10.1111/j.1467-789X.2011.00894.x. [DOI] [PubMed] [Google Scholar]
- 62.Schmidt SL, Kealey EH, Horton TJ, VonKaenel S, Bessesen DH. The effects of short-term overfeeding on energy expenditure and nutrient oxidation in obesity-prone and obesity-resistant individuals. Int J Obes (Lond). 2013;37(9):1192–1197. doi: 10.1038/ijo.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown WH, et al. Fatty acid amide hydrolase ablation promotes ectopic lipid storage and insulin resistance due to centrally mediated hypothyroidism. Proc Natl Acad Sci U S A. 2012;109(37):14966–14971. doi: 10.1073/pnas.1212887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Buddenborg C, Daudel D, Liebrecht S, Greune L, Humberg V, Schmidt MA. Development of a tripartite vector system for live oral immunization using a gram-negative probiotic carrier. Int J Med Microbiol. 2008;298(1–2):105–114. doi: 10.1016/j.ijmm.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 65.Majander K, et al. Extracellular secretion of polypeptides using a modified Escherichia coli flagellar secretion apparatus. Nat Biotech. 2005;23(4):475–481. doi: 10.1038/nbt1077. [DOI] [PubMed] [Google Scholar]
- 66.Blight MA, Holland IB. Heterologous protein secretion and the versatile Escherichia coli haemolysin translocator. Trends Biotechnol. 1994;12(11):450–455. doi: 10.1016/0167-7799(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 67.Huibregtse IL, et al. Genetically modified Lactococcus lactis for delivery of human interleukin-10 to dendritic cells. Gastroenterol Res Pract. 2012;2012:639291. doi: 10.1155/2012/639291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mileykovskaya E, et al. Phosphatidic acid and N-acylphosphatidylethanolamine form membrane domains in Escherichia coli mutant lacking cardiolipin and phosphatidylglycerol. J Biol Chem. 2009;284(5):2990–3000. doi: 10.1074/jbc.M805189200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang JH, Yun SI, Park MH, Park JH, Jeong SY, Park HO. Anti-obesity effect of Lactobacillus gasseri BNR17 in high-sucrose diet-induced obese mice. PLoS One. 2013;8(1):e54617. doi: 10.1371/journal.pone.0054617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fak F, Backhed F. Lactobacillus reuteri prevents diet-induced obesity, but not atherosclerosis, in a strain dependent fashion in Apoe–/– mice. PLoS One. 2012;7(10):e46837. doi: 10.1371/journal.pone.0046837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee HY, et al. Human originated bacteria, Lactobacillus rhamnosus PL60, produce conjugated linoleic acid and show anti-obesity effects in diet-induced obese mice. Biochim Biophys Acta. 2006;1761(7):736–744. doi: 10.1016/j.bbalip.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 72.An HM, et al. Antiobesity and lipid-lowering effects of Bifidobacterium spp. in high fat diet-induced obese rats. Lipids Health Dis. 2011;10:116. doi: 10.1186/1476-511X-10-116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andersson U, et al. Probiotics lower plasma glucose in the high-fat fed C57BL/6J mouse. Benef Microbes. 2010;1(2):189–196. doi: 10.3920/BM2009.0036. [DOI] [PubMed] [Google Scholar]
- 74.Capone KA, Dowd SE, Stamatas GN, Nikolovski J. Diversity of the human skin microbiome early in life. J Invest Dermatol. 2011;131(10):2026–2032. doi: 10.1038/jid.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Swanson KS, et al. Phylogenetic and gene-centric metagenomics of the canine intestinal microbiome reveals similarities with humans and mice. ISME J. 2011;5(4):639–649. doi: 10.1038/ismej.2010.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.DeSantis TZ, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.