

Abstract
Alzheimer's disease is the most prevalent cause of dementia and is associated with accumulation of amyloid-β peptide (Aβ), particularly the 42-amino acid Aβ1-42, in the brain. Aβ1-42 levels can be decreased by γ-secretase modulators (GSM), which are small molecules that modulate γ-secretase, an enzyme essential for Aβ production. BMS-869780 is a potent GSM that decreased Aβ1-42 and Aβ1-40 and increased Aβ1-37 and Aβ1-38, without inhibiting overall levels of Aβ peptides or other APP processing intermediates. BMS-869780 also did not inhibit Notch processing by γ-secretase and lowered brain Aβ1-42 without evidence of Notch-related side effects in rats. Human pharmacokinetic (PK) parameters were predicted through allometric scaling of PK in rat, dog, and monkey and were combined with the rat pharmacodynamic (PD) parameters to predict the relationship between BMS-869780 dose, exposure and Aβ1-42 levels in human. Off-target and safety margins were then based on comparisons to the predicted exposure required for robust Aβ1-42 lowering. Because of insufficient safety predictions and the relatively high predicted human daily dose of 700 mg, further evaluation of BMS-869780 as a potential clinical candidate was discontinued. Nevertheless, BMS-869780 demonstrates the potential of the GSM approach for robust lowering of brain Aβ1-42 without Notch-related side effects.
1. Introduction
Alzheimer's disease (AD) is the most prevalent cause of dementia. More than 35 million people have dementia worldwide and the prevalence is expected to double in the next 20 years [1]. Medicines are available for treatment of symptoms but provide limited benefit and do not prevent AD [2]. The cause of AD is not completely understood, but a widely held view of its pathogenesis is based on the amyloid hypothesis. Accumulation and aggregation of the toxic amyloid-β peptide (Aβ), particularly the 42-amino acid form Aβ1-42 [3], initiates neuronal dysfunction that eventually leads to brain atrophy, dementia, and death [4, 5]. Aβ is naturally produced in the brain by proteolytic processing of a type I transmembrane protein, the amyloid-β precursor protein (APP). APP is processed by the β-site APP cleaving enzyme (BACE), releasing a secreted ectodomain and a membrane-anchored C-terminal fragment (APP-CTFβ). Subsequent cleavage of APP-CTFβ within the transmembrane domain by γ-secretase then produces a cytosolic intracellular domain (AICD) and Aβ, which is secreted. In addition, a fraction of APP is cleaved by α-secretase at a site within the Aβ sequence to produce APP-CTFα, which is subsequently cleaved by γ-secretase to produce nonamyloidogenic peptides [6]. Compounds targeting either BACE or γ-secretase have been tested in clinical trials, but adequate Aβ-lowering at tolerated doses in patients has been a challenge [7].
γ-Secretase is a lipid bilayer-embedded aspartyl protease consisting of four core subunits; nicastrin, Aph-1, Pen-2, and presenilin. Presenilin carries the active site aspartyl residues, whereas the other subunits play ancillary roles in enzymatic activity and maturation [6, 8, 9]. Structural studies of γ-secretase using electron micrographic image analysis and biochemical methods to map locations of amino acid residues suggest a compact structure with the active site contained within a hydrophilic chamber surrounded by transmembrane domains [10–14]. High resolution structure of γ-secretase has not been reported, but, based on analogy to X-ray crystallography of a presenilin homolog [15], it seems likely that the active site aspartates would be located within an intramembrane pore surrounded by the transmembrane domains of presenilin. The biological role of γ-secretase involves the proteolytic cleavage of transmembrane domains of at least 80 different protein substrates, including APP and the Notch family [16]. Although the physiological significance of substrate processing is unknown in most cases, γ-secretase cleavage of Notch and production of the Notch intracellular domain (NICD) are critical for adult cell differentiation in the immune system and gastrointestinal tract [17, 18]. Inhibitors of γ-secretase (GSI) can be effective for Aβ lowering; however Notch inhibition likely contributes to dose-limitation [19].
A class of small molecules that avoid Notch inhibition are the γ-secretase modulators (GSM), which, like GSIs, target presenilin [20–24]. In contrast to GSIs, which inhibit Aβ production, GSMs have relatively little overall effect on Aβ production. Instead, GSMs change the lengths of Aβ peptides produced, causing decreased amounts of longer peptides, such as Aβ1-40 and Aβ1-42, and increased amounts of shorter peptides, such as Aβ1-37 and Aβ1-38 [25]. GSMs have analogous effects on other γ-secretase substrates, including Notch, causing a shift from the longer Nβ1-25 to the shorter Nβ1-21 Notch-derived peptides [26, 27]. The shift from longer to shorter Aβ peptides was first described for several nonsteroidal anti-inflammatory drugs (NSAID), which have low potency GSM activity [28], and was subsequently found in other small molecules and natural products [25]. The effect of GSMs is thought to result from allosteric stimulation of the stepwise cleavage mechanism of γ-secretase. The stepwise mechanism initiates with an endopeptidic cleavage of APP-CTF near the cytosolic face of the lipid bilayer, at either position 48 or position 49 (using the conventional amino acid numbering starting at position 1 for the aspartyl residue at the N-terminus of Aβ). Subsequently, γ-secretase carries out a series of carboxypeptidase-like cleavages at three or four amino acid intervals thereby producing tripeptides or tetrapeptides and Aβ peptides of different lengths [29, 30]. Typically, the 40-amino acid long Aβ1-40 is the major product, but lesser amounts of other Aβ peptides such as Aβ1-38 and Aβ1-42 are also produced. For example, Aβ1-40 biogenesis appears to require four cycles of APP-CTF cleavage at positions 49, 46, 43, and 40, resulting in the release of three tripeptides and one AICD (amino acids 50–99) for each Aβ1-40 peptide produced. Likewise, production of the Aβ1-42 peptide is associated with a series of cleavages that start at position 48 of APP-CTF [30, 31]. In the presence of a GSM, γ-secretase carries out an increased number of carboxypeptidase cycles per molecule of APP-CTF substrate, resulting in shorter Aβ peptides without substantially affecting the overall amount of Aβ produced [27, 32].
Despite having low potency, NSAID GSMs such as flurbiprofen were reported to lower brain Aβ1-42 in rodents [33, 34]. However, in clinical trials, flurbiprofen (tarenflurbil) was found to have no effect on Aβ1-42 in cerebrospinal fluid even at high doses [35]. High potency GSMs have also entered early stage clinical trials, but effects on Aβ1-42 in cerebrospinal fluid have not been reported [36, 37]. Many further GSMs have been evaluated in vitro and in animal studies, which, like the NSAID GSMs, do not inhibit Notch or other γ-secretase substrates [25, 38–51]. In addition, chronic dosing of GSMs has been reported to ameliorate plaque pathology [39, 45, 52] and enhance cognition in APP transgenic mice [48, 52, 53]. Thus, GSMs are capable of lowering brain Aβ1-42 and are expected to avoid the dose limitations of GSIs that are due to Notch-related side effects. A number of studies have reported at least some of the systemic exposure data associated with brain Aβ1-42 lowering, thus giving an idea of active exposures and doses for GSM [39–42, 44–46, 52]. However, preclinical predictions relating to safety margins or human dose have not been reported. Furthermore, the active exposures and doses in rodents are high, making it a matter of conjecture whether or not any of the currently published GSMs are likely to exhibit adequate safety margins or clinically acceptable doses. Here we describe the pharmacological properties of the GSM BMS-869780, a potent bicyclic triazole GSM [54, 55], and the quantitative predictions of human dose and off-target side effects. While these outcomes prevented the development of BMS-869780, they put in perspective the extent of further enhancements in drug-like properties that would be necessary to justify clinical testing of a future GSM.
2. Materials and Methods
2.1. Compounds
The GSM, BMS-869780 [54, 55], and the GSIs BMS-299897 [56] and BMS-433796 [57] have been reported previously. The GSI BMS-698861 is described in a BMS patent [58]. Chemical structures are shown in Figure 1(a).
Figure 1.
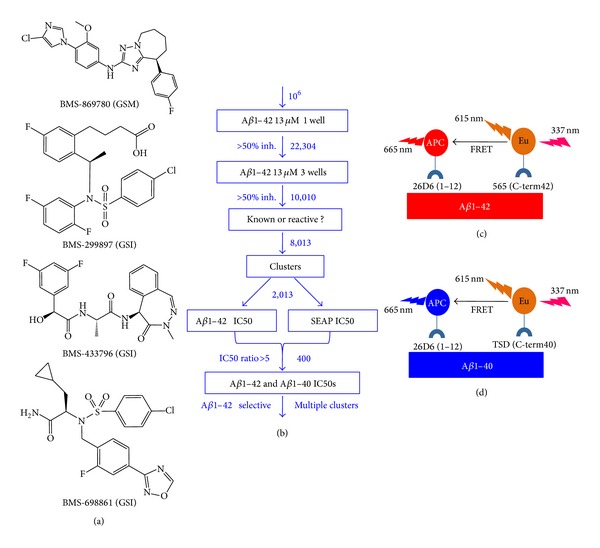
(a) Chemical structures of the compounds used in this study are shown. (b) Overview of the HTS and subsequent triage of compounds summarizes experimentation steps in boxes, with outcomes indicated beside the arrows. Costs of reagents and disposables were a major consideration in the design, particularly the initial screen of 106 samples. (c) Principle of the Aβ1-42 immunoassay; simultaneous binding of monoclonal antibody conjugates 252-APC and 565-Eu (specific for C-terminus of Aβ1-42) to Aβ1-42 leads to FRET-based emission at 665 nm. The ratio of emission at 665 nm to fluorescence at 615 nm represents the level of Aβ1-42 in the sample. (d) Principle of the Aβ1-40 immunoassay; same as described above for the Aβ1-42 immunoassay, except that the monoclonal antibody conjugate TSD-Eu (specific for C-terminus of Aβ1-40) was used in place of 565-Eu.
2.2. Cell Cultures
H4-APPsw cell cultures were maintained on Dulbecco's modified Eagle's medium (DMEM) supplemented with L-glutamine (2 mM), fetal bovine serum (10%), and G418 (100 μg/mL). For IC50 determinations, cells were harvested, resuspended in DMEM supplemented with 0.0125% bovine serum albumin, and dispensed into 384-well plates (1.5 × 104 cells per well). Aβ1-42 and Aβ1-40 assays, and Notch inhibition assays, were carried out as described previously [59]. Mouse embryonic fibroblasts deficient in PS1 and PS2 (MEF dKO) [60] were passaged twice per week in DMEM/F-12 medium, consisting of a 1 : 1 mixture of DMEM and F-12 nutrient mixture supplemented with 10% fetal bovine serum, penicillin, and streptomycin. For expression of human presenilin-1 in MEF dKO cells, the full length human presenilin-1 cDNA open reading frame was cloned between the BamHI and XhoI sites of the vector pcDNA5/FRT (Invitrogen), placing presenilin-1 expression under the control of the CMV promoter. The M146V mutated allele was introduced into the presenilin-1 expression construct by polymerase chain reaction using the oligonucleotide primers 5′-CAGTGTCATTGTTGTCGTGACTATCCTCCTGGTGG-3′ and 5′-CCACCAGGAGGATAGTCACGACAACAATGACACTG-3 (QuikChange kit, Invitrogen). MEF dKO cultures were cotransfected with DNA constructs expressing APP-CTF, encoding the C-terminal 99 amino acids of APP [61] and either the human presenilin-1 or presenilin-1 M146V allele. Transfected cultures were incubated overnight at 37°C in 5% CO2 atmosphere and then harvested, resuspended in DMEM/F-12 medium, dispensed into 96-well culture plates, and incubated for 6 hours at 37°C in 5% CO2 atmosphere. Culture medium was then replaced with Ultraculture serum free medium (Lonza, Rockland, ME) with or without compounds at a range of concentrations and incubated overnight.
2.3. Aβ Antibodies and Conjugates
Anti-Aβ monoclonal antibodies and their epitopes used in this study were 4G8 (Aβ17-24; Covance), 252Q6 (rodent Aβ1-12; Invitrogen), D2A6H (Aβ37 C-terminal; Cell Signaling, catalog number 12356BF), TSD (Aβ40 C-terminal), 26D6 (human Aβ1-12), and 565 (Aβ42 C-terminal; Bristol-Myers Squibb). The covalent antibody-fluorophore conjugates were made at Perkin-Elmer, including TSD-Europium cryptate (TSD-Eu), 565-Europium cryptate (565-Eu), and 26D6-allophycocyanin (26D6-APC). Streptavidin-horseradish peroxidase (SA-HRP) and 4G8-biotin conjugates were from Covance. Horseradish peroxidase (HRP) conjugates of 565, 26D6 and 252Q6 (565-HRP, 26D6-HRP and 252Q6-HRP) were made using preactivated HRP (Easylink, Zymed/Invitrogen). The 6E10-sulfo-tag conjugate was from Mesoscale Discovery (catalog number K15148E-1), and the 252Q6-sulfo-tag conjugate was made using a kit (Mesoscale Discovery catalog number R91AN-1).
2.4. Immunoassays
IC50 determinations for Aβ1-42 and Aβ1-40 in H4-APPsw cultures in 384-well format were determined using homogeneous time-resolved fluorescence immunoassays as previously described [59]. The principle of these assays is illustrated in Figures 1(c) and 1(d). In other experiments using H4-APPsw cultures, Aβ was quantified by ELISA; for Aβ1-42 the combination of monoclonal antibodies was 26D6 and 565-HRP; for Aβ1-40 it was TSD and 26D6-HRP, and for Aβ1-x it was 4G8 and 26D6-HRP. In some experiments a novel 4-plex Aβ electrochemiluminescence immunoassay was used (Mesoscale Discovery catalog number N45ZA-1). Briefly, the 4-plex was carried out in 96-well format, with 4 separate spots of capture antibodies in each well. The 96-well plates were prepared by the manufacturer, with spots of monoclonal antibodies for Aβ1-42, Aβ1-40, and Aβ1-38, and an additional fourth spot of streptavidin in each well. Plates were initially incubated with blocking buffer (5% BSA in phosphate buffered saline, 200 μL per well) for 2 hours at ambient temperature and then with D2A6H-biotin conjugate (50 ng/mL in 1% BSA, phosphate buffered saline, 25 μL per well) for 1 hour. Plates were then rinsed with phosphate buffered saline before addition of experimental samples for determination of Aβ1-42, Aβ1-40, Aβ1-38, and Aβ1-37 levels, following the manufacturer's instructions as for the Aβ 3-plex kit (catalog number K15148E-1). Rat brain extracts for use in the 4-plex assay were made in 0.2% diethylamine, as previously described [62]. For detection of rat Aβ peptides in the 4-plex (Figure 5), 252Q6-sulfo-tag conjugate was used, and for detection of human Aβ peptides from cell cultures 6E10-sulfo-tag conjugate was used. For Aβ in transiently transfected PS1/PS2 dKO fibroblasts, Aβ1-42 was quantified using an ELISA kit (WACO), and Aβ1-40 was quantified by ELISA as described above for H4-APPsw cultures. For triple transgenic mice (3xTg; [63]), human transgenic Aβ1-42 was assayed in brain homogenates using an ELISA kit (WACO). For the rat and mouse experiments illustrated in Figures 7 and 8, brain samples were prepared by solid phase extraction [64], and endogenous rat brain Aβ1-42 and Aβ1-40 were quantified by ELISA as previously described for wild type mice [65]. For brain extracts made using solid phase extraction, calibration of Aβ was relative, based on the approximately linear response of the assay in the range tested. For brain extracts made in 0.2% diethylamine, Aβ1-42, Aβ1-40, Aβ1-38, and Aβ1-37 concentrations in brain and cell culture samples were determined by fitting the results of immunoassays against calibration curves derived from a range of dilutions of the corresponding synthetic peptides on each assay plate using a quadratic curve fit (Graphpad Prizm 5.0). Aβ1-x was calibrated in the same way against synthetic Aβ1-40 peptide. Results were expressed in units of pM, corrected for sample dilution.
Figure 5.

BMS-869780 modulated Aβ but did not cause accumulation of βCTF or αCTF in rat brain. Rats were given oral doses of BMS-869780, and levels of brain Aβ, βCTF, and αCTF were determined 24 hours later. For comparison, BMS-698861 was dosed in a separate experiment and samples were taken 5 hours later. (a) Brain levels of Aβ1-42 (red), Aβ1-40 (green), Aβ1-38 (blue), and Aβ1-37 (purple) are shown as bars stacked upon one another. The total height of each bar therefore represents the sum of the four peptides. (b) Aβ1-42 (red—left Y axis) and Aβ1-x (grey—right Y axis). The same results for Aβ1-42 are plotted in both (a) and (b). (c) Rat brain βCTF was detected by western blotting of immunoprecipitates from samples of the same rat brains used for Aβ determinations. V, vehicle groups; results from rats dosed with 1.9, 22, 100, and 235 mg/kg of BMS-869780 and 10 mg/kg BMS-698861 (GSI) are indicated. (d) Western blots of immunoprecipitated αCTF from the same rat brain samples. (e) and (f) quantification of western blots shown in (c) and (d), respectively, expressed relative to percent of average level of CTF in vehicle-treated rats. Actual doses of BMS-869780 were determined by analysis of concentrations in left-over dosing solutions.
Figure 7.
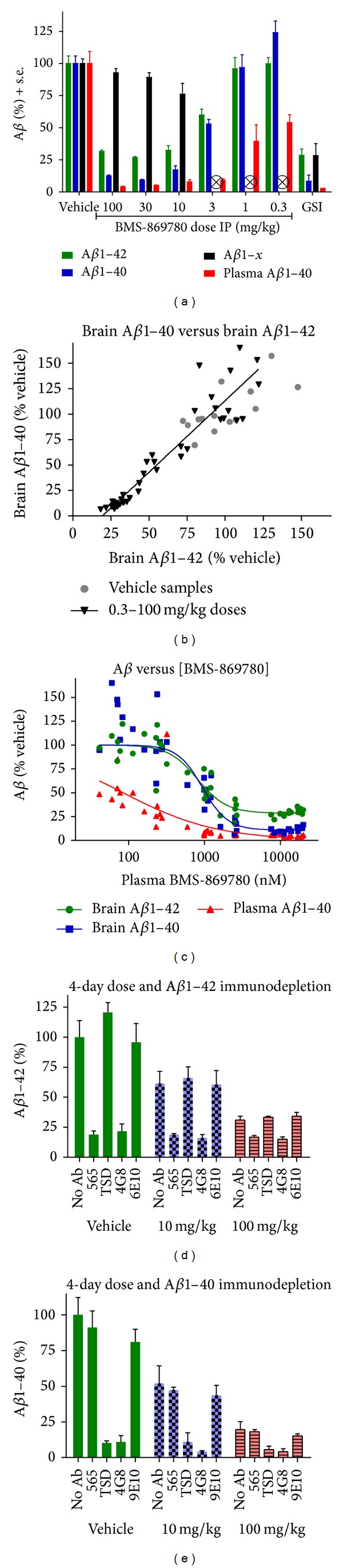
BMS-869780 dose response and evaluation of residual Aβ levels in rat brain. (a) Groups of rats received intraperitoneal (IP) injections of vehicle or BMS-869780 at doses of 100, 30, 10, 3, 1, and 0.3 mg/kg. Additional rats were dosed with GSI BMS-698861 at 30 mg/kg as a positive control for Aβ-lowering. Group sizes were seven rats for each dose of BMS-869780 and 14 rats for vehicle and GSI dose groups. Brain and plasma were harvested 5 hours after dosing. Brain Aβ1-42 (green), brain Aβ1-40 (blue), brain Aβ1-x (black), and plasma Aβ1-40 (red) were determined. Values are expressed as % relative to vehicle group mean. Whiskers represent standard error. ⊗Aβ1-x was not determined in the groups dosed at 3, 1, and 0.3 mg/kg. (b) Brain Aβ1-40 was plotted against brain Aβ1-42 for each rat dosed with BMS-869780 (black ▼) and for each rat dosed with vehicle (grey ●). Values are expressed as % relative to vehicle group mean. Whiskers represent standard error. (c) Brain Aβ1-42 (●), brain Aβ1-40 (■), and plasma Aβ1-40 (▲) were plotted against plasma concentration of BMS-869780 and the data were evaluated by fit to a four-parameter dose response curve. The top of the dose response curve was defined by vehicle group mean (100%), and the apparent IC50 values in terms of the plasma BMS-869780 concentration obtained for brain Aβ1-42, brain Aβ1-40, and plasma Aβ1-40 were 807 nM, 943 nM, and 84 nM, respectively. The respective 95% confidence intervals were 618–1053 nM, 704–1264 nM, and 44–158 nM. (d) Rats were dosed once daily with BMS-869780 for 4 days at 10 and 100 mg/kg or vehicle, plasma, and brain samples were taken 5 hours after the last dose, and immunodepletion of brain extracts was carried out prior to Aβ1-42 ELISA assays. Specific monoclonals used were 565 (Aβ1-42 selective), TSD (Aβ1-40 selective), 4G8 (binds both Aβ1-42 and Aβ1-40), and 6E10 (does not bind rat Aβ). After immunodepletion, Aβ1-42 was assayed by ELISA. (e) Same as described in (d), except that Aβ1-40 ELISA was carried out following the immunodepletion.
Figure 8.

Analysis of the PK/PD relationship for BMS-869780 in rats and mice. Rats were dosed orally with BMS-869780 at 10 mg/kg, and triple transgenic mice were orally dosed at 30 mg/kg and 100 mg/kg. Additional groups of rats and mice were dosed with vehicle alone. Brain and plasma were harvested at 3, 5, 8, 16, and 24 hours after dosing for determination of brain Aβ1-42 and plasma BMS-869780 concentration. Group sizes were 5 rats or 4 mice. Whiskers represent standard error. (a) Plasma concentrations of BMS-869780 were determined for the mice dosed at 100 mg/kg (▲) and 30 mg/kg (■) and for the rats dosed at 10 mg/kg (▼). The data were fit to a one-compartment PK model and the predicted plasma BMS-869780 concentrations are shown for mouse (solid lines) and rat (broken line). (b) Brain Aβ1-42 levels were determined for the mice dosed with BMS-869780 at 100 mg/kg (▲), 30 mg/kg (■), or vehicle alone (●). The data were fitted using the indirect pharmacodynamic response model and predicted values are shown (solid lines). (c) Brain Aβ1-42 levels were determined for the rats dosed with BMS-869780 at 10 mg/kg (▼) or vehicle alone (●). The data were fitted using the indirect pharmacodynamic response model and predicted values are shown (dashed line). The values of the PK and PD parameters determined from these experiments are summarized in Table 2. (d) Brain Aβ1-40 levels determined in the same rats as illustrated in (c).
2.5. Immunodepletion of Aβ
For immunodepletion of rat brain Aβ, solid phase extracts [64] were pooled within treatment groups. The pools were divided into equal aliquots and incubated with or without monoclonals 565, TSD, 4G8, or 6E10 (10 μg) at 4°C overnight. Protein G beads (50 μL; EZview, Sigma) were added, and incubation was continued with agitation for 1 hour. The beads were removed by centrifugation, and Aβ1-40 and Aβ1-42 in the unbound fraction were quantified by ELISA, as described above.
2.6. Western Blotting
For western blotting of Aβ peptides from H4-APPsw cell cultures, Aβ was immunoprecipitated directly from the cell culture medium and was eluted from the protein G beads by addition of lithium dodecyl sulfate (LDS) electrophoresis sample buffer (Invitrogen). Aβ peptides were separated by gel electrophoresis in the presence of 8 M urea [66], transferred to PVDF membrane, and detected by western blotting using monoclonal 26D6-HRP conjugate.
For western blotting of APP-CTF in cell lysates, H4-APPsw cell cultures in T-75 flasks were rinsed with DPBS, harvested, isolated by centrifugation, and stored at −80°C until needed. Cells were suspended in SDS sample buffer (20,000 cells/μL), boiled for 10 min, and centrifuged 3000 ×g for 5 min. Total protein content was determined using an assay kit (EZQ, Invitrogen cat number R33200). Proteins were separated by electrophoresis on Bis-Tris 16% polyacrylamide gels and transferred to nitrocellulose filters. APP-CTFβ was detected using 26D6-HRP, and APP-CTFα was detected using ct695 polyclonal (Invitrogen, cat number 51-2700) and secondary goat anti-rabbit horseradish peroxidase conjugate (Zymed, catalog number 62-6120). Chemiluminescence images were captured and quantified using an imaging station (Fuji model number LAS-3000). The ct695 western blots also show APP-CTFβ, which migrates as a fainter band above APP-CTFα under these conditions. To confirm the consistency of sample loading, the APP-CTF western blots were reprobed for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) using monoclonal 1D4 (Enzo Life Sciences, cat number CSA-335).
For immunoprecipitation and western blotting of APP-CTFβ and APP-CTFα from rat brain, weighed sagittal brain halves were homogenized using a rotary homogenizer (Polytron) in 5 volumes of RIPA buffer (Sigma R-0278; 150 mM NaCl, 1.0% IGEPAL CA-630, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing protease inhibitors (Roche complete cat number 11836145001) and centrifuged at 25,000 ×g for 30 min. All steps were carried out on ice or at 4°C. The pellet from centrifugation was discarded. Total protein concentration in the supernatant was determined using BCA protein assay (Pierce number 23227), and all samples were adjusted to a concentration of 18 mg/mL by addition of RIPA buffer. For APP-CTFβ immunoprecipitation, 5 μg of 252Q6 was added to 1 mL of homogenate and incubated on ice for 1 hour, and then 50 μL of magnetic protein A/G beads (Thermo Scientific cat number 88803) was added, and incubation was continued overnight with mixing. Using a magnet to isolate the beads, beads were washed once with 1 mL RIPA and twice with 1 mL of Tris saline pH 7.5 (50 mM Tris pH 7.5, 15 mM NaCl). The beads were resuspended in 40 μL of SDS sample buffer, boiled 5 mins, and removed using the magnet before gel electrophoresis and western blotting using ct695 polyclonal, as described above. For APP-CTFα, 5 μg of 4G8 was added to homogenates previously used for immunoprecipitation of APP-CTFβ, and immunoprecipitation using magnetic beads followed by western blotting using ct695 polyclonal was carried out as described above.
2.7. Mass Spectrometry of Aβ Peptides from Cell Cultures
H4-APPsw cell cultures were grown in T-75 flasks until 75% confluent and rinsed with Dulbecco's phosphate buffered saline (DPBS; Gibco cat number 14109), and 20 mL of DMEM supplemented with L-glutamine (2 mM), geneticin, penicillin, streptomycin, and DMSO with or without BMS-869780 was added (final concentration of DMSO was 0.2%). After incubation for 24 hours at 37°C in 5% CO2, culture medium was removed, centrifuged to remove cells, and frozen in aliquots at −80°C. For immunoprecipitation, aliquots of cell medium (4 mL) were thawed, followed by the addition of protease inhibitor cocktail (Sigma P-8340) to a final concentration of 1%, 60 ng of [15N]-Aβ1-40 synthetic peptide (rPeptide, 1101-1, Athens, GA), 30 μg of monoclonal 26D6, and 15 μg of monoclonal 4G8. After incubation for 20 min on ice, 80 μL of protein G agarose beads (Pierce Rockford, IL) was added, and incubation was continued overnight at 4°C. Beads were isolated by centrifugation and washed three times by centrifugation in 1 mL of ice cold phosphate buffered saline and then washed a final time in 1 mL 10 mM Tris-HCl pH 8.0. Aβ was eluted from the beads using 30 μL of 70% acetonitrile/0.1% formic acid. MALDI-TOF MS analysis was conducted using a Bruker Ultraflex III TOF/TOF mass spectrometer (Billerica, MA). A mix of Aβ standard peptides (AnaSpec, Fremont, CA) Aβ1-37 ([M+H]+ = m/z 4071.5), Aβ1-38 ([M+H]+ = m/z 4128.5), Aβ1-39 ([M+H]+ = m/z 4227.7), Aβ1-40 ([M+H]+ = m/z 4326.9), Aβ1-42 ([M+H]+ = m/z 4511.2), and [15N]Aβ1-40 ([M+H]+ = m/z 4378.4) was prepared to a final concentration of 1 ng/μL in 50% : 50% acetonitrile : water (v : v). Both the standard peptide sample and the samples from H4-APPsw cell medium following immunoprecipitation (IP) were further processed by mixing 5 μL sample with 5 μL of 7 mg/mL MALDI matrix (α-cyano-4-hydroxycinnamic acid, CHCA, from Sigma-Aldrich, St. Louis, MO) in 70% : 30% acetonitrile : water (v : v) with 0.1% trifluoroacetic acid (v : v). 0.5 μL of sample was spotted to an Anchorchip 384-well target plate (Bruker Daltonics, Billerica, MA) and allowed to dry in air before analysis. Analysis of various Aβ peptide isoforms from the standard peptides and the cell samples was performed in positive linear mode, accumulating 2000 spectra. Intensities of each analyte were normalized against a MALDI matrix peak (m/z 824.8) and the internal standard peak ([15N]Aβ1-40, m/z 4378.4).
2.8. Notch Signaling and Processing Assays
Inhibition of Notch signaling in cultured cells using a mouse-derived truncated Notch1 transgene, mNotch-ΔE [67], has been described in detail previously [59]. Western blots for mNotchΔ1865 and NICD using 9E10 anti-c-myc monoclonal were carried out as previously described [68].
2.9. Animals and Dosing
All experimental procedures with animals followed National Institutes of Health guidelines and were authorized by and in compliance with policies of the Bristol-Myers Squibb Animal Use and Care Committee. Mice and rats were housed with a 6:00 AM to 6:00 PM light/dark cycle and allowed free access to food and water. For 3xTg mice [63], BMS-869780 was dosed in three-month-old females by oral gavage at 6 mL/kg in vehicle consisting of 84% polyethylene glycol average molecular weight of 400 (PEG-400), 15% EtOH, and 1% Tween-80 (w/w/w). The compound was dissolved in EtOH and then diluted with PEG-400 and Tween, after which the vial was sealed, vortexed, and sonicated at 56°C for 1 hour. Animals were euthanized by asphyxiation in CO2. Blood was collected by cardiac puncture and placed into ethylene-diaminetetraacetic acid microtainer tubes for the preparation of plasma. The cerebellum was collected for the determination of compound concentration, and the remaining brain was separated into left and right halves before freezing in liquid nitrogen. For the rat time course study, 8- to 12-week-old female Sprague-Dawley rats were obtained from Charles River Laboratories (Wilmington, MA), BMS-869780 was dosed orally at 4 mL/kg, and brain and plasma samples were collected as described for the 3xTg mice. For the four day repeat dose rat study, BMS-869780 was given by oral gavage in vehicle consisting of PEG-400, PEG-200, D-α-tocopheryl polyethylene glycol succinate, Solutol HS 15, in the ratio 80 : 10 : 5 : 5 (w/w/w/w). Methods for the pharmacokinetics studies in rat, dog, and monkey are described below.
2.10. Pharmacokinetic to Pharmacodynamic (PK/PD) Relationship in Rodents
The data generated in the mouse and rat time course experiments were analyzed sequentially by nonlinear regression (WinNonlin Pharsight Corporation, Mountain View, CA). The pharmacokinetic data were best described by a 1-compartment linear model with first-order absorption and elimination. Subsequently, the pharmacodynamic parameters were estimated from the BMS-869780 plasma concentrations and the observed reductions of brain Aβ1-42 or Aβ1-40 by fitting to the equation for inhibition of synthesis [69]. The goodness-of-fit was determined by visual inspection, Akaike Information Criterion, Schwartz Criterion, examination of the residuals and the coefficient of variation of the parameter estimates.
2.11. Pharmacokinetics
Male Sprague-Dawley (SD) rats (300–350 g) were fasted overnight, and three per group received BMS-869780 either as an intravenous (IV) infusion in PEG400 : ethanol (90 : 10 w/w) at 1 mg/kg over 5 min via the jugular vein or as nanosuspension (d50 ca. 300 nm) by oral gavage at 5 mg/kg in Povidone K-30 : sodium lauryl sulfate : water (2.5 : 0.12 : 97.38 w/w/w/w). For the IV infusion, serial blood samples were obtained before dose and at 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 hours after dose. For the PO nanosuspension, serial blood samples were obtained before dose and at 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 h after dose. Blood samples, ~0.3 mL, for all studies were collected from the jugular vein into K3EDTA-containing tubes and then centrifuged at 4°C (1500–2000 ×g) to obtain plasma, which was stored at −20°C until analysis by LC/MS/MS. In male beagle dogs, the PK of BMS-869780 was evaluated in a cross-over study design with a one-week washout between treatments. Dogs were fasted overnight, and three animals (9.5 to 10.7 kg) received BMS-869780 by IV infusion at 1 mg/kg over 5 minutes in PEG400 : ethanol (90 : 10 w/w) or as nanosuspension (d50 ca. 300 nm) by oral gavage at 5 mg/kg in Povidone K-30 : sodium lauryl sulfate : water (2.5 : 0.12 : 97.38 w/w/w/w). Serial blood samples (~0.3 mL) were collected from a saphenous vein into K3EDTA-containing tubes before dose and at 0.083, 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 hours after IV dose, and 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 hours after oral dose, followed by centrifugation at 4°C (1500 to 2000 ×g) to obtain plasma. Samples were stored at −20°C until analysis of BMS-869780 levels by LC-MS/MS. In male cynomolgus monkeys, the PK of BMS-869780 was evaluated in a cross-over study design with a 1-week washout between treatments. Following an overnight fast, three animals (4.5 to 8 kg) received BMS-869780 by IV infusion via a femoral vein at 1 mg/kg over 10 minutes in PEG400 : ethanol (90 : 10), or by oral gavage at 5 mg/kg in Povidone K-30 : sodium lauryl sulfate : water (2.5 : 0.12 : 97.38). Serial blood samples, ~0.3 mL, were collected from a femoral artery into K3EDTA-containing tubes before dose and at 0.083, 0.17, 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 hours after IV dose, and 0.25, 0.5, 0.75, 1, 2, 4, 6, 8, and 24 hours after oral dose, followed by centrifugation at 4°C (1500 to 2000 ×g) to obtain plasma. Samples were stored at −20°C until analysis of BMS-869780 levels by LC-MS/MS. The PK parameters of BMS-869780 were obtained by noncompartmental analysis of plasma concentration versus time data (WinNonlin software, Version 5.0; Pharsight Corporation, Mountain View, CA). The peak concentration (C max) and time for C max (T max) were recorded directly from experimental observations. The area under the curve from time zero to the last sampling time (AUC0-T) and the area under the curve from time zero to infinity (AUCINF) were calculated using a combination of linear and log trapezoidal summations. The total plasma clearance (CLTp), steady-state volume of distribution (Vss), apparent elimination half-life (T 1/2), and mean residence time (MRT) were estimated after IV administration. The absolute oral bioavailability (F) was estimated as the ratio of dose-normalized AUC values following oral and IV doses.
2.12. Prediction of Human Dose
Interspecies allometric scaling adjusted for brain weight was used to predict human CLTp [70], while simple allometric scaling was used to predict Vss [71]. Briefly, the Vss and brain weight adjusted CLTp values from nonclinical species were plotted against body weight on a log-log scale to yield estimates of Vss and CLTp × brain weight in humans. The estimated CLTp × brain weight for human was adjusted for the brain weight of humans to yield a predicted CLTp. All of the nonclinical species demonstrated mono- or biexponential plasma concentration-time profiles; therefore, the MRT method was used to simulate the human pharmacokinetic profile as described below. Noncompartmental analysis was performed using WinNonlin software (version 5.0; Pharsight Corporation, Mountain View, CA). The k a for each species was obtained from the IV and PO (nanosuspensions) of rat, dog, and cynomolgus monkey by deconvolution of plasma concentration-time data using Kinetica (version 5.0; Seattle, WA). The bioavailability was estimated from the nanosuspensions from rat, dog, and cynomolgus monkeys. The average k a and F, along with the Vss and CLTp from allometric scaling, were incorporated into a two-compartment model to predict the human oral plasma concentration-time profiles. Human steady state doses to achieve plasma AUCs comparable to those in rats which produced 25% ABEC (area between baseline and Aβ effect-time curve) reductions were estimated. The results are summarized in Tables 3 and 4.
Table 3.
Relationship between dose, plasma AUC, and Aβ ABEC.
| Dose | Plasma AUC0–24 hr | Brain Aβ ABEC24 hr | |
|---|---|---|---|
| (μM·h) | Aβ1-42 (%) | Aβ1-40 (%) | |
| Mouse 30 mg/kg PO solution (observed 0–24 hours) | 45.9 | 30.9 | n.d. |
| Mouse 100 mg/kg PO solution (observed 0–24 hours) | 102 | 55.1 | n.d. |
| Rat 10 mg/kg PO solution (observed 0–24 hours) | 46.3 | 47.0 | 46.1 |
| Rat 3 mg/kg PO solution (predicted steady state after 3 daily doses)∗ | 18.6 | 26.9 | |
| Human 700 mg PO suspension (predicted steady state)∗ | 17.8 | 25 | |
*Dose predicted to achieve Aβ42 ABEC of ca. 25%.
Table 4.
Summary pharmacokinetics observed in three species and predicted in human.
| Rat | Dog | Monkey | Human predicted | ||
|---|---|---|---|---|---|
| IV 1 mg/kg |
Clearance (mL/min/kg) | 24.3 ± 1.7 | 5.7 ± 1.7 | 15.2 ± 5.9 | 5.6 |
| AUClast (nM·h) | 1484 ± 121 | 6640 ± 2257 | 2632 ± 926 | ||
| Half-life (hour) | 2.0 ± 0.3 | 5.2 ± 1.4 | 4.0 ± 0.5 | 13 | |
| MRT (hour) | 1 ± 0.2 | 6.6 ± 2.0 | 3.8 ± 0.7 | ||
| Vss (L/kg) | 1.8 ± 0.5 | 2.2 ± 0.8 | 3.7 ± 1.3 | 5.2 | |
|
| |||||
| PO suspension 5 mg/kg | AUClast (nM·h) | 3995 ± 964 | 4546 ± 2157 | 2031 ± 1038 | |
| C max (nM) | 674 ± 227 | 487 ± 155 | 219 ± 94 | ||
| T max (hour) | 3.3 ± 1.2 | 1.7 ± 0.6 | 2.0 ± 0 | ||
| F (%) | 54% | 13% | 16% | 28% | |
MRT-mean residence time; AUClast-area under the plasma concentration-time curve from zero time until the last quantifiable concentration.
2.13. Determination of BMS-869780 Concentrations
Plasma and brain samples were analyzed using an ultraperformance liquid chromatography-tandem mass spectrometry (UPLC-MS-MS) method. The UPLC MS-MS system consisted of a Waters Aquity Ultra Performance LC Sample Organizer, Solvent Manager and Sample Manager, a Waters BEH C18, 1.7 u 50 × 2.1 mm, column operated at 60°C, and a SCIEX API 4000 Q trap mass spectrometer. The mobile phase consisted of (A) water with 0.1% formic acid and (B) acetonitrile with 0.1% formic acid, delivered at 600 μL/min using a gradient program. The initial elution condition was 5% B which was maintained for 0.2 min and increased to 95% B in 0.5 min and maintained for 0.4 min. It was then returned to 5% B in 0.1 min and maintained for 0.2 min. The MS-MS analysis was performed using the heated nebulizer under positive ion mode with the source temperature at 400°C. The capillary voltage was 5000 eV and the collision energy 49 eV. The mass-to-charge ratios of 453 (precursor ion) and 438 (product ion) were used for multiple reaction mode monitoring of BMS-869780. The quantitation range for BMS-869780 was 1 to 5000 nM. Plasma samples were deproteinized and extracted with four portions of acetonitrile. Brain samples (0.1 g) were homogenized in 0.4 mL of acetonitrile.
2.14. Other Methods
Determination of IC50 values for inhibition of secreted alkaline phosphatase (SEAP) was carried out using an H4 cell line stably expressing SEAP. Cells were treated overnight with compounds in 384-well format, and SEAP accumulation in the culture media was quantified using a chemiluminescence substrate. The pregnane-X-receptor transactivation assay (PXR-TA) was based on the methods of Goodwin et al. [72] as described previously [59]. In the 4-day rat study with BMS-869780, tissues were fixed in 10% neutral buffered formalin, embedded in paraffin, sectioned, stained with hematoxylin and eosin, and examined by light microscopy.
3. Results
3.1. Identification of BMS-869780
The first compounds reported to have GSM activity were NSAIDs that exhibited selective inhibition of Aβ1-42 production [28]. A high throughput screen (HTS) was therefore carried out using FRET-based immunoassays for Aβ1-42 and Aβ1-40 levels in H4-APPsw cultures. Approximately 106 compound samples were incubated for 24 hours in H4-APPsw cell cultures, one compound per well in 384-well format, at a single concentration of 13 μM. The 22,304 samples exhibiting greater than 50% inhibition of Aβ1-42 were subsequently retested in triplicate, that is, three additional wells at 13 μM. This yielded 10,010 samples for which the average inhibition of Aβ1-42 in the four test wells was greater than 50%. After elimination of some samples due to previously known Aβ inhibition or chemical reactivity, 8,013 compounds were organized into clusters of related structures based on structural similarities [73], and 2,013 representative compounds were chosen for further evaluation. To rule out nonspecific effects on production or secretion of Aβ, dose response curves were determined for secreted alkaline phosphatase (SEAP) and for Aβ1-42 to compare the IC50 values. This yielded 409 samples that were relatively selective for Aβ1-42 (using a cutoff of ≥5-fold SEAP/Aβ1-42 IC50 ratio). To assess possible selectivity for Aβ1-42 lowering, IC50 values were determined in parallel for Aβ1-42 and Aβ1-40 in ca. 400 samples, and a series of compounds was identified from which BMS-869780 was subsequently derived through iterative improvements in potency and off-target profiles [54, 55]. An outline of the screening tiers and results obtained is shown in Figure 1(b), and the principle of the Aβ assays is shown in Figures 1(c) and 1(d).
3.2. Potency of BMS-869780 and Related Compounds for Aβ1-42 and Aβ1-40-Lowering
While many of the ca. 400 samples showed wide separations between the Aβ1-42 and Aβ1-40 IC50 values, BMS-869780 itself showed only a four-fold separation between the IC50 values (Figure 2(a)), presenting a minimal contrast with GSIs such as BMS-299897 (Figure 2(b)) and BMS-433796 (see summary of IC50 values in Table 1). Nevertheless, as a group, compounds chemically related to BMS-869780 showed limited overlap with GSIs based on the separation of Aβ1-42 and Aβ1-40 IC50 values, as illustrated for 236 GSMs and 688 GSIs (Figure 2(c)). This implies different Aβ-lowering mechanisms between the two groups. The ratios between Aβ1-42 and Aβ1-40 IC50 values in the GSM group ranged from little more than two-fold to almost 250-fold, with a trend toward lower ratios for compounds with lower IC50 values (Figure 2(d)). Anticipating subsequent experiments in the 3xTg mouse, the effect of the presenilin M146V FAD mutant on BMS-869780 potency was evaluated. MEF cell cultures lacking endogenous presenilins were therefore cotransfected with human APP-CTFβ and human presenilin, either wild type or M146V allele. The IC50 for Aβ1-42 was ca. 3-fold higher in cultures expressing the M146V allele, relative to cells expressing wild type presenilin. Likewise, Aβ1-40 IC50 values were ca. 3-fold shifted, although the IC50 appeared lower for Aβ1-40 in the wild type MEF cell cultures than in the H4-APPsw cultures. Thus, BMS-869780 appeared to be a little less potent in the context of the presenilin-1 M146V allele. IC50 values are summarized in Table 1.
Figure 2.

In vitro potency of BMS-869780 in the HTS assay. (a) H4-APPsw cultures were treated overnight with BMS-869780 (a GSM) at a range of concentrations and the relative levels of Aβ1-42 (red ●) and Aβ1-40 (blue ♦) were determined for calculation of IC50 values (summarized in Table 1). (b) H4-APPsw cultures were treated overnight with BMS-299897 (a representative GSI) as described for BMS-869780 in panel (a). IC50 values are summarized in Table 1. (c) IC50 values for Aβ1-42 and Aβ1-40 lowering were determined for 236 GSM (red ●) and for 688 GSI that were mostly of the type containing the aryl sulfonamide core (blue ▲). For some compounds, the Aβ1-40 IC50 value was greater than 30 μM, the highest concentration tested in the Aβ1-40 assay (arrow). (d) The ratio of the Aβ1-40 IC50 to Aβ1-42 IC50 was plotted against Aβ1-42 IC50 for the same 236 GSM illustrated in (c).
Table 1.
Aβ and Notch IC50 values.
| Assay (cell line) | BMS-869780 (GSM) | BMS-299897 (GSI ) | BMS-433796 (GSI) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| IC50 (nM) | sd | n | IC50 (nM) | sd | n | IC50 (nM) | sd | n | |
| Aβ1-42 (H4-APPsw) | 5.1 | 2.2 | 13 | 0.47 | 0.61 | 322 | 0.16 | 0.08 | 256 |
| Aβ1-40 (H4-APPsw) | 24.1 | 7.7 | 7 | 1.3 | 1.2 | 93 | 0.19 | 0.14 | 20 |
| Aβ1-42 (MEF PS1wt) | 7.7 | 2.3 | 2 | ||||||
| Aβ1-40 (MEF PS1wt) | 11 | 4 | 2 | ||||||
| Aβ1-42 (MEF M146V) | 22 | 1.4 | 2 | ||||||
| Aβ1-40 (MEF M146V) | 34 | 9 | 2 | ||||||
| mNotch1ΔE (luciferase) | >10000 | 3 | 340 | 108 | 147 | 2.1 | 1.0 | 256 | |
| mNotch1ΔE/Aβ1-42 | >1785x | 723x | 13x | ||||||
3.3. Evidence for the Noninhibitory Mechanism of BMS-869780 In Vitro
BMS-869780 showed only a 4-fold shift in IC50 values between Aβ1-42 and Aβ1-40. The effect of BMS-869780 on Aβ1-37 and Aβ1-38 was therefore evaluated, as a more diagnostic test of the GSM mechanism [25]. H4-APPsw cultures were treated overnight with BMS-869780, and Aβ peptides were evaluated by mass spectrometry and western blotting. Using MALDI mass spectrometry, Aβ1-37, Aβ1-38, and Aβ1-40 were readily detected in vehicle-treated cultures, although Aβ1-42 levels were near the limit of quantitation. After treatment with BMS-869780 (100 nM), Aβ1-37 and Aβ1-38 were dramatically increased, whereas Aβ1-40 and Aβ1-42 were essentially undetectable (Figure 3(a)). The same conclusion was reached in experiments using a western blot method that separates different forms of Aβ. Increased levels of the shorter Aβ1-37 and Aβ1-38 peptides, which migrate more slowly than Aβ1-40 or Aβ1-42 by this method, were observed in cultures treated with BMS-869780 at 100 nM and 10 nM (Figure 3(b), lanes 3 and 4, resp.). Thus, taking the experimental data illustrated in Figures 2 and 3 together, BMS-869780 increased Aβ1-37 and Aβ1-38, while decreasing Aβ1-40 and Aβ1-42. This suggested that BMS-869780 would have a minimal effect, if any, on APP-CTFα and APP-CTFβ turnover. H4-APPsw cell cultures were therefore treated at high concentrations, relative to the IC50s, of BMS-869780 and the GSI BMS-299897. For APP-CTFα, BMS-299897 caused an 8-fold increase in APP-CTFα, whereas BMS-869780 showed a 1.4-fold increase, averaged across doses (Figure 4(a)). The two compounds also showed a dramatic contrast in their effects on Aβ under these conditions. Whereas the GSI BMS-299897 dramatically reduced all Aβ1-x peptides, including Aβ1-42 and Aβ1-40, BMS-869780 selectively lowered Aβ1-42 and Aβ1-40, without any decrease in the overall levels of Aβ1-x (Figure 4(b)). In contrast to the result for APP-CTFα, APP-CTFβ levels were not affected in this experiment by either compound (Figure 4(a)), suggesting that γ-secretase was not a major pathway for APP-CTFβ turnover under these conditions. Indeed, it was recently reported that APP-CTFβ turnover in H4 cells occurs largely through proteasomal and lysosomal pathways, in contrast to APP-CTFα turnover which is more dependent on γ-secretase [74]. Thus, the effect of BMS-869780 on APP-CTFβ could not be directly evaluated in the H4-APPsw cell line under these conditions. To address the effect of BMS-869780 on APP-CTFβ, experiments were carried out in the context of the intended target organ, that is, in the brain of rats given oral doses of BMS-869780. The GSI BMS-698861 was dosed for comparison. BMS-869780 decreased Aβ1-40 and Aβ1-42 and increased Aβ1-37 and Aβ1-38 in rat brain. The sum total of Aβ1-40, Aβ1-42, Aβ1-38, and Aβ1-37 suggested no significant change in overall Aβ levels (Figure 5(a)). This was consistent with results obtained in the Aβ1-x assay, which showed no significant decrease despite the robust decrease in Aβ1-42 (Figure 5(b)). In contrast, BMS-698861 decreased levels of all the peptides, Aβ1-40, Aβ1-42, Aβ1-38, Aβ1-37, and Aβ1-x (Figures 5(a) and 5(b)). APP-CTFβ and APP-CTFα in samples of the same rat brains were evaluated by immunoprecipitation and western blotting. Neither APP-CTFβ nor APP-CTFα levels were affected in rats given BMS-869780, whereas levels of both peptides increased several-fold in rats given the GSI BMS-698861 (Figures 5(c)–5(f)). Thus, in brain, inhibition of γ-secretase by BMS-698861 resulted in APP-CTFβ and APP-CTFα accumulation, whereas modulation of Aβ by BMS-869780 had no effect on APP-CTFβ or APP-CTFα levels.
Figure 3.

BMS-869780 increased the levels of the shorter peptides Aβ1-38 and Aβ1-37. (a) Top panel: an equimolar mix of synthetic peptides Aβ1-37, Aβ1-38, Aβ1-39, Aβ1-40, [15N]-Aβ1-40, and Aβ1-42 was evaluated by MALDI-TOF mass spectrometry. H4-APPsw cell cultures were treated with vehicle (0.1% DMSO—middle panel) or BMS-869780 (100 nM—bottom panel), [15N]-Aβ1-40 was added, and Aβ peptides were immunoprecipitated and evaluated by MALDI-TOF mass spectrometry. (b) H4-APPsw cell cultures were treated with BMS-869780 or DMSO vehicle. Aβ peptides were separated by gel electrophoresis in the presence of urea and detected by western blotting. Under these conditions, higher molecular weight Aβ peptides exhibit greater gel mobility. Lane 1: an equimolar mix of synthetic peptides Aβ1-38, Aβ1-40, and Aβ1-42. Lane 2: DMSO vehicle-treated cell culture. Lanes 3–6: Cell cultures treated with BMS-869780 at final concentrations of 100, 10, 1 and 0.1 nM, respectively.
Figure 4.

BMS-869780 had minimal effect on APP-CTF accumulation in vitro. H4-APPsw cell cultures were treated overnight with the indicated concentrations of BMS-869780, BMS-299897 or vehicle (0.1% DMSO). (a) Cells were harvested and analyzed by western blotting for APP-CTFα, APP-CTFβ, and GAPDH. Lane 1; culture treated with vehicle 0.1% DMSO. Lanes 2–5; cultures treated with BMS-869780 at 100 nM, 300 nM, 1000 nM, or 3000 nM, respectively. Lanes 6–8; cultures treated with BMS-299897 at 30 nM, 100 nM, or 300 nM, respectively. (b) Levels of Aβ1-42 (red; left Y-axis), Aβ1-40 (green; right Y-axis), and Aβ1-x (grey; right Y-axis) were quantified.
3.4. BMS-869780 Does Not Inhibit Notch Processing
The effect of BMS-869780 on Notch processing was evaluated using transcriptional reporter assays and western blotting of NICD levels. HeLa cell cultures were transfected with mNotch1ΔE and CBF1-luciferase reporter constructs and treated with BMS-869780. In most replicates of this experiment, inhibition of luciferase reporter occurred with IC50 > 10 μM. In contrast, the GSIs BMS-299897 and BMS-433796 robustly inhibited luciferase activation, with IC50 = 340 nM and 2.1 nM, respectively (Figure 6(a)). The IC50 values for Notch-dependent luciferase expression are summarized in Table 1. To further evaluate the effect of BMS-869780 on Notch processing, western blots of cell cultures were carried out using mNotch1Δ1865, a truncated version of mNotch1ΔE that facilitates separation of NICD product from mNotch1 substrate on western blots [68]. HeLa cell cultures were transfected with mNotch1Δ1865 and treated overnight with compounds before western blotting. BMS-869780 had no effect on NICD levels at concentrations up to 3 μM, whereas the GSIs, BMS-299897, and BMS-433796, greatly reduced NICD (Figure 6(b)). Furthermore, the GSIs caused an increased level of mNotch1Δ1865 substrate, most likely due to inhibition of its turnover by γ-secretase, as previously noted by Blat et al. [68]. In contrast, 10 μM BMS-869780 did not increase mNotch1Δ1865 levels (Figure 6(b)), suggesting that the concomitant loss of NICD at 10 μM was due to a nonspecific effect, rather than inhibition of mNotch1Δ1865 turnover. This was also consistent with the observation of detached and dead cells in the presence of 10 μM BMS-869780.
Figure 6.

BMS-869780 did not inhibit Notch cleavage in vitro. (a) HeLa cell cultures were transfected with mNotchΔE and CBF1-luciferase reporter constructs, treated overnight with BMS-869780 (●), BMS-299897 (▲), or BMS-433796 (■), and luciferase assays were carried out. (b) HeLa cell cultures were transfected with mNotchΔ1865, treated with compounds overnight, and cell extracts were evaluated by western blot using anti-c-myc-HRP conjugate. Lanes 1 and 9: DMSO (0.1%) vehicle. Lane 2: BMS-299897 at 1 μM. Lane 3: BMS-433796 at 0.3 μM. Lanes 4–8: BMS-869780 at 0.1, 0.3, 1, 3, and 10 μM, respectively.
3.5. Evaluation of BMS-869780 PK/PD and Residual Levels of Aβ
To evaluate BMS-869780 PK/PD, rats were given BMS-869780 intraperitoneally at a range of doses from 0.3 mg/kg to 100 mg/kg. The GSI BMS-698861 was dosed at 30 mg/kg for comparison. Five hours after dosing, brain Aβ1-42 and brain Aβ1-40 exhibited dose-dependent lowering with ca. 50% lowering at 3 mg/kg, and plasma Aβ1-40 lowering was dose dependent with ca. 50% lowering at 0.3 mg/kg (Figure 7(a)). In contrast, brain Aβ1-x levels were not affected by BMS-869780 (Aβ1-x was evaluated only in samples from the highest three doses). For comparison, the GSI, BMS-698861, caused robust lowering in all Aβ assays, including Aβ1-x (Figure 7(a)). The concentration of BMS-869780 in plasma associated with 50% lowering of brain Aβ1-42 was ca. 1 μM (Figure 7(c)). Aβ1-42 appeared to be less completely inhibited than Aβ1-40. The linear correlation of Aβ1-42 with Aβ1-40 showed an intercept of 19.4% on the Aβ1-42 axis (Figure 7(b)), indicating a higher assay signal for Aβ1-42 relative to Aβ1-40 under conditions of maximal inhibition. Furthermore, at high plasma concentrations of BMS-869780, the best fit curve suggested a residual 29% Aβ1-42 and 11% Aβ1-40 assay signal remaining (Figure 7(c)). To investigate the residual assay signal further, brain Aβ1-42 and brain Aβ1-40 were evaluated in rats given four daily doses of BMS-869780 to achieve high sustained exposures, and residual levels of brain Aβ1-42 and Aβ1-40 were evaluated by immunodepletion. Despite the high exposures achieved (Table 4) and the extended time period in this experiment, residual ELISA signals for Aβ1-42 of 31% and for Aβ1-40 of 19% were observed (Figures 7(d) and 7(e)). Immunodepletion of the samples prior to ELISA was carried out to determine how much of the assay signal was due to residual Aβ1-42 or Aβ1-40. Specific monoclonals 565 and TSD selectively depleted Aβ1-42 and Aβ1-40, respectively, whereas monoclonal 4G8, which binds both, depleted both Aβ1-42 and Aβ1-40. The monoclonal 6E10, which selectively binds human Aβ relative to rat Aβ, depleted neither Aβ1-42 nor Aβ1-40. After immunodepletion, there was a residual signal of 16% for Aβ1-42 and 4% for Aβ1-40, averaged across depleted samples in both dose groups (Figures 7(d) and 7(e)). Thus, the residual signal appeared to be a combination of nonspecific background signal and small residual pools of ca. 15% Aβ1-42 and 7% Aβ1-40. This residual Aβ1-42 may represent either a slow turnover pool of Aβ1-42 or a source of Aβ1-42 production that is not readily inhibited by γ-secretase-targeted compounds.
To evaluate the pharmacokinetic to pharmacodynamic (PK/PD) relationship for BMS-869780 in more detail, time courses of Aβ lowering and plasma BMS-869780 concentration were carried out in mice and rats. The PK and PK/PD were then modeled sequentially using 1-compartment PK and indirect response PK/PD models, respectively. Triple transgenic mice [63] were dosed orally at 30 mg/kg or 100 mg/kg, and brain and plasma were taken from groups of animals after 3, 5, 8, 16, and 24 hours. Rats were dosed orally at 10 mg/kg, and brain and plasma were taken at the same time points as for mice. Plasma BMS-869780 showed increasing concentration until 4–8 hours then decreased (Figure 8(a)). Brain Aβ1-42 in the mouse, and both Aβ1-42 and Aβ1-40 in the rat, decreased until 8–16 hours relative to vehicle-treated groups (Figures 8(b)–8(d)). The in vivo plasma IC50 values for brain Aβ1-42 were 1.9 μM and 4.0 μM for rat and mouse, respectively. After taking account of the protein bound fractions (99.5% and 99.6% in rat and mouse, resp.), these are within 2-fold factors of the in vitro IC50 values of 5.1 nM and 22 nM for wild type and presenilin-1 M146V alleles, respectively (Table 1). Estimates of the PK and PD parameters are summarized in Table 2. Using the PK and PK/PD models for the rat data, steady-state assuming linear PK was predicted to occur after three daily doses, and a 3 mg/kg oral dose of BMS-869780 was predicted to yield AUC24 h = 18.6 μM·h with corresponding Aβ1-42 ABEC0-24 h = 26.9% at steady state. Observed and predicted values of dose, AUC, and ABEC in rat and mouse are summarized in Table 3.
Table 2.
PK/PD model values from mouse and rat studies.
| Parameter | Units | Mouse | CV (%) | Rat | CV (%) |
|---|---|---|---|---|---|
| PK parameters | |||||
| Kel | h−1 | 0.28 | 44.1 | 0.10 | 48.4 |
| V/F | L/kg | 1.94 | 48.3 | 1.94 | 43.8 |
| Ka1 (10 mg/kg) | h−1 | 0.19 | 56.0 | ||
| Ka1 (30 mg/kg) | h−1 | 0.11 | 29.8 | ||
| Ka2 (100 mg/kg) | h−1 | 0.046 | 30.0 | ||
|
| |||||
| PD parameters | |||||
| K OUT | h−1 | 0.72 | 24.9 | 0.48 | 51.4 |
| IC50
|
nM | 3979 | 9.16 | 1892 | 26.4 |
| R0 | % | 100 | Fixed | 100 | Fixed |
|
I
max
|
1 | Fixed | 1 | Fixed | |
|
K
in
|
%/h | 72 | Calculated | 48 | Calculated |
Kel-first order rate constant for drug elimination; V-volume of distribution; Ka rate constant for drug absorption; K OUT-first order rate constant for degradation of Aβ1-42 or Aβ1-40; IC50-plasma concentration required for 50% inhibition of Aβ1-42 or Aβ1-40 production; R is the response in Aβ1-42 or Aβ1-40 levels, assumed 100% at time zero, R0; I max-range of the response of Aβ1-42 or Aβ1-40 levels; K in-zero order constant for Aβ1-42 or Aβ1-40 production.
3.6. Pharmacokinetics and Human Dose Prediction
To make a prediction of human PK for BMS-869780, the PK profiles for solution IV and nanoparticle suspension PO dosing of BMS-869780 were determined in rat, dog, and cynomolgus monkey (Figure 9; Table 4). Plasma exposure was readily detectable for 24 hours after dosing, and the average bioavailability of the nanoparticle suspension for the three species was 28%. Allometric scaling of the observed animal PK parameters was used to predict human PK parameters (Table 4) [70, 71]. From the predicted human PK, in combination with the PK/PD parameters from rat, a 10 mg/kg once daily dose (700 mg total) was predicted to achieve a steady-state AUC = 17.8 μM·h, C max = 1.27 μM, and corresponding brain Aβ1-42 ABEC = 25% (Table 3).
Figure 9.

Pharmacokinetics (PK) of BMS-869780 across species for prediction of human PK. Rats, dogs, and cynomolgus monkeys were dosed with BMS-869780 intravenously (IV ▲) at 1 mg/kg or oral nanosuspension (PO ●) at 5 mg/kg. Plasma concentrations of BMS-869780 were determined for up to 24 hours after the dose. (a) Rat. (b) Dog. (c) Cynomolgus monkey. The derived PK parameters are summarized in Table 4.
3.7. Off-Targets and Safety
BMS-869780 was evaluated in a range of in vitro off-target activity assays. In one of these assays, pregnane-X-receptor transactivation (PXR-TA), BMS-869780 exhibited robust activity. In further experiments, BMS-869780 was shown to increase CYP3A4 mRNA expression in primary human hepatocytes. In both the PXR-TA and the primary human hepatocytes, transcription was activated at concentrations of 0.3 μM and above (Figure 10(a)), suggesting potential activation of CYP3A4 metabolism and risk for drug-drug interactions in human at the exposures predicted to lower Aβ1-42.
Figure 10.

BMS-869780 caused PXR activation in vitro and lipidosis in rat liver. (a) PXR activation in the presence of BMS-869780 was evaluated in HepG2 cell cultures using a luciferase transcriptional reporter construct (●), or by assay of CYP3A4 mRNA levels in primary human hepatocyte (PHH) cultures from two individual donors (▲ and ▼). Activation in both assays is expressed as % relative to activation in the presence of rifampicin, 10 μM, in parallel cultures. (b) Liver section from vehicle-dosed rats. (c) Liver section from rats given 4 daily doses of BMS-869780 at 100 mg/kg. A summary of the plasma BMS-869780 exposures from this experiment is shown in Table 5.
As a preliminary evaluation of safety in vivo, male and female rats were given 10, 30, and 100 mg/kg BMS-869780 orally once daily for 4 days. Endpoints included brain Aβ1-42, as described above (Figures 7(d) and 7(e)), plasma PK on day 1 and day 3 (Table 4), and histopathology for duodenum, liver, and kidney. There was no loss of exposure between day 1 and day 3, consistent with a lack of autoinduction via PXR and/or lack of BMS-869780 metabolism by CYP3A in the rat. Duodenum and kidney histology were unchanged at all dose levels. However, liver exhibited macro- and microvesicular vacuolar degeneration consistent with a fatty acid change, likely lipidosis, at all doses (Figures 10(b) and 10(c)). In the 10 mg/kg dose groups, the lowest mean AUC = 17.5 μM·h, and C max = 1.9 μM, were commensurate with the target exposures determined for Aβ1-42 lowering (see Table 3). In contrast, even at the highest dose of 100 mg/kg, where exposures in excess of 200 μM·h were achieved, there was no Notch-related effect on differentiation in the duodenum.
4. Discussion
The discovery and evaluation of BMS-869780 started with a high-throughput screen of the BMS compound inventory to identify selective inhibitors of Aβ1-42 in cell cultures and ended with predictions for human dose and exposure margins for off-target activity and safety. BMS-869780 is a GSM that decreased production of the longer peptides, Aβ1-40 and Aβ1-42, and increased production of the shorter peptides, Aβ1-37 and Aβ1-38. BMS-869780 did not significantly inhibit overall levels of Aβ production, APP-CTF processing, or Notch processing.
4.1. Potency and Mechanism of BMS-869780
BMS-869780 exhibited high potency (IC50 = 5.6 nM) for Aβ1-42 lowering in cell cultures, greater or equal in potency to the most potent GSMs reported [39, 43, 75, 76], and was robustly active in vivo, capable of lowering brain Aβ1-42 and Aβ1-40 in mice and rats by 75% or more. At the same time, levels of Aβ1-37 and Aβ1-38 were increased by BMS-869780, such that overall levels of Aβ remained essentially unchanged. The effect of BMS-869780 on γ-secretase therefore does not appear to involve inhibition. To evaluate the mechanism of BMS-869780 further, APP processing intermediates were evaluated both in H4-APPsw cell cultures and in rat brain. Levels of APP-CTFβ and APP-CTFα were not affected by BMS-869780, in contrast to the GSIs BMS-698861 and BMS-299897, which caused robust accumulation. An unexpected observation emerged in the H4-APPsw cell line when it was found that GSI treatment, while causing robust APP-CTFα accumulation, did not result in APP-CTFβ accumulation. This appears to be a quirk of the H4-APPsw cell line, possibly resulting from APP-CTFβ degradation taking place predominantly through the proteasomal and lysosomal pathways, as recently reported [74]. In rat brain, γ-secretase inhibition caused robust increases in both APP-CTFα and APP-CTFβ. In contrast, while BMS-869780 caused robust decreases in Aβ1-42 and Aβ1-40, there was no effect on APP-CTFα and APP-CTFβ levels in rat brain. Thus, in the target organ, brain, BMS-869780 was demonstrated to act solely as a GSM, without inhibitory effects on γ-secretase.
The in vivo potency of BMS-869780 for brain Aβ1-42 lowering in rat and 3xTg mouse was evaluated in single dose time course experiments using a PK/PD indirect response model. This yielded in vivo IC50 values that were within a factor of 2-fold of the IC50 values for Aβ1-42 determined in vitro, when plasma protein binding was taken into account. In the 3xTg mouse, the in vivo and in vitro IC50 values were also within a factor of 2-fold when the effect of the presenilin-1 M146V allele on potency was additionally taken into account. The potency of BMS-869780 for Aβ1-42 lowering was ca. 3-fold less in cell cultures expressing the presenilin-1 M146V allele, consistent with previous reports that presenilin FAD mutants can affect the potency of GSMs [77, 78]. Thus, the in vivo activity of BMS-869780 in rodents corresponded well with its potency determined in vitro, suggesting that rodents would be predictive of activity in human.
4.2. Lack of Notch Inhibition by BMS-869780
The lack of effect of BMS-869780 on Notch processing was demonstrated using three approaches. First, a luciferase transcriptional reporter assay for Notch1 signaling was tested in cell cultures. For BMS-869780, the ratio of Notch to Aβ1-42 IC50s could not be precisely determined because of its weak activity in the Notch assays but was >1785-fold, based on an IC50 value >10 μM for Notch. In the same assays, GSIs exhibited a wide range of Notch/Aβ1-42 IC50 ratios, with values of 13 and 723 for BMS-433796 and BMS-299897, respectively (Table 1). For GSIs, it has been shown that the absolute values of Notch/Aβ1-42 IC50 ratios for GSIs are strongly affected by APP substrate expression levels [61], and therefore cell culture data do not translate directly to Notch/Aβ1-42 margins in vivo. For example, the relatively Notch-selective GSI avagacestat exhibited a Notch/Aβ1-40 IC50 ratio of 193 [59], but the doses that achieved Aβ lowering without Notch-related side effects were more limited in vivo [19]. For BMS-869780, the lack of Notch-related side effects is expected based not only on the large in vitro Notch/Aβ1-42 IC50 ratio, but also on its noninhibitory mechanism of γ-secretase modulation. In rats, BMS-869780 did not cause any histological change in duodenum, specifically a lack of goblet cell metaplasia, even after four days of dosing at high exposures. The plasma exposure of BMS-869780 achieved in this experiment was more than 12-fold above the exposure required for lowering Aβ1-42 by 25% (Aβ1-42 ABEC = 25%). A wide variety of evidence from human genetics and transgenic APP mouse models suggests lowering of Aβ1-42 by 25% would be beneficial in AD [7]. Taken together, these data support the idea that GSMs such as BMS-869780 do not cause Notch-related side effects at doses predicted to cause sufficient Aβ1-42 lowering.
4.3. Human Predictions for BMS-869780
To predict human dose, off-target, and safety margins, the PK/PD relationship determined in rodents was used as a guide, and a target Aβ1-42 ABEC = 25% was chosen, based on evidence from rodent models that 25% might ultimately translate to a significant effect in AD [7]. The assumption that human and rat PK/PD would be similar was supported by the concordance of the human in vitro IC50 with the in vivo IC50s determined in rat and mouse, as discussed above. The human PK parameters were then predicted through allometric scaling of PK in three species; rat, dog, and monkey, using the average bioavailability (F = 28%) achieved with a nanosuspension. The nanosuspension was chosen as a clinically relevant formulation with potential to enhance bioavailability. A dose of 700 mg was calculated to achieve brain Aβ1-42 ABEC = 25%, with associated AUC = 17.6 μM·h and C max = 1.27 μM. These AUC and C max values were then used as benchmarks to compare the Aβ1-42 lowering activity against in vivo side effects and in vitro off-target activities. BMS-869780 did not cause duodenal neoplasia, the characteristic Notch-related side effect observed in rats given GSIs, even after four days dosing that achieved AUC = 316 μM·h and C max = 15.5 μM (Table 5). This predicted a safety margin, specifically related to duodenal toxicity, including Notch-related side effects in rat, of at least 12-fold above the Aβ1-42 lowering exposure benchmarks. On the other hand, lipidosis in the liver was observed after four daily doses of 10 mg/kg with mean C max = 1.9 μM and AUC = 17.5 μM (Table 5), indicating no separation of hepatotoxicity from the Aβ1-42 lowering exposure benchmarks. Subsequent studies with other potent GSMs (not shown) did not exhibit hepatotoxicity under these conditions, suggesting an off-target mechanism of hepatotoxicity. BMS-869780 was evaluated in a wide range of in vitro off-target activity assays. In the case of the human PXR transcriptional reporter assay, BMS-869780 was found to be active at concentrations of 0.3 μM and above. Further experiments confirmed the activation of CYP3A4 transcription in primary human hepatocyte cultures at similar concentrations, raising the possibility of metabolic induction and risk of drug-drug interactions at exposures required for Aβ1-42 lowering in human [79]. An overview of how experimental data were combined to determine off-target and safety margins is shown in Figure 11. In general, many GSMs exhibit poor drug-like properties, in particular high lipophilicity resulting in high active exposures and risk of systemic toxicity. Nevertheless, approaches for further optimization have been proposed, and the potential to improve drug-like properties has been demonstrated [80]. Alternatively, identification of new structural scaffolds might eventually lead to compounds with improved properties. Whether by optimization of current leads or new scaffolds, potentially the most useful guide for future compound design would be the availability of high resolution structures for GSM binding to γ-secretase.
Table 5.
Summary of exposure in rat 4-day dosing experiment.
| Dose (mg/kg) |
Male | Female | ||
|---|---|---|---|---|
| Day 1 | Day 3 | Day 1 | Day 3 | |
| C max (μM) | ||||
| 10 | 2.0 (±0.4) | 1.9 (±0.46) | 2.9 (±0.4) | 2.6 (±0.06) |
| 30 | 6.1 (±1.8) | 10.4 (±2.0) | 8.2 (±1.7) | 14.4 (±1.0) |
| 100 | 7.7 (±0.9) | 13.2 (±1.5) | 10.6 (±1.0) | 15.5 (±2.3) |
|
| ||||
| AUC (0–24 h) (μM·h) | ||||
| 10 | 20.2 (±4.8) | 17.5 (±5.7) | 41.0 (±5.6) | 25.0 (±1.5) |
| 30 | 95.2 (±19.9) | 128.6 (±65.0) | 152.2 (±21.0) | 255.1 (±28.5) |
| 100 | 167.5 (±14.5) | 216.9 (±80.5) | 205.9 (±43.2) | 316.4 (±77.0) |
|
| ||||
| T max (h) | ||||
| 10 | 5.0 (±2.0) | 5.0 (±2.0) | 7.0 (±2.0) | 5.0 (±2.0) |
| 30 | 8.0 (±0.0) | 3.0 (±1.0) | 5.0 (±2.0) | 3.0 (±1.0) |
| 100 | 19.0 (±9.0) | 7.0 (±2.0) | 24 (±0.0) | 4.0 (±4.0) |
|
| ||||
| Dose (mg/kg) |
Terminal concentration day 4 (μM) | |||
| Male | Female | |||
|
| ||||
| 10 | 2.4 (±0.6) | 3.0 (±0.3) | ||
| 30 | 8.9 (±1.9) | 12.0 (±4.6) | ||
| 100 | 9.2 (±3.2) | 13.7 (±0.9) | ||
Figure 11.
An overview of the characterization of BMS-869780 illustrates only the key steps in integration of data. Additional off-target and pharmaceutics evaluations necessary for decisions on individual compounds are not represented in this diagram.
In conclusion, BMS-869780 demonstrated the potential of the GSM approach, namely, the high potency, the robust translation of activity and mechanism in vivo, and the absence of a Notch-related side effect after multiple days of dosing at high sustained exposures. While liver toxicity and the high predicted dose of 700 mg caused studies of BMS-869780 to be discontinued, there was no evidence to suggest that liver toxicity or PXR activation were intrinsic to the GSM mechanism. In principle, therefore, an optimal combination of sufficient potency, PK, pharmaceutical properties, and off-target profile is within reach for testing a future GSM in Alzheimer's disease.
Acknowledgments
The authors thank their colleagues at Bristol-Myers Squibb for their assistance: Meredith Ferrante, Lizbeth Gallagher,and Chu Ngo, Lead Discovery; Lynn Balanda, Suki Jayachandra, Jane Palmer, and Barbara Robertson, Cell culture Resource Group; Emily Greenough, Sandra Matson, and Jay Stevenson, Compound Management; Lisa Johnson for database curation; Antara Majumdar, Biostatistics; Kurt Sims, Discovery Toxicology; Sharon Aborn, James Brennan, Corey Cirillo, Heidi Dulac, Zuzanna Haarhof, Laurie Henson, Jinwen Huang, John Light, Tami Orcutt, Jennifer Pizzano, Megan Quitko, and Cammie Symonowicz, Veterinary Sciences.
Abbreviations
- ABEC:
Area between baseline and Aβ effect-time curve
- AD:
Alzheimer's disease
- APP:
β-Amyloid precursor protein
- APP-CTF:
β-Amyloid precursor protein C-terminal fragment
- AUC:
Area under the plasma concentration-time curve
- CYP3A4:
Cytochrome P450 3A4 isozyme
- DMEM:
Dulbecco's modified Eagle's medium
- ELISA:
Enzyme-linked immunosorbent assay
- FAD:
Familial Alzheimer's disease
- FRET:
Förster resonance energy transfer
- GAPDH:
Glyceraldehyde-3-phosphate dehydrogenase
- GSM:
γ-Secretase modulator
- GSI:
γ-Secretase inhibitor
- HTS:
High-throughput screen
- KO:
Knockout (gene ablation)
- dKO:
Double knockout (gene ablation)
- MEF:
Mouse embryonic fibroblast
- MRT:
Mean residence time
- NICD:
Notch intracellular domain
- PD:
Pharmacodynamics
- PK:
Pharmacokinetics
- PK/PD:
Pharmacokinetic/pharmacodynamic relationship
- PXR-TA:
Pregnane-X-receptor transactivation assay
- SEAP:
Secreted alkaline phosphatase.
Conflict of Interests
All the authors are employees or former employees of Bristol-Myers Squibb.
References
- 1.Wimo A, Prince M. The global economic impact of dementia. Alzheimer’s Disease International World Alzheimer Report 2010. 2010
- 2.Prince M, Bryce R, Ferri C. The benefits of early diagnosis and intervention. Alzheimer’s Disease International World Alzheimer Report 2011. 2011
- 3.Findeis MA. The role of amyloid β peptide 42 in Alzheimer’s disease. Pharmacology & Therapeutics. 2007;116(2):266–286. doi: 10.1016/j.pharmthera.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 4.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 5.Karran E, Mercken M, de Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature Reviews Drug Discovery. 2011;10(9):698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 6.Haass C. Take five—BACE and the γ-secretase quartet conduct Alzheimer’s amyloid β-peptide generation. The EMBO Journal. 2004;23(3):483–488. doi: 10.1038/sj.emboj.7600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Toyn JH, Ahlijanian MK. Interpreting Alzheimer’s disease clinical trials in light of the effects on amyloid-β . Alzheimer's Research & Therapy. 2014;6, article 14 doi: 10.1186/alzrt244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and γ-secretase activity. Nature. 1999;398(6727):513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- 9.Lundqvist J, Näslund J. γ-Secretase: a complex target for Alzheimer’s disease. Current Opinion in Pharmacology. 2007;7:112–118. doi: 10.1016/j.coph.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 10.Lazarov VK, Fraering PC, Ye W, Wolfe MS, Selkoe DJ, Li H. Electron microscopic structure of purified, active γ-secretase reveals an aqueous intramembrane chamber and two pores. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(18):6889–6894. doi: 10.1073/pnas.0602321103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osenkowski P, Li H, Ye W, et al. Cryoelectron microscopy structure of purified γ-secretase at 12 Å resolution. Journal of Molecular Biology. 2009;385(2):642–652. doi: 10.1016/j.jmb.2008.10.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Renzi F, Zhang X, Rice WJ, et al. Structure of γ-secretase and its trimeric pre-activation intermediate by single-particle electron microscopy. The Journal of Biological Chemistry. 2011;286(24):21440–21449. doi: 10.1074/jbc.M110.193326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sato C, Morohashi Y, Tomita T, Iwatsubo T. Structure of the catalytic pore of γ-secretase probed by the accessibility of substituted cysteines. The Journal of Neuroscience. 2006;26(46):12081–12088. doi: 10.1523/JNEUROSCI.3614-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato T, Diehl TS, Narayanan S, et al. Active γ-secretase complexes contain only one of each component. The Journal of Biological Chemistry. 2007;282(47):33985–33993. doi: 10.1074/jbc.M705248200. [DOI] [PubMed] [Google Scholar]
- 15.Li X, Dang S, Yan C, Gong X, Wang J, Shi Y. Structure of a presenilin family intramembrane aspartate protease. Nature. 2013;493(7430):56–61. doi: 10.1038/nature11801. [DOI] [PubMed] [Google Scholar]
- 16.Haapasalo A, Kovacs DM. The many substrates of presenilin/γ-secretase. Journal of Alzheimer’s Disease. 2011;25(1):3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milano J, McKay J, Dagenais C, et al. Modulation of Notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicological Sciences. 2004;82(1):341–358. doi: 10.1093/toxsci/kfh254. [DOI] [PubMed] [Google Scholar]
- 18.Wong GT, Manfra D, Poulet FM, et al. Chronic treatment with the γ-secretase inhibitor LY-411,575 inhibits γ-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. The Journal of Biological Chemistry. 2004;279(13):12876–12882. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 19.Albright CF, Dockens R, Meredith JE, Jr., et al. Pharmacodynamics of selective inhibition of γ-secretase by avagacestat. Journal of Pharmacology and Experimental Therapeutics. 2013;344(3):686–695. doi: 10.1124/jpet.112.199356. [DOI] [PubMed] [Google Scholar]
- 20.Crump CJ, Fish BA, Castro SV, et al. Piperidine acetic acid based γ-secretase modulators directly bind to presenilin-1. ACS Chemical Neuroscience. 2011;2(12):705–710. doi: 10.1021/cn200098p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ebke A, Luebbers T, Fukumori A, et al. Novel γ-secretase enzyme modulators directly target presenilin protein. The Journal of Biological Chemistry. 2011;286(43):37181–37186. doi: 10.1074/jbc.C111.276972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ohki Y, Higo T, Uemura K, et al. Phenylpiperidine-type γ-secretase modulators target the transmembrane domain 1 of presenilin 1. The EMBO Journal. 2011;30(23):4815–4824. doi: 10.1038/emboj.2011.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jumpertz T, Rennhack A, Ness J, et al. Presenilin is the molecular target of acidic γ-secretase modulators in living cells. PLoS ONE. 2012;7(1, article e30484) doi: 10.1371/journal.pone.0030484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pozdnyakov N, Murrey HE, Crump CJ, et al. γ-Secretase modulator (GSM) photoaffinity probes reveal distinct allosteric binding sites on presenilin. The Journal of Biological Chemistry. 2013;288(14):9710–9720. doi: 10.1074/jbc.M112.398602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tate B, McKee TD, Loureiro RMB, et al. Modulation of gamma-secretase for the treatment of Alzheimer’s disease. International Journal of Alzheimer's Disease. 2012;2012:10 pages. doi: 10.1155/2012/210756.210756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okochi M, Fukumori A, Jiang J, et al. Secretion of the Notch-1 Aβ-like peptide during Notch signaling. The Journal of Biological Chemistry. 2006;281(12):7890–7898. doi: 10.1074/jbc.M513250200. [DOI] [PubMed] [Google Scholar]
- 27.Okochi M, Steiner H, Fukumori A, et al. Presenilins mediate a dual intramembranous γ-secretase cleavage of Notch-1. The EMBO Journal. 2002;21(20):5408–5416. doi: 10.1093/emboj/cdf541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weggen S, Eriksen JL, Das P, et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414(6860):212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- 29.Takami M, Nagashima Y, Sano Y, et al. γ-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of β-carboxyl terminal fragment. The Journal of Neuroscience. 2009;29(41):13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takami M, Funamoto S. γ-Secretase-dependent proteolysis of transmembrane domain of amyloid precursor protein: successive tri-and tetrapeptide release in amyloid β-protein production. International Journal of Alzheimer's Disease. 2012;2012:7 pages. doi: 10.1155/2012/591392.591392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sato T, Dohmae N, Qi Y, et al. Potential link between amyloid β-protein 42 and C-terminal fragment γ 49-99 of β-amyloid precursor protein. The Journal of Biological Chemistry. 2003;278(27):24294–20301. doi: 10.1074/jbc.M211161200. [DOI] [PubMed] [Google Scholar]
- 32.Chávez-Gutiérrez L, Bammens L, Benilova I, et al. The mechanism of γ-secretase dysfunction in Alzheimer’s disease. The EMBO Journal. 2012;31(10):2261–2274. doi: 10.1038/emboj.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eriksen JL, Sagi SA, Smith TE, et al. NSAIDs and enantiomers of flurbiprofen target γ-secretase and lower Aβ42 in vivo . The Journal of Clinical Investigation. 2003;112(3):440–449. doi: 10.1172/JCI18162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kukar T, Prescott S, Eriksen JL, et al. Chronic administration of R-flurbiprofen attenuates learning impairments in transgenic amyloid precursor protein mice. BMC Neuroscience. 2007;8, article 54 doi: 10.1186/1471-2202-8-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galasko DR, Graff-Radford N, May S, et al. Safety, tolerability, pharmacokinetics, and Aβ levels after short-term administration of R-flurbiprofen in healthy elderly individuals. Alzheimer Disease & Associated Disorders. 2007;21(4):292–299. doi: 10.1097/WAD.0b013e31815d1048. [DOI] [PubMed] [Google Scholar]
- 36.Nakano-Ito K, Fujikawa Y, Hihara T, et al. E2012-induced cataract and its predictive biomarkers. Toxicological Sciences. 2014;137(1):249–258. doi: 10.1093/toxsci/kft224. [DOI] [PubMed] [Google Scholar]
- 37.Yu Y, Logovinsky V, Schuck E, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the novel γ-secretase modulator, E2212, in healthy human subjects. The Journal of Clinical Pharmacology. 2014;54(5):528–536. doi: 10.1002/jcph.249. [DOI] [PubMed] [Google Scholar]
- 38.Caldwell JP, Bennett CE, McCracken TM, et al. Iminoheterocycles as γ-secretase modulators. Bioorganic & Medicinal Chemistry Letters. 2010;20(18):5380–5384. doi: 10.1016/j.bmcl.2010.07.121. [DOI] [PubMed] [Google Scholar]
- 39.Kounnas MZ, Danks AM, Cheng S, et al. Modulation of γ-secretase reduces β-amyloid deposition in a transgenic mouse model of Alzheimer’s disease. Neuron. 2010;67(5):769–780. doi: 10.1016/j.neuron.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stanton MG, Hubbs J, Sloman D, et al. Fluorinated piperidine acetic acids as γ-secretase modulators. Bioorganic & Medicinal Chemistry Letters. 2010;20(2):755–758. doi: 10.1016/j.bmcl.2009.11.034. [DOI] [PubMed] [Google Scholar]
- 41.Hawkins J, Harrison DC, Ahmed S, et al. Dynamics of Aβ42 reduction in plasma, CSF and brain of rats treated with the γ-secretase modulator, GSM-10h. Neurodegenerative Diseases. 2011;8(6):455–464. doi: 10.1159/000324511. [DOI] [PubMed] [Google Scholar]
- 42.Hussain I, Harrison DC, Hawkins J, et al. TASTPM mice expressing amyloid precursor protein and presenilin-1 mutant transgenes are sensitive to γ-secretase modulation and amyloid-β42 lowering by GSM-10h. Neurodegenerative Diseases. 2011;8(1-2):15–24. doi: 10.1159/000313903. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Huang Y, Jin S, et al. γ-Secretase modulators do not induce Aβ-rebound and accumulation of β-C-terminal fragment. Journal of Neurochemistry. 2012;121(2):277–286. doi: 10.1111/j.1471-4159.2011.07560.x. [DOI] [PubMed] [Google Scholar]
- 44.Peng H, Talreja T, Xin Z, et al. Discovery of BIIB042, a potent, selective, and orally bioavailable γ-secretase modulator. ACS Medicinal Chemistry Letters. 2011;2(10):786–791. doi: 10.1021/ml200175q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Broeck B, Chen J-M, Tréton G, et al. Chronic treatment with a novel γ-secretase modulator, JNJ-40418677, inhibits amyloid plaque formation in a mouse model of Alzheimer’s disease. British Journal of Pharmacology. 2011;163(2):375–389. doi: 10.1111/j.1476-5381.2011.01207.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wan Z, Hall A, Sang Y, et al. Pyridine-derived γ-secretase modulators. Bioorganic & Medicinal Chemistry Letters. 2011;21(16):4832–4835. doi: 10.1016/j.bmcl.2011.06.042. [DOI] [PubMed] [Google Scholar]
- 47.Borgegard T, Juréus A, Olsson F, et al. First and second generation γ-secretase modulators (GSMs) modulate amyloid-β (Aβ) peptide production through different mechanisms. The Journal of Biological Chemistry. 2012;287(15):11810–11819. doi: 10.1074/jbc.M111.305227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mitani Y, Yarimizu J, Saita K, et al. Differential effects between γ-secretase inhibitors and modulators on cognitive function in amyloid precursor protein-transgenic and nontransgenic mice. The Journal of Neuroscience. 2012;32(6):2037–2050. doi: 10.1523/JNEUROSCI.4264-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Z-Y, Asberom T, Bara T, et al. Cyclic hydroxyamidines as amide isosteres: discovery of oxadiazolines and oxadiazines as potent and highly efficacious γ-secretase modulators in vivo . Journal of Medicinal Chemistry. 2012;55(1):489–502. doi: 10.1021/jm201407j. [DOI] [PubMed] [Google Scholar]
- 50.Huang Y, Li T, Eatherton A, et al. Orally bioavailable and brain-penetrant pyridazine and pyridine-derived γ-secretase modulators reduced amyloidogenic Aβ peptides in vivo . Neuropharmacology. 2013;70:278–286. doi: 10.1016/j.neuropharm.2013.02.003. [DOI] [PubMed] [Google Scholar]
- 51.Loureiro RM, Dumin JA, McKee TD, et al. Efficacy of SPI-1865, a novel gamma-secretase modulator, in multiple rodent models. Alzheimer’s Research & Therapy. 2013;5(2, article 19) doi: 10.1186/alzrt173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rogers K, Felsenstein KM, Hrdlicka L, et al. Modulation of γ-secretase by EVP-0015962 reduces amyloid deposition and behavioral deficits in Tg2576 mice. Molecular Neurodegeneration. 2012;7(1, article 61) doi: 10.1186/1750-1326-7-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mitani Y, Yarimizu J, Akashiba H, Shitaka Y, Ni K, Matsuoka N. Amelioration of cognitive deficits in plaque-bearing Alzheimer’s disease model mice through selective reduction of nascent soluble Aβ 42 without affecting other Aβ pools. Journal of Neurochemistry. 2013;125(3):465–472. doi: 10.1111/jnc.12125. [DOI] [PubMed] [Google Scholar]
- 54.Marcin LR, Thompson LA, III, Boy KM, et al. Bicyclic compounds for the reduction of beta-amyloid production. Bristol-Myers Squibb. 2010;(US20110015175, WO2010083141)
- 55.Marcin LR, Higgins MA, Lentz KA, et al. Bicyclic triazoles as modulators of γ-secretase for the treatment of Alzheimer’s disease. Proceedings of the 243rd ACS National Meeting & Exposition; March 2012; San Diego, Calif, USA. [Google Scholar]
- 56.Barten DM, Guss VL, Corsa A, et al. Dynamics of β-amyloid reductions in brain, cerebrospinal fluid, and plasma of β-amyloid precursor protein transgenic mice treated with a γ-secretase inhibitor. Journal of Pharmacology and Experimental Therapeutics. 2005;312(2):635–643. doi: 10.1124/jpet.104.075408. [DOI] [PubMed] [Google Scholar]
- 57.Prasad CVC, Zheng M, Vig S, et al. Discovery of (S)-2-((S)-2-(3,5-difluorophenyl)-2-hydroxyacetamido)-N-((S,Z)-3-methyl-4-oxo-4,5-dihydro-3H-benzo[d][1,2]diazepin-5-yl)propanamide (BMS-433796): A γ-secretase inhibitor with Aβ lowering activity in a transgenic mouse model of Alzheimer’s disease. Bioorganic & Medicinal Chemistry Letters. 2007;17(14):4006–4011. doi: 10.1016/j.bmcl.2007.04.082. [DOI] [PubMed] [Google Scholar]
- 58.Gilligan PJ. Sulfonamide bicyclic compounds. Bristol-Myers Squibb. 2005;(US20067144894, WO2006034480)
- 59.Gillman K, Starrett JE, Jr., Parker MF, et al. Discovery and evaluation of BMS-708163, a potent, selective and orally bioavailable γ-secretase inhibitor. ACS Medicinal Chemistry Letters. 2010;1(3):120–124. doi: 10.1021/ml1000239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Herreman A, Hartmann D, Annaert W, et al. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(21):11872–11877. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Burton CR, Meredith JE, Barten DM, et al. The amyloid-β rise and γ-secretase inhibitor potency depend on the level of substrate expression. The Journal of Biological Chemistry. 2008;283(34):22992–23003. doi: 10.1074/jbc.M804175200. [DOI] [PubMed] [Google Scholar]
- 62.Sankaranarayanan S, Price EA, Wu G, et al. In vivoβ-secretase 1 inhibition leads to brain Aβ lowering and increased α-secretase processing of amyloid precursor protein without effect on neuregulin-1. Journal of Pharmacology and Experimental Therapeutics. 2008;324(3):957–969. doi: 10.1124/jpet.107.130039. [DOI] [PubMed] [Google Scholar]
- 63.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39(3):409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 64.Lanz TA, Schachter JB. Demonstration of a common artifact in immunosorbent assays of brain extracts: development of a solid-phase extraction protocol to enable measurement of amyloid-β from wild-type rodent brain. Journal of Neuroscience Methods. 2006;157(1):71–81. doi: 10.1016/j.jneumeth.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 65.Toyn JH, Lin X-A, Thompson MW, et al. Viable mouse gene ablations that robustly alter brain Aβ levels are rare. BMC Neuroscience. 2010;11, article 143 doi: 10.1186/1471-2202-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klafki H-W, Wiltfang J, Staufenbiel M. Electrophoretic separation of βA4 peptides (1–40) and (1–42) Analytical Biochemistry. 1996;237(1):24–29. doi: 10.1006/abio.1996.0195. [DOI] [PubMed] [Google Scholar]
- 67.Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(4):1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blat Y, Meredith JE, Wang Q, et al. Mutations at the P1′ position of Notch1 decrease intracellular domain stability rather than cleavage by γ-secretase. Biochemical and Biophysical Research Communications. 2002;299(4):569–573. doi: 10.1016/s0006-291x(02)02705-5. [DOI] [PubMed] [Google Scholar]
- 69.Dayneka NL, Garg V, Jusko WJ. Comparison of four basic models of indirect pharmacodynamic responses. Journal of Pharmacokinetics and Biopharmaceutics. 1993;21(4):457–478. doi: 10.1007/BF01061691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mahmood I. Application of allometric principles for the prediction of pharmacokinetics in human and veterinary drug development. Advanced Drug Delivery Reviews. 2007;59(11):1177–1192. doi: 10.1016/j.addr.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 71.Mordenti J, Chappell W. Toxicokinetics and Drug Development. New York, NY, USA: Pergamon Press; 1989. The use of interspecies scaling in toxicokinetics; pp. 42–96. [Google Scholar]
- 72.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Molecular Pharmacology. 1999;56(6):1529–1539. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 73.Li W. A fast clustering algorithm for analyzing highly similar compounds of very large libraries. Journal of Chemical Information and Modeling. 2006;46(5):1919–1923. doi: 10.1021/ci0600859. [DOI] [PubMed] [Google Scholar]
- 74.Bustamante HA, Rivera-Diccter A, Cavieres VA, et al. Turnover of C99 is controlled by a crosstalk between ERAD and ubiquitin-independent lysosomal degredation in human neuroglioma cells. PLoS ONE. 2013;8(12, article e83096) doi: 10.1371/journal.pone.0083096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Borghys H, Tuefferd M, van Broeck B, et al. A canine model to evaluate efficacy and safety of γ-secretase inhibitors and modulators. Journal of Alzheimer’s Disease. 2012;28(4):809–822. doi: 10.3233/JAD-2011-111475. [DOI] [PubMed] [Google Scholar]
- 76.Qin J, Zhou W, Huang X, et al. Discovery of a potent pyrazolopyridine series of γ-secretase modulators. ACS Medicinal Chemistry Letters. 2011;2(6):471–476. doi: 10.1021/ml2000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Page RM, Baumann K, Tomioka M, et al. Generation of Aβ38 and Aβ42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and γ-secretase modulation. The Journal of Biological Chemistry. 2008;283(2):677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- 78.Kretner B, Fukumori A, Gutsmiedl A, et al. Attenuated Aβ42 responses to low potency γ-secretase modulators can be overcome for many pathogenic presenilin mutants by second-generation compounds. The Journal of Biological Chemistry. 2011;286(17):15240–15251. doi: 10.1074/jbc.M110.213587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sinz MW. Pregnane X receptor: “prediction and attenuation of human CYP3A4 enzyme induction and drug-drug interactions’. Annual Reports in Medicinal Chemistry. 2008;43:405–418. [Google Scholar]
- 80.Gijsen HJM, Mercken M. γ-Secretase modulators: can we combine potency with safety? International Journal of Alzheimer’s Disease. 2012;2012:10 pages. doi: 10.1155/2012/295207.295207 [DOI] [PMC free article] [PubMed] [Google Scholar]