

Abstract
BACKGROUND. Hepatitis C virus (HCV) infects approximately 170 million people worldwide and may lead to cirrhosis and hepatocellular carcinoma in chronically infected individuals. Treatment is rapidly evolving from IFN-α–based therapies to IFN-α–free regimens that consist of directly acting antiviral agents (DAAs), which demonstrate improved efficacy and tolerability in clinical trials. Virologic relapse after DAA therapy is a common cause of treatment failure; however, it is not clear why relapse occurs or whether certain individuals are more prone to recurrent viremia.
METHODS. We conducted a clinical trial using the DAA sofosbuvir plus ribavirin (SOF/RBV) and performed detailed mRNA expression analysis in liver and peripheral blood from patients who achieved either a sustained virologic response (SVR) or relapsed.
RESULTS. On-treatment viral clearance was accompanied by rapid downregulation of IFN-stimulated genes (ISGs) in liver and blood, regardless of treatment outcome. Analysis of paired pretreatment and end of treatment (EOT) liver biopsies from SVR patients showed that viral clearance was accompanied by decreased expression of type II and III IFNs, but unexpectedly increased expression of the type I IFN IFNA2. mRNA expression of ISGs was higher in EOT liver biopsies of patients who achieved SVR than in patients who later relapsed.
CONCLUSION. These results suggest that restoration of type I intrahepatic IFN signaling by EOT may facilitate HCV eradication and prevention of relapse upon withdrawal of SOF/RBV.
TRIAL REGISTRATION. ClinicalTrials.gov NCT01441180.
FUNDING. Intramural Programs of the National Institute of Allergy and Infectious Diseases, National Institutes of Health Clinical Center, and National Cancer Institute; German Research Foundation.
Introduction
Until recently, therapeutic eradication of chronic HCV infection has required the use of injectable IFN-α formulations, which are difficult to tolerate and frequently unsuccessful (1). IFN-free treatment of HCV infection is now possible with recently developed directly acting antiviral agents (DAAs) that inhibit the function of viral proteins such as the NS3/4A viral protease, the NS5B RNA polymerase, and the NS5A nonstructural protein (1–3). IFN-free DAA combination therapies for HCV are oral regimens that are favorably tolerated, and typically induce rapid and sustained viral suppression in all patients in clinical trials (1–3). However, some patients experience treatment failure due to on-treatment viral breakthrough or relapse after cessation of therapy, possibly due to unknown host factors, unappreciated viral resistance, or viral reservoirs (1–3). Relapse after cessation of therapy is the major cause of treatment failure with regimens containing SOF, a recently approved HCV-specific NS5B RNA polymerase inhibitor (4–8).
In chronic HCV infection, continued activation of host-mediated inflammation in the liver is, in part, driven by endogenous IFNs, and this response is thought to contribute to the pathologic changes that lead to progressive hepatic fibrosis (9). There are 3 IFN families (type I, II, and III), which signal through distinct receptor complexes, but it is unclear which members of these families drive the hepatic response in chronic HCV infection (10–12). An endogenous IFN response is activated in all patients during acute and chronic HCV infection, but is ineffective in eradicating HCV once chronic infection is established (13). More robust preactivation of the endogenous IFN system, as measured by mRNA expression of ISGs in liver biopsies, is associated with treatment failure for IFN-α–based regimens; patients with elevated pretreatment ISG expression levels have a blunted response to exogenous IFN-α and lower odds of achieving SVR (10, 14–17). Biological correlates of SVR or relapse in patients receiving IFN-free DAA therapy have not been studied in detail, and the relevance of the endogenous IFN system for IFN-free therapy treatment outcome is unknown.
Here we report changes in blood and hepatic gene expression during IFN-free DAA therapy with sofosbuvir plus ribavirin (SOF/RBV) in treatment-naive, chronic HCV genotype-1 patients and describe associations with treatment outcome. Although patients in this trial had a high prevalence of traditional unfavorable treatment predictors to IFN-α–based therapy, including African-American race (83%) and advanced liver fibrosis (23%), 69% of patients who completed this clinical trial achieved SVR (defined by absence of detectable virus in serum 24 weeks after treatment cessation), while 31% relapsed within 12 weeks of treatment completion (8). Based on the differential treatment outcome observed in this study, we sought to explore biologic correlates of treatment response in patients who achieved SVR versus those who relapsed.
Results
We first assessed changes in hepatic gene expression during IFN-free therapy with SOF/RBV using whole-liver tissue biopsies obtained prior to treatment or at end of treatment (EOT). Microarray gene expression profiling was performed on paired biopsies available from 8 patients, 7 of whom achieved SVR and 1 who relapsed (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI75938DS1). Ingenuity Pathway Analysis (IPA) of the top 1% of differentially expressed genes (Supplemental Table 2) identified IFN signaling and antigen presentation pathways as being downregulated with treatment (Figure 1A). Of the 109 genes that met filtering and significance criteria (Figure 1B), 65 were classified as ISGs (18), known to be induced by endogenous IFNs. Reduced activation at EOT versus pretreatment was predicted for type I (IFNA2, IFNA1, and IFNB1), type II (IFNG), and type III (IFNL1) IFNs (Figure 1C).
Figure 1. Microarray mRNA expression analysis reveals downregulation of endogenous IFN signaling in liver after SOF/RBV treatment.

(A) Top canonical pathways identified by IPA as changing over the course of treatment in paired liver biopsies (n = 8). Ratios represent the number of genes identified among the top 1% of differentially expressed genes assigned to specific pathways, relative to the total number of genes in the respective pathway. The cutoff for significance of these enrichments was –logP of 1.3 (P = 0.05). Genes downregulated at EOT relative to pretreatment are shown in bold font. (B) Heatmap of genes differentially expressed after SOF/RBV treatment. The top 1% of differentially expressed genes were filtered using cutoffs of >1.2 for fold difference and unadjusted P < 0.05. Microarray expression data were used to create a hierarchical clustering of samples using a Euclidean dissimilarity measure with average linkage. Data from all 8 paired liver biopsies are shown. Red color indicates higher relative expression. Asterisks denote data from the patient who relapsed. (C) IPA upstream analysis of activation state of proteins annotated as cytokines by IPA, comparing EOT versus pretreatment activity levels. Negative z scores predict a less active state of cytokines at EOT compared with pretreatment. Scores of >2 or <–2 were considered significant.
Quantitative RT-PCR (qRT-PCR) analysis validated downregulation of a panel of ISGs, including members of the ISGF3 complex (STAT1, STAT2, and IRF9), essential for induction of ISG expression by type I/III IFNs (Figure 2A and refs. 12, 19). Also downregulated were ISGs with anti-HCV activity (IRF1, IFIH1, IRF7, PLSCR1, IFIT3, TRIM14, and IFITM1) (11, 20, 21), genes associated with antigen presentation (PSMB8, PSMB9, HLAB, and B2M), an ISG that inhibits IFN-α signaling and is elevated in chronic HCV (USP18) (22), and ligands for the chemokine receptor CXCR3 (CXCL9, CXCL10, and CXCL11) (Figure 2A and ref. 23). The patient who relapsed showed reductions in expression that were among the largest for most tested ISGs (Figure 2A).
Figure 2. qRT-PCR and ISH confirm downregulation of ISGs in liver biopsies.

(A and B) qRT-PCR expression profiling of ISGs (A) and other genes (B) in paired pretreatment and EOT liver biopsies from patients who achieved SVR (n = 7; black) and who later relapsed (n = 1; red). Gene expression was measured using 1 ng total RNA per reaction, and fold changes are for EOT versus pretreatment expression levels after normalization to GAPDH. Shown are individual fold changes with a group median and interquartile range. All genes shown met significance criteria (Wilcoxon matched-pairs signed rank test; P < 0.05 considered significant). (C) Detection of IFI44 mRNA expression in pretreatment and EOT liver biopsies, performed in FFPE tissue samples from 4 patients: 2 achieved SVR, 1 relapsed (REL) with EOT biopsy prior to relapse, and 1 had relapse detected at the time of EOT liver biopsy (biopsy and relapse were 4 weeks after EOT, with undetectable viral load 2 weeks after EOT). Original magnification, ×40. Scale bar: 100 μm.
Genes involved in modulation of T cell activation (CTLA4 and CD244), lipid metabolism (APOL3, MTTP, and LEPR), and extracellular matrix formation and integrity (TIMP1, LGALS3, LGALS3BP, KRT18, and MYL12A) were downregulated (Figure 2B and ref. 24), whereas expression of other matrix-associated genes (COL1A1, COL1A2, and SPP1) and immunoregulatory genes (IL10 and TGFB1) was not affected (data not shown). In situ hybridization (ISH) in liver biopsies confirmed reduced expression of an ISG mRNA transcript, IFI44, during treatment, regardless of SVR or relapse status, followed by reactivation of its expression at the time of viral relapse (Figure 2C and Supplemental Figure 1).
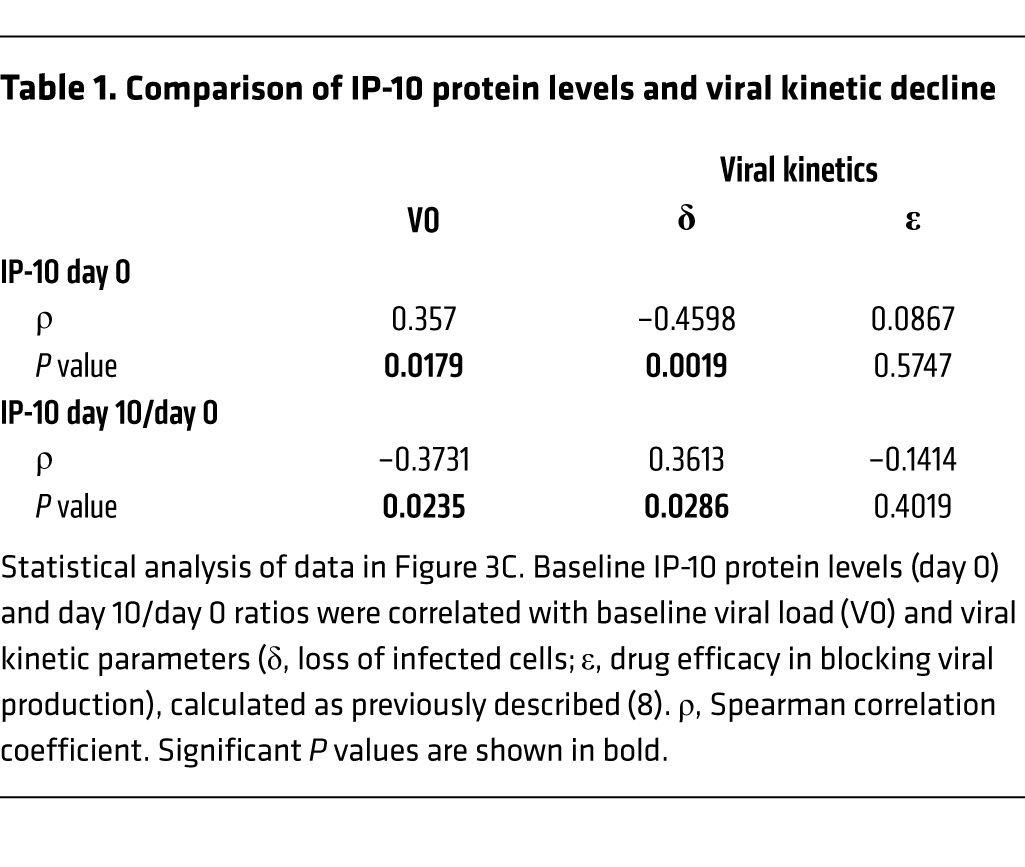
We next evaluated on-treatment serum protein levels of select chemokines and cytokines and observed similar expression at baseline and during treatment comparing patients who achieved SVR versus those who relapsed (Supplemental Table 3). Serum levels of the IFN-inducible cytokine IP-10, the protein product of the CXCL10 gene that was downregulated in liver (Figure 2A), correlated significantly with baseline viral load (Figure 3A). Expression decreased rapidly on-treatment, regardless of treatment outcome, and increased with relapse (Figure 3B). Viral kinetic and IP-10 decline were significantly correlated (Figure 3C and Table 1). IL-10 and IFN-γ decreased modestly during treatment, while expression of most other proteins did not change, including TGF-β1 and TIMP1, which are associated with hepatic fibrosis (Supplemental Table 3 and ref. 25).
Figure 3. Serum IP-10 levels normalize rapidly with SOF/RBV treatment and correlate with viral kinetic decline.

(A) Correlation between baseline viral load and serum IP-10 protein levels. Data are from patients who achieved SVR (n = 28; black) or who later relapsed (n = 14; red). Statistical analysis was by nonparametric Spearman correlation (r) test including all data. (B) Serum IP-10 protein levels decreased rapidly upon treatment, regardless of outcome. Results are displayed by outcome (SVR, black; relapse, red; n as indicated). Shown are individual measurements with a group median and interquartile range. Statistical analysis using a mixed model with fixed effects for group, time, and group-by-time interaction showed a significant change over time regardless of treatment outcome (P < 0.0001). The pattern over time differed significantly by treatment outcome, but this difference disappeared if week 36 data were excluded. Data from 5 healthy volunteers are shown for comparison, but were not analyzed statistically. (C) Serum IP-10 levels at baseline and during treatment mirrored changes in viral load. Shown are median values with interquartile range for viral load (red) and median values with 95% confidence intervals for IP-10 (black). LLOD, lower limit of detection for viral load. See Table 1 for statistical analysis. Data are from 44 patients with available data.
Table 1.
Comparison of IP-10 protein levels and viral kinetic decline
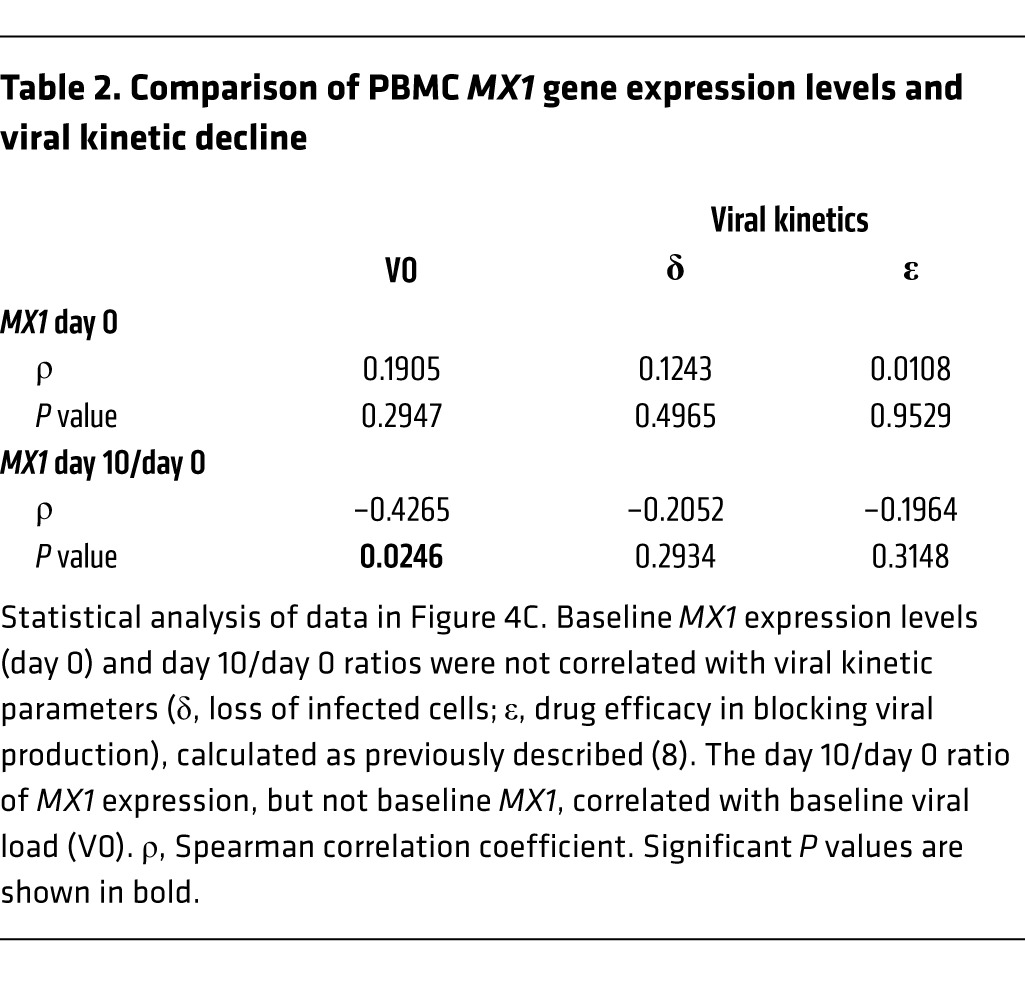
To assess whether a similar pattern of gene expression changes could be observed in the periphery, we performed microarray mRNA analysis in PBMCs collected before treatment, early in treatment (day 6–11), and at EOT (week 24) and identified a significant decrease of IFN signaling during treatment (Supplemental Figure 2 and Supplemental Table 4). qRT-PCR analysis in PBMCs confirmed rapid and sustained downregulation of ISGs, several of which declined to EOT expression levels by day 6–11 of therapy (Figure 4A). There were no significant gene expression differences by treatment outcome during therapy, but expression of ISGs increased at relapse (Supplemental Figure 3). Unlike IP-10 protein levels, gene expression of a representative ISG, MX1, in PBMCs did not correlate with baseline viral load (Figure 4B). Furthermore, whereas viral load and PBMC ISG expression both declined early on-treatment, the magnitude of gene expression changes and viral kinetic decline were not significantly correlated in a group analysis (Figure 4C and Table 2).
Figure 4. Rapid treatment-induced downregulation of endogenous IFN signaling in peripheral blood.

(A) Rapid decrease of gene expression of 6 selected ISGs in PBMCs. Shown are individual measurements (n as indicated) with group median and interquartile range. A linear mixed-effects model showed a significant decrease in expression of all 6 genes over the course of treatment (P < 0.0001). (B) MX1 gene expression in blood did not correlate with baseline viral load. Data points are from patients who achieved SVR (n = 22; black) and from patients who later relapsed (n = 10; red). Statistical analysis was by nonparametric Spearman correlation (r) including all data. (C) Viral kinetic decline during treatment did not correlate with MX1 expression in PBMCs. Shown are median values with interquartile range for viral load (red) and median values with 95% confidence intervals for MX1 (black). LLOD, lower limit of detection for viral load. Data are from the 32 patients with available data. See Table 2 for statistical analysis.
Table 2.
Comparison of PBMC MX1 gene expression levels and viral kinetic decline
It is unclear which IFNs drive expression of ISGs in liver and blood during chronic HCV infection (11). To address this question, we measured gene expression of select type I, II, and III IFNs and their receptors using RNA isolated from paired liver biopsies. In the 7 patients who achieved SVR, total expression of all measured IFNs decreased over the course of treatment (Figure 5A), consistent with the observed reductions in ISG expression (Figures 1 and 2). Interestingly, expression of type II IFNs and several type III IFNs decreased in parallel with ISGs, while expression of type I IFNs unexpectedly increased (IFNA2) or remained unchanged (IFNB1) at EOT (Figure 5B). Furthermore, the relative contribution of IFNA2 and IFNB1 to total measured IFN expression increased by EOT (Figure 5C). IFNA1 expression was not detected in any liver sample before or after treatment. Although IFNA2 gene expression increased in the liver over the course of treatment, IFN-α2 protein was undetectable in serum at all time points tested (Supplemental Table 3). In liver, mRNA expression of receptors for IFN-γ (IFNGR2) and IFN-λ (IFNLR1) was downregulated, while expression of other IFN receptors was not affected (Figure 5D). In the patient who relapsed, there was a similar pattern of decreased hepatic type II and III IFN expression during therapy; however, expression of IFNA2 was not detected at EOT (Supplemental Figure 4). The correlation of hepatic type III, rather than type I, IFN expression with ISG expression in chronic HCV was consistent with previous findings (26–28), but ours is the first report based on longitudinal hepatic assessment in patients treated with an IFN-free DAA regimen.
Figure 5. Altered balance of IFN expression in liver during treatment of patients achieving SVR.

(A) Total IFN expression decreased during treatment in all SVR patients (n = 7). qRT-PCR for individual IFNs (shown in B) was performed using 5–15 ng RNA per reaction with technical duplicates. The sum of relative gene expression values of all measured IFNs at each time point is shown for each patient who achieved SVR. (B) qRT-PCR for individual IFNs was performed using 5–15 ng RNA per reaction with technical duplicates. Shown are individual measurements with group median and interquartile range for the 7 patients with paired liver biopsies who achieved SVR. P values were determined by Wilcoxon matched-pairs signed rank test. The functional allele for IFNL4 (IFNL4-ΔG, which produces the IFN-λ4 protein; ref. 31) was shown only for the 5 patients who carried this allele. LLOD, lower limit of detection. (C) Relative ratio of individual IFNs within the total pool of measured IFNs. Median and interquartile range is shown for patients achieving SVR (n = 7). P values compare EOT vs. pretreatment (Wilcoxon signed-rank test). (D) On-treatment changes in IFN receptor mRNA expression. Data and analysis are as in B.
To explore a potential mechanism of IFNA2 induction, we performed immunohistochemistry analysis for plasmacytoid DCs (pDCs) in paired biopsies from 12 patients (n = 9 [SVR]; 3 [relapse]). While a human tonsil positive control had readily detectable pDCs, we did not detect any pDCs in the 12 paired pretreatment and EOT liver biopsies tested (data not shown). This negative finding might be due to the low relative numbers of these cells in hepatic tissue in the setting of small sampling size afforded by core needle biopsies.
Although we observed a rapid decline in ISG expression in PBMCs (Figure 4A), peripheral IFN gene expression did not change during treatment, regardless of treatment outcome, and expression of IFN-λ genes was not detected in PBMCs (Figure 6A and data not shown). There was a significant change in PBMC gene expression of IFN receptor genes during treatment (Figure 6B).
Figure 6. Treatment-induced changes in expression of IFNs and IFN receptors in PBMCs.

Total RNA from PBMCs of 22 patients (12 SVR, black; 10 relapse, red) was analyzed for gene expression of IFNs (A) and IFN receptors (B) by qRT-PCR using 2 ng RNA per assay with technical duplicates. Group median and interquartile range is shown. A linear mixed-effects model was used to determine P for change in gene expression over the course of treatment, with significant P values displayed. Gene expression of IFNL1, IFNL2, IFNL3, and IFNL4-ΔG was not detected using equal amounts of RNA (not shown).
Together, these results demonstrated that reduction of intrahepatic expression of type II and III IFNs and their receptors paralleled the decrease in hepatic and blood ISG expression during HCV suppression by SOF/RBV. The selective increase in hepatic IFNA2 expression in patients achieving SVR suggested that reactivation of hepatic type I IFN signaling at EOT could be important for achieving favorable treatment outcome.
To assess this possibility, we performed microarray mRNA expression profiling in unpaired liver biopsies obtained at EOT from 17 patients who achieved SVR and 8 who relapsed (Supplemental Table 5), all of whom received EOT liver biopsies prior to relapse. Ranking of the top 1% of differentially expressed genes (Supplemental Table 6) identified IFN signaling as a top pathway distinguishing SVR from relapse, with lower expression of an IFN gene signature at EOT in patients who relapsed (Figure 7A). Of the 88 differentially expressed genes that met filtering criteria (Supplemental Figure 5), 41 were identified as ISGs (18). All IFN types (I–III) were predicted to have lower activity in patients who relapsed (Figure 7B). Gene expression analysis of the 46 ISGs tested in the paired liver biopsies (Figure 1A) showed that in EOT liver biopsies, expression of OAS1, RIGI, and USP18 was lower in patients who subsequently relapsed (Figure 7C). While expression of most individual genes did not differ significantly by outcome, combined analysis suggested lower overall gene expression of this set of ISGs in patients who relapsed (Figure 7D). Relative expression of IFNs or their receptors in EOT liver biopsies did not differ by treatment outcome (Figure 8, A–D), although this analysis was limited by the absence of pretreatment liver biopsies, precluding assessment of longitudinal changes.
Figure 7. Patients who relapse have lower expression of an IFN gene signature at EOT in liver.

(A) Top IPA canonical pathways differing between SVR and relapse patients from EOT liver biopsy microarray mRNA expression data. Ratios represent genes among the top 1% of genes differentially expressed by outcome assigned to specific pathways, relative to the total number of genes in the respective pathway. Genes with lower expression at EOT in relapse vs. SVR patients are denoted by bold font. (B) IPA upstream analysis of predicted activation state of proteins annotated as cytokines in EOT liver biopsies, comparing SVR and relapse patients. Negative activation z scores predict lower activation at EOT in patients who relapsed. Scores of >2 were considered significant. (C) qRT-PCR validation of select ISGs. qRT-PCR analysis was performed using 2.5–5 ng RNA per reaction. Expression of IRF9 and OAS1 was measured in technical duplicates (n = 16 [SVR; black]; 8 [relapse; red]), whereas other ISGs were tested as single replicates due to sample limitations (n = 12 [SVR; black]; 8 [relapse; red]). Statistical analysis was by Mann-Whitney test. Shown are individual measurements with group median and interquartile range. (D) qRT-PCR expression analysis of 46 ISGs in EOT liver biopsies from patients with SVR or relapse on SOF/RBV treatment. Median relative expression for individual ISGs was determined for the 20 patients with sufficient RNA available for analysis (n = 12 [SVR]; 8 [relapse]). For each ISG, the percent of patients in each outcome group with expression above the group median is plotted.
Figure 8. No difference in IFN or receptor expression between by outcome in unpaired EOT liver biopsies.

(A) Sum of total expression of IFNs, including IFNA2, IFNB1, IFNG, IFNL3, and IFNL4-ΔG. (B) Expression of individual IFN genes. (C) Relative expression of individual IFN genes as a percentage of total IFN expression. (D) Expression of IFN receptors. Data are from SVR (n = 16; black) and relapse (n = 8; red) patients in unpaired EOT liver biopsies. All assays were performed with technical duplicates using 2.5–5 ng RNA per reaction, with the exception of IFNA2 (25 ng). Shown are individual measurements with group median and interquartile range. Expression of IFNL1 and IFNL2 was not detected in any sample. No P values met criteria for significance (P < 0.05, Mann-Whitney test).
We genotyped all samples (n = 33) for 3 genetic variants associated with HCV clearance: the intronic rs12979860-C/T (29, 30) and exonic rs368234815-TT/ΔG (31, 32) of IFNL4 and the 3′ untranslated region (UTR) rs4803217-T/G of IFNL3 (Supplemental Tables 1 and 5 and ref. 33). The majority of patients enrolled in the study were African-Americans, who are less likely to have the favorable genotypes CC, TT/TT, and GG at the rs12979860, rs368234815, and rs4803217 loci, respectively. Only 2 of 8 paired biopsies and 4 of 25 unpaired EOT liver biopsies were from patients with the favorable IFNL4 rs368234815-TT/TT genotype, the strongest marker associated with HCV clearance in African-Americans (31, 34). Due to high linkage disequilibrium between these markers, the genotypes for the 2 other markers were correlated. The small number of samples with favorable genotype at each marker did not allow us to further explore changes in gene expression in relation to IFNL3 and IFNL4 genotypes.
Discussion
Our present results demonstrate for the first time that HCV clearance achieved during IFN-free treatment with the DAA regimen of SOF/RBV is accompanied by hepatic downregulation of type II and III IFNs, their receptors, and ISGs. Downregulation of ISGs was associated with on-treatment viral suppression and occurred regardless of treatment outcome, since all patients achieved virologic suppression on therapy. However, patients who achieved SVR unexpectedly had higher intrahepatic expression of ISGs at EOT compared with patients who relapsed. The increase in expression of IFNA2, but not other types of IFNs, observed in paired liver biopsies from patients who achieved SVR suggested that the higher EOT ISG expression could be driven by an enhanced type I IFN response. By inference, these data suggest a model whereby aberrant expression of hepatic ISGs triggered by HCV and/or type II/III IFNs decreases early during treatment. After prolonged viral suppression by SOF/RBV, higher activation of ISG expression at EOT may be induced by type I IFNs, and this in turn could promote elimination of residual virus. Data from paired biopsies were available from only 1 patient who relapsed, and showed no induction of IFNA2, consistent with this model.
As is recognized in other conditions characterized by prolonged immune stimulation, resolution of infection and inflammation may be followed by a period of relative immune suppression (35). Conceivably, HCV patients able to reestablish IFN homeostasis by EOT with SOF/RBV may be more likely to achieve an SVR, whereas patients who fail to restore homeostasis may be more prone to viral relapse. This could account for the higher rate of relapse on SOF/RBV observed in previous null responders to IFN-α/RBV therapy (7), who typically have an elevated endogenous IFN response prior to treatment, and thus may have a higher barrier to restoration of immune and IFN homeostasis. Hence, the ability to mount an endogenous, intrahepatic type I IFN response could be important for achieving SVR, even with IFN-α–free DAA regimens.
In this clinical trial in which patients were randomized to weight-based or low-dose RBV, treatment with low-dose RBV was not associated with an increased risk of treatment relapse in a bivariable model of treatment outcome, a conclusion limited by the small sample size of the clinical study (8). Because RBV is known to sensitize the host to the effect of IFN and reduce hepatic transaminases (17, 36–38), it is possible that the IFN signature described herein could be partially attributed to interindividual differences in responsiveness to RBV. To explore this possibility, our present findings should be assessed in future IFN- and RBV-free DAA clinical trials.
Intrahepatic upregulation of inhibitors of IFN-α signaling, such as USP18, has previously been demonstrated in chronic HCV patients, while type III IFN signaling is not affected by USP18 protein (11, 22, 39, 40). Chronic upregulation of such inhibitors could explain, at least in part, the lack of clinical response to injectable IFN-α formulations observed in some patients, in particular those with high endogenous hepatic expression of ISGs. In the present study, we observed downregulation of USP18 during treatment (Figure 2A), which may allow restoration of sensitivity to endogenous IFN-α. Interpretation of the higher USP18 expression observed in EOT liver biopsies of patients achieving SVR is complicated, as USP18 is involved in inhibition of IFN-α signaling, but is also an ISG induced by active IFN signaling (39). Relative differences in host sensitivity to endogenous IFNs after DAA-induced viral suppression could explain the association of ISG expression with treatment outcome observed here.
Recently, the interdependence and counterregulation of different families of IFNs has been described in the setting of HCV infection. For example, IFN-α produced by pDCs was necessary for expression of IFN-γ from NK cells upon coculture of PBMCs with JFH-1/Huh7.5 cells, a process that was augmented by the presence of monocytes (41). NK-derived IFN-γ contributed to enhanced ISG expression and DC maturation in this coculture model (41). In the same coculture model, BDCA+ myeloid DCs in PBMCs were found to be the major producers of type III IFNs through a TLR-3–dependent mechanism (42). Understanding cell type–specific IFN production in the liver in the setting of DAA therapy is an important area for future research, and use of longitudinally collected liver tissue affords an opportunity to assess dynamic changes in cell populations over time (14). In the present study, we were unable to detect the presence of pDCs as a potential source of IFNA2 induction during treatment by immunohistochemistry in pretreatment or EOT liver. Analysis of cell type–specific IFN production in situ was limited by tissue availability, but this analysis should be considered in subsequent DAA clinical trials as an invaluable tool for investigating the differential roles of diverse innate immune cells in the liver (43).
Genotypes of IFNL4 variants (intronic rs12979860-C/T, previously known as the IFNL3 [IL28B] marker and exonic rs368234815-TT/ΔG) and a 3′ UTR IFNL3 variant (rs4803217-T/G) have been shown to affect treatment outcome for HCV infection and correlate with intrahepatic ISG expression, transcript stability of IFNL3, and balance of the innate immune system in response to HCV infection (28, 31, 33, 44–46). Although we genotyped these markers in our subjects (Supplemental Tables 1 and 5), the incidence of favorable IFNL3 and IFNL4 genotypes was too infrequent to explore differential IFN regulation based on host genotype. In addition, while high viral load and advanced liver disease were both associated with treatment relapse in a bivariable model of the clinical trial results (8), we lacked the power in the subset of patients with liver biopsies to explore an effect of these variables on the biological correlates measured here. High viral load was common and advanced liver disease was uncommon in patients with EOT liver biopsies available (Supplemental Tables 1 and 5).
Our study is unique as the first attempt to comprehensively analyze expression changes in biological samples in an IFN-free clinical trial. The limited availability of paired liver biopsies, particularly in patients who relapsed, precluded more definitive longitudinal analysis of the correlation of tissue expression of IFNs with clinical outcome, and we acknowledge the role of chance in affecting the findings, given the small sample size. The strength of our conclusions is also tempered by the absence of a difference in IFNA2 expression in EOT liver biopsies between patients who achieved SVR versus those who relapsed; importantly, longitudinal changes could not be assessed in these patients without pretreatment tissue for comparison. Although the pattern of endogenous induction of IFNA2 (paired biopsies) and higher EOT ISG expression in patients achieving SVR (unpaired EOT biopsies) was consistently observed, we did not have enough samples to prove a lack of induction of IFNA2 in patients who experienced treatment relapse. Lack of data addressing mechanisms of relapse is an inherent problem of treatments with high therapeutic efficacy, such as that with SOF/RBV described herein, and biological samples — particularly liver biopsies — from relapsing patients are unlikely to be available in large numbers.
In conclusion, we showed that for the DAA regimen of SOF/RBV, changes in hepatic IFN expression profiles are associated with treatment outcome. The ability to restore intrahepatic type I IFN signaling may be biologically important for eradication of residual virus in the setting of prolonged HCV suppression with IFN-free therapy. Knowledge of expression variability in components of the IFN system could help determine the optimal duration of therapy for patients, especially in resource-limited settings, a concept that will be pursued in future studies. As HCV therapy evolves from IFN-based to DAA-only regimens, our study fosters a role for the host in determining favorable treatment outcome in chronically infected patients treated with SOF/RBV.
Methods
Clinical trial and sample analysis
60 treatment-naive, chronic HCV genotype-1 patients were treated with SOF (400 mg daily; provided by Gilead Sciences) and low (600 mg daily) or weight-based (<75 kg, 400 mg QAM with 600 mg QPM; >75 kg, 600 mg BID) RBV for 24 weeks, as previously described (8). 10 patients enrolled in part 1 of the trial received open label treatment with SOF and weight-based RBV. 50 patients enrolled in part 2 were randomized to receive SOF with either low or weight-based RBV. Plasma HCV RNA levels were measured using a qRT-PCR HCV assay (Abbott), with a lower limit of quantification (LLOQ) of 12 IU/ml and a lower limit of detection (LLOD) of 3 IU/ml. Histopathological assessments of liver biopsies were performed by a single pathologist in a nonblinded fashion at the time of biopsy and staged according to the Knodell histological activity index (Knodell-HAI) and ISHAK scoring systems (47, 48). Clinical samples chosen for biological analysis were selected based on availability and treatment outcome. AST and ALT were measured in the clinical laboratory of the National Institutes of Health Clinical Center.
RNA isolation and microarrays
Paired liver biopsies.
Core liver biopsies obtained transcutaneously with an 18-gauge needle were immediately placed in RNAlater (Ambion) and stored at –80°C until processing. Total RNA was extracted with the TissueRuptor and RNeasy kit (Qiagen), with an on-column elimination of residual DNA by DNAseI digestion (Qiagen). Microarray expression analysis was performed using 50–100 ng total RNA that was amplified, biotinylated, and hybridized to the Affymetrix Human Genome U-219 array, composed of over 530,000 probes representing more than 36,000 transcripts, using a GeneAtlas Personal Microarray System (Affymetrix). Gene expression results were normalized using the RMA method, which did not identify technical outliers. No probe sets with significant differences in expression levels were identified using a 2-way ANOVA with a multiple test correction by the false discovery rate (FDR) step-up method (49). An ascending rank order approach was implemented to equally factor in the individual ranks of fold change, probe design, signal intensity above background, and P values for each probe set. The top 1% of differentially expressed genes was used for IPA. A subset of genes with fold change > 1.2 and unadjusted P < 0.05 were chosen for representation by hierarchical clustering in a heatmap.
Pretreatment liver biopsies were obtained within 1.2 years prior to starting therapy, and EOT biopsies were obtained within 10 days of completing treatment, but prior to serologic relapse. ISG designation was determined using the Interferome v2.0 database (18) considering both human and mouse genes, with no filtering applied to search criteria. The molecule activator pathway of IPA was used for predicted activation state of cytokines, with values >2 considered significant (http://ingenuity.force.com/ipa/IPATutorials?id=kA2500000008ZBCCA2).
PBMCs.
Blood samples were collected prior to or at enrollment, within 6–11 days of treatment initiation, and at EOT. Samples from patients who achieved SVR were used for analysis (n = 4 [day 0]; 6 [day 6–11]; 12 [week 24]). PBMCs were obtained from whole blood by Ficoll gradient separation on the day of collection and resuspended in RPMI containing 10% FCS and 7.5% DMSO for storage in liquid nitrogen. Frozen cells were rescued at 37°C and washed twice with PBS plus 10% FCS, and total RNA was extracted using the Ambion PureLink RNA kit with on-column DNAseI treatment. Microarray analysis was performed as above using the Affymetrix U-219 array, and a rank order list of the top 1% of differentially expressed genes was generated as above.
EOT liver biopsies.
Liver biopsies collected in RNAlater as above were thawed and then homogenized in TRIzol (Thermofisher Scientific) using the FastPrep Green lysing matrix (MP Biomedicals), combined with 200 μl of 1-bromo-3-chloropropane (Sigma-Aldrich), and centrifuged at 4°C at 16,000 g for 15 minutes. The RNA-containing aqueous phase was passed through a Qiashredder column (Qiagen) at 21,000 g for 2 minutes. RNA was extracted using the RNeasy 96 kit according to manufacturer’s recommendations, including an on-column DNAseI treatment. RNA quality was determined on the 2100 Bioanalyzer (Agilent Technologies) using the Agilent RNA 6000 Pico kit. The average RNA Integrity Number (RIN) value for liver biopsy samples was 7.0, ranging from 4.1 to 8.3. DNA microarray probes were synthesized using the Ovation Pico WTA system (Nugen Inc.) and used for hybridization on the Affymetrix GeneChip Human Gene 2.0 ST array, covering 40,716 RefSeq transcripts. Gene expression results were normalized, and a gene list was created by a rank order approach as described above for the paired liver biopsies.
Accession number.
Microarray data were deposited in GEO (accession no. GSE51699).
qRT-PCR
Expression analysis of select genes was performed with predesigned or custom (for IFNL3 and IFNL4; ref. 31) TaqMan assays individually or assembled into custom-designed 96-well plates or 384-well microfluidic cards (Life Technologies). Total RNA isolated from liver or PBMCs was reverse transcribed using random primers with the High Capacity cDNA Reverse Transcriptase Kit (Life Technologies). 1–25 ng RNA was used for each qRT-PCR reaction. Taqman expression assays were run with technical duplicates except where indicated. Gene expression was determined as Ct based on 40 PCR cycles. For statistical analysis, undetectable expression was assigned a minimal detectable level with Ct of 40. Expression of GAPDH was used as an endogenous control, with GAPDH Ct values for all samples being distributed between 20 and 25, without clear differences between groups of samples. Relative expression of targets normalized by GAPDH expression (ΔCt) was calculated as CtGAPDH – Cttarget, with conversion and display relative to GAPDH expression by 2ΔCt. ΔΔCt values, used to calculate changes in expression between samples or groups of samples, were calculated as ΔCtsample A – ΔCtsample B, then converted to fold change by 2–ΔΔCt. Relative expression of individual IFNs in liver and blood was also calculated as a percent of the sum of total measured IFNs ([2ΔCt of each individual IFN/ total 2ΔCt of all measured IFNs] × 100). Expression reactions in 96-well plates were run on a 7500 Real-Time PCR System (IFNs and receptors). 384-well microfluidic cards were run on a 7900HT Fast Real-Time PCR System (Life Technologies).
mRNA ISH
Liver biopsy specimens were processed into formalin-fixed, paraffin-embedded (FFPE) cores per NIH Clinical Center protocols. Paired pretreatment and EOT cores from 4 patients were chosen for analysis: 2 achieved SVR, 1 relapsed after EOT liver biopsy, and 1 had an EOT liver biopsy at the time of relapse (biopsy and relapse were both 4 weeks after EOT after having undetectable HCV RNA 2 weeks after EOT). RNA ISH for IFI44 mRNA was performed manually using the RNAscope 2.0 FFPE Reagent Kit (Advanced Cell Diagnostics) (50). The RNAscope probe was designed specific to the sequence regions spanning nucleotides 187–1,170 of IFI44 mRNA (NM_006417). Briefly, 5-μm tissue sections were pretreated with heat and protease prior to hybridization with the target oligonucleotide probes. Pretreatment was modified from standard conditions to increase boiling time from 15 to 30 minutes (50). Preamplifier, amplifier, and HRP-labeled oligonucleotides were then hybridized sequentially, followed by chromogenic precipitate development with DAB. Each sample was quality controlled for RNA integrity with an RNAscope probe for cyclophilin B (PPIB) RNA (positive control) and for nonspecific background with a probe for bacterial dapB RNA (negative control). Specific RNA staining signal was identified as brown, punctate dots. Samples were counterstained with Gill’s hematoxylin, and brightfield images were acquired with a Leica SCN400 digital slide scanner using a ×40 objective.
Immunohistochemistry
Immunohistochemistry for BDCA2 (also known as CD303) to identify pDCs was performed with anti-BDCA2 mouse monoclonal antibody (clone 10E6.1; Millipore) in paired liver biopsies from 12 patients (n = 9 [SVR]; 3 [relapse]). FFPE slides were deparaffinized and subjected to antigen retrieval with a pressure cooker in citrate buffer (pH 9). Antibody was diluted 1:100, then applied for 2 hours at room temperature, and chromagen detection was mediated with Dako Envision+ and DAB for 10 minutes. A positive control of human tonsil was performed concurrently.
Genotyping
Genotyping of IFNL4 variants rs12979860-T/C and rs368234815-TT/ΔG (originally designated as ss469415590) was performed using genomic DNA samples and custom-designed TaqMan assays as previously described (31, 32). rs4803217-T/G was genotyped with a custom-designed TaqMan assay: forward primer, CTGTGTGTCTGACCCTTCCG; reverse primer, TCCTGGAGGTGAGTTGGATTTAC; probe for allele T, CAATAAATTAAGACAAGTGGCTA (VIC, MGB); probe for allele G, ATAAATTAAGCCAAGTGGCTA (FAM, MGB). The assay was validated by Sanger sequencing in HapMap samples. All markers were genotyped using the same experimental conditions as described previously (31).
Viral kinetic modeling
Longitudinal serum viral loads were obtained and analyzed in a viral kinetic model as described previously (8). Correlations between serum IP-10 protein level and PBMC MX1 gene expression with viral kinetic parameters (loss of infected cells and drug efficacy in blocking viral production) were determined.
Cytokine and chemokine ELISA
Whole blood was allowed to clot in SST tubes for at least 60 minutes prior to centrifugation at 1,700 g for 15 minutes. Serum was collected under sterile conditions and aliquoted prior to freezing at –80°C. After thawing, serum was centrifuged at 1,450 g for 10 minutes, and the supernatant was used for ELISA. Quantitation was performed with multiplex (Chemokine 9-plex, Proinflammatory 9-plex) or single-plex (IFN-α2a, TIMP-1, and TGF-β1) assays (MesoScale Discovery). Plate-to-plate variability of pooled samples was used for normalization across experiments. All samples were run as technical duplicates, and averages were used for data analysis.
Statistics
Mann-Whitney or Wilcoxon matched-pairs signed rank test was used for 2-group comparisons, and a linear mixed-effects model was used for multigroup longitudinal comparisons with fixed effects for group (SVR versus relapse), time, and group/time interaction. A significant group/time interaction indicated that the 2 groups had different patterns over time. A class variable was used for time to allow an arbitrary average time pattern within a group. In addition to fixed effects summarizing average group responses over time, the model accommodated random, patient-specific deviations in intercept and time effects. Correlations were assessed by nonparametric Spearman rank correlation. Spotfire S+ 8.2 (TIBCO) and Prism 6.0 software (GraphPad) were used for statistical analysis and data presentation. P values less than 0.05 were considered significant.
Study approval
All patients provided written or oral informed consent approved by the NIAID/NIH Institutional Review Board prior to inclusion in the study.
Supplementary Material
Acknowledgments
We thank Michael Proschan, Jing Qin, and Jeff Skinner for statistical assistance. We thank Gilead Sciences for provision of sofosbuvir. This project has been funded in part with federal funds from the intramural programs of the National Institute of Allergy and Infectious Diseases, the National Cancer Institute (contract no. HHSN261200800001E), the Division of Cancer Epidemiology and Genetics of the National Cancer Institute, and the National Institutes of Health Clinical Center. The study was also supported in part by the German Research Foundation (DFG) by the clinical research unit KFO 129.
Footnotes
Conflict of interest: John McHutchison is an employee of Gilead Sciences, manufacturer of sofosbuvir.
Reference information:J Clin Invest. 2014;124(8):3352–3363. doi:10.1172/JCI75938.
References
- 1.Liang TJ, Ghany MG. Current and future therapies for hepatitis C virus infection. N Engl J Med. 2013;368(20):1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Casey LC, Lee WM. Hepatitis C virus therapy update 2013. Curr Opin Gastroenterol. 2013;29(3):243–249. doi: 10.1097/MOG.0b013e32835ff972. [DOI] [PubMed] [Google Scholar]
- 3.Shah N, Pierce T, Kowdley KV. Review of direct-acting antiviral agents for the treatment of chronic hepatitis C. Expert Opin Investig Drugs. 2013;22(9):1107–1121. doi: 10.1517/13543784.2013.806482. [DOI] [PubMed] [Google Scholar]
- 4.Kowdley KV, et al. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet. 2013;381(9883):2100–2107. doi: 10.1016/S0140-6736(13)60247-0. [DOI] [PubMed] [Google Scholar]
- 5.Jacobson IM, et al. Sofosbuvir for hepatitis C genotype 2 or 3 in patients without treatment options. N Engl J Med. 2013;368(20):1867–1877. doi: 10.1056/NEJMoa1214854. [DOI] [PubMed] [Google Scholar]
- 6.Lawitz E, et al. Sofosbuvir for previously untreated chronic hepatitis C infection. N Engl J Med. 2013;368(20):1878–1887. doi: 10.1056/NEJMoa1214853. [DOI] [PubMed] [Google Scholar]
- 7.Gane EJ, et al. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. Engl J Med. 2013;368(1):34–44. doi: 10.1056/NEJMoa1208953. [DOI] [PubMed] [Google Scholar]
- 8.Osinusi A, et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: a randomized clinical trial. JAMA. 2013;310(8):804–811. doi: 10.1001/jama.2013.109309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rehermann B. Pathogenesis of chronic viral hepatitis: differential roles of T cells and NK cells. Nat Med. 2013;19(7):859–868. doi: 10.1038/nm.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosen HR. Emerging concepts in immunity to hepatitis C virus infection. J Clin Invest. 2013;123(10):4121–4130. doi: 10.1172/JCI67714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heim MH. 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat Rev Immunol. 2013;13(7):535–542. doi: 10.1038/nri3463. [DOI] [PubMed] [Google Scholar]
- 12.Donnelly RP, Dickensheets H, O’Brien TR. Interferon-lambda and therapy for chronic hepatitis C virus infection. Trends Immunol. 2011;32(9):443–450. doi: 10.1016/j.it.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wieland S, et al. Simultaneous detection of hepatitis C virus interferon stimulated gene expression in infected human liver. Hepatology. 2014;59(6):2121–2130. doi: 10.1002/hep.26770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lau DT, et al. Innate immune tolerance and the role of kupffer cells in differential responses to interferon therapy among patients with HCV genotype 1 infection. Gastroenterology. 2013;144(2):402–413. doi: 10.1053/j.gastro.2012.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McGilvray I, et al. Hepatic cell-type specific gene expression better predicts HCV treatment outcome than IL28B genotype. Gastroenterology. 2012;142(5):1122–1131. doi: 10.1053/j.gastro.2012.01.028. [DOI] [PubMed] [Google Scholar]
- 16.Sarasin-Filipowicz M, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci U S A. 2008;105(19):7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feld JJ, et al. Hepatic gene expression during treatment with peginterferon and ribavirin: Identifying molecular pathways for treatment response. Hepatology. 2007;46(5):1548–1563. doi: 10.1002/hep.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rusinova I, et al. Interferome v2.0: an updated database of annotated interferon-regulated genes. Nucleic Acids Res. 2013;41(Database issue):D1040–D1046. doi: 10.1093/nar/gks1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131(6):1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 20.Wilkins C, et al. IFITM1 is a tight junction protein that inhibits hepatitis C virus entry. Hepatology. 2013;57(2):461–469. doi: 10.1002/hep.26066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Metz P, et al. Identification of type I and type II interferon-induced effectors controlling hepatitis C virus replication. Hepatology. 2012;56(6):2082–2093. doi: 10.1002/hep.25908. [DOI] [PubMed] [Google Scholar]
- 22.Dill MT, et al. Interferon-γ-stimulated genes, but not USP18, are expressed in livers of patients with acute hepatitis C. Gastroenterology. 2012;143(3):777–786. doi: 10.1053/j.gastro.2012.05.044. [DOI] [PubMed] [Google Scholar]
- 23.Groom JR, Luster AD. CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol Cell Biol. 2011;89(2):207–215. doi: 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diamond DL, et al. Proteome and computational analyses reveal new insights into the mechanisms of hepatitis C virus-mediated liver disease posttransplantation. Hepatology. 2012;56(1):28–38. doi: 10.1002/hep.25649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wynn TA, Ramalingam TR. Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat Med. 2012;18(7):1028–1040. doi: 10.1038/nm.2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thomas E, et al. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology. 2012;142(4):978–988. doi: 10.1053/j.gastro.2011.12.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Diegelmann J, et al. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS One. 2010;5(12):e15200. doi: 10.1371/journal.pone.0015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Honda M, et al. Hepatic interferon-stimulated genes are differentially regulated in the liver of chronic hepatitis C patients with different interleukin-28B genotypes. Hepatology. 2014;59(3):828–838. doi: 10.1002/hep.26788. [DOI] [PubMed] [Google Scholar]
- 29.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 30.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prokunina-Olsson L, et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat Genet. 2013;45(2):164–171. doi: 10.1038/ng.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meissner EG, Bon D, Prokunina-Olsson L, Tang W, Masur H, O’Brien TR, Herrmann E, Kottilil S, Osinusi A. IFNL4-ΔG genotype is associated with slower viral clearance in hepatitis C, genotype-1 patients treated with sofosbuvir and ribavirin. J Infect Dis. 2014;209(11):1700–1704. doi: 10.1093/infdis/jit827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McFarland AP, et al. et al. The favorable IFNL3 genotype escapes mRNA decay mediated by AU-rich elements and hepatitis C virus-induced microRNAs. Nat Immunol. 2014;15(1):72–79. doi: 10.1038/ni.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aka PV, et al. Association of the IFNL4-ΔG allele with impaired spontaneous clearance of hepatitis C virus. J Infect Dis. 2014;209(3):350–354. doi: 10.1093/infdis/jit433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. The resolution of inflammation. Nat Rev Immunol. 2013;13(1):59–66. doi: 10.1038/nri3362. [DOI] [PubMed] [Google Scholar]
- 36.Testoni B, Levrero M, Durantel D. Mechanism of action of ribavirin in anti-HCV regimens: new insights for an age-old question? Gut. 2014;63(1):3–4. doi: 10.1136/gutjnl-2013-304528. [DOI] [PubMed] [Google Scholar]
- 37.Feld JJ, et al. Ribavirin improves early responses to peginterferon through improved interferon signaling. Gastroenterology. 2010;139(1):154–162. doi: 10.1053/j.gastro.2010.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevenson NJ, Murphy AG, Bourke NM, Keogh CA, Hegarty JE, O’Farrelly C. Ribavirin enhances IFN-α signalling and MxA expression: a novel immune modulation mechanism during treatment of HCV. PLoS One. 2011;6(11):e27866. doi: 10.1371/journal.pone.0027866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Makowska Z, Duong FH, Trincucci G, Tough DF, Heim MH. Interferon-β and interferon-λ signaling is not affected by interferon-induced refractoriness to interferon-α in vivo. Hepatology. 2011;53(4):1154–1163. doi: 10.1002/hep.24189. [DOI] [PubMed] [Google Scholar]
- 40.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol. 2014;14(1):36–49. doi: 10.1038/nri3581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S, Saha B, Kodys K, Szabo G. IFN-γ production by human natural killer cells in response to HCV-infected hepatoma cells is dependent on accessory cells. J Hepatol. 2013;59(3):442–449. doi: 10.1016/j.jhep.2013.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang S, Kodys K, Li K, Szabo G. Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology. 2013;144(2):414–425. doi: 10.1053/j.gastro.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jenne CN, Kubes P. Immune surveillance by the liver. Nat Immunol. 2013;14(10):996–1006. doi: 10.1038/ni.2691. [DOI] [PubMed] [Google Scholar]
- 44.Sheahan T, et al. Interferon λ alleles predict innate antiviral immune responses and hepatitis C virus permissiveness. Cell Host Microbe. 2014;15(2):190–202. doi: 10.1016/j.chom.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Amanzada A, Kopp W, Spengler U, Ramadori G, Mihm S. Interferon-λ4 (IFNL4) transcript expression in human liver tissue samples. PLoS One. 2013;8(12):e84026. doi: 10.1371/journal.pone.0084026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holmes JA, Desmond PV, Thompson AJ. Does IL28B genotyping still have a role in the era of direct-acting antiviral therapy for chronic hepatitis C infection? J Viral Hepat. 2012;19(10):677–684. doi: 10.1111/jvh.12003. [DOI] [PubMed] [Google Scholar]
- 47.Ishak K, et al. Histological grading and staging of chronic hepatitis. J Hepatol. 1995;22(6):696–699. doi: 10.1016/0168-8278(95)80226-6. [DOI] [PubMed] [Google Scholar]
- 48.Knodell RG, et al. Formulation and application of a numerical scoring system for assessing histological activity in asymptomatic chronic active hepatitis. Hepatology. 1981;1(5):431–435. doi: 10.1002/hep.1840010511. [DOI] [PubMed] [Google Scholar]
- 49.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 50.Wang F, et al. RNAscope: a novel in situ RNA analysis platform for formalin-fixed, paraffin-embedded tissues. J Mol Diagn. 2012;14(1):22–29. doi: 10.1016/j.jmoldx.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.