

Abstract
Background. Dilated cardiomyopathy (DCM) is one of the leading causes of heart failure with high morbidity and mortality. Although more than 40 genes have been reported to cause DCM, the role of genetic testing in clinical practice is not well defined. Mutations in the troponin T (TNNT2) gene represent an important subset of known disease-causing mutations associated with DCM. Therefore, the aim of the present study was to determine the genetic variations in TNNT2 and the associations of those variations with DCM in Chinese patients. Methods. An approximately 4 kb fragment of the TNNT2 gene was isolated from 103 DCM patients and 192 healthy controls and was analyzed by DNA sequence analysis for genetic variations. Results. A total of 6 TNNT2 mutations were identified in 99 patients, including a G321T missense mutation (Leu84Phe) and 5 novel intronic mutations. Alleles of two novel SNPs (c.192 + 353 C>A, OR = 0.095, 95% CI: 0.013–0.714, P = 0.022; c.192 + 463 G>A, OR = 0.090, 95% CI: 0.012–0.675, P = 0.019) and SNP rs3729843 (OR = 1.889, 95% CI: 1.252–2.852; P = 0.002) were significantly correlated with DCM. Conclusions. These results suggest that the missense mutation (Leu84Phe) and two novel SNPs (c.192 + 353 C>A, c.192 + 463 G>A) in TNNT2 gene might be associated with DCM in the Chinese population.
1. Background
Idiopathic or primary dilated cardiomyopathy (DCM) is one of the leading causes of heart failure with high morbidity and mortality [1, 2]. The prevalence of DCM is 36.5 cases per 100,000 individuals, and 30–50% of all cases are diagnosed as a familial form of DCM [2–4]. Recent studies have reported that more than 40 genes, including 2 X-linked genes, are associated with DCM [5, 6]. In the vast majority of cases, these genes encode for sarcomeric contractile proteins such as troponin T (TNNT2), troponin I (TNNI3), and cardiac α-actin (ACTC) [7, 8].
The TNNT2 gene (OMIM number ∗191045) encodes the thin-filament contractile protein cardiac troponin T, which links the troponin complex to tropomyosin in the sarcomere [9]. TNNT2 contains 16 exons, is located on chromosome 1q32, and comprises 25 kb of the genome. Recent data have indicated that TNNT2 mutations are associated with DCM and that the overall frequency of TNNT2 mutations in familial DCM is approximately 3–6% [10, 11]. In our laboratory, we found a significant association between tagging SNPs rs3729547, rs3729843 of TNNT2 and DCM in the Chinese Han population [12]. In the present study, we further explored additional variants in DCM patients by sequencing an approximately 4 kb (3992 bp) DNA fragment covering the rs3729843 and rs3729547 mutations. We identified 6 novel TNNT2 mutations in DCM patients, including a G12026T (c.252 G>T) missense mutation (Leu84Phe), 5 novel mutations in introns, and 2 novel single-nucleotide polymorphisms (SNP) (c.192 + 353 C>A, c.192 + 463 G>A).
2. Materials and Methods
2.1. Subjects and Selection of Tagging SNPs
This case-control study enrolled 103 unrelated DCM patients from the Fuwai Hospital. The clinical diagnosis of DCM was made in accordance with revised criteria [2]. Primary DCM was defined as systolic dysfunction (left ventricular (LV) ejection fraction <50%) with or without LV dilation in the absence of an apparent secondary cause of cardiomyopathy (such as congenital heart disease, ischemic heart disease, uncontrolled hypertension, significant valvular disease, glycogen storage disease, arrhythmogenic right ventricular cardiomyopathy, or concomitant complex congenital heart disease). Coronaroangiography was performed in all patients and patients with coronary artery disease were excluded. A total of 192 healthy, unrelated individuals from a routine health survey were enrolled as controls. This study was approved by the Ethics Committee of Fuwai Hospital. Subjects were informed of the study aims and provided written, informed consent prior to participating.
2.2. Samples
Blood samples were collected via puncture of the cubital vein. Tubes with ethylene diamine tetra acetic acid were used for DNA analysis. Blood samples were stored at 4°C and DNA isolation was performed by a modified salting out procedure [13] within a week of collection.
2.3. DNA Sequencing
Based on the SNP and linkage disequilibrium analysis of TNNT2 in our recent study in DCM patients [12], an approximately 4-kilo-base (kb) fragment of TNNT2 located at chr.201333582-201337484 was selected, and the fragment included exons 6, 7, 8, 9, and 10 and introns 5, 6, 7, 8, 9, and 10. The TNNT2 fragment was amplified by PCR with primers (primer sequences available on request). Products were then sequenced using an ABI 3730 DNA Sequencer (Applied Biosystems, Foster City, CA). The DNA sequence was viewed and analyzed using the Sequencher computer program (Gene Codes Corporation, Ann Arbor, MI).
2.4. Statistical Analysis
Differences in the distributions of selected variables and TNNT2 genotypes between cases and controls were evaluated using the chi-square (χ 2) test and continuous variables were analyzed using the independent-samples Student's t-test. The correlation between the TNNT2 genotype and the risk of DCM was estimated by using logistic regression analysis to compute odds ratios (ORs) and 95% confidence intervals (CIs). The χ 2 test was used to determine Hardy-Weinberg equilibrium among control subjects. All statistical analyses were performed with SPSS 16.0 (SPSS Inc., Chicago, IL, USA) and a P value of <0.05 was considered statistically significant.
3. Results
3.1. Characteristics of the Study Cohort
The study included 295 subjects, consisting of 103 patients with DCM and 192 healthy control subjects; 4 patients and 3 control subjects were excluded from analyses due to indeterminate sequence results. A total of 79.4% of control subjects were males with a mean age of 54.0 ± 3.6 years. Patients with DCM were of a similar age to controls (51.6 ± 12.0 years; t = 1.700, P = 0.092) and were made up of a similar percentage of males (77.3%; χ 2 = 0.160, P = 0.689). In addition, 43.3% of DCM patients were smokers. There were no pregnant or peripartum subjects in this study. DCM patients were defined as having systolic dysfunction (LV ejection fraction <50%), LV dilation detected by echocardiogram, and no apparent secondary cause of cardiomyopathy. In DCM patients, the mean LV ejection fraction was 32.0 ± 8.4%, LV diameter was 67.7 ± 8.6 mm, and left atrium diameter was 42.7 ± 7.6 mm. The genotypic frequencies of each SNP in control subjects fit the assumption of Hardy-Weinberg equilibrium (Table 1).
Table 1.
The sequences of the primers used to amplify the 4 kb region in TNNT2.
| Markers | Sequences of PCR primers | Tm | Length |
|---|---|---|---|
| TNNT2-1F | CATGTTCTGTGGTGCCAGAC | 57 | 899 |
| TNNT2-1R | TGCCACCAAGTTCTGTCCTC | 58 | |
| TNNT2-2F | AGGAGGCTGAAGGTAAGGAT | 55 | 691 |
| TNNT2-2R | ACACTCACGCAGTGTGGAAC | 56 | |
| TNNT2-3F | AGGCCTTGTCACTGTGAAGC | 58 | 573 |
| TNNT2-3R | TGCACGATTGGTGATGGAGT | 59 | |
| TNNT2-4F | TGTGTACTGCACAAGCGTCTC | 59 | 514 |
| TNNT2-4R | GTGCACAAGAGGCCAGGAAG | 61 | |
| TNNT2-5F | ATAGGCATGGCGGCTTCA | 60 | 862 |
| TNNT2-5R | ACAGCCACCGCTTACATCAA | 59 | |
| TNNT2-6F | GGCAGTGCTGGAAGATTCTC | 57 | 629 |
| TNNT2-6R | GGCCATCAGAGAATGTTAGG | 55 | |
| TNNT2-7F | AATGAGTTGCAGGCGCTGAT | 61 | 454 |
| TNNT2-7R | CTCCAGCCAGAGCAGCATGT | 61 | |
| TNNT2-8F | CCTTGACTGCCAGAGCTGAG | 58 | 559 |
| TNNT2-8R | GAGAAGGTGACATCGCAGGTA | 59 |
3.2. Identification of Mutations
The screening of the 3992-nucleotide fragment of TNNT2 led to the identification of one novel missense mutation. A single-nucleotide variant consisting of a G>T transversion (TTG>TTT) at nucleotide 12026 (G12026T, c.252 G>T) (Figure 1) in exon 9 was found in one DCM patient and would substitute a phenylalanine for the normal leucine at residue 84 (Leu84Phe). This patient was diagnosed at the age of 43 and was clinically symptomatic of heart failure, with LV 60 mm and LV ejection fraction 31%. Additionally, this patient had a familial history of coronary heart disease. In addition, we also identified 5 mutations in introns 6, 7, 9, and 10 in a subset of the DCM patients (Figure 1). Their clinical data are shown in Table 2.
Figure 1.
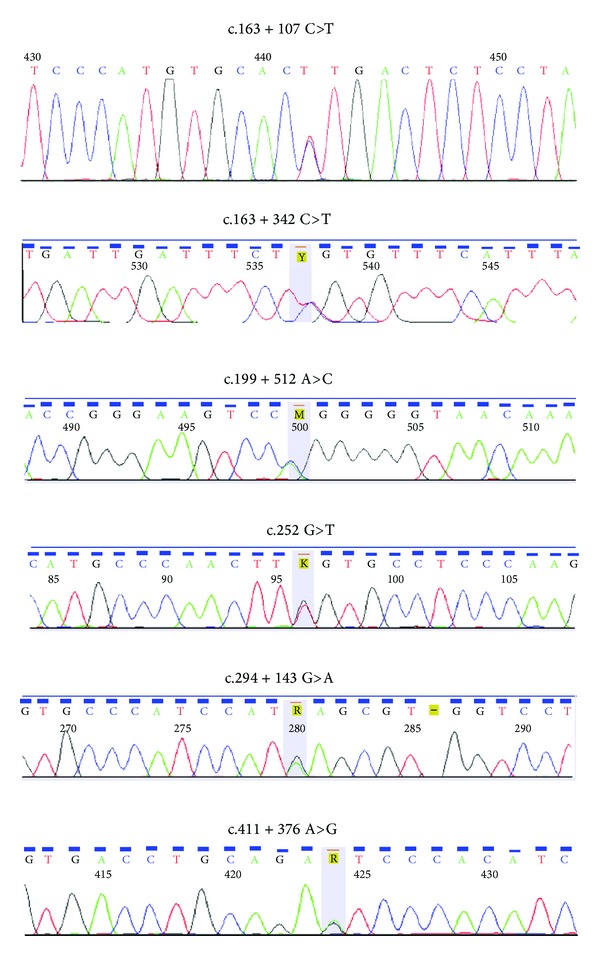
Sequences of TNNT2. Six novel variants in TNNT2 gene in DCM patients, including a missense mutation (G12026T, c.252 G>T, Leu84Phe), five novel variants in introns, respectively.
Table 2.
Mutations in TNNT2 in DCM patients.
| Markers | Exon/intron | Location of nucleotide change | Amino acid change | Number of patients | Gender | Age at diagnosis (years) | Family history | LVEDD (mm) | LVEF (%) |
|---|---|---|---|---|---|---|---|---|---|
| c.163 + 107 | Intron 6 | 201337183 C>T | Noncoding | 1 | Male | 37 | No | 82 | 18 |
| c.163 + 342 | Intron 6 | 201336948 C>T | Noncoding | 1 | Male | 30 | No | 81 | 31 |
| c.199 + 512 | Intron 7 | 201336387 A>C | Noncoding | 1 | Male | 59 | Suspensive coronary heart disease | 60 | 45 |
| c.252 | Exon 9 | 201334780 G>T | Leu 84 Phe | 1 | Male | 43 | Suspensive coronary heart disease |
60 | 31 |
| c.294 + 143 | Intron 9 | 201334595 G>A | Noncoding | 1 | Male | 45 | No | 58 | 40 |
| c.411 + 376 | Intron 10 | 201333948 A>G | Noncoding | 1 | Male | 62 | Suspensive cardiovascular disease | 80 | 36 |
Note: LVEDD: left ventricular end-diastolic diameter; LVEF: left ventricular ejection fraction.
3.3. Identification of Polymorphisms
During sequence analysis, 11 known SNPs and two novel SNPs (c.192 + 353 C>A, c.192 + 463 G>A) were identified in both DCM patients and control samples. We compared the genotype and allele frequencies of the TNNT2 SNPs between the DCM patients and control subjects. Our results showed that the allele of the two novel SNPs (c.192 + 353 C>A, OR = 0.095, 95% CI = 0.013–0.714, P = 0.022; c.192 + 463 G>A, OR = 0.090, 95% CI = 0.012–0.675, P = 0.019) and SNP rs3729843 (OR = 1.889, 95% CI = 1.252–2.852, P = 0.002) were significantly correlated with DCM. The allele and genotype frequencies of the 13 SNPs identified in both DCM patients and control subjects are shown in Table 3, with corresponding results from statistical analysis.
Table 3.
Genotype and allele frequencies of the SNPs in TNNT2 in the DCM patients and control subjects.
| Marker | Genotype | χ 2, P value | Allele | P value | OR (95% CI) | Power | |||
|---|---|---|---|---|---|---|---|---|---|
| rs45576939 | A/A | A/G | G/G | A | G | 0.173 | 0.235 (0.029, 1.889) | 28% | |
| Patients | 0 (0) | 1 (0.01) | 97 (0.99) | χ 2 = 2.231 | 1 (0.01) | 195 (0.99) | |||
| Controls | 0 (0) | 8 (0.043) | 179 (0.957) | P = 0.135 | 8 (0.021) | 366 (0.979) | |||
| rs3729842 | C/C | C/T | T/T | C | T | 0.865 | 1.042 (0.651, 1.667) | 5.4% | |
| Patients | 74 (0.755) | 16 (0.163) | 8 (0.082) | χ 2 = 4.282 | 164 (0.837) | 32 (0.163) | |||
| Controls | 135 (0.722) | 45 (0.241) | 7 (0.037) | P = 0.118 | 315 (0.842) | 59 (0.158) | |||
| rs3729843 | A/A | A/G | G/G | A | G | 0.002 | 1.889 (1.252, 2.852) | 85.2% | |
| Patients | 11 (0.112) | 33 (0.337) | 54 (0.551) | χ 2 = 8.315 | 55 (0.281) | 141 (0.719) | |||
| Controls | 7 (0.037) | 50 (0.267) | 130 (0.695) | P = 0.004 | 64 (0.171) | 310 (0.829) | |||
| rs1892028 | A/A | A/G | G/G | A | G | 0.140 | 0.770 (0.544, 1.090) | 31.3% | |
| Patients | 31 (0.313) | 54 (0.5) | 14 (0.143) | χ 2 = 4.719 | 116 (0.586) | 82 (0.414) | |||
| Controls | 55 (0.294) | 86 (0.460) | 47 (0.251) | P = 0.094 | 196 (0.521) | 180 (0.479) | |||
| rs10800775 | C/C | C/T | T/T | C | T | 0.152 | 0.766 (0.533, 1.103) | 29.7% | |
| Patients | 46 (0.465) | 45 (0.455) | 9 (0.090) | χ 2 = 2.142 | 137 (0.685) | 63 (0.315) | |||
| Controls | 73 (0.388) | 89 (0.473) | 26 (0.138) | P = 0.343 | 235 (0.625) | 141 (0.375) | |||
| rs7544061 | T/T | T/C | C/C | T | C | 0.612 | 0.821 (0.383, 1.761) | 73% | |
| Patients | 0 (0) | 10 (0.101) | 89 (0.899) | χ 2 = 0.274 | 10 (0.051) | 188 (0.949) | |||
| Controls | 0 (0) | 23 (0.116) | 166 (0.834) | P = 0.601 | 23 (0.061) | 355 (0.939) | |||
| rs3767545 | A/A | A/G | G/G | A | G | 0.294 | 1.680 (0.638, 4.424) | 20.3% | |
| Patients | 0 (0) | 8 (0.081) | 91 (0.919) | χ 2 = 1.160 | 8 (0.04) | 190 (0.960) | |||
| Controls | 0 (0) | 9 (0.049) | 175 (0.951) | P = 0.282 | 9 (0.024) | 359 (0.976) | |||
| rs3729547 | C/C | T/C | T/T | C | T | 0.033 | 0.679 (0.475, 0.970) | 56.9% | |
| Patients | 9 (0.092) | 51 (0.520) | 38 (0.388) | χ 2 = 5.679 | 69 (0.352) | 127 (0.648) | |||
| Controls | 37 (0.196) | 94 (0.497) | 58 (0.307) | P = 0.058 | 168 (0.444) | 210 (0.556) | |||
| rs3767546 | A/A | A/T | T/T | A | T | 0.457 | 0.751 (0.353, 1.597) | 10.6% | |
| Patients | 0 (0) | 10 (0.102) | 89 (0.908) | χ 2 = 0.816 | 10 (0.051) | 188 (0.949) | |||
| Controls | 1 (0.005) | 23 (0.122) | 165 (0.873) | P = 0.665 | 25 (0.067) | 353 (0.934) | |||
| rs117962659 | C/C | C/A | A/A | C | A | 0.186 | 1.920 (0.730, 5.049) | 12.4% | |
| Patients | 0 (0) | 6 (0.061) | 93 (0.939) | χ 2 = 0.895 | 6 (0.030) | 192 (0.970) | |||
| Controls | 1 (0.005) | 15 (0.080) | 172 (0.915) | P = 0.639 | 17 (0.045) | 359 (0.955) | |||
| rs2275860 | A/A | A/G | G/G | A | G | 0.448 | 0.745 (0.351, 1.588) | 10.8% | |
| Patients | 1 (0.010) | 8 (0.081) | 90 (0.909) | χ 2 = 3.564 | 10 (0.051) | 188 (0.949) | |||
| Controls | 0 (0) | 25 (0.133) | 163 (0.867) | P = 0.168 | 25 (0.066) | 351 (0.934) | |||
| c.192 + 353 C/A | C/C | C/A | A/A | C | A | 0.022 | 0.095 (0.013, 0.714) | 87.2% | |
| Patients | 98 (0.990) | 1 (0.010) | 0 (0) | χ 2 = 8.333 | 197 (0.995) | 1 (0.005) | |||
| Controls | 168 (0.898) | 19 (0.102) | 0 (0) | P = 0.004 | 355 (0.949) | 19 (0.051) | |||
| c.192 + 463 G/A | G/G | A/G | A/A | G | A | 0.019 | 0.090 (0.012, 0.675) | 89.7% | |
| Patients | 98 (0.99) | 1 (0.01) | 0 (0) | χ 2 = 8.925 | 197 (0.995) | 1 (0.005) | |||
| Controls | 167 (0.893) | 20 (0.107) | 0 (0) | P = 0.003 | 354 (0.947) | 20 (0.053) | |||
Note: OR: odds ratio, at which the minor allele was viewed as an exposure factor in this case-control study.
4. Discussion
In our study, we analyzed a 3992-nucleotide sequence of the TNNT2 gene containing exons 6, 7, 8, 9, and 10 and introns 5, 6, 7, 8, 9, 10, and 11. We identified 6 novel TNNT2 mutations in DCM patients, including a missense mutation (G12026T, c.252 G>T, Leu84Phe) and 5 novel intronic mutations. We also found that the genotype of two novel single-nucleotide polymorphisms (c.192 + 353 C>A, c.192 + 463 G>A) and the previously identified rs3729843 [12] were correlated with DCM. These data indicate that TNNT2 variants are associated with DCM in Chinese population. To our knowledge, the two novel SNPs and the six mutations, in which five are in intron and one is in exon, have not been reported in dilated cardiomyopathy before.
Dilated cardiomyopathy is an important cause of heart failure and the leading indication for cardiac transplantation worldwide [14]. With careful evaluation of relatives, familial disease can be identified in approximately 30% of patients with idiopathic DCM [15]. Genetic characterization of DCM has been a challenging task owing to incomplete knowledge of the genes involved in the disease as well as difficulties in determining the clinical significance of DNA variants identified in patients. More than 40 different genes have been implicated in the pathophysiology of DCM [5, 6], and TNNT2 mutations in familial DCM are approximately 3–6% [10, 11].
Cardiac troponin (cTn) is made up of three distinct subunits, each named according to its function: cardiac troponin I (cTnI) can inhibit the actomyosin ATPase activity independently of the other Tn subunits; cardiac troponin C (cTnC) binds Ca2+ to a low affinity Ca2+-specific binding site, relieving the cTnI inhibition; and cardiac troponin T (cTnT) binds the entire cTn complex to tropomyosin (Tm) [16, 17]. Recent studies have suggested that cardiac TnT is essential not only for the structural integrity of the troponin complex but also for sarcomere assembly and cardiac contractility [18]. The troponin complex is a calcium sensor that regulates the contraction of striated muscle and modulates actomyosin ATPase activity and force [19]. Over the past decade, mutations in the TNNT2 gene have been found to be associated with familial HCM and DCM [3, 10, 11, 20–22]. Many studies in reconstituted systems have provided valuable information on the functional effects of disease-associated mutations in TnT [23–28].
Independent laboratories have consistently observed that changes in Ca2+-sensitivity, ATPase activity, or force development due to cTn mutations decrease in DCM [23, 24]. All DCM mutations in cTnT (R131W, R141W, R205L, and ΔK210) showed decreases in ATPase activation [25–28]. Mutant transgenic mice expressing cTnT I79N show increased Ca2+ sensitivity of force development and decreased force per cross-sectional area [24]. The observed decrease in force per cross-sectional area by this mutation has been reported by different groups [29, 30]. Lin and colleagues [31] demonstrated that transgenic mice carrying either cTnT mutations F110I or R278C displayed significant increases in energy cost. Alterations in the energy cost could be explained as an increase in the rate of dissociation of myosin cross bridges and/or a decrease in the average force per cross bridge. The functional effects of a cTnT mutation (ΔK210) linked to DCM have been explored in reconstituted systems and in animal models. In vitro studies showed that cTnT ΔK210 exchanged in skinned porcine cardiac fibers decreased the relative maximal force [27, 29].
In the present study, we found a missense mutation (G12026T, c.252 G>T, Leu84Phe) in one DCM patient. This mutation was first reported in DCM patients and resulted in the substitution of a phenylalanine for the normal leucine at residue 84 in TnT. We used the website http://www.proteinmodelportal.org/query/pdb to query the crystal structure of the 52 kDa domain of human TNNT2; however, the crystal structure around the 84th amino acid in the protein is not reported up to now. Therefore, we estimate the damage of TNNT2 Leu 84 Phe transition using website http://genetics.bwh.harvard.edu/pph/. The result indicated that the mutation is predicted to be probably damaging with a score of 0.992 (total score is equal to one; sensitivity 0.7; specificity 0.97). The 84th amino acid in TNNT2 is a very conserved site in human, rat, mice, and zebrafish. The 84th amino acid was substituted by phenylalanine which function and structure with a benzene ring is different from leucine. Therefore, this mutation might contribute to the occurrence of dilated cardiomyopathy.
In addition, we found 5 intronic mutations in DCM patients as well as a significant association between the genotypes of 2 novel SNPs and rs3729843 with DCM. Although there were no encoding amino acid changes in these mutations and polymorphisms, numerous functional roles for introns have been elucidated; these functions include augmenting proteome diversity by enabling alternative splicing, enhancing gene expression, and harboring various cis- and trans-regulatory elements [32–37]. In addition, some studies also showed that some polymorphisms in noncoding regions may influence the expression level of the encoded protein, thus resulting in a more subtle variation in the associated phenotype [38]. These results suggest that TNNT2 mutations and polymorphisms may play an important role in DCM in the Chinese Han population. However, additional functional analyses are needed to confirm the role of these variants in DCM pathogenesis.
In conclusion, based on the sequence analysis of TNNT2 in DCM patients, the present study identified 6 novel TNNT2 variants, a missense mutation (G12026T, c.252 G>T, Leu84Phe), 5 mutations in introns, SNP rs3729843, and 2 novel single-nucleotide polymorphisms (c.192+353 C>A, c.192+463 G>A). These TNNT2 variants might be associated with DCM in the Chinese population.
5. Limitations of the Study
There are clear limitations to the feasibility and practicality of screening for mutations in only one gene. Genetic analysis as performed in this study, which is limited to only a small number of potentially involved exons, does not seem to be applicable. It was impossible to screen all other genes associated with DCM, so we cannot exclude the possibility that some patients were compound heterozygotes. Functional analyses of mutations are required to confirm the findings in this study. In addition, most of patients and many SNPs are the same as those in the previous study [12] although the detecting method is different.
Acknowledgments
This study was supported in part by Grants from the National Natural Science Foundation of China (nos. 81000104, 81160141), the China Postdoctoral Science Foundation (2012M510356, 2013M542120), and the Postdoctoral Fellows Foundation of the Chinese Academy of Medical Sciences (2011-XH-9).
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the european society of cardiology working group on myocardial and pericardial diseases. European Heart Journal. 2008;29(2):270–276. doi: 10.1093/eurheartj/ehm342. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113(14):1807–1816. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 3.Kamisago M, Sharma SD, DePalma SR, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. The New England Journal of Medicine. 2000;343(23):1688–1696. doi: 10.1056/NEJM200012073432304. [DOI] [PubMed] [Google Scholar]
- 4.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2005;45(7):969–981. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 5.Dellefave L, McNally EM. The genetics of dilated cardiomyopathy. Current Opinion in Cardiology. 2010;25(3):198–204. doi: 10.1097/HCO.0b013e328337ba52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hershberger RE, Siegfried JD. Update 2011: clinical and genetic issues in familial dilated cardiomyopathy. Journal of the American College of Cardiology. 2011;57(16):1641–1649. doi: 10.1016/j.jacc.2011.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.García-Castro M, Coto E, Reguero JR, et al. Mutations in Sarcomeric Genes MYH7, MYBPC3, TNNT2, TNNI3, and TPM1 in Patients With Hypertrophic Cardiomyopathy. Revista Espanola de Cardiologia. 2009;62(1):48–56. [PubMed] [Google Scholar]
- 8.Landstrom AP, Adekola BA, Bos JM, Ommen SR, Ackerman MJ. PLN-encoded phospholamban mutation in a large cohort of hypertrophic cardiomyopathy cases: summary of the literature and implications for genetic testing. American Heart Journal. 2011;161(1):165–171. doi: 10.1016/j.ahj.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.García-Castro M, Reguero JR, Batalla A, et al. Hypertrophic cardiomyopathy: low frequency of mutations in the β-myosin heavy chain (MYH7) and cardiac troponin T (TNNT2) genes among Spanish patients. Clinical Chemistry. 2003;49(8):1279–1285. doi: 10.1373/49.8.1279. [DOI] [PubMed] [Google Scholar]
- 10.Chang AN, Parvatiyar MS, Potter JD. Troponin and cardiomyopathy. Biochemical and Biophysical Research Communications. 2008;369(1):74–81. doi: 10.1016/j.bbrc.2007.12.081. [DOI] [PubMed] [Google Scholar]
- 11.Hershberger RE, Pinto JR, Parks SB, et al. Clinical and functional Characterization of TNNT2 mutations identified in patients with dilated cardiomyopathy. Circulation: Cardiovascular Genetics. 2009;2(4):306–313. doi: 10.1161/CIRCGENETICS.108.846733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li X, Wang H, Luo R, et al. TNNT2 gene polymorphisms are associated with susceptibility to idiopathic dilated cardiomyopathy in the Han Chinese population. BioMed Research International. 2013;2013:6 pages. doi: 10.1155/2013/201372.201372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Research. 1988;16(3, article 1215) doi: 10.1093/nar/16.3.1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor DO, Edwards LB, Boucek MM, et al. Registry of the international society for heart and lung transplantation: twenty-fourth official adult heart transplant report—2007. Journal of Heart and Lung Transplantation. 2007;26(8):769–781. doi: 10.1016/j.healun.2007.06.004. [DOI] [PubMed] [Google Scholar]
- 15.Mahon NG, Murphy RT, MacRae CA, Caforio ALP, Elliott PM, McKenna WJ. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Annals of Internal Medicine. 2005;143(2):108–115. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- 16.Solaro RJ, Rarick HM. Troponin and tropomyosin: Proteins that switch on and tune in the activity of cardiac myofilaments. Circulation Research. 1998;83(5):471–480. doi: 10.1161/01.res.83.5.471. [DOI] [PubMed] [Google Scholar]
- 17.Farah CS, Reinach FC. The troponin complex and regulation of muscle contraction. The FASEB Journal. 1995;9(9):755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 18.Sehnert AJ, Huq A, Weinstein BM, Walker C, Fishman M, Stainier DYR. Cardiac troponin T is essential in sarcomere assembly and cardiac contractility. Nature Genetics. 2002;31(1):106–110. doi: 10.1038/ng875. [DOI] [PubMed] [Google Scholar]
- 19.Potter JD, Sheng Z, Pan B, Zhao J. A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. The Journal of Biological Chemistry. 1995;270(6):2557–2562. doi: 10.1074/jbc.270.6.2557. [DOI] [PubMed] [Google Scholar]
- 20.Mogensen J, Kubo T, Duque M, et al. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. Journal of Clinical Investigation. 2003;111(2):209–216. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thierfelder L, Watkins H, MacRae C, et al. α-tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell. 1994;77(5):701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 22.Townsend PJ, Farza H, MacGeoch C, et al. Human cardiac troponin T: identification of fetal isoforms and assignment of the TNNT2 locus to chromosome 1q. Genomics. 1994;21(2):311–316. doi: 10.1006/geno.1994.1271. [DOI] [PubMed] [Google Scholar]
- 23.Szczesna D, Zhang R, Zhao J, Jones M, Guzman G, Potter JD. Altered regulation of cardiac muscle contraction by troponin T mutations that cause familial hypertrophic cardiomyopathy. The Journal of Biological Chemistry. 2000;275(1):624–630. doi: 10.1074/jbc.275.1.624. [DOI] [PubMed] [Google Scholar]
- 24.Miller T, Szczesna D, Housmans PR, et al. Abnormal contractile function in transgenic mice expressing a familial hypertrophic cardiomyopathy-linked troponin T (I79N) mutation. The Journal of Biological Chemistry. 2001;276(6):3743–3755. doi: 10.1074/jbc.M006746200. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto S, Lu Q-W, Harada K, et al. Ca 2+-desensitizing effect of a deletion mutation ΔK210 in cardiac troponin T that causes familial dilated cardiomyopathy. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(2):913–918. doi: 10.1073/pnas.022628899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robinson P, Mirza M, Knott A, et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2002;277(43):40710–40716. doi: 10.1074/jbc.M203446200. [DOI] [PubMed] [Google Scholar]
- 27.Venkatraman G, Gomes AV, Kerrick WGL, Potter JD. Characterization of troponin T dilated cardiomyopathy mutations in the fetal troponin isoform. Journal of Biological Chemistry. 2005;280(18):17584–17592. doi: 10.1074/jbc.M409337200. [DOI] [PubMed] [Google Scholar]
- 28.Venkatraman G, Harada K, Gomes AV, Kerrick WG, Potter JD. Different functional properties of troponin T mutants that cause dilated cardiomyopathy. The Journal of Biological Chemistry. 2003;278(43):41670–41676. doi: 10.1074/jbc.M302148200. [DOI] [PubMed] [Google Scholar]
- 29.Sweeney HL, Feng HS, Yang Z, Watkins H. Functional analyses of troponin T mutations that cause hypertrophic cardiomyopathy: Insights into disease pathogenesis and troponin function. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(24):14406–14410. doi: 10.1073/pnas.95.24.14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hernandez OM, Szczesna-Cordary D, Knollmann BC, et al. F110I and R278C troponin T mutations that cause familial hypertrophic cardiomyopathy affect muscle contraction in transgenic mice and reconstituted human cardiac fibers. The Journal of Biological Chemistry. 2005;280(44):37183–37194. doi: 10.1074/jbc.M508114200. [DOI] [PubMed] [Google Scholar]
- 31.Lin D, Bobkova A, Homsher E, Tobacman LS. Altered cardiac troponin T in vitro function in the presence of a mutation implicated in familial hypertrophic cardiomyopathy. Journal of Clinical Investigation. 1996;97(12):2842–2848. doi: 10.1172/JCI118740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Graveley BR. Alternative splicing: Increasing diversity in the proteomic world. Trends in Genetics. 2001;17(2):100–107. doi: 10.1016/s0168-9525(00)02176-4. [DOI] [PubMed] [Google Scholar]
- 33.Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Molecular and Cellular Biology. 1991;11(6):3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Le Hir H, Nott A, Moore MJ. How introns influence and enhance eukaryotic gene expression. Trends in Biochemical Sciences. 2003;28(4):215–220. doi: 10.1016/S0968-0004(03)00052-5. [DOI] [PubMed] [Google Scholar]
- 35.Valencia P, Dias AP, Reed R. Splicing promotes rapid and efficient mRNA export in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(9):3386–3391. doi: 10.1073/pnas.0800250105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rearick D, Prakash A, McSweeny A, Shepard SS, Fedorova L, Fedorov A. Critical association of ncRNA with introns. Nucleic Acids Research. 2011;39(6):2357–2366. doi: 10.1093/nar/gkq1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mattick JS, Makunin IV. Non-coding RNA. Human Molecular Genetics. 2006;15:R17–R29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 38.Ho E, Bhindi R, Ashley EA, Figtree GA. Genetic analysis in cardiovascular disease: a clinical perspective. Cardiology in Review. 2011;19(2):81–89. doi: 10.1097/CRD.0b013e318207ffac. [DOI] [PubMed] [Google Scholar]