

Abstract
DYNC1H1 encodes the heavy chain of cytoplasmic dynein 1, a motor protein complex implicated in retrograde axonal transport, neuronal migration, and other intracellular motility functions. Mutations in DYNC1H1 have been described in autosomal dominant Charcot-Marie-Tooth type 2 and in families with distal spinal muscular atrophy (SMA) predominantly affecting the legs (SMA-LED). Recently, defects of cytoplasmic dynein 1 were also associated with a form of mental retardation and neuronal migration disorders. Here we describe two unrelated patients presenting a combined phenotype of congenital motor neuron disease associated with focal areas of cortical malformation. In each patient we identified a novel de novo mutation in DYNC1H1: c.3581A>G (p.Gln1194Arg) in one case and c.9142G>A (p.Glu3048Lys) in the other. The mutations lie in different domains of the dynein heavy chain, and are deleterious to protein function as indicated by assays for Golgi recovery after nocodazole washout in patient fibroblasts. Our results expand the set of pathological mutations in DYNC1H1, reinforce the role of cytoplasmic dynein in disorders of neuronal migration and provide evidence for a syndrome including spinal nerve degeneration and brain developmental problems.
Keywords: DYNC1H1, distal SMA, SMA-LED, abnormal cortical development
Dyneins are a family of ATP-driven motor proteins that move toward the minus ends of microtubules [Vallee et al., 2012]. Cytoplasmic dynein 1 (hereafter “cytoplasmic dynein” or “dynein”) transports diverse intracellular cargoes and generates forces involved in cell division. In the nervous system it has been implicated in retrograde axonal transport and neuronal migration. Cytoplasmic dynein consists of heavy, intermediate, light intermediate, and light chains. The heavy chain contains a long N-terminal tail domain, responsible for dimerization, binding of other subunits and recruitment of cellular cargoes. The C-terminal region of the heavy chain contains the motor domain, arranged as a ring of six globular AAA modules, which hydrolyze ATP to generate force along microtubules [Carter et al., 2011]. Dynein can interact directly with cargo through its accessory subunits, or indirectly through its accessory complexes, which each also regulates dynein motor activity. Two major regulatory complexes are dynactin and NudE- or NudE-LIS1 which act via the dynein intermediate chain [Vallee et al., 2012]. Mutations in NudE, NudEL, and LIS1 cause alterations in mouse, rat, and human brain development, leading to microcephaly and lissencephaly [Dobyns et al., 1993; Feng et al., 2000; Sasaki et al., 2000; Tsai et al., 2005; Alkuraya, et al., 2011]. In contrast, mutations in dynactin result in various motor neuron diseases including amyotrophic lateral sclerosis (ALS) and hereditary motor neuropathy type VIIB (HMNVIIB) [Puls et al., 2003].
Loss-of-function mutations in DYNC1H1 (MIM# 600112) were initially recognized as a cause of spontaneous murine phenotypes: Legs at odd angles (Loa), Cramping 1 (Cra1) [Hafezparast et al., 2003] and Sprawling (Swl) [Chen et al., 2007] mice. These mice display progressive loss of locomotor ability and limb deformity, which in the case of Loa and Cra1 mice is due to a deficiency in the number of spinal cord motor neurons [Hafezparast et al., 2003]. Interestingly, the brains of Loa mice also show cortical disorganization and reduction in the rate of radial migration of bipolar neurons [Ori-McKenney KM and Vallee RB, 2011]. These results provided evidence that mutations in dynein can alter cortical lamination and result in a syndrome of combined peripheral neurodegeneration and brain developmental defects.
Recently mutations in DYNC1H1 have been associated with three human phenotypes: an axonal form of Charcot-Marie-Tooth (CMT-2O) [Weedon et al., 2011], a type of distal spinal muscular atrophy (distal SMA) predominantly affecting the lower extremities (SMA-LED) [Harms et al., 2010, Harms et al., 2012, Tsurusaki et al., 2012] and hereditary mental retardation with cortical neuronal migration defects [Vissers et al., 2010, Willemsen et al., 2012].
While writing this report further 11 patients carrying mutations in DYNC1H1 mainly presenting with severe brain abnormalities and focal or generalised epilepsy were also described [Poirer et al., 2013]. In three patients, common signs of a possible peripheral neuropathy were detected (but demonstrated with nerve conduction study in one case).
Equally, selected CMT or distal SMA patients presented with cognitive impairment, though this was not followed up by brain imaging [Weedon et al., 2011, Harms et al., 2012]. (see Supp. Table S1)
Assuming that the combination of neuropathy and brain malformation might be the hallmark of a dynein defect, we tested for DYNC1H1 mutations in four patients presenting with both distal SMA and abnormal cortical gyration and identified two novel missense mutations in DYNC1H1: c.3581A>G (p.Gln1194Arg) and c.9142G>A (p.Glu3048Lys), from two unrelated patients of Italian origin.
The first case is a 19-year-old male, born to healthy parents, and who presented at birth with congenital foot deformity. EMG indicated signs of muscle denervation, and a muscle biopsy at age five confirmed neurogenic damage. A brain MRI at age 14 revealed bilateral widening of sylvian fissures with perisylvian abnormal gyration and polymicrogyria (PMG)-like cortical malformation (Figure 1A–B). Mild cognitive impairment was detected, especially in verbal skills with a verbal IQ of 44 on the Wechsler Intelligence Scale for Children-III at age 15. At last neurological examination he showed pes cavus, lower limb weakness and atrophy with waddling gait (Supp. Figure S1). Hyperlordosis and right curve scoliosis were also present. An EEG showed non-specific abnormalities, and the patient never manifested seizures. No sensory problems were reported and nerve conduction studies were normal.
Figure 1.
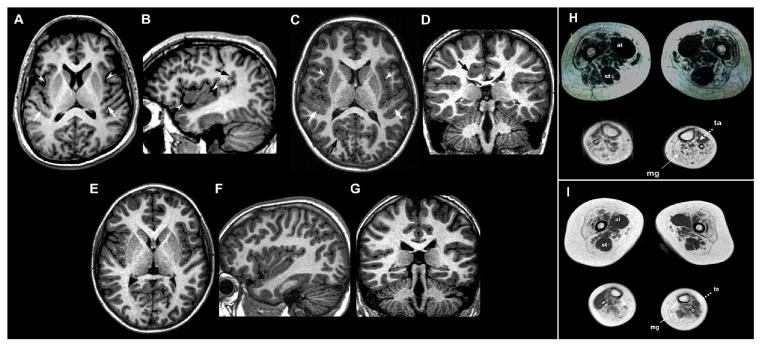
Brain MRI examinations of Patient 1 (A–B) and Patient 2 (C–D), with corresponding normal control images (E–G) and muscle MRI examination of Patient 1 (H) and Patient 2 (I) at thigh and calf level
Axial A) and right parasagittal B) 3D SPGR T1-weighted images show abnormal gyration and delicate PMG-like cortical malformation involving the insulae (white arrowheads) and Heschle gyri (white arrows) with abnormal vertical orientation of the posterior part of sylvian fissure (black arrow). C) Axial 3D TFE T1-weighted image reveals bilateral perisylvian PMG-like cortical malformation involving the insulae (white arrowheads) and Heschle gyri (white arrows). Note an additional area of abnormal cortical gyration with infolding (black arrows) in the right occipital lobe. D) Coronal 3D TFE T1-weighted image depicts another area of abnormal cortical gyration with infolding (black arrow) in the right posterior cingulum slightly deforming the right lateral ventricle.
In muscle MRIs of lower limbs patients present diffuse atrophy and fat replacement of the thigh muscles which appear more prominent in Patient 2 (I), who accordingly displays the most severe phenotype. A selective similar sparing with compensatory hypertrophy of the adductor longus (al) and semitendinosus (st) muscles is evident to a similar extent in both cases. At leg level, MRI showed severe fat replacement of medial gastrocnemii (mg) with relative preservation of anterior tibialis (ta).
The second case is a 9-year-old male, born to healthy parents, and who displayed bilateral foot deformity at birth and delayed motor milestones with floppiness. EMG and muscle biopsy performed at 18 months detected neurogenic alterations of lower limb muscles. Nerve studies displayed normal motor and sensory conductions. A brain MRI at age 4 revealed multiple areas of cortical gyration anomalies and PMG-like cortical malformations involving the perisylvian/insular regions, the right occipital lobe and right posterior cingulum (Figure 1C–D). Upon most recent neurological examination, the patient could walk with bilateral support from orthoses. Muscle bulk was reduced in the legs, and mild weakness was noted in the shoulder girdle muscles. Deep tendon reflexes were globally absent. While standing the patient demonstrated hyperlordosis of the spine (Supp. Figure S1). Mild cognitive impairment was present with attention defect.
A lower extremity muscle MRI was performed in both patients at 19 and 9 years old, respectively. Proximal to the knee, the muscle scans displayed a pattern of diffuse atrophy and fat replacement of the thigh muscles, however there was selective hypertrophy of the adductor longus and semitendinosus muscles (Figure 1 H–I). Distal to the knee, the MRI showed preferential involvement of the posterior compartment with severe fat replacement of both gastrocnemii and relative preservation of anterior tibialis (Figure 1H–I). These findings are largely consistent with previous reports of distal SMA of idiopathic genetic origin [Mercuri et al., 2004], but also suggest a pattern of muscle damage distinctively linked to defective dynein. In particular, the calf muscle involvement is apparently different from that described in TRPV4 mutated distal SMA patients [Astrea et al., Neurology 2012].
Genetic analysis ruled out deletion of SMN1, a common cause of SMA, and mutations in HSPB1, HSPB8, BSCL2, SETX and TRPV4, all known genes implicated in distal SMA or hereditary motor neuropathies [Rossor et al., 2012]. Adopting a “reverse” candidate gene approach, we used skin fibroblast RNA as template and amplified the coding region of DYNC1H1 (NM_001376.4) by RT- PCR and identified two novel missense variants. Both mutations were then confirmed on peripheral blood DNA using specific genomic primers.
In the remaining 2 cases no pathogenetic mutations of DYNC1H1 were identified. The patients were female, ages 2.6 and 4, presenting severe muscular atrophy of neurogenic origin with foot malformation and abnormalities of the central nervous system, including cortical dysplasia. The older patient also had ocular malformation and involvement of upper limbs, whereas the younger patient developed dysphagia and respiratory defect.
In patient 1 we identified a novel heterozygous variant c.3581A>G (p.Gln1194Arg) in exon 16. This de novo mutation was not detected in other family members, in 500 ethnically-matched control chromosomes, nor in large SNP databases. c.3581A>G (p.Gln1194Arg) replaces a highly conserved (Supp. Figure S1 C) hydrophobic glutamine with a hydrophilic arginine at residue 1194, which is positioned in the small flexible “neck domain” (aa 1150-1300) that has been proposed to participate in interactions between the AAA ring of the motor domain and the large “linker” domain connected to the dynein tail. As such the neck domain may have a role in dynein step size or force production. [Roberts et al., 2012]. In silico predictions indicated that the mutation is deleterious (Condel score of 0.948; Polyphen-2 score of 1) and disease causing (Mutation Taster, score of 0.999). Accordingly, structural modeling of the tail region (Supp. Methods), suggested that p.Q1194R alters the conformation of the region lowering the global stability of this domain (Figure 2A).
Figure 2.

A–B Protein modeling
A) Cartoon representation of secondary structure elements of the tail/linker region of cytoplasmic dynein 1 heavy chain 1 protein. The model exhibits the typical three helix fold of the spectrin repeat-domain (pfam00435) with the. Gln1194 residue (highlighted in red) located in the α-helix B. The box is shows a magnification of the region of interest with the predicted consequences of the replacement of Arginine for Glutamine at residue 1194. The effect of mutation on protein structure and stability, as predicted by Rosetta Backrub server and FoldX, is that it determines a change in the conformation of the α-helix B and decreases the global stability of the domain (ΔΔG: 4.66 ± 0.18 kcal/mol).
B) Cartoon (left) and electrostatic surface (right) representations of wild-type (upper) and c.9142G>A (p.Glu3048Lys) (lower) AAA4-α/β submodule of human DYNC1H1 (residues 2870–3096). The AAA4-α/β domain exhibits the Rossmann-type fold with the Glu3048 residue located in the pre-Sensor I region (colored in red), the characteristic insert that extends from α-helix 3 and β-sheet 4 of the AAA4 module. The c.9142G>A (p.Glu3048Lys) mutation slightly decreases the global stability of the domain (ΔΔG: 1.06±0.08 kcal/mol) but severely alters the electrostatic surface of the pre-Sensor I region of AAA4 domain. Electrostatic surface potentials were colored according to charge with blue denoting positive charge (+5 kT/e-) and red, negative charge (−5 kT/e-).
C–E Nocodazole washout experiments with cultured skin fibroblasts from patients 1 and 2 (yellow and green bars respectively) or a control individual (cyan bars) lacking the dynein mutations. Results are shown as percentage of cells from each cell line with bracketed numbers representing the number of cells examined. C) For all cell lines, at the time of nocodazole exposure, nearly 100% of cells had a fully disrupted Golgi. D) At 30 minutes after nocodazole washout, a higher percentage of cells from patients 1 and 2, as compared to control cells, showed a fully disrupted Golgi. E) At 90 minutes after nocodazole washout, the percentage of cells with a fully disrupted Golgi was higher in patient cells compared to control cells. This was particularly evident in patient 2, where the number of cells retaining a fully disrupted Golgi was nearly 50% of the total. Results are taken from at least 3 Independent experiments. Error bars=standard deviation; n= number of cells. Unpaired t-test: *, p<0.05 **, p<0.001.
F. Representative images of cultured skin fibroblasts with intact (upper panel), partially disrupted (middle panel) and fully disrupted (lower panel) Golgi are shown. Golgi is stained in red. Scale bars=5μm.
The missense mutation detected in patient 2 (also heterozygous), c. 9142G>A (p.Glu3048Lys), lies in exon 47 of DYNC1H1 and replaces a highly conserved (Supp. Figure S1 D) glutamic acid with lysine. This mutation was not found in the parents of the patient nor in 200 control chromosomes and in silico predictions indicated that it is deleterious (Condel score of 0.782; Polyphen-2 score of 0.998), and disease causing (Mutation Taster score of 0.999). Unlike most of the previously described mutations in DYNC1H1, c.9142G>A (p.Glu3048Lys) is located in the motor domain of the protein in the unique helical insert (pre-Sensor I) that extends from α-helix 3 (H3) and β-sheet 4 (β4) of the AAA4 module. The mutation is not expected to affect the overall structure of the AAA4 module, but might decrease the global stability of the domain since the mutation drastically changes electrostatic surface potential of the entire module (Figure 2B). According to the crystal structures of dynein [Kon et al., 2012, Carter et al. 2011], the pre-Sensor I region of the AAA4 domain (as well as the H3/β4 inserts that lie in AAA2, AAA3 modules) is a secondary binding site for the linker domain during the mechanochemical cycle of the motor. In this context, the decrease of negative surface charge of the pre-Sensor I region caused by the mutation might alter interactions with the linker domain and impair dynein activity.
To further assess the pathological consequences of the c.3581A>G (p.Gln1194Arg) and c. 9142G>A (p.Glu3048Lys) mutations, we assayed for dynein function in the patients’ cultured skin fibroblasts. We did not detect any obvious defects in the typical perinuclear distribution of lysosomes or the Golgi apparatus, as occurs under conditions of severe dynein inhibition [Burkhardt et al., 1997]. Most lysosomes were immobile, making live analysis of motor-mediated transport difficult. To test for more subtle changes in motor behavior, we examined Golgi reassembly following microtubule depolymerization by nocodazole and subsequent drug washout, as performed with cultured fibroblasts from homozygous Loa mice [Hafezparast et al. 2003] and dynactin mutant human fibroblasts [Levy et al. 2006]. We observed clear delays in Golgi reassembly at 60 and 90 minutes when compared to control fibroblasts. The effect was more severe in Patient 2 than in Patient 1 (Figure 2C–E). We also tested c.3581A>G (p.Gln1194Arg) and c.9142G>A (p.Glu3048Lys) mutant dynein from fibroblast cytosolic lysates for its biochemical behavior. We observed apparently normal behavior, with the protein sedimenting as a symmetric peak at ~20S, as judged by immunoblotting of sucrose gradient fractions (Supp. Figure S2). Microtubule (MT) binding of the mutant dynein from the cytosolic lysates and its release from microtubules by ATP also appeared normal (Supp. Figure S2).
Our study characterizes two novel DYNC1H1 variants and confirms that mutations in either tail or motor portions of the protein can give rise to a similar clinical and cellular phenotype, providing evidence for the importance of the two dynein heavy chain regions in normal physiological function. Mutation c.3581A>G (p.Gln1194Arg) is the first within the newly described dynein neck region, and offers the first test of its physiological significance. Notably this locus is close to the mouse Cra1 mutation, which also has neurodegenerative consequences, though effects on brain development have not been assessed [Hafezparast et al 2003]. In addition, the neck domain may interact with the AAA4 ring of the motor domain, which interestingly, is the region containing the other mutation investigated here, c.9142G>A (p.Glu3048Lys). Thus both mutations conceivably affect a common motor-tail interaction mechanism, which is further suggested by similar cellular and physiological effects observed in the two patients from this study.
From a clinical prospective the current work is also the first to report the association of cortical malformation with effects on spinal nerves and to provide a distinctive pattern of brain and muscle pathologies as visualized by MRI. In particular, the PMG-like cortical malformation identified in the patients is clinically mild and is distinct from classic PMG comprising cobblestone-like features, such as over-migration of neurons through gaps in the leptomeninges. The latter should be more correctly classified as a cobblestone-like malformation, according to current nomenclature [Barkovich et al. 2012]. Conversely, the distal involvement of motor neurons is the predominate and most disabling feature, and the patients could be either classified as distal SMA or more specifically as SMA-LED, if the prevalent lower limb weakness is taken into account. Importantly, the combined features of cortical malformation and spinal motor neuron defect in our cases recall the dual involvement of the peripheral and central nervous systems in mice with mutated DYNC1H1 and delineate a similar human pathology, representing a possible continuum of clinical presentation.
Acknowledgments
We wish to thank the families of patients for their kind cooperation in this work. RBV receives support from NIH (NIH grant GM102347).
Footnotes
All authors declare no conflict of interest.
The variants described have been submitted to a public database (www.lovd.nl/DYNC1H1).
References
- Asai DJ, Koonce MP. The dynein heavy chain: structure, mechanics and evolution. Trends Cell Biol. 2001;11:196–202. doi: 10.1016/s0962-8924(01)01970-5. [DOI] [PubMed] [Google Scholar]
- Astrea G, Brisca G, Fiorillo C, Valle M, Tosetti M, Bruno C, Santorelli FM, Battini R. Muscle MRI in TRPV4-related congenital distal SMA. Neurology. 2012;78(5):364–5. doi: 10.1212/WNL.0b013e318245295a. [DOI] [PubMed] [Google Scholar]
- Alkuraya FS, Cai X, Emery C, Mochida GH, Al-Dosari MS, Felie JM, Hill RS, Barry BJ, Partlow JN, Gascon GG, Kentab A, Jan M, Shaheen R, Feng Y, Walsh CA. Human mutations in NDE1 cause extreme microcephaly with lissencephaly [corrected] Am J Hum Genet. 2011;88(5):536–47. doi: 10.1016/j.ajhg.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain. 2012;135:1348–1369. doi: 10.1093/brain/aws019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, Echeverri CJ, Nilsson T, Vallee RB. Overexpression of the dynamitin (p50) subunit of the dynactin complex disrupts dynein-dependent maintenance of membrane organelle distribution. J Cell Biol. 1997;139(2):469–84. doi: 10.1083/jcb.139.2.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Cho C, Jin L, Vale RD. Crystal structure of the dynein motor domain. Science. 2011;331:1159–1165. doi: 10.1126/science.1202393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen XJ, Levedakou EN, Millen KJ, Wollmann RL, Soliven B, Popko B. Proprioceptive sensory neuropathy in mice with a mutation in the cytoplasmic Dynein heavy chain 1 gene. J Neurosci. 2007;27(52):14515–24. doi: 10.1523/JNEUROSCI.4338-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobyns WB, Reiner O, Carrozzo R, Ledbetter DH. Lissencephaly. A human brain malformation associated with deletion of the LIS1 gene located at chromosome 17p13. JAMA. 1993;270(23):2838–42. doi: 10.1001/jama.270.23.2838. Review. [DOI] [PubMed] [Google Scholar]
- Feng Y, Olson EC, Stukenberg PT, Flanagan LA, Kirschner MW, Walsh CA. LIS1 regulates CNS lamination by interacting with mNudE, a central component of the centrosome. Neuron. 2000;28(3):665–79. doi: 10.1016/s0896-6273(00)00145-8. [DOI] [PubMed] [Google Scholar]
- Faulkner NE, Dujardin DL, Tai CY, Vaughan KT, O’Connell CB, Wang Y, Vallee RB. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat Cell Biol. 2000;2:784–791. doi: 10.1038/35041020. [DOI] [PubMed] [Google Scholar]
- Gennerich A, Carter AP, Reck-Peterson SL, Vale RD. Force-induced bidirectional stepping of cytoplasmic dynein. Cell. 2007;131:952–965. doi: 10.1016/j.cell.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafezparast M, Klocke R, Ruhrberg C, Marquardt A, Ahmad-Annuar A, Bowen S, Lalli G, Witherden AS, Hummerich H, Nicholson S, Morgan PJ, Oozageer R, et al. Mutations in dynein link motor neuron degeneration to defects in retrograde transport. Science. 2003;300:808–812. doi: 10.1126/science.1083129. [DOI] [PubMed] [Google Scholar]
- Harms MB, Allred P, Gardner R, Jr, Fernandes Filho JA, Florence J, Pestronk A, Al-Lozi M, Baloh RH. Dominant spinal muscular atrophy with lower extremity predominance: linkage to 14q32. Neurology. 2010;75:539–546. doi: 10.1212/WNL.0b013e3181ec800c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms MB, Ori-McKenney KM, Scoto M, Tuck EP, Bell S, Ma D, Masi S, Allred P, Al-Lozi M, Reilly MM, Miller LJ, Jani-Acsadi A, et al. Mutations in the tail domain of DYNC1H1 cause dominant spinal muscular atrophy. Neurology. 2012;78:1714–1720. doi: 10.1212/WNL.0b013e3182556c05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höök P, Vallee RB. The dynein family at a glance. J Cell Sci. 2006;119:4369–4371. doi: 10.1242/jcs.03176. [DOI] [PubMed] [Google Scholar]
- Kon T, Oyama T, Shimo-Kon R, Imamula K, Shima T, Sutoh K, Kurisu G. The 2.8 Å crystal structure of the dynein motor domain. Nature. 2012;484(7394):345–50. doi: 10.1038/nature10955. [DOI] [PubMed] [Google Scholar]
- Levy JR, Sumner CJ, Caviston JP, Tokito MK, Ranganathan S, Ligon LA, Wallace KE, LaMonte BH, Harmison GG, Puls I, Fischbeck KH, Holzbaur EL. A motor neuron disease-associated mutation in p150Glued perturbs dynactin function and induces protein aggregation. J Cell Biol. 2006;172(5):733–45. doi: 10.1083/jcb.200511068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercuri E, Messina S, Kinali M, Cini C, Longman C, Battini R, Cioni G, Muntoni F. Congenital form of spinal muscular atrophy predominantly affecting the lower limbs: a clinical and muscle MRI study. Neuromuscul Disord. 2004;14:125–129. doi: 10.1016/j.nmd.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Ori-McKenney KM, Xu J, Gross SP, Vallee RB. A cytoplasmic dynein tail mutation impairs motor processivity. Nat Cell Biol. 2010;12(12):1228–34. doi: 10.1038/ncb2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ori-McKenney KM, Vallee RB. Neuronal migration defects in the Loa dynein mutant mouse. Neural Dev. 2011;6:26. doi: 10.1186/1749-8104-6-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poirier K, Lebrun N, Broix L, Tian G, Saillour Y, Boscheron C, Parrini E, Valence S, Pierre BS, Oger M, Lacombe D, Geneviève D, et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat Genet. 2013;45(6):639–47. doi: 10.1038/ng.2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33(4):455–6. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Roberts AJ, Malkova B, Walker ML, Sakakibara H, Numata N, Kon T, Ohkura R, Edwards TA, Knight PJ, Sutoh K, Oiwa K, Burgess SA. ATP-driven remodeling of the linker domain in the dynein motor. Structure. 2012;20(10):1670–80. doi: 10.1016/j.str.2012.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossor AM, Kalmar B, Greensmith L, Reilly MM. The distal hereditary motor neuropathies. J Neurol Neurosurg Psychiatry. 2012;83(1):6–14. doi: 10.1136/jnnp-2011-300952. [DOI] [PubMed] [Google Scholar]
- Sasaki S, Shionoya A, Ishida M, Gambello MJ, Yingling J, Wynshaw-Boris A, Hirotsune S. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron. 2000;28:681–696. doi: 10.1016/s0896-6273(00)00146-x. [DOI] [PubMed] [Google Scholar]
- Tsurusaki Y, Saitoh S, Tomizawa K, Sudo A, Asahina N, Shiraishi H, Ito J, Tanaka H, Doi H, Saitsu H, Miyake N, Matsumoto N. A DYNC1H1 mutation causes a dominant spinal muscular atrophy with lower extremity predominance. Neurogenetics. 2012;13(4):327. doi: 10.1007/s10048-012-0337-6. [DOI] [PubMed] [Google Scholar]
- Vallee RB, McKenney RJ, Ori-McKenney KM. Multiple modes of cytoplasmic dynein regulation. Nat Cell Biol. 2012;14(3):224–30. doi: 10.1038/ncb2420. [DOI] [PubMed] [Google Scholar]
- Vissers LE, de Ligt J, Gilissen C, Janssen I, Steehouwer M, de Vries P, van Lier B, Arts P, Wieskamp N, del Rosario M, van Bon BW, Hoischen A, et al. A de novo paradigm for mental retardation. Nature Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- Weedon MN, Hastings R, Caswell R, Xie W, Paszkiewicz K, Antoniadi T, Williams M, King C, Greenhalgh L, Newbury-Ecob R, Ellard S. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am J Hum Genet. 2011;89:308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen MH, Vissers LE, Willemsen MA, van Bon BW, Kroes T, de Ligt J, de Vries BB, Schoots J, Lugtenberg D, Hamel BC, van Bokhoven H, Brunner HG, et al. Mutations in DYNC1H1 cause severe intellectual disability with neuronal migration defects. J Med Genet. 2012;49:179–183. doi: 10.1136/jmedgenet-2011-100542. [DOI] [PubMed] [Google Scholar]