

Abstract
miRNAs are important regulators of biological processes in many tissues, including the differentiation and function of brown and white adipocytes. The endoribonuclease dicer is a major component of the miRNA-processing pathway, and in adipose tissue, levels of dicer have been shown to decrease with age, increase with caloric restriction, and influence stress resistance. Here, we demonstrated that mice with a fat-specific KO of dicer develop a form of lipodystrophy that is characterized by loss of intra-abdominal and subcutaneous white fat, severe insulin resistance, and enlargement and “whitening” of interscapular brown fat. Additionally, KO of dicer in cultured brown preadipocytes promoted a white adipocyte–like phenotype and reduced expression of several miRNAs. Brown preadipocyte whitening was partially reversed by expression of miR-365, a miRNA known to promote brown fat differentiation; however, introduction of other miRNAs, including miR-346 and miR-362, also contributed to reversal of the loss of the dicer phenotype. Interestingly, fat samples from patients with HIV-related lipodystrophy exhibited a substantial downregulation of dicer mRNA expression. Together, these findings indicate the importance of miRNA processing in white and brown adipose tissue determination and provide a potential link between this process and HIV-related lipodystrophy.
Introduction
Adipose tissue is a heterogeneous tissue. White adipose tissue (WAT) is the major site of energy storage and is mainly located in subcutaneous and intra-abdominal locations, whereas brown adipose tissue (BAT) is involved in energy utilization through the expression of uncoupling protein-1 (UCP1) (1–5). In rodents, the largest BAT depot is localized in the interscapular region, whereas in humans, BAT depots are localized to the neck and the supraclavicular and mediastinal regions (6). In addition, there is a pool of inducible brown adipocytes mixed in with WAT depots that is sometimes referred to as beige or brite fat (7–10).
Alterations in fat mass and distribution can have a major impact on whole-body metabolism. Both obesity (increased WAT) and lipodystrophy (abnormal fat accumulation) lead to increased risk of type 2 diabetes and cardiovascular disease — 2 major causes of mortality and morbidity worldwide (11–13). Different WAT depots play different roles in these metabolic defects. Accumulation of visceral, i.e., intra-abdominal, WAT is associated with the metabolic syndrome, whereas accumulation of subcutaneous WAT presents little risk and may even offer some protection against metabolic and cardiovascular complications (14). Recent studies have indicated that adult humans have active BAT (1–3), and studies in rodents have suggested that this constitutive BAT and the inducible beige/brite fat may be potential targets for promoting weight loss and improving glucose metabolism (15–18).
miRNAs play important roles in many biological processes (19, 20) and have been proposed to play a major role in the differentiation and function of both white and brown fat (21). The miR-193b-365 cluster of miRNAs and miR-155 participate in brown adipogenesis (22, 23), while miR-143 and miR-103 have been implicated in white adipocyte differentiation and insulin resistance (24–26). Likewise, miR-133 acts to repress Prdm16 in common adipocyte/muscle precursor cells to favor the myogenic over the brown adipogenic program (27, 28), while miR-196a, which is expressed primarily in WAT progenitor cells in response to cold or adrenergic stimulation, can suppress the white fat gene HoxC8 post-transcriptionally to favor brown adipogenesis over white adipogenesis (29).
Recently, we have shown that many miRNAs are markedly downregulated in the WAT of mice as they age (30). These changes are due to a decrease in expression of the miRNA-processing enzyme dicer and can be reversed by caloric restriction, a condition that is known to prolong lifespan. They can also be mimicked in preadipocytes in culture by exposure to oxidative and UV stress. A decline in dicer and miRNA processing with age is also observed in cultured human preadipocytes and in C. elegans, where it is linked to an increased susceptibility of the organism to stress. Aging and caloric restriction also impact adipocyte differentiation and function and through this and other mechanisms affect the risk of development of metabolic diseases (31, 32).
In the present study, we show that mice with a tissue-specific KO of dicer in adipose tissue develop a form of lipodystrophy characterized by decreased WAT mass and an increase and “whitening” of the interscapular BAT mass. These phenotypes are accompanied by insulin resistance, adipose tissue inflammation, dyslipidemia, and other systemic features that resemble the human HIV–associated lipodystrophy syndrome. Consistent with this observation, dicer mRNA expression is reduced in the adipose tissue of HIV patients. Taken together, our data indicate an essential role for adipose tissue dicer and miRNA processing in control of brown and white fat determination and whole-body metabolism, and this may contribute to the pathogenesis of HIV-related lipodystrophy.
Results
Fat-specific dicer-KO mice exhibit abnormal fat accumulation.
To explore the role of dicer and miRNAs in adipose tissue in vivo, we created mice with conditional inactivation of the dicer allele in adipose tissue. To this end, we bred mice carrying the dicer floxed allele with mice carrying the aP2-Cre transgene (33), however, this resulted in a high degree of early postnatal lethality, and those mice that did survive the first week of life were runted and died by 1 month of age. The low yield and poor health of these aP2-Cre–induced dicer-KO mice is consistent with the previous report by Mudhasani et al. (34) and is almost certainly due to the early, off-target expression of the aP2-Cre transgene, i.e., expression in tissues other than adipose, as was recently reported (35). Indeed, this lethality could be overcome by creating a fat-directed dicer KO using the Cre recombinase gene driven by the adiponectin promoter (Adicer-KO). Different from the aP2-Cre dicerfl/fl mice, the Adicer-KO mice were born at normal Mendelian ratios, had no gross abnormalities, and were viable up to more than 1 year of age. We performed quantitative RT-PCR (RT-qPCR) in tissues from 3-month-old mice, which revealed reductions of dicer mRNA by 61%, 70%, and 81% in subcutaneous WAT, perigonadal WAT, and BAT, respectively. On tissue fractionation, this decrease was entirely due to decreases in dicer in the adipocyte fraction, with no change in dicer levels in the stromovascular cells (30). There was also no reduction in dicer levels in other nonadipose tissues, including muscle and liver.
On a chow diet, Adicer-KO mice had body weight (Figure 1A) and length (Supplemental Figure 1, A and B; supplemental material available online with this article; doi:10.1172/JCI73468DS1) similar to those of Lox controls over the first 8 months of life; however, this was accompanied by a significant change in body composition. Dual-energy x-ray absorptiometry (DEXA) at 8 months of age revealed a 71% decrease in fat mass and a 28% increase in lean mass of the Adicer-KO mice (Figure 1B and Table 1). Despite the decrease in overall fat mass, MRI indicated enlargement of the interscapular fat depot (Figure 1C and Supplemental Figure 1C). These changes in fat mass and distribution were confirmed at sacrifice, which revealed 41% and 73% decreases in weight of the subcutaneous (flank) and intra-abdominal fat pads, but a greater than 2-fold increase in the mass of the interscapular brown fat pad (Figure 1, D and E, and Supplemental Figure 1D).
Figure 1. Partial lipodystrophy in fat-specific Adicer-KO mice.

(A) Ten-week-old Adicer-KO and Lox control mice were maintained on a chow diet for 5 months, and body weight was monitored (n = 5–12 animals per group). (B) Eight-month-old mice on a chow diet were subjected to a DEXA scan, and fat and lean mass was calculated (n = 5–6 mice per group). (C) MRI was performed in 4-month-old mice fed a chow diet. Images show a sagittal plane representative of 1 mouse of 2 per genotype. Fat is indicated by the red demarcation. (D–H) Ten-week-old Adicer-KO and Lox control mice were put on an HFD or maintained on a chow diet for 5 months prior to sacrifice (n = 6–12 animals per group). (D) Images of representative fat tissues. (E) Tissue weight at time of sacrifice. (F) Body weight was assessed weekly. (G) Dorsal view of mice showing a “buffalo hump” after 5 months on an HFD. (H) H&E staining of fat samples. Representative images (original magnification, ×200) of at least 3 animals per group. *P < 0.05. Values are the mean ± SEM. SQ (flank), subcutaneous WAT of the flank region; SQ (SS), subcutaneous WAT of the suprascapular region; BAT, interscapular BAT. Epididymal, mesenteric, and perirenal represent different depots of intra-abdominal WAT.
Table 1.
Body composition of Adicer-KO mice
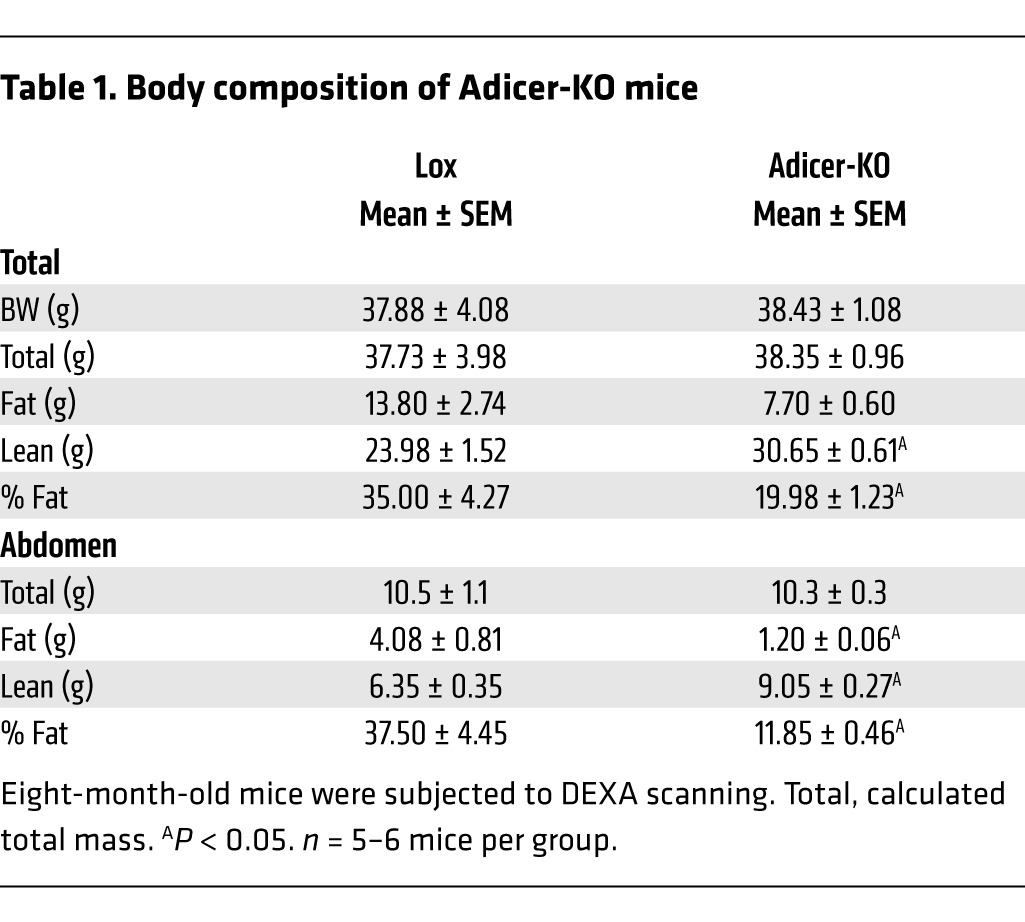
When placed on a 60% high-fat diet (HFD), the alterations in fat distribution and body composition in Adicer-KO mice were further exaggerated. Thus, Adicer-KO mice did not gain as much weight as their littermate controls (Figure 1F) and demonstrated a clearly abnormal fat distribution, with relatively little accumulation of subcutaneous and intra-abdominal fat (Figure 1, D and E), but clear enlargement of the interscapular and suprascapular fat pads (Figure 1, D, E, and G). After 5 months on an HFD, the control mice showed 2- to 3-fold increases in the weight of all fat pads when compared with those of mice of the same age on a chow diet (Figure 1E). In contrast, the weights of white fat depots (epididymal, flank, mesenteric, and perirenal) in Adicer-KO mice did not change or even decreased after an HFD, with a reduction of 62% to 94% in comparison with those of controls on an HFD. On the other hand, there was an increase in fat accumulation in the interscapular region of Adicer-KO mice on an HFD, with a 4.5-fold increase in the interscapular brown fat pad when compared with that of controls on the same diet (Figure 1E). Interestingly, the suprascapular white adipose depot adjacent to the interscapular BAT, which is between the classical white and brown depots, showed no significant increase or decrease. We observed no differences in other organ weights (Figure 1E). Liver histology was similar between Adicer-KO and Lox mice (Supplemental Figure 1E). We found that hepatic (Supplemental Figure 1F) and muscle (Supplemental Figure 1G) triglyceride content was only slightly higher in Adicer-KO than in Lox mice (neither reached statistical significance), suggesting that BAT is the primary site for fat accumulation in Adicer-KO mice.
Another striking phenotype of the brown fat depot of Adicer-KO mice was its color. While control interscapular adipose tissue had a classical brown-red color, the same fat pads in Adicer-KO mice were cream colored (Figure 1D). Histological analysis of this tissue revealed adipocytes with mixed white-brown characteristics with both unilocular and multilocular fat droplets (Figure 1H). WAT depots in Adicer-KO mice showed large, unilocular adipocytes similar to those of controls, but in the epididymal depot, there was increased fibrosis and inflammation-related crown-like structures (Figure 1H, right panels, and Supplemental Figure 1E). Consistent with this, we observed increased macrophage infiltration in the epididymal WAT of Adicer-KO mice, as evidenced by Cd68 expression (Supplemental Figure 2A) and F4/80 immunostaining (Supplemental Figure 2B).
Adicer-KO mice have signs of the human HIV–related partial lipodystrophy syndrome.
Abnormal fat accumulation and increased inflammation in adipose tissue are usually associated with insulin resistance and are also observed in some patients with lipodystrophy (36–39). Indeed, following insulin stimulation in vivo, while control mice showed robust AKT and ERK phosphorylation in white and brown fat depots, insulin-stimulated AKT and ERK phosphorylation was severely impaired in the Adicer-KO mice (Figure 2A and Supplemental Figure 3A). In addition, we found that genes involved in glucose (Glut4) or lipid (Srebp1c, Fas, Atgl, Hsl, and Lpl) metabolism and the genes encoding leptin and adiponectin were downregulated by 59% to 83% in the white fat of chow-fed Adicer-KO mice and were even more downregulated in the HFD-fed animals (Figure 2B and Supplemental Figure 3B). By contrast, in interscapular BAT, with the exception of Glut4, these mRNAs did not show significant differences (Supplemental Figure 3C). Consistent with decreased insulin signaling and the altered gene expression pattern, basal and insulin-induced lipogenesis in WAT and BAT was decreased by greater than 75% and by greater than 90%, respectively (Figure 2C and Supplemental Figure 3D). Isoproterenol-induced lipolysis was also impaired in the WAT of Adicer-KO mice, with a 63% reduction (Figure 2D and Supplemental Figure 3E).
Figure 2. Adicer-KO mice are insulin resistant.

(A) Four-month-old mice fed a chow diet were i.v. injected with either saline or insulin (10 U), and AKT and ERK phosphorylation were assessed by Western blotting in the flank subcutaneous adipose tissue. (B, E, and F) Ten-week-old Adicer-KO and Lox control mice were put on an HFD or maintained on a chow diet for 5 months prior to sacrifice (6–12 animals per group). (B) Gene expression analysis in the flank subcutaneous adipose tissue. (C) Lipogenesis and (D) lipolysis were assessed in isolated adipocytes of the subcutaneous adipose tissue of the flank region of 4-month-old mice on a chow diet (n = 8 per group). *P < 0.05. (E) An i.p. ITT was performed using 1 U insulin/kg body weight injection and expressed as a percentage of basal blood glucose level. (F) An i.p. glucose tolerance test using 2 g glucose/kg body weight. *P < 0.05 versus Lox; #P < 0.05 versus chow diet. (G–I) Hyperinsulinemic-euglycemic clamps were performed in conscious 5-month-old Adicer-KO and Lox control mice (6 animals per group). (G) Glucose infusion rate represents the amount of exogenous glucose required to maintain blood glucose levels during a hyperinsulinemic state. (H) Hepatic glucose production was calculated based on the endogenous production of glucose during basal or hyperinsulinemic (clamp) states. (I) Glucose uptake by indicated tissues during hyperinsulinemic state. Values are the mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001.
White fat secretes adipokines that control whole-body metabolism and disease susceptibility (40, 41). In Adicer-KO mice, we observed a 57%–74% decrease in circulating adiponectin (P < 0.05) and an approximately 50% reduction in serum leptin in mice on chow and HFDs, although the latter did not quite reach statistical significance. Despite the increase in inflammation in adipose tissue, serum IL-6 levels did not change, but other adipokines associated with cardiovascular disease risk, such as plasminogen activator inhibitor-1 (PAI-1) and monocyte chemotactic protein-1 (MCP-1), were increased in Adicer-KO mice, especially in mice on an HFD (Table 2). Adicer-KO mice also showed signs of dyslipidemia with high serum triglycerides and FFAs and a 4.5-fold increase in circulating insulin levels (Table 2). Consistent with hyperinsulinemia, Adicer-KO mice on a chow diet exhibited increased basal and glucose-stimulated insulin secretion (Supplemental Figure 3F) and insulin resistance by insulin tolerance testing (ITT) (Figure 2E and Supplemental Figure 3G). In these mice, the increased insulin balanced the insulin resistance, so that glucose tolerance in the chow-fed animals was normal (Figure 2F).
Table 2.
Serum profile of Adicer-KO mice
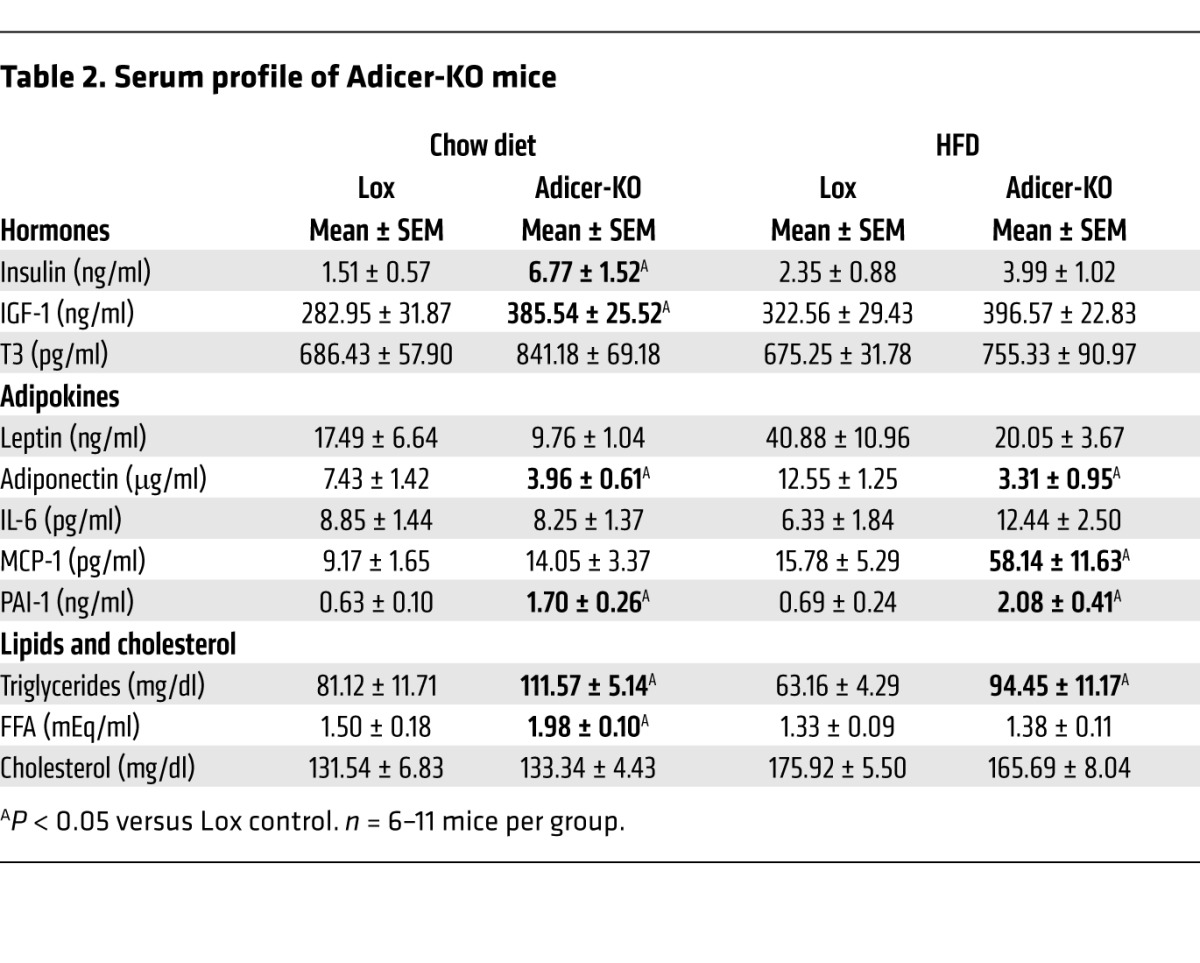
To explore the causes of insulin resistance in Adicer-KO mice, these mice were subjected to hyperinsulinemic-euglycemic clamps in the conscious state. Consistent with the ITT data, we found that the glucose infusion and whole-body glucose turnover rates during the clamp were decreased in Adicer-KO mice (Figure 2G and Supplemental Table 1). This was associated with an inability of insulin to suppress hepatic glucose production (Figure 2H) and marked decreases in glucose uptake in BAT (72% reduction) and WAT (39% reduction) in Adicer-KO mice as compared with that in controls (Figure 2I). We also observed a trend toward decreased glucose uptake in skeletal muscle and a significant reduction in heart glucose uptake in Adicer-KO mice (Supplemental Table 1). This insulin resistance was further exacerbated by an HFD in Adicer-KO mice (Figure 2E), resulting in clear hyperglycemia in the fed state (0 time point of the ITT in Supplemental Figure 3G). The Adicer-KO mice also had mild elevations of fasting glucose, but were somewhat more glucose tolerant than were the controls (Figure 2F), reflecting the combined effects of reduced fat, increased lean mass, and increased insulin levels. Together, these findings indicate that dicer KO in adipose tissue results in a form of partial lipodystrophy that resembles features of the human HIV–related partial lipodystrophy syndrome. These features include redistribution of fat depots, insulin resistance, adipose tissue inflammation, dyslipidemia, and markers of cardiovascular disease risk.
Increased resting energy expenditure despite whitening of adipose tissue in Adicer-KO mice.
In metabolic cage assessment of female Adicer-KO mice on a chow diet, we found that food intake and activity were similar (Supplemental Figure 4A and B), but there was a mild increase in O2 consumption (Figure 3A), heat production (Supplemental Figure 4C), and respiratory exchange ratio (Supplemental Figure 4D) between Adicer-KO mice and controls fed a chow diet, indicating an increase in resting energy expenditure. Male mice also had an increase in resting energy expenditure, but this was accompanied by a mild decrease in spontaneous activity (Supplemental Figure 4, E and F).
Figure 3. Bioenergetic profile and brown fat whitening in Adicer-KO mice.

(A) Six-month-old female mice were subjected to comprehensive lab animal monitoring system (CLAMS) analysis, in which O2 consumption was measured (n = 4 mice per group). Mice were fasted during the last 24 hours of the experiment. Values were normalized by lean mass. (B) UCP1 immunohistochemistry (IHC) of interscapular BAT from 4-month-old mice at room temperature (23°C). UCP1 staining (brown, cytoplasmic); hematoxylin counterstaining (blue, nuclear). Images (original magnification, ×400) are representative of at least 3 mice per group. (C and d) Four-month-old mice were subjected to 4-day chronic exposure to 6°C (n = 5 mice per group). (C) UCP1 expression in interscapular BAT from mice at room temperature (RT) or after chronic cold exposure (Cold) as assessed by Western blotting. β-Tubulin was used as the loading control. (D) Markers of brown and white adipocytes were assessed in the interscapular BAT by qPCR. (E and F) Five-month-old mice were injected with CL-316,243, and (E) O2 consumption and (F) rectal temperature were measured (n = 4–8 mice per group). (G and H) Rectal temperature of 4-month-old or 2-month-old mice was measured during exposure to 6°C (G, n = 5 mice per group; H, n = 5–6 mice per group). (I) Shivering time of 2-month-old mice during a 1-minute period after 36 hours of cold (6°C) exposure (n = 5–6 mice per group). (J) UCP1 IHC of inguinal WAT from 4-month-old mice subjected to 4 days of cold exposure (n = 5 mice per group). UCP1 staining with hematoxylin counterstaining. Representative images (original magnification, ×400) of at least 3 mice per group. *P < 0.05; ***P < 0.001. Values are the mean ± SEM.
Histological studies and measurement of brown and white fat markers in the interscapular BAT were consistent with whitening of this depot in the Adicer-KO mice. Thus, while the “brown” adipocytes showed a markedly increased size of their lipid droplets, often resembling white adipocytes, all of the cells in the interscapular fat pad stained positively for UCP1, although the level appeared reduced (Figure 3B). This decrease in UCP1 expression was confirmed by Western blotting (Figure 3C) and qPCR (Figure 3D) and was accompanied by an increased expression of WAT markers such as leptin and HoxC9 (Figure 3D). Other brown fat markers, such as Elovl3 and Cidea mRNAs, were also reduced in the BAT of Adicer-KO mice, while levels of Dio2, Prdm16, and Pgc1a mRNAs were unchanged (Figure 3D). Despite the low basal levels of Ucp1 and Elovl3 in the Adicer-KO mice, both the KO and control mice showed similar fold increases in these mRNAs in response to chronic cold exposure (Figure 3D), and this was confirmed at the protein level for UCP1 (Figure 3C). Consistent with whitening of the brown fat, the increases in O2 consumption (Figure 3E) and temperature (Figure 3F) following injection with the β3-adrenergic receptor agonist CL-316,243 were blunted in Adicer-KO mice. However, Adicer-KO mice were able to maintain their body temperature during cold exposure as well as, or even better than, controls (Figure 3G), indicating that these mice develop a compensatory response to overcome their brown fat deficiency. Indeed, even in young (6- to 8-week-old) mice, when no differences in white fat distribution or lipodystrophy were observed (Supplemental Table 2 and Supplemental Figure 5A), Adicer-KO mice were more thermotolerant than or as thermotolerant as Lox controls during acute (Figure 3H) and chronic cold exposure (Supplemental Figure 5B), despite having decreased BAT mass and decreased Ucp1 and Elovl3 mRNA expression in both BAT and flank subcutaneous WAT after the chronic cold exposure (Supplemental Figure 5, C–E). This appeared to be due to a marked increase in shivering in Adicer-KO mice during cold exposure (Figure 3I). Thus, Adicer-KO mice compensate their impaired brown adipose function with increased shivering, a high energy–demanding process that contributes to increased resting energy expenditure and helps maintain body temperature. Also, despite these alterations in energy expenditure and the evolving features of metabolic syndrome, at both 6 and 16 months of age, the Adicer-KO mice were able to withstand a challenge with the oxidative toxin paraquat as well as or slightly better than did controls (Supplemental Figure 5, F and G), consistent with a link between adipose tissue miRNA processing and longevity and stress resistance (30).
As noted above, in addition to the interscapular preformed BAT in mice, there is a pool of inducible brown adipocytes, referred to as beige or brite adipocytes, mixed in with white adipose depots (7, 9, 10, 42, 43). In both the Adicer-KO and control mice, we observed that subcutaneous WAT had very low levels of UCP1, which increased in response to cold exposure, but in the Adicer-KO mice, this increase in UCP1, i.e., recruitment of beige adipocytes, was impaired (Figure 3J and Supplemental Figure 5E). Thus, normal miRNA processing is required for the development and function of interscapular BAT as well as for browning of WAT.
Downregulation of dicer in adulthood leads to fat redistribution and insulin resistance.
To determine whether the effects of dicer deficiency in adipocytes were exclusively a developmental phenomenon or could be acquired during adulthood, we created a second model in which dicer was knocked down in fat using a tamoxifen-induced aP2-Cre transgene (iaP2dicer-KO). Although the inducible aP2-Cre does not produce as effective a recombination as the adiponectin-Cre (35), this avoided the early lethality of the aP2-Cre dicerfl/fl mice and allowed us to use tamoxifen induction in the mice from 3 months until 8 months of age. In this model, we found an approximately 40% decrease in dicer mRNA in the fat depots of the dorsocervical region (i.e., interscapular brown and suprascapular white), but no significant changes in dicer expression levels in other subcutaneous or intra-abdominal depots (Figure 4A). Although there was no change in the size of most fat pads or nonadipose tissues in this mouse, consistent with the Adicer-KO phenotype, the iaP2dicer-KO mice exhibited approximately 70% enlargement of interscapular BAT (Figure 4, B–D). This was accompanied by a parallel decrease in UCP1 levels in the interscapular BAT (Figure 4E) and systemic insulin resistance as measured by a fall in glucose during i.p. ITT (Figure 4F). These mice also had increased lean mass (Supplemental Table 3) and showed a trend toward hyperinsulinemia and hypertriglyceridemia (Supplemental Table 4), also mimicking features of the Adicer-KO mice. Thus, downregulation of dicer in adulthood can lead to abnormal fat distribution with a partial loss of brown adipocyte identity, increased lean mass, and development of some features of the metabolic syndrome.
Figure 4. Characterization of inducible fat–specific Adicer-KO (iaP2dicer-KO) mice.

Mice carrying the Cre recombinase gene driven by a tamoxifen-induced aP2 promoter or their littermate Lox controls were given 3 mg tamoxifen weekly from 3 to 8 months of age (n = 6–7 mice per group). (A) Expression of dicer mRNA as assessed by qPCR. (B–D) Tissue weights normalized to body weight for (B) interscapular BAT, (C) WAT depots, and (D) nonadipose tissues. (E) UCP1 expression in interscapular BAT. β-Tubulin was used to control for protein concentration. The β-tubulin immunoblot was performed with samples run on a separate gel. (F) ITT using i.p. injection of 1 U insulin/kg body weight. *P < 0.05. Values are the mean ± SEM. SQ fat, subcutaneous WAT of the flank region; SS fat, subcutaneous WAT of the suprascapular region; BAT, interscapular BAT; PG fat, perigonadal (epididymal) WAT; RP fat, retroperitoneal WAT; Quad, quadriceps skeletal muscle.
Whitening of dicer-KO brown adipocytes in vitro.
To explore the mechanisms underlying the change in brown adipocyte identity in the dicer-KO mice and to determine whether the effect was cell autonomous, preadipocytes were isolated from the interscapular adipose tissue of mice carrying the floxed dicer allele (dicerfl/fl), immortalized using SV-40-large T antigen, and subjected to gene recombination ex vivo by infection with Cre- or GFP-bearing adenoviruses. This resulted in a greater than 95% decrease in dicer expression at the mRNA level (Supplemental Figure 6A). In vitro, these dicer-KO preadipocytes differentiated into adipocytes as well as the control cells did, as evidenced by the similar levels of the pan-adipocyte markers Pparg, Cebpa, and aP2 (Figure 5A) and by the Oil Red O staining for lipid accumulation (Figure 5, B and C). However, there appeared to be fewer dicer-KO cells overall after differentiation, and this was confirmed by a 28% decrease of DNA content in the plate (Supplemental Figure 6B). This is consistent with a role for dicer and mRNA processing in cell replication and stress response (30). More importantly, after differentiation, dicer-KO brown preadipocytes expressed lower levels of the brown adipocyte–specific genes Ucp1 and Elovl3 and higher levels of the white adipocyte marker leptin (Figure 5A). As in vivo, dicer-KO adipocytes were responsive to β-adrenergic and cyclic AMP (cAMP) stimulation, but even under these conditions the level of Ucp1 mRNA remained at about 60% of that of the control cells (Figure 5D). When compared with the Lox control cells, these cells also had an approximately 25% reduction in the level of uncoupled respiration rate, as assessed in a Seahorse Flux Analyzer (Figure 5E), but showed no alterations in basal respiration, maximal respiratory capacity, or nonmitochondrial respiration (Supplemental Figure 7). Thus, brown preadipocytes with ablation of dicer in vitro show a partial shift in identity from brown to white, i.e., the effect was cell autonomous, but as in vivo, these cells preserved some characteristics of normal brown adipocytes, including their response to upregulate UCP1 following adrenergic stimulation.
Figure 5. Brown to white identity switch upon dicer KO in adipocytes.

Immortalized interscapular brown preadipocytes of dicerfl/fl mice were transduced with adenoviruses harboring GFP (Lox) or Cre recombinase (dicer KO, denoted in the figure as KO), and 4 days later (day 0, D0), cells were differentiated in vitro into adipocytes (D8). (A) Adipocyte markers were assessed by qPCR (n = 3 per group). (B and C) Lipid accumulation during differentiation was measured using Oil Red O staining on day 8. Visualization at (B) lower (original magnification, ×1 scanning the bottom of the plate with an optical scanner) and (C) higher magnifications (original magnification, ×200 using an optical microscope). Representative images of 3 independent cell lines per group. (D) Differentiated cells were treated for 4 hours with 500 μM cAMP or 10 μM isoproterenol. Ucp1 mRNA expression was measured by qPCR, and fold induction versus the nonstimulated control was calculated (n = 3 per group). (E) Uncoupled respiration was measured using a Seahorse Bioscience XF24-3 Extracellular Flux Analyzer (n = 5 per group). Values represent the amount of O2 consumed by cells when incubated with the ATP synthase inhibitor oligomycin A. (F and G) Three days after adenoviral transduction, Lox and dicer-KO preadipocytes were transfected with miRNA mimics or a scramble, nonsilencing control (NS). Adipocyte differentiation started on the following day (D0), and (F) Ucp1 and (G) Cebpb mRNAs were measured by qPCR when cells were fully differentiated (D8) (n = 3 per group). In F and G, gene expression was normalized by aP2 mRNA levels to avoid small differences in adipocyte differentiation. *P < 0.05. Values are the mean ± SEM.
To determine which miRNAs might play a role in the dicer-KO phenotype, we examined the mRNAs upregulated in dicer-KO preadipocytes (GEO GSE24683) and asked whether there were enriched miRNA targets among them. Using gene set enrichment analysis (GSEA), we found targets of the let-7 family, miR-346, miR-365, miR-154, miR-455, the miR-25 and miR-26 families, and miR-362 to be significantly upregulated in dicer-KO cells (Supplemental Table 5). As expected, we found that all these miRNAs were significantly reduced in the dicer-KO preadipocytes compared with those in the Lox control preadipocytes (Supplemental Figure 8A), with the exception of miR-25, which was not above the level of detection in preadipocytes. While reintroduction of miR-154, miR-346, and miR-362 had no significant effect on Ucp1 mRNA expression in the KO cells, expression of miR-365 was able to increase levels of Ucp1 mRNA by 20-fold in the KO cells, reversing the low levels, and also increased levels of Ucp1 in the Lox cells by almost 3-fold (Figure 5F). This occurred with no effect on adipogenesis as measured by the ability of the cells to accumulate lipids (Supplemental Figure 8B). The differential expression of HoxC9 mRNA, a WAT marker, between KO and control cells also tended to diminish with transfection of the miR-365 mimic, but was not restored to normal levels (Supplemental Figure 8C), and other markers of brown and white adipocyte identity, like Elovl3 and leptin, were not modified by the reexpression of miR-365 (Supplemental Figure 8C). Reexpression of miR-346 and miR-362 in the dicer-KO cells, on the other hand, partially reversed the decrease in Cebpb, a key switch in the brown adipogenic gene expression program (ref. 29 and Figure 5G). These results are consistent with the notion that the cell-autonomous phenotypes of dicer-KO cells are likely a consequence of the regulation of multiple miRNAs and their target genes, rather than of a single miRNA.
HIV-related lipodystrophy is associated with dicer downregulation in human adipose tissue.
Many of the phenotypes observed in mice lacking dicer in adipose tissue, including loss of white fat, appearance of increased interscapular fat, insulin resistance, dyslipidemia, increased resting energy expenditure, and increased markers of cardiovascular disease risk, are often observed in humans with partial lipodystrophy, especially in patients with HIV-related lipodystrophy (36–39, 44–47). To determine whether the latter might be linked to dicer downregulation, we analyzed biopsies of subcutaneous fat depots from the dorsocervical and abdominal regions of 2 independent cohorts of controls and HIV patients undergoing antiretroviral treatment (Supplemental Tables 6 and 7). One cohort was from Boston and had biopsies of the dorsocervical region (48); the other was from Finland and had biopsies from subcutaneous abdominal fat (49). In the Boston cohort, dicer mRNA expression was reduced by over 75% in the dorsocervical adipose tissue of HIV patients compared with that in normal controls (Figure 6A). This was true whether subjects displayed more fat loss (subcutaneous lipoatrophy) or fat gain (visceral lipohypertrophy) (48). Indeed, in this cohort, none of the patients with HIV lipodystrophy had a dicer mRNA level as high as that of the lowest control. In the larger Finnish cohort, dicer mRNA expression was also significantly reduced in the abdominal subcutaneous fat of HIV patients, although the magnitude of decrease in this area was less than that in the dorsocervical region (Figure 6B). Again, this decrease was independent of whether the patients had systemic lipoatrophy. Thus, downregulation of dicer in fat is a feature of humans with HIV, and this downregulation is more severe in certain adipose depots, such as the dorsocervical region, in which fat distribution is often abnormal.
Figure 6. Expression of dicer mRNA in adipose tissue of humans with HIV.

(A) Expression of dicer mRNA in human dorsocervical subcutaneous adipose tissue of HIV-positive lipodystrophy patients and controls (Boston cohort: n = 6–14 per group). Symbols represent the size of the tissue in relation to body weight. (B) Expression of dicer mRNA in human abdominal subcutaneous adipose tissue of HIV-positive lipodystrophy patients and controls (Finnish cohort: n = 27–60 per group). Symbols represent the size of the tissue in relation to body weight. (C) Schematic model of factors that influence dicer expression in fat and its pathophysiological consequences. ***P < 0.001. Values are the mean ± SEM.
Discussion
Brown and white adipocyte identity is defined during development but can be modified by a myriad of complex events including genetic factors, epigenetic factors, hormonal factors, and other environmental challenges that might occur during adipocyte differentiation (50–52). miRNAs play a major role in maintaining cell identity by fine-tuning cell-specific transcriptional networks (19, 20) and have been shown to play important roles in both brown and white fat differentiation (22, 24–29). miRNAs in adipose tissue change in response to obesity, insulin resistance, or cold exposure (24, 29), and there is an even more global change in the miRNA profile in fat with aging due to age-dependent downregulation of dicer (30). The latter phenomenon is conserved in species from worms to humans and can be rescued by caloric restriction, suggesting it plays an important role in the regulation of longevity. Here, we created 2 mouse models to explore the role of the decline in dicer with age specifically in adipose tissue. We found that whether initiated during development or in adult animals, KO of dicer in fat in mice impaired adipocyte determination, resulting in abnormal fat distribution, whitening of constitutive and inducible brown adipocytes, increased lean mass, and features of the metabolic syndrome. The change in BAT can be reproduced in vitro, i.e., it is cell autonomous and is partly due to changes in levels of miRNAs such as miR-365, miR-346, and miR-362. Thus, downregulation of dicer and miRNAs in adipocytes results in partial loss of adipocyte identity, bringing forth an important mechanism of regulation of adipocyte function and whole-body metabolism.
With the recent discovery that adult humans have brown fat and the potential to use this tissue to promote weight loss, much effort has been put forth to define how brown adipocytes are formed and the exact nature of their relationship to white adipocytes. Considerable data have been accumulated, indicating that brown, beige, and white adipocytes originate from different cell lineages and require distinct adipogenic stimuli to be formed (7, 53). However, some recent studies suggest that the functional identity of adipocytes can also be set, at least in part, post-developmentally. Sanchez-Gurmaches and colleagues have shown that adipocytes from the same depot might derive from different developmental origins but still share a singular identity, while adipocytes from different depots might share the same precursor lineage but differ in terms of function and morphology (8). Likewise, Rosenwald and colleagues have demonstrated that white and beige adipocytes interconvert bidirectionally in the WAT of mice in response to different environmental cues (54). These results are consistent with the potential for transdifferentiation to occur in at least some fraction of the adipocyte pool (55). Our results suggest that this post-developmental switch may involve reprogramming by miRNAs, since in mice, KO of dicer in mature adipocytes using adiponectin or aP2 promoter–directed recombination (both of which are turned on in mid to late differentiation; ref. 56) resulted in a marked adipocyte identity switch.
Creation of fat-specific dicer-KO mice also illustrates some of the important caveats of tissue-selective recombination in fat, as previously reported by us and others (35, 57). Thus, in our first attempt to determine the role of dicer in adipose tissue, we created KO mice using aP2-Cre, and, as observed by Mudhasani et al. (34), these aP2-Cre dicerfl/fl mice die shortly after birth and are much smaller and more fragile than their littermate controls. This is almost certainly due to expression of the aP2-Cre transgene in nonadipose tissues, including in germline cells and endothelial cells of the heart (35), where it has been shown that dicer KO is lethal (58, 59). We therefore created and characterized 2 other mouse models of fat-specific dicer KO, which avoided the limitations brought by the aP2-Cre transgene. One model used the tamoxifen-inducible aP2-Cre transgene, which allowed study of the effects of dicer KO in adult mice, but was limited in its effectiveness of recombination; the other model used an adiponectin-Cre transgene, which we have previously shown is truly exclusive to fat in its distribution and highly effective in inducing recombination (35).
Both of these latter models exhibited a clear phenotype of abnormal fat distribution with features of the HIV partial lipodystrophy syndrome. This included a decrease in white fat in both the subcutaneous and intra-abdominal depots, enlargement of fat with characteristics that are intermediate between brown and white fat in the interscapular region, and signs of metabolic syndrome including insulin resistance, adipose tissue inflammation, dyslipidemia, increased resting energy expenditure, and increased markers of cardiovascular disease risk (36, 48). Interestingly, and consistent with this, dicer mRNA was significantly downregulated in adipose tissue of patients with HIV and HIV-related partial lipodystrophy, linking dicer expression in fat to at least 1 syndrome of abnormal fat distribution in humans.
While changes in glucose and lipid homeostasis are expected in lipodystrophy, there are other systemic consequences of dicer downregulation in adipose tissue, including increased lean body mass, which were unexpected. While the exact mechanism of the latter is unknown at present, muscular hypertrophy is observed in some patients with both generalized and partial lipodystrophy (60–62). KO of dicer in fat also changes sensitivity to oxidative stress at the whole-body level in interesting ways. In very young mice, dicer KO increases sensitivity to paraquat (30), whereas in the present study, we found no change or perhaps mild resistance to paraquat toxicity in middle-aged and older mice. The changing relation of paraquat sensitivity with aging may reflect the fact that in the control mice, dicer expression in fat decreases with age, making the difference between the control and KO mice less pronounced. Clearly, the exact mechanisms underlying these cell-nonautonomous effects of loss of dicer in fat will require further study.
Given the role of adipose tissue dicer in stress resistance and aging, it is important to consider how the physiological changes of our fat-specific dicer-KO models may be linked to the downregulation of dicer expression in adipose tissue that has been observed in mice with aging (30) and in humans with HIV (this study). We have shown that oxidative stress downregulates dicer and that dicer KO leads to premature senescence in preadipocytes in vitro (30). Increased oxidative stress and premature senescence in adipose tissue can also lead to accelerated aging and lipodystrophy (63, 64). We now find that dicer is downregulated in the adipose tissue of patients with HIV and HIV-related partial lipodystrophy, and these individuals may exhibit signs of premature aging, metabolic syndrome (diabetes and dyslipidemia), cardiovascular disease, and increased sensitivity to stress (38). Whether patients with other forms of congenital or acquired lipodystrophy also have alterations in dicer expression in fat remains to be determined.
In summary, our findings indicate that dicer and miRNA processing play a role in brown and white adipocyte determination. Our data also suggest that aging and metabolic diseases can act through downregulation of dicer in adipose tissue to shift fat determination toward insulin resistant, metabolically impaired white adipocytes, leading to features of the metabolic syndrome and, in some cases, a partial lipodystrophy syndrome (Figure 6C). Furthermore, our previous study indicates that caloric restriction can prevent the age-related decline in dicer expression and in miRNA processing in fat (30), and caloric restriction is known to reverse many features of the metabolic syndrome and improve longevity. These findings raise the possibility of using agents to increase dicer or specific miRNA expression in adipose tissue to treat features of the metabolic syndrome and some forms of the partial lipodystrophy syndrome.
Methods
Mice.
Adipose-specific dicer-KO mice were generated by breeding dicerfl/fl mice with mice carrying an adiponectin promoter Cre transgene (30) or Cre recombinase driven by a tamoxifen-induced aP2 promoter (65). For the iaP2dicer-KO, tamoxifen (Sigma-Aldrich) treatment (3 mg once per week by gavage) was started in 3-month-old mice and continued until the mice were 8 months of age. All mice were maintained on a normal chow diet (Mouse Diet 9F; PharmaServ). Males were used for this study unless otherwise indicated. For some experiments, 8-week-old mice were given a 60% HFD (D12492; Research Diets) for 20 weeks. At sacrifice, tissues were collected, snap-frozen in N2, and stored at –80°C. For insulin stimulation, mice were anesthetized with Avertin and injected i.v. with either saline or 10 U of insulin (Humalog; Lilly). After 5 minutes, mice were sacrificed, and tissues were collected. Paraquat was injected i.p. at 6.5 mg/kg body weight, and survival was monitored periodically.
MRI.
Mice were anesthetized using isoflurane/O2 (2.5/2.0%) 10 minutes prior to and during the experiments. MR images were acquired on an Aspect M2 1T tabletop MRI scanner (Aspect Imaging).
Histological analyzes.
Tissues were fixed in 10% formalin. Slides were stained with H&E or immunostained with UCP1 antibody (sc-6528; Santa Cruz Biotechnology Inc.) or F4/80 antibody (ab6640; Abcam) and counterstained as described (53).
Metabolism studies.
Glucose tolerance, insulin tolerance, and glucose-stimulated insulin secretion tests were performed as previously described (66). Hyperinsulinemic-euglycemic clamps were performed in conscious mice as reported elsewhere (67). Serum parameters were determined by the Joslin Diabetes Center’s DERC assay core. Triglyceride content was measured in liver and quadriceps muscle using the Triglyceride Quantification kit (Abcam) and normalized by protein levels as determined by the Bradford reagent. Fat and lean mass were measured by DEXA scanning. Activity, food intake, O2 consumption, and CO2 production were measured using the Oxymax OPTO-M3 system (Columbus Instruments). O2 uptake (VO2) levels were normalized by lean mass as assessed by DEXA scanning. For cold exposure, mice were housed in a cold room at 6°C, and the temperature was measured periodically using a RET-3 rectal probe (Physitemp). Shivering was measured during a period of 1 minute while the mouse was handled in the cold room. The assessment was subjective and based on the motion sensing of the operator while handling the animal. These assessments were always performed in triplicate by the same operator in a blinded fashion. For β3-adrenergic receptor stimulation in vivo, mice were injected i.p. with 1 mg/kg CL-316,243 (Sigma-Aldrich). Adipocyte isolation, lipogenesis, and lipolysis assays were performed as previously described (66, 68).
Western blotting and qPCR.
Cells or tissues were harvested and homogenized in radioimmunoprecipitation assay (RIPA) buffer containing protease and phosphatase inhibitors (Pierce Biotechnology). Lysates (10–30 μg) were subjected to SDS-polyacrylamide gel electrophoresis, transferred to a PVDF membrane (Thermo Fisher Scientific), and immunoblotted with the appropriate antibodies (66). miRNA expression was determined using the miRNome miRNA PCR Array (SABiosciences, QIAGEN), and RT-qPCR was performed as previously described (30). The nucleotide sequences of the primers are shown in Supplemental Table 8.
Cell culture and adipocyte differentiation.
Immortalized dicerfl/fl brown preadipocytes were transduced with an adenoviral Cre recombinase (30) and differentiation induced using a cocktail containing 20 nM insulin, 1 nM triiodothyronine, 0.5 mM isobutylmethylxanthine, 1 μM dexamethasone, and 0.125 mM indomethacin (66). Reintroduction of miRNA mimics in dicer-KO cells was performed according to a previously published protocol (30). For stimulation of Ucp1 mRNA, differentiated cells were incubated with 500 μM dibutyrul cAMP or 10 μM isoproterenol for 4 hours. Cellular respiration of preadipocytes was assessed using the Seahorse Bioscience XF24-3 Extracellular Flux Analyzer as previously described (22).
GSEA.
We used microarray expression data of dicer-KO and Lox immortalized preadipocytes available in the Gene Expression Omnibus (GEO) database (GEO GSE24683) (30). Enriched targets of miRNAs were sought among the genes upregulated in KO cells using the GSEA bioinformatics tool (Broad Institute).
Human studies.
Two cohorts of patients were studied: a Boston cohort and a Finnish cohort. In the Boston cohort, sampling of dorsocervical fat was performed from patients with HIV recruited from the Beth Israel Deaconess Medical Center, the Joslin Diabetes Center, and the Massachusetts General Hospital (all in Boston), as previously described (48). In the Finnish cohort, biopsies of abdominal subcutaneous fat were obtained from patients recruited from the HIV outpatient clinic of the Helsinki University Central Hospital in Helsinki, Finland, as previously described (49). The samples were immediately frozen in liquid nitrogen and stored at –80°C until analysis. RNA was extracted using an RNeasy Mini Kit (QIAGEN). cDNA was prepared as described above for the dorsocervical samples and using a SuperScript VILO cDNA synthesis kit (Life Technologies) for the abdominal samples. For the dorsocervical samples, dicer expression was assessed by TaqMan qPCR (Applied Biosystems) and normalized to FAM-TBP (Applied Biosystems) (30). For the abdominal samples, dicer expression was measured using SYBR Green qPCR and normalized to the geometric mean of TBP and 36B4.
Statistics.
Results are expressed as the mean ± SEM. For mouse studies, we used a 2-tailed Student’s t test for statistical analysis between 2 groups and ANOVA when comparing more than 2 groups. The human study groups were compared using Fisher’s exact test for categorical variables and an unpaired t test or a Mann-Whitney U test, as appropriate, for continuous variables. Statistical significance was set at P < 0.05.
Study approval.
Protocols for animal use were approved by the IACUCs of the Joslin Diabetes Center and Brandeis University and were in accordance with NIH guidelines. The human studies were approved by the IRBs of Beth Israel Deaconess Medical Center, Joslin Diabetes Center, Massachusetts General Hospital, and Helsinki University. All patients or their guardians provided written informed consent.
Supplementary Material
Acknowledgments
We thank M. Merkenschlager and H.M. Kronenberg for the dicerfl/fl mice, E. Rosen for the adiponectin-Cre mice, Pierre Chambon for the iaP2-Cre mice, and Lei Sun for reagents and helpful discussions. We thank the Longwood Small Animal Imaging Core Facility and the Joslin Histology, Media, and Physiology Core Facilities for help with experiments. This study was supported by NIH grants DK082659, DK033201, AI060354, and DK040561, as well as by grants from the Ellison Foundation, the Joslin Diabetes and Endocrinology Research Center cores (DK036836), the Mary K. Iacocca Professorship, the Academy of Finland, the Sigrid Juselius Foundation, and the Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP; 2010/52557-0). The clamp study was performed at the UMass Mouse Metabolic Phenotyping Center and supported by NIH grant U24-DK093000.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(8):3339–3351. doi:10.1172/JCI73468.
References
- 1.Cypess AM, et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360(15):1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.van Marken Lichtenbelt WD, et al. Cold-activated brown adipose tissue in healthy men. N Engl J Med. 2009;360(15):1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 3.Virtanen KA, et al. Functional brown adipose tissue in healthy adults. N Engl J Med. 2009;360(15):1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- 4.Frontini A, Cinti S. Distribution and development of brown adipocytes in the murine and human adipose organ. Cell Metab. 2010;11(4):253–256. doi: 10.1016/j.cmet.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Inokuma K, et al. Indispensable role of mitochondrial UCP1 for antiobesity effect of beta3-adrenergic stimulation. Am J Physiol Endocrinol Metab. 2006;290(5):E1014–E1021. doi: 10.1152/ajpendo.00105.2005. [DOI] [PubMed] [Google Scholar]
- 6.Cypess AM, et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat Med. 2013;19(5):635–639. doi: 10.1038/nm.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu J, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150(2):366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab. 2012;16(3):348–362. doi: 10.1016/j.cmet.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walden TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am J Physiol Endocrinol Metab. 2012;302(1):E19–E31. doi: 10.1152/ajpendo.00249.2011. [DOI] [PubMed] [Google Scholar]
- 10.Qiang L, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell. 2012;150(3):620–632. doi: 10.1016/j.cell.2012.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brookheart RT, Michel CI, Schaffer JE. As a matter of fat. Cell Metab. 2009;10(1):9–12. doi: 10.1016/j.cmet.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samuel VT, Shulman GI. Mechanisms for insulin resistance: common threads and missing links. Cell. 2012;148(5):852–871. doi: 10.1016/j.cell.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carobbio S, Rodriguez-Cuenca S, Vidal-Puig A. Origins of metabolic complications in obesity: ectopic fat accumulation. The importance of the qualitative aspect of lipotoxicity. Curr Opin Clin Nutr Metab Care. 2011;14(6):520–526. doi: 10.1097/MCO.0b013e32834ad966. [DOI] [PubMed] [Google Scholar]
- 14.Tran TT, Yamamoto Y, Gesta S, Kahn CR. Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 2008;7(5):410–420. doi: 10.1016/j.cmet.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whittle AJ, Lopez M, Vidal-Puig A. Using brown adipose tissue to treat obesity - the central issue. Trends Mol Med. 2011;17(8):405–411. doi: 10.1016/j.molmed.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Tseng YH, Cypess AM, Kahn CR. Cellular bioenergetics as a target for obesity therapy. Nat Rev Drug Discov. 2010;9(6):465–482. doi: 10.1038/nrd3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bostrom P, et al. A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481(7382):463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stanford KI, et al. Brown adipose tissue regulates glucose homeostasis and insulin sensitivity. J Clin Invest. 2013;123(1):215–223. doi: 10.1172/JCI62308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ebert MS, Sharp PA. Roles for microRNAs in conferring robustness to biological processes. Cell. 2012;149(3):515–524. doi: 10.1016/j.cell.2012.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christodoulou F, et al. Ancient animal microRNAs and the evolution of tissue identity. Nature. 2010;463(7284):1084–1088. doi: 10.1038/nature08744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trajkovski M, Lodish H. MicroRNA networks regulate development of brown adipocytes. Trends Endocrinol Metab. 2013;24(9):442–450. doi: 10.1016/j.tem.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun L, et al. Mir193b-365 is essential for brown fat differentiation. Nat Cell Biol. 2011;13(8):958–965. doi: 10.1038/ncb2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, et al. miR-155 regulates differentiation of brown and beige adipocytes via a bistable circuit. Nat Commun. 2013;4:1769. doi: 10.1038/ncomms2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie H, Lim B, Lodish HF. MicroRNAs induced during adipogenesis that accelerate fat cell development are downregulated in obesity. Diabetes. 2009;58(5):1050–1057. doi: 10.2337/db08-1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trajkovski M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474(7353):649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- 26.Jordan SD, et al. Obesity-induced overexpression of miRNA-143 inhibits insulin-stimulated AKT activation and impairs glucose metabolism. Nat Cell Biol. 2011;13(4):434–446. doi: 10.1038/ncb2211. [DOI] [PubMed] [Google Scholar]
- 27.Trajkovski M, Ahmed K, Esau CC, Stoffel M. MyomiR-133 regulates brown fat differentiation through Prdm16. Nat Cell Biol. 2012;14(12):1330–1335. doi: 10.1038/ncb2612. [DOI] [PubMed] [Google Scholar]
- 28.Yin H, et al. MicroRNA-133 controls brown adipose determination in skeletal muscle satellite cells by targeting Prdm16. Cell Metab. 2013;17(2):210–224. doi: 10.1016/j.cmet.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mori M, Nakagami H, Rodriguez-Araujo G, Nimura K, Kaneda Y. Essential role for miR-196a in brown adipogenesis of white fat progenitor cells. PLoS Biol. 2012;10(4):e1001314. doi: 10.1371/journal.pbio.1001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori MA, et al. Role of microRNA processing in adipose tissue in stress defense and longevity. Cell Metab. 2012;16(3):336–347. doi: 10.1016/j.cmet.2012.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das M, Gabriely I, Barzilai N. Caloric restriction, body fat and ageing in experimental models. Obes Rev. 2004;5(1):13–19. doi: 10.1111/j.1467-789X.2004.00115.x. [DOI] [PubMed] [Google Scholar]
- 32.Rogers NH, Landa A, Park S, Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. 2012;11(6):1074–1083. doi: 10.1111/acel.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carvalho E, et al. GLUT4 overexpression or deficiency in adipocytes of transgenic mice alters the composition of GLUT4 vesicles and the subcellular localization of GLUT4 and insulin-responsive aminopeptidase. J Biol Chem. 2004;279(20):21598–21605. doi: 10.1074/jbc.M312269200. [DOI] [PubMed] [Google Scholar]
- 34.Mudhasani R, Puri V, Hoover K, Czech MP, Imbalzano AN, Jones SN. Dicer is required for the formation of white but not brown adipose tissue. J Cell Physiol. 2011;226(5):1399–1406. doi: 10.1002/jcp.22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee KY, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62(3):864–874. doi: 10.2337/db12-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang-Doran I, Sleigh A, Rochford JJ, O’Rahilly S, Savage DB. Lipodystrophy: metabolic insights from a rare disorder. J Endocrinol. 2010;207(3):245–255. doi: 10.1677/JOE-10-0272. [DOI] [PubMed] [Google Scholar]
- 37.Tershakovec AM, Frank I, Rader D. HIV-related lipodystrophy and related factors. Atherosclerosis. 2004;174(1):1–10. doi: 10.1016/S0021-9150(03)00246-6. [DOI] [PubMed] [Google Scholar]
- 38.Caron-Debarle M, Lagathu C, Boccara F, Vigouroux C, Capeau J. HIV-associated lipodystrophy: from fat injury to premature aging. Trends Mol Med. 2010;16(5):218–229. doi: 10.1016/j.molmed.2010.03.002. [DOI] [PubMed] [Google Scholar]
- 39.Stanley TL, Grinspoon SK. Body composition and metabolic changes in HIV-infected patients. J Infect Dis. 2012;205(suppl 3):S383–S390. doi: 10.1093/infdis/jis205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ahima RS, Osei SY. Adipokines in obesity. Front Horm Res. 2008;36:182–197. doi: 10.1159/000115365. [DOI] [PubMed] [Google Scholar]
- 41.Mori MA, et al. A systems biology approach identifies inflammatory abnormalities between mouse strains prior to development of metabolic disease. Diabetes. 2010;59(11):2960–2971. doi: 10.2337/db10-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xue B, Coulter A, Rim JS, Koza RA, Kozak LP. Transcriptional synergy and the regulation of Ucp1 during brown adipocyte induction in white fat depots. Mol Cell Biol. 2005;25(18):8311–8322. doi: 10.1128/MCB.25.18.8311-8322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanchez-Gurmaches J, Hung CM, Sparks CA, Tang Y, Li H, Guertin DA. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab. 2012;16(3):348–362. doi: 10.1016/j.cmet.2012.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fitch KV, et al. Decreased respiratory quotient in relation to resting energy expenditure in HIV-infected and noninfected subjects. Metabolism. 2009;58(5):608–615. doi: 10.1016/j.metabol.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kazakos K, et al. Familial clustering strongly suggests that the phenotypic variation of the 8344 A>G lys mitochondrial tRNA mutation is encoded in cis. Ann Hum Genet. 2012;76(4):296–300. doi: 10.1111/j.1469-1809.2012.00711.x. [DOI] [PubMed] [Google Scholar]
- 46.Ampollini L, Carbognani P. Images in clinical medicine. Madelung’s disease. N Engl J Med. 2011;364(5):465. doi: 10.1056/NEJMicm1003531. [DOI] [PubMed] [Google Scholar]
- 47.Pivonello R, et al. Pathophysiology of diabetes mellitus in Cushing’s syndrome. Neuroendocrinology. 2010;92(suppl 1):77–81. doi: 10.1159/000314319. [DOI] [PubMed] [Google Scholar]
- 48.Torriani M, et al. Deiodinase 2 expression is increased in dorsocervical fat of patients with HIV-associated lipohypertrophy syndrome. J Clin Endocrinol Metab. 2012;97(4):E602–E607. doi: 10.1210/jc.2011-2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sevastianova K, et al. Comparison of dorsocervical with abdominal subcutaneous adipose tissue in patients with and without antiretroviral therapy-associated lipodystrophy. Diabetes. 2011;60(7):1894–1900. doi: 10.2337/db11-0075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131(2):242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 51.Seale P, et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454(7207):961–967. doi: 10.1038/nature07182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schulz TJ, et al. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature. 2013;495(7441):379–383. doi: 10.1038/nature11943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tseng YH, et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature. 2008;454(7207):1000–1004. doi: 10.1038/nature07221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Bi-directional interconversion of brite and white adipocytes. Nat Cell Biol. 2013;15(6):659–667. doi: 10.1038/ncb2740. [DOI] [PubMed] [Google Scholar]
- 55.Himms-Hagen J, Melnyk A, Zingaretti MC, Ceresi E, Barbatelli G, Cinti S. Multilocular fat cells in WAT of CL-316243-treated rats derive directly from white adipocytes. Am J Physiol Cell Physiol. 2000;279(3):C670–C681. doi: 10.1152/ajpcell.2000.279.3.C670. [DOI] [PubMed] [Google Scholar]
- 56.Park BH, Qiang L, Farmer SR. Phosphorylation of C/EBPbeta at a consensus extracellular signal-regulated kinase/glycogen synthase kinase 3 site is required for the induction of adiponectin gene expression during the differentiation of mouse fibroblasts into adipocytes. Mol Cell Biol. 2004;24(19):8671–8680. doi: 10.1128/MCB.24.19.8671-8680.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Martens K, Bottelbergs A, Baes M. Ectopic recombination in the central and peripheral nervous system by aP2/FABP4-Cre mice: implications for metabolism research. FEBS Lett. 2010;584(5):1054–1058. doi: 10.1016/j.febslet.2010.01.061. [DOI] [PubMed] [Google Scholar]
- 58.Chen JF, et al. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008;105(6):2111–2116. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bernstein E, et al. Dicer is essential for mouse development. Nat Genet. 2003;35(3):215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- 60.Senior B. Lipodystrophic muscular hypertrophy. Arch Dis Child. 1961;36:426–431. doi: 10.1136/adc.36.188.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wildermuth S, Spranger S, Spranger M, Raue F, Meinck HM. Kobberling-Dunnigan syndrome: a rare cause of generalized muscular hypertrophy. Muscle Nerve. 1996;19(7):843–847. doi: 10.1002/(SICI)1097-4598(199607)19:7<843::AID-MUS5>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 62.Garg A, Stray-Gundersen J, Parsons D, Bertocci LA. Skeletal muscle morphology and exercise response in congenital generalized lipodystrophy. Diabetes Care. 2000;23(10):1545–1550. doi: 10.2337/diacare.23.10.1545. [DOI] [PubMed] [Google Scholar]
- 63.Peinado JR, et al. Proteomic profiling of adipose tissue from Zmpste24–/– mice, a model of lipodystrophy and premature aging, reveals major changes in mitochondrial function and vimentin processing. Mol Cell Proteomics. 2011;10(11):M111 008094. doi: 10.1074/mcp.M111.008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baker DJ, et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011;479(7372):232–236. doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hensler M, et al. The inhibition of fat cell proliferation by n-3 fatty acids in dietary obese mice. Lipids Health Dis. 2011;10:128. doi: 10.1186/1476-511X-10-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boucher J, et al. Impaired thermogenesis and adipose tissue development in mice with fat-specific disruption of insulin and IGF-1 signalling. Nat Commun. 2012;3:902. doi: 10.1038/ncomms1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hong EG, et al. Cardiac expression of human type 2 iodothyronine deiodinase increases glucose metabolism and protects against doxorubicin-induced cardiac dysfunction in male mice. Endocrinology. 2013;154(10):3937–3946. doi: 10.1210/en.2012-2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Macotela Y, Boucher J, Tran TT, Kahn CR. Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes. 2009;58(4):803–812. doi: 10.2337/db08-1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.