

Abstract
Many plants possess specialized structures that are involved in the production and secretion of specific low molecular weight compounds and proteins. These structures are almost always localized on plant surfaces. Among them are nectaries or glandular trichomes. The secreted compounds are often employed in interactions with the biotic environment, for example as attractants for pollinators or deterrents against herbivores.
Glands that are unique in several aspects can be found in carnivorous plants. In so-called pitcher plants of the genus Nepenthes, bifunctional glands inside the pitfall-trap on the one hand secrete the digestive fluid, including all enzymes necessary for prey digestion, and on the other hand take-up the released nutrients. Thus, these glands represent an ideal, specialized tissue predestinated to study the underlying molecular, biochemical, and physiological mechanisms of protein secretion and nutrient uptake in plants. Moreover, generally the biosynthesis of secondary compounds produced by many plants equipped with glandular structures could be investigated directly in glands.
In order to work on such specialized structures, they need to be isolated efficiently, fast, metabolically active, and without contamination with other tissues. Therefore, a mechanical micropreparation technique was developed and applied for studies on Nepenthes digestion fluid. Here, a protocol is presented that was used to successfully prepare single bifunctional glands from Nepenthes traps, based on a mechanized microsampling platform. The glands could be isolated and directly used further for gene expression analysis by PCR techniques after preparation of RNA.
Keywords: Plant Biology, Issue 82, Plant, Plant Preparations, Plant Physiological Processes, Plant Pathology, micropreparation, mechanical dissection, glands, carnivory, Nepenthes, PCR, RNA
Introduction
Plants produce a wide huge variety of compounds with different industrial and pharmaceutical applications, ranging from small molecular weight molecules to polymers. Such compounds are frequently produced and secreted by highly specialized structures, many of which are localized at the surface of the plants such as glandular trichomes or nectaries, or internally such as idioblasts or latex and resin ducts, respectively. However, relatively little is known about the biology of these specialized secretory structures, although all types of those glands display many features that are indicative of active metabolism. In particular, glands of carnivorous plants such as Nepenthes can be seen as model systems in plant cell biology1.
Carnivorous plants are the object of several studies since 1875 when Charles Darwin's 'Insectivorous Plants' was published2. However, only in the last few years research on the molecular level has been done and still our knowledge is limited. For example, the protein composition of the digestion fluid in the pitcher plants of the genus Nepenthes is still not completely known. Only recently several hydrolytic proteins have been identified and a few of the corresponding genes as well3. In the pitfall-traps of Nepenthes (Figure 1), multicellular glands are localized at the inner bottom of the pitchers. These glands do both production and secretion of the digestive enzymes into the pitcher fluid and absorption of dissolved nutrients from the pitcher fluid4. Hence, these bifunctional glands represent a unique microtissue with a central position and function in the carnivory of Nepenthes that is worth to be studied in detail.
 Figure 1. Nepenthes alata pitcher. The lower part of the pitcher contains the digestive fluid and glands on the inside. Click here to view larger image.
Figure 1. Nepenthes alata pitcher. The lower part of the pitcher contains the digestive fluid and glands on the inside. Click here to view larger image.
In plant sciences, many experimental approaches rely on 'bulk material' because no defined tissue is investigated but entire organs. As a consequence, their homogenization and analysis can only provide results that are diluted and proportionately with respect to individual tissues and cells5. To solve this problem, micropreparation techniques, for example the so-called laser capture microdissection (LCM), were developed and successfully used to harvest specific plant tissues and analyze their individual contents6. However, LCM is often limited if unstable and/or fast degrading cell components are targeted such as RNA. In such a case, the need of previous tissue fixation that is time consuming and often accompanied by degradation of the biomolecules, is of disadvantage. Moreover, due to the high water content of many plant tissues and the strength of cell walls, LCM often failed to prepare straight from fresh tissue7. Any approach to investigate cellular components that are present in secretory glands from genes, mRNA, and proteins to secondary compounds and their biosynthesis needs alternative techniques for the particular preparation. Thus, specific techniques to isolate single cells or even multicellular glands that remain intact after preparation are necessary.
Here a method is presented, employing a mechanized microsampling platform that features a direct microscopic visualization. This technique is both rapid and efficient in the preparation of individual glands from the pitchers of Nepenthes species. These multicellular glands are bifunctional, i.e. they are involved in secretion as well as in uptake processes, they are sessile, and not exposed cell layers are impregnated to form an endodermis. Entire glands can be isolated without neighboring tissue and deposited in a well-defined manner into a PCR tube. This technique is applied to study specific genes as well as gene expression in the glands' tissues.
Protocol
1. Plant Material Preparation
All Nepenthes species were grown either in the greenhouses of the MPI for Chemical Ecology or the Botanical Garden in Jena, Germany.
For the preparation of secreting tissue harvest a pitcher and remove the upper one third including lid, peristome, and slippery zone. The pitcher can be used either freshly or frozen in liquid nitrogen and stored at -80 °C.
2. Microdissection of Pitcher Tissue
The microsampling platform consists of a stereomicroscope and a micromanipulator, which is directly attached to the microscope body and is operated by a 3D joystick. Here, an early prototype of aureka was used as documented (Figure 2A).
For the dissection of secretory glands from Nepenthes pitchers use a microforceps and attach it to the manipulator (Figures 2A and 2B).
For RNA extraction, treat all instruments (razor blades, tweezers, micromanipulator, microforceps, object slide) with RNaseZAP.
- Prepare the collection tubes by adding 9 μl nuclease-free water and 1 μl RNase inhibitor in a PCR tube for RNA extraction. For direct PCR from tissue samples use only nuclease-free water (10 μl).
- Take the fresh or frozen pitcher and remove a 1-2 cm2 piece of tissue containing the secretory glands by using a razor blade and tweezers. Wash the tissue with water; add 0.1% diethylpyrocarbonate (DEPC) if RNA will be extracted.
- Fix the plant material with double-faced adhesive tape on an object slide (the gland covered surface should face upwards, Figures 2C and 2D) and secure the slide with adhesive tape on a universal stage, which should be applied with a moveable table.
- For extracting nucleic acids the preparation should be done on ice. Therefore take a flat Styrofoam box, place the universal stage containing the sample inside and fill the box with ice. Afterwards place the box under the microscope.
- For preparation the positioning of the glands is very important. The secretory glands are partial covered by a protective hood (Figure 3) and only accessible by the forceps from one side. Furthermore the angle of the sample to the forceps has to be adjusted by tilting the table of the universal stage approximately 135°, depending on the Nepenthes species.
- The microdissection is done under visual control. For gland isolation, use a non-UV filter, to limit the degradation of DNA and RNA.
- Move micromanipulator and forceps via the control panel to the selected gland. When reached the gland, place each arm of the forceps on either side of the gland. Remove the gland from the remaining tissue by closing the forceps (Figure 4A).
- Direct the gland attached to the forceps away from the object and to the collection tube. Bring the very tips of forceps in contact with the solution in the tube (Figure 4B). The release of the gland by opening the forceps has to be well observed, because the glands tend to stick on the tool tip. The tissue can be stored at -80 °C until PCR or RNA extraction.
3. Direct PCR Amplification from Nepenthes Tissue
To prove the efficiency of the preparation method, a PCR approach directly from the gland tissue without a DNA extraction step can be performed.
- For the PCR amplification use gene specific primers; here encoding for the sequence of a thaumatin-like protein (TLP) (forward: 5'-CAATGAGCCAATTCATAAAATTCATTG-3', reverse: 5'-CAGTTATACTTTAAGGGCAAAACACAAC-3').
- Collect 1-5 glands in a PCR tube containing 10 μl of nuclease-free water. For a 50 μl reaction add 5 μl of 10x Taq buffer, 1 μl 10 mM deoxynucleoside triphosphates (dNTPs), 3 μl 25 mM MgCl2, 0.4 μl (1.25 U) Taq DNA polymerase, 1 μl (100 pmol) of each primer, and 28.6 μl of nuclease-free water.
- Carry out PCR reactions in a thermocycler with the following program: initial denaturation 94 °C for 3 min; 35 cycles of denaturation (94 °C for 30 sec), annealing (59 °C for 30 sec), and extension (72 °C for 1 min); and a final extension step of 10 min at 72 °C.
- Prove the amplification of the PCR product by using agarose gel electrophoresis. Therefore use 1.5% (w/v) agarose and dissolve it in 1x TAE (40 mM Tris base, 20 mM glacial acetic acid, 1 mM EDTA, pH 8) buffer. Heat the solution in a microwave until the agarose is dissolved. To 100 ml agarose solution, add 1 μl ethidium bromide (10 μg/μl) for downstream visualization. Run the gel in 1x TAE buffer at 300 V, 115 mA for approximately 20 min in an electrophoresis chamber. The correct size of the TLP amplicon must be at 690 bp.
4. RNA Extraction from Small Tissue Samples and Reverse Transcription
Perform the following steps on ice. For the extraction of RNA two extraction methods were used, a Micro Kit for the extraction of total RNA and a mRNA isolation kit applied with magnetic beads.
Collect at least 30 secretory glands by micropreparation in a PCR tube containing 1 μl RNase inhibitor and 9 μl of nuclease-free water. Afterwards lyse the tissue in a thermocycler for 10 min at 75 °C.
- Extraction of total RNA.
- After preparation add 90 μl of lysis solution to the sample and incubate for 10 min at 75 °C. Afterwards load the reaction mix on the column (provided with the kit) and proceed with the washing steps as described in the manual of the manufacturer.
- Elute the RNA two times with 10 μl Elution Solution respectively and continue with the DNase I treatment.
- To 20 μl extracted RNA add 2 μl of 10x DNase I buffer and 1 μl DNase I (provided with RNA extraction kit) and incubate 1 hr at 37 °C. Stop the reaction by adding 2 μl of DNase Inactivation Reagent and incubate for 2 min at room temperature. After a centrifugation step for 1.5 min at maximum speed, the RNA can be transferred to a fresh RNase-free tube. Determine the RNA concentration of each sample using a spectrophotometer. The samples should be stored at -80 °C.
- Extraction of mRNA using magnetic beads.
- Adjust the sample volume to 100 μl with lysis/binding buffer (supplied in the kit) and incubate for 10 min at 75 °C. Add 20 μl of magnetic beads supplied with an Oligo (dT)25 overhang and mix the reaction for 5 min at room temperature. The mRNA binds by the poly(A) tail to the Oligo (dT)25 overhang of the beads.
- Perform the washing steps of the mRNA bound to the magnetic beads by following the manufacturer's protocol. Solutions can be removed by using a magnet. Resuspend the beads in 20 μl 10 mM Tris-HCl buffer (pH 7.5).
- Remove contaminations of genomic DNA by treating the sample with 2U (1 μl) of DNase (2 μl 10x DNase buffer and 17 μl nuclease-free water) for 1 hr at 37 °C. Afterwards the enzyme solution is removed by the magnet and the beads are resuspended in 20 μl of 10 mM Tris-HCl buffer. It is not possible to measure the RNA concentration since it is attached to the beads. The samples should be stored at -80 °C.
- For the reverse transcription use a cDNA Synthesis kit. Reactions were performed by following the manufacturer's protocol.
- For the reverse transcription of total RNA add 1 μl (100 pmol) Oligo (dT)20 primer or 1 μl (0.4 μg) Random Hexamers, 1 μl RNase inhibitor, and 1 μl 10 mM dNTP Mix to 1 μg of RNA and adjust the volume to 14 μl with nuclease-free water. Incubate at 65 °C for 5 min and afterwards place the samples on ice. Add 1 μl 0.1M DTT, 1 μl reverse transcriptase, and 4 μl of 5x RT buffer. The reactions are incubated for 55 min at 50 °C, followed by an inactivation step at 70 °C for 10 min. After reverse transcription samples can be stored at -20 °C.
- For mRNA bound to magnetic beads, replace the Tris-HCl buffer with the first reaction mix (1 μl RNase inhibitor, 1 μl 10 mM dNTP Mix, and 12 μl nuclease-free water) and incubate for 5 min at 65 °C. Because of the Oligo (dT)25 overhang of the beads it is not necessary to add an Oligo (dT)20 primer or Random Hexamers to the samples. Afterwards follow the protocol above (step 4.5.1).
- Measure the cDNA concentration with a spectrophotometer. Store the samples at -20 °C.
5. Real-time PCR
For the analyses with Real-time PCR use total RNA extracted with the Micro Kit and a SYBR Green QPCR Master Mix. To ensure an equal amount of transcripts, adjust the cDNA concentration of each sample to the same value after the reverse transcription.
In addition to the samples insert control reactions. Use a nontemplate control to screen for contamination in the reagents and to check the quality of your primers. To verify the absence of genomic DNA run a non-RT control. Both reactions should be negative (no fluorescence signal) in the Real-time PCR.
To guaranty an equal transcript level between the different samples use at least one reference gene. Here, designed primer pairs encoding for an actin (forward: 5'-CTCTTAACCCCAAAGCAAACAGG-3', reverse: 5'-GTGAGAGAACAGCCTGGATG-3') and an 18SrRNA (forward: 5'-CTTGATTCTATGGGTGGTGGTG-3', reverse: GTTAGCAGGCTGAGGTCTC-3') were used. As a target gene the TLP (forward: 5'- GTGGGCAACAATTGGACCCAG-3', reverse: 5'-CATTCGATCCGGAAAGGCTAC-3') was chosen. The size of each amplified fragment was about 100bp.
For a 25 μl reaction add 12.5 μl 2x SYBR Green QPCR Master Mix, cDNA (≥20 ng), 400 nM of each primer, and 30 nM of the passive reference dye ROX (provided with kit). Adjust the sample volume to 25 μl with nuclease-free water.
Reactions were performed in a QPCR thermocycler with the following program: initial denaturation at 95 °C for 10 min to active the polymerase; 40 cycles of denaturation (95 °C for 30 sec), annealing (61 °C for 60 sec), and extension (72 °C for 1 min); followed by the dissociation curve (95 °C for 60 sec, 55 °C for 30 sec, 95 °C for 30 sec). The dissociation curve is necessary to determine the absence of primer dimers, which can be detected by an additional peak.
Determine the efficiency (E) of each PCR reaction by using the method of Liu and Saint8. The efficiency can be deflected from the linear part of the amplification curve. Here the relative amplicon number (Rn, A) of one cycle point (CPA) is linked to the amount of amplicons (Rn, B) of another cycle (CPB).
Analyze the Real-time PCR results with the Relative Expression Software Tool (REST 2009, http://www.gene-quantification.de/rest-2009.html) or a similar software. Use the program to calculate the expression ratio of a gene in a treated sample compared to a control.
Representative Results
For the preparation of single glands from Nepenthes pitcher tissue, a mechanized microdissection technique was developed and applied. The micropreparation platform needs to be equipped with microforceps, attached to the micromanipulator, which in turn are operated by a control panel. Individual glands, visible under the microscope, could be targeted with the forceps that grasped the gland tissue and removed it mechanically (Figure 4A).
The glandular tissue was transferred directly into a PCR tube for subsequent analysis (Figure 4B). The success of gland removal could be observed because it uncovered the plant's vascular system below the glands (Figure 4A).
In order to further prove the quality and efficiency of the preparation method and to make sure that this is a useful method, a PCR approach was performed. As a candidate gene a thaumatin-like protein (TLP) from Nepenthes gracilis (NCBI Acc. No DQ352144) was selected and amplified successfully (Figure 4C). However, not only single genes could be detected but, in addition, a direct isolation of both total- and mRNA was effectively performed and used for cDNA synthesis. By using TLP again, the amplification of that cDNA by PCR was demonstrated (Figure 4D). Furthermore, the cDNA was introduced in various Real-time PCR experiments. Therefore glands and fragments of the epidermal tissue, which is surrounding the glands, were isolated by the micropreparation technique from closed and open pitchers of N. alata and N. mirabilis. In a Real-time PCR reaction the TLP expression was analyzed. Using this method it was shown that the TLP transcript level is higher in the secretory glands compared to the epidermal tissue. After prey trapping (simulated by feeding the plant with Drosophila melanogaster, fruit flies) the epidermal tissue exhibited a higher TLP expression (Figure 4D-1). In addition, the inducibility of gene expression by the presence of prey was analyzed. Here, the TLP transcript level of glands and epidermal tissue in a control (nonfed) and treated (addition of fruit flies) pitcher of N. mirabilis was compared (Figure 4D-2). The upregulation of TLP expression in both tissue types indicates that the production of pitcher fluid proteins can indeed be induced by prey.
 Figure 2. Microsampling platform.(A) aureka platform consisting of a stereomicroscope and a micromanipulator, equipped with microforceps. (B) Detailed view of the microforceps; in the yellow oval, the universal stage with a moveable table to present the sample can been seen. (C) The same as in (B) with an object slide presenting the plant tissue. (D) Inner tissue of a Nepenthes pitcher spotted with glands. For photography, an UV-filter with an excitation wavelength of 365 nm was used. Click here to view larger image.
Figure 2. Microsampling platform.(A) aureka platform consisting of a stereomicroscope and a micromanipulator, equipped with microforceps. (B) Detailed view of the microforceps; in the yellow oval, the universal stage with a moveable table to present the sample can been seen. (C) The same as in (B) with an object slide presenting the plant tissue. (D) Inner tissue of a Nepenthes pitcher spotted with glands. For photography, an UV-filter with an excitation wavelength of 365 nm was used. Click here to view larger image.
 Figure 3. Individual glands of different Nepenthes species, covered by a protective hood. Note the strong autofluorescence of the cell walls in the glands of N. thorelii (1), N. superba (2), N.mirabilis (7), and N. superpum (9), which is not present in the glands of the other species (3, 4, 5, 6, and 8). Click here to view larger image.
Figure 3. Individual glands of different Nepenthes species, covered by a protective hood. Note the strong autofluorescence of the cell walls in the glands of N. thorelii (1), N. superba (2), N.mirabilis (7), and N. superpum (9), which is not present in the glands of the other species (3, 4, 5, 6, and 8). Click here to view larger image.
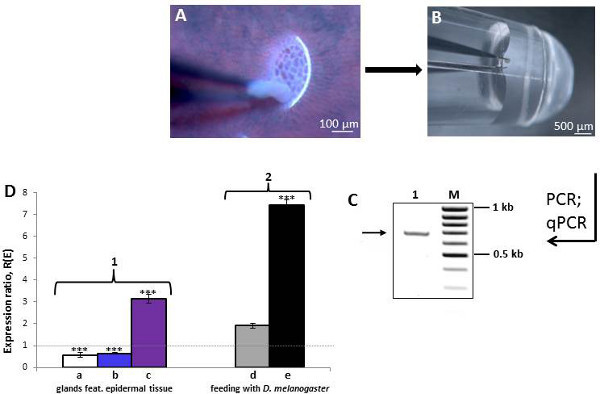 Figure 4. Micropreparation of individual Nepenthes glands.(A) Forceps taking gland tissue, thereby uncovering the vascular system below the gland. (B) Direct transfer of an isolated gland into a PCR tube. For photo-documentation the usage of an UV-filter was necessary (excitation at 365 nm). (C) Individual gene-expression analysis. Here, five glands were combined and prepared by lysis for 20 min at 70 °C for mRNA extraction and reverse transcription of the mRNA. Genomic DNA was digested with DNase. The cDNA was used for the subsequent PCR-reaction to amplify the gene sequence of interest, indicated by the arrow (lane 1; M: DNA-ladder). (D) Analyses of TLP gene expression by Real-time PCR. Secretory glands and fragments of the glands surrounding epidermal tissue of closed and open pitchers (treated with Drosophila melanogaster) were microdissected. Total RNA was extracted from all samples. After digestion of genomic DNA and reverse transcription, the cDNA was used in various Real-time PCR experiments. (1) The TLP expression of glands (control) and epidermal tissue (treatment) of closed pitchers from N. alata (a) and N. mirabilis (b) was compared and showed a higher TLP transcript level in the secretory glands. After feeding D. melanogaster to an open N. mirabilis (c) pitcher the TLP transcript level was 3-fold higher in the epidermal tissue compared to the glands. (2) Analysis of TLP expression of two open N. mirabilis pitchers after feeding with D. melanogaster. The level of TLP transcripts in glands (d) and epidermal tissue (e) of the control (nonfed) and the treated (fed with D. melanogaster) pitcher was compared. The TLP-mRNA level in both tissue types was higher after feeding with fruit flies, which indicates the inducibility of the gene by prey; (expression ratio R(E) (R(E) < 1: downregulation, R(E) > 1: upregulation); ***, indicates a significant difference between groups compared, P(H1) <0.001). Click here to view larger figure.
Figure 4. Micropreparation of individual Nepenthes glands.(A) Forceps taking gland tissue, thereby uncovering the vascular system below the gland. (B) Direct transfer of an isolated gland into a PCR tube. For photo-documentation the usage of an UV-filter was necessary (excitation at 365 nm). (C) Individual gene-expression analysis. Here, five glands were combined and prepared by lysis for 20 min at 70 °C for mRNA extraction and reverse transcription of the mRNA. Genomic DNA was digested with DNase. The cDNA was used for the subsequent PCR-reaction to amplify the gene sequence of interest, indicated by the arrow (lane 1; M: DNA-ladder). (D) Analyses of TLP gene expression by Real-time PCR. Secretory glands and fragments of the glands surrounding epidermal tissue of closed and open pitchers (treated with Drosophila melanogaster) were microdissected. Total RNA was extracted from all samples. After digestion of genomic DNA and reverse transcription, the cDNA was used in various Real-time PCR experiments. (1) The TLP expression of glands (control) and epidermal tissue (treatment) of closed pitchers from N. alata (a) and N. mirabilis (b) was compared and showed a higher TLP transcript level in the secretory glands. After feeding D. melanogaster to an open N. mirabilis (c) pitcher the TLP transcript level was 3-fold higher in the epidermal tissue compared to the glands. (2) Analysis of TLP expression of two open N. mirabilis pitchers after feeding with D. melanogaster. The level of TLP transcripts in glands (d) and epidermal tissue (e) of the control (nonfed) and the treated (fed with D. melanogaster) pitcher was compared. The TLP-mRNA level in both tissue types was higher after feeding with fruit flies, which indicates the inducibility of the gene by prey; (expression ratio R(E) (R(E) < 1: downregulation, R(E) > 1: upregulation); ***, indicates a significant difference between groups compared, P(H1) <0.001). Click here to view larger figure.
Discussion
For a long time, studies of plant secretory structures, in particular trichomes have been performed essentially on anatomy and were purely descriptive. There is a large body of publications of this type as summarized in reviews by9-11. Our biochemical and molecular knowledge of plant secretory structures is still limited in spite of some early remarkable work e.g. on mint glandular trichomes showing that glandular trichomes are the site of biosynthesis of monoterpenoids which are secreted into the subcuticular space12. Thus, it is necessary to create tools and to develop techniques for the isolation of such specialized organs in order to study them independently. Direct access to these glands would offer the possibility to directly investigate and understand both physiological and molecular processes in secretory systems of plants. The function of genes playing a key role in the biosynthesis and the transport of the secreted compounds could be addressed as well as the question how development of such highly specialized organs is regulated.
It should be emphasized that the production and secretion capacities of many secreting glands identified them as prime targets for metabolic engineering in plants with potential applications in a wide range of areas including human health or durable crop protection13,14. In addition, secretions by these different structures play important roles in the interactions of the plants with their abiotic and biotic environment in particular with insects. Secreted compounds can serve as attractants to pollinating insects, as in the case of volatile monoterpenes such as linalool produced by glandular trichomes, and sugars secreted by flower nectaries15. Resistance against insect pests is another important aspect as illustrated by various reports on this topic16,17.
In order to isolate single multicellular glands from the pitcher tissue of carnivorous Nepenthes species a fast and efficient micropreparation technique was developed and employed. This method is an alternative to the widespread but, in terms of applications, limited technique of laser capture dissection. The advantage of the new methods is due to the fact that no previous fixation of plant material is necessary and, instead, the freshly harvested plant tissue can be used immediately. Experiments where PCR studies of individual genes were performed18, as well as successful RNA extraction combined with subsequent gene expression analysis showed that the new preparation method is highly efficient. Thus, its application is suitable for many other molecular and biochemical studies.
However, also some drawbacks exit using the mechanical micropreparation. In particular, it often happens that the prepared samples stick to the forceps and it is hard to stripe off the preparations. That will cause a rapid drying-out and, as a consequence, a loss of the samples. Thus, experience with the technique is important to be fast and precise in any step, starting from positioning of the forceps to transferring the sample into the target tubes. Automation of such steps might be helpful. In addition, for any work with very small tissue samples it is necessary to provide a (micro)-environment that guarantees nondestructive conditions. That means, for example, high humidity, low temperatures, and low UV-irradiation in order to keep subcellular structures (DNA, RNA, proteins) intact.
The technique presented appears to be adaptable to a wide range of plants that carry multicellular glands, offering the ability to study the different secretory systems. However, preparation of glands or other microstructures from living tissues is only the first step providing the samples of interest for further analyses. The subsequent analytical techniques that will be applied in the particular studies have different requirements in terms of the nature and quality of the sample. This might require individual methodical developments for different samples such as newly designed forceps or the use of anti-adhesive materials.
In the long term, this technique will create a series of subsequent approaches which are relevant to understand plant secretory structures, including deep sequencing of cDNAs, proteomics or transcription promoter analyses specific to glandular cells could be performed in various plant species, not only carnivorous plants such as Nepenthes or Drosera but also, for example, from Artemisia, Mentha or Cistus or many others. In general, applications beyond plant gland physiology are easily conceivable such as single cell analysis in biological and medicinal sciences or even forensic.
Disclosures
We have nothing to disclose.
Acknowledgments
We thank the greenhouse team at the MPI-CE at the Botanical Garden of the Friedrich Schiller University, Jena, for cultivating plants and Wilhelm Boland and the Max Planck Society for support.
References
- Adlassnig W, Peroutka M, Land I, Lichtscheidl IK. Glands of carnivorous plants as a model system in cell biological research. Acta Bot. Gall. 2005;152:111–124. [Google Scholar]
- Darwin C. Insectivorous plants. London: John Murray; 1875. [Google Scholar]
- Mithöfer A. Carnivorous pitcher plants: Insights in an old topic. Phytochemistry. 2011;72:1678–1682. doi: 10.1016/j.phytochem.2010.11.024. [DOI] [PubMed] [Google Scholar]
- Owen Jr. TP, Lennon KA, Santo MJ, Anderson AN. Pathways for nutrient transport in the pitchers of the carnivorous plant. Nepenthes alata Ann. Bot. 1999;84:459–466. [Google Scholar]
- Brandt SP. Microgenomics: gene expression analysis at the tissue-specific and single-cell levels. J. Exp. Bot. 2005;56:495–505. doi: 10.1093/jxb/eri066. [DOI] [PubMed] [Google Scholar]
- Hölscher D, Schneider B. Application of laser-assisted microdissection for tissue and cell-specific analysis of RNA, proteins, and metabolites. Prog. Bot. 2008;69:141–167. [Google Scholar]
- Rottloff S, Müller U, Kilper R, Mithöfer A. Micropreparation of single secretory glands from the carnivorous plant Nepenthes. Anal. Biochem. 2009;394:135–137. doi: 10.1016/j.ab.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Liu WH, Saint DH. A new quantitative method of real time reverse transcription polymerase chain reaction assay based on simulation of polymerase chain reaction kinetics. Anal. Biochem. 2002;302:52–59. doi: 10.1006/abio.2001.5530. [DOI] [PubMed] [Google Scholar]
- Werker E. Trichome diversity and development. Adv. Bot. Res. Inc. Adv. Plant Pathol. 2000;31:1–35. [Google Scholar]
- Fahn A. Structure and function of secretory cells. Adv. Bot. Res. Inc. Adv. Plant Pathol. 2000;31:37–75. [Google Scholar]
- Juniper BE, Robins RJ, Joel DM. The carnivorous plants. London: Academic Press; 1989. [Google Scholar]
- Gershenzon J, McCaskill D, Rajaonarivony J, Mihaliak C, Karp F, Croteau R. Isolation of secretory-cells from plant glandular trichomes and their use in biosynthetic-studies of monoterpenes and other gland products. Anal. Biochem. 1992;200:130–138. doi: 10.1016/0003-2697(92)90288-i. [DOI] [PubMed] [Google Scholar]
- Rontein D, Onillon S, et al. CYP725A4 from yew catalyzes complex structural rearrangement of taxa-4(5),11(12)-diene into the cyclic ether 5(12)-oxa-3(11)-cyclotaxane. J. of Biol. Chem. 2008;283(5):6067–6075. doi: 10.1074/jbc.M708950200. [DOI] [PubMed] [Google Scholar]
- Aharoni A, Jongsma MA, Bouwmeester HJ. Volatile science? Engineering of terpenoid biosynthesis in plants. Trends Plant Sci. 2005;10:594–602. doi: 10.1016/j.tplants.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Mithöfer A, Boland W, Maffei ME. Chemical ecology of plant-insect interactions. Ann. Plant Rev. 2009;34:261–291. [Google Scholar]
- Kennedy GG. Tomato pests, parasitoids, and predators: Tritrophic interactions involving the genus Lycopersicon. Ann. Rev. Entomol. 2003;48:51–72. doi: 10.1146/annurev.ento.48.091801.112733. [DOI] [PubMed] [Google Scholar]
- Mithöfer A, Boland W. Plant defense against herbivores: Chemical aspects. Ann. Rev. Plant Biol. 2012;63:431–450. doi: 10.1146/annurev-arplant-042110-103854. [DOI] [PubMed] [Google Scholar]
- Rottloff S, Stieber R, Maischak H, Turini FG, Heubl G, Mithöfer A. Functional characterization of a class III acid endochitinase from the traps of the carnivorous pitcher plant genus, Nepenthes. J. Exp. Bot. 2011;62:4649–4647. doi: 10.1093/jxb/err173. [DOI] [PMC free article] [PubMed] [Google Scholar]