

Abstract
Niemann-Pick type C (NPC) disease is a lysosomal storage disease in which endocytosed cholesterol becomes sequestered in late endosomes/lysosomes (LEs/Ls) because of mutations in either the NPC1 or NPC2 gene. Mutations in either of these genes can lead to impaired functions of the NPC1 or NPC2 proteins and progressive neurodegeneration as well as liver and lung disease. NPC1 is a polytopic protein of the LE/L limiting membrane, whereas NPC2 is a soluble protein in the LE/L lumen. These two proteins act in tandem and promote the export of cholesterol from LEs/Ls. Consequently, a defect in either NPC1 or NPC2 causes cholesterol accumulation in LEs/Ls. In this review, we summarize the molecular mechanisms leading to NPC disease, particularly in the CNS. Recent exciting data on the mechanism by which the cholesterol-sequestering agent cyclodextrin can bypass the functions of NPC1 and NPC2 in the LEs/Ls, and mobilize cholesterol from LEs/Ls, will be highlighted. Moreover, the possible use of cyclodextrin as a valuable therapeutic agent for treatment of NPC patients will be considered.
Keywords: neurodegenerative disease, lysosomal storage disease, Purkinje cells, gangliosides, neurons, astrocytes, microglia, cholesterol homeostasis, endoplasmic reticulum
Niemann-Pick type C (NPC) (1) disease is a progressive inherited disease in which unesterified cholesterol and other lipids accumulate in late endosomes/lysosomes (LEs/Ls) of all cells and tissues. The underlying cause of NPC disease is distinct from that of Niemann-Pick disease types A/B, which are caused by defects in the lysosomal acid sphingomyelinase gene (1). NPC disease is a recessive, autosomal disorder that affects ∼1/150,000 live births and causes premature death due to relentless neurodegeneration as well as lung and liver dysfunction (reviewed in Refs. 2, 3). Because neuropathological changes in NPC disease start long before the onset of symptoms, early diagnosis of the disease is crucial. However, clinical diagnosis of NPC disease in childhood can be difficult as the early symptoms are typical of many other neurological disorders, and therapeutic intervention is largely restricted to symptomatic treatment. Furthermore, access of therapeutic agents to the CNS is often problematic because the blood-brain barrier is impermeable to many molecules and prevents their passage into the brain from the peripheral circulation. In this review, we will summarize the molecular mechanisms that lead to NPC disease and discuss recent exciting data indicating that the cholesterol-sequestering agent, cyclodextrin, might be a valuable therapeutic agent for treatment of NPC disease.
BIOCHEMICAL AND CELL BIOLOGICAL IMPLICATIONS OF NPC DEFICIENCY
In mammalian cells, cholesterol is a key component of cellular membranes and is a precursor of steroid hormones and bile acids. Cells acquire cholesterol from endogenous synthesis in the endoplasmic reticulum (ER) and from receptor-mediated uptake of exogenous LDLs via LDL receptors (4). The underlying cellular defect in NPC disease is an impaired egress of endocytosed, LDL-derived unesterified cholesterol from LEs/Ls, resulting in the accumulation of unesterified cholesterol in LEs/Ls (5). This cellular defect was first revealed by Pentchev and coworkers (6) who demonstrated that the esterification of LDL-derived cholesterol was profoundly diminished in fibroblasts from NPC patients. However, the activity of the cholesterol esterification enzyme, ACAT, as well as the receptor-mediated uptake of LDL and the lysosomal hydrolysis of LDL-derived cholesteryl esters were normal. Thus, these observations indicated that LDL-derived cholesterol was sequestered in LEs/Ls because export of unesterified cholesterol from LEs/Ls was disrupted. Consequently, the amount of cholesterol reaching the ER and plasma membrane from LEs/Ls was reduced (6–9). In contrast, the transport of newly synthesized cholesterol from the ER to the plasma membrane occurs normally in fibroblasts from NPC patients (8, 10).
Homozygous mutations in either the NPC1 gene (11, 12) or the NPC2 gene (13, 14) were subsequently identified as being causative for NPC disease. Most (∼95%) identified cases of NPC disease are due to mutations in the NPC1 gene, whereas the remaining 5% of cases are caused by mutations in the NPC2 gene. The NPC1 gene encodes an LE/L membrane protein that contains 1,254 amino acids, 13 presumed transmembrane domains, a leucine zipper, a lysosomal targeting motif, and a sterol-sensing motif (12). The latter motif is similar to a sequence that has been identified in several proteins involved in cholesterol metabolism. The NPC2 gene (originally called HE1) encodes a 151-amino-acid soluble protein of the LE/L lumen (14, 15). The NPC1 and NPC2 proteins both bind cholesterol (16–19). The N-terminal domain of NPC1 is exposed to the LE/L lumen and binds cholesterol with high affinity in a 1:1 molar ratio (18–22). Fluorescence dequenching assays show that NPC2 rapidly transfers cholesterol, but not glycosphingolipids, ceramide, phospholipids, or fatty acids (16, 19, 21), between phospholipid liposomes in vitro. This transfer is promoted by an acidic environment and is markedly stimulated by the presence of bis(monoacylglycerol) phosphate (16, 23), a phospholipid that is highly enriched in lumenal multivesicular bodies of LEs/Ls and accumulates in NPC-deficient cells (24). X-ray crystallographic studies showed that orientation of the cholesterol molecule attached to NPC2 is opposite from that of cholesterol bound to the hydrophobic pocket of NPC1 (17, 18, 20–22).
Importantly, homozygous mutations in either NPC1 or NPC2 cause essentially the same clinical and cellular phenotypes (25, 26). Thus, a model was proposed for the concerted transfer of cholesterol from NPC2 to NPC1 within LEs/Ls (Fig. 1). In this model, endocytosed unesterified cholesterol is delivered to LEs/Ls and then transferred via NPC2 from membranes of the internal vesicles of LEs/Ls to NPC1 in the limiting membrane (19). An important feature of this model is that the hydrophobic cholesterol molecule can be exported without direct contact with the aqueous lumen of LEs/Ls. In support of this model, a direct interaction between NPC2 and a lumenal domain of NPC1 was demonstrated (27). This protein-protein interaction occurs only at acidic pH similar to that of the LE/L lumen and requires that cholesterol be bound to NPC2. The molecular mechanisms by which cholesterol is exported from NPC1 on the LE/L limiting membrane to the ER and plasma membrane have not yet been elucidated, although vesicular transport involving Rab proteins (28, 29), transport via carrier proteins such as oxysterol binding proteins (30, 31), and direct membrane contact have been proposed.
Fig. 1.
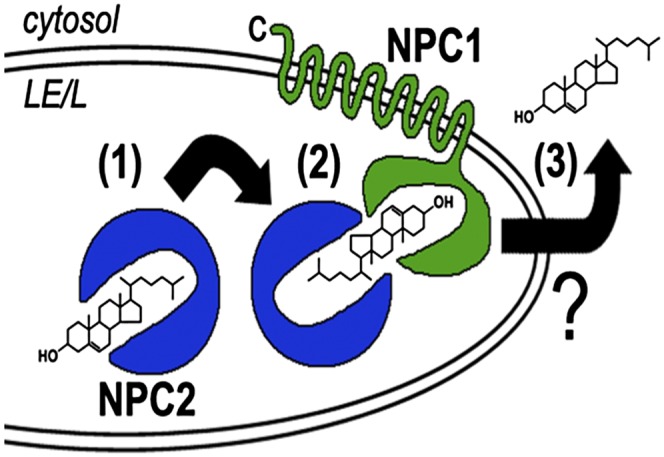
Proposed mechanism for the concerted action of NPC1/NPC2 in cholesterol export from LEs/Ls. NPC2 (blue), a soluble protein in the LE/L lumen, binds endocytosed cholesterol in the LE/L lumen (1). Subsequently, NPC2 shuttles the cholesterol to the cholesterol-binding pocket of NPC1 (green), a polytopic protein in the LE/L limiting membrane, without direct contact of the hydrophobic cholesterol molecule with the aqueous LE/L lumen (2). The mechanism by which NPC1-associated cholesterol is exported from the outer LE/L membrane to the ER and plasma membrane (3) has not been established. Reproduced from Ref. 165 with permission.
Because the sequestration of cholesterol in LEs/Ls of NPC-deficient cells impairs cholesterol delivery to the ER (6–8), the amount of cholesterol in the ER, as a molar % of total ER lipids, can fall below a critical threshold (32). Thus, although cholesterol accumulates in LEs/Ls, there is a deficit of cholesterol in the ER so that the sterol response element-binding protein (SREBP) pathway (33) is enhanced, and the synthesis and uptake of cholesterol are increased (9, 34). The impaired trafficking of cholesterol in NPC-deficient cells profoundly affects multiple cellular functions such as regulation of lysosomal calcium homeostasis (35), oxidative stress (36, 37), and vesicle trafficking pathways mediated by Rab proteins (28, 29), as well as fusion of LEs/Ls (38). NPC1 deficiency is particularly detrimental for the functioning of LEs/Ls in the brain (39). Intriguingly, cholesterol transport from LEs/Ls to mitochondria is not defective in NPC1-deficient cells (40). Indeed, the cholesterol content of mitochondria isolated from brains is increased, not decreased, by NPC1 deficiency (40, 41). Several properties of mitochondria such as ATP production, oxidative stress, and possibly mitophagy are altered by NPC1 deficiency (41–43), probably because of the increase in mitochondrial cholesterol. In contrast to deficiency of NPC1, however, the movement of endosomal cholesterol to mitochondria is disrupted by NPC2 deficiency (44). Moreover, mutants of NPC2 that bind cholesterol, but cannot transfer cholesterol to NPC1, can restore cholesterol trafficking to mitochondria in NPC2-deficient cells (44). Because NPC2 transfers cholesterol directly to membranes (23), a possible explanation for this difference between NPC1 and NPC2 is that NPC2 transfers cholesterol from the LE/L lumen directly to the perimeter membrane of LEs/Ls, as well as to transmembrane proteins such as MLN64 and NPC1.
CLINICAL MANIFESTATIONS OF NPC DEFICIENCY
The clinical manifestations of NPC disease usually become apparent in early childhood, but the age of onset and rate of disease progression vary over a wide range (2, 45, 46). In both humans and mice, the most severe consequence of NPC deficiency is neurodegeneration, particularly of Purkinje neurons in the cerebellum, resulting in progressive motor impairment (47, 48). In other brain regions, neuronal abnormalities, such as axonal swelling and degeneration and ectopic dendrite formation, also occur and are often more prominent than neuron death (49–52). These impairments contribute to the reduced weight gain, cognitive decline, and premature death that characterize NPC disease. Severe liver disease is also prevalent in many NPC-deficient patients (2) and mice (53–56). The majority (∼80%) of circulating LDLs are cleared by the liver via receptor-mediated endocytosis (57, 58). Consequently, cholesterol accumulation in the LEs/Ls of NPC1-deficient patients and mice is more pronounced in liver than in any other tissue (58, 59). The lung is another tissue whose function is impaired in NPC disease (58, 60–62), and lipid-laden macrophages are abundant in the lungs of NPC-deficient mice (63). Moreover, age-related retinal degeneration occurs in Npc1−/− mice (64). Two recent studies have reported that levels of mRNAs and proteins of several enzymes of energy metabolism are altered and that lactate levels are increased in NPC1-deficient cerebella. Moreover, pyruvate oxidation is decreased and anaerobic glycolysis is increased during progression to the symptomatic stage of NPC disease (65, 66). Thus, glucose metabolism appears to be affected at early stages of NPC disease. Markers of oxidative stress are also increased in brains of human NPC patients and animal models (36, 37, 67).
MODELS USED FOR STUDYING NPC DISEASE
Several cell and animal models of NPC disease are available and have provided key information on the mechanism by which deficiency of NPC1 or NPC2 leads to the disease phenotype. The cell biological and biochemical defects in NPC-deficient cells were initially discovered using NPC1-deficient fibroblasts (mutant Chinese hamster ovary cells and fibroblasts from NPC patients) (6, 7, 10, 34, 68, 69). Although the class II amphiphilic amine, U18666, also causes cholesterol sequestration in LEs/Ls and has been widely used to mimic the NPC defect in cultured cells, this drug produces side effects including inhibition of cholesterol synthesis (70, 71). The most widely used model of NPC disease is the Balb/cNctr-NpcN/+ mouse that completely lacks NPC1 protein (72). These mice are asymptomatic at birth but develop ataxia and die within ∼3 months. A more recent mouse model carries the D1005G mutation in NPC1 (73). This mutation is in the cysteine-rich loop of NPC1, the site at which the most common mutations occur in human NPC patients, and these mice have a more slowly progressing disease than that of NPC1-null mice (73). NPC1 expression has also been reduced in mouse liver by antisense technology (74). Hypomorphic NPC2-deficient mice (that express only 0.4% of the normal amount of NPC2 protein) (26) and NPC2-null mice (75) have also been generated. The phenotype of these mice is similar to, but slightly less severe than, that of Npc1−/− mice (26, 75). More recently, NPC1 was eliminated specifically in certain cell types (76) and, reciprocally, was expressed only in specific cells of Npc1−/− mice (77). These studies have been particularly useful for understanding in which cells of the brain loss of NPC function is the most detrimental (see Loss of NPC Function in Cells of the Brain). As an alternative to mouse models of NPC disease, an NPC1-deficient feline model was established (78, 79) and has provided valuable information relevant to human NPC disease. Other models that have been used to elucidate pathways defective in NPC disease are mutants of yeast (80) and Drosophila (81, 82). An advantage of these nonmammalian models is that genetic manipulation is relatively straightforward. Recently, studies have been initiated in human stem cells isolated from individual NPC patients, particularly stem cells that were differentiated into neuron-like cells (43, 83–85). It is likely that these cells will be useful for evaluation of drug candidates for NPC disease.
LIPID ACCUMULATION IN THE LEs/Ls
Cholesterol is not the only lipid that accumulates abnormally in LEs/Ls of NPC-deficient cells. Sphingomyelin (86), gangliosides (particularly GM2 and GM3) (87–89), sphingosine (35), and bis(monoacylglycerol) phosphate (24, 90) also accumulate. Compelling evidence indicates that the primary offending molecule is cholesterol and that the accumulation of the other lipids is secondary to the sequestration of cholesterol. First, NPC1 and NPC2 each bind cholesterol with high affinity, but not gangliosides or sphingosine (17, 18, 91). In addition, a mutation in the cholesterol-binding site of NPC1 abolished the binding of cholesterol, and expression of this mutant protein in mice induced the typical NPC phenotype (92). Second, the storage of gangliosides was genetically reduced by crossing Npc1−/− mice with mice that lacked the galactosyltransferase responsible for synthesis of GM2 ganglioside or with mice lacking GM3 synthase (93). In these mice, ganglioside storage in LEs/Ls was eliminated, but cholesterol still accumulated and the neurodegeneration was not improved (93–95). Third, because lysosomal acid sphingomyelinase activity is inhibited when the amount of cholesterol in LE/L membranes is increased, the accumulation of sphingomyelin in LEs/Ls of NPC-deficient cells also appears to be secondary to cholesterol sequestration (86, 96). Fourth, although sphingosine accumulation was reported prior to cholesterol accumulation (35), these studies were not performed in NPC1-deficient cells but in cells treated with the amphiphile U18666A, which also inhibits cholesterol synthesis. Fifth, 2-hydroxypropyl-β-cyclodextrin (CYCLO), which forms an inclusion complex with cholesterol (reviewed in Ref. 97), dramatically reduced cholesterol storage in tissues of NPC-deficient mice, thereby attenuating the neurodegeneration and extending life span (98–100) (see Cyclodextrin as a Therapy for NPC Disease). These data provide convincing evidence that cholesterol sequestration in LEs/Ls of NPC-deficient cells is the primary event responsible for NPC disease.
LOSS OF NPC FUNCTION IN CELLS OF THE BRAIN
Although NPC1 and NPC2 appear to be expressed in all cells and tissues, the most devastating consequences of NPC deficiency typically occur in the brain. Cholesterol metabolism in the brain is compartmentalized from that in the periphery by the blood-brain barrier (101, 102). Thus, essentially all cholesterol in the brain is synthesized within the brain rather than being delivered from plasma lipoproteins. The most striking consequence of NPC1 deficiency in the brains of mice and humans is a profound loss of Purkinje neurons in the cerebellum (103–105). In other regions of the brain, such as the cerebral cortex and thalamus, neuronal apoptosis also occurs but is less common than axonal swelling and ectopic dendrite formation (51, 52, 106–109). The reason why Purkinje neurons are the most sensitive neurons to NPC deficiency is not clear (39).
Interestingly, although cholesterol accumulates in NPC1-deficient peripheral tissues, such as the liver, the cholesterol content of NPC-deficient brains does not increase, but even decreases, with age. The reason for this apparent anomaly is that myelin contains ∼75% of cholesterol in the brain, and extensive demyelination occurs in NPC-deficient brains (58, 110). In addition, genetic deletion of NPC1 in oligodendrocytes of mice reduces myelin formation (111, 112). Cholesterol does, however, accumulate in LEs/Ls of NPC-deficient neurons, such as Npc1−/− mouse sympathetic neurons (113), hippocampal neurons (Fig. 2) (114), and cerebellar granule neurons (115). In Npc1−/− sympathetic neurons, cholesterol accumulates in LEs/Ls of the cell bodies, whereas the cholesterol content of distal axons is less than that of Npc1+/+ neurons (113). In addition, cholesterol transport from cell bodies to distal axons is impaired by lack of NPC1 (116). Although the accumulation of cholesterol in cell bodies of NPC-deficient neurons is generally considered to be the major cause of neurodegeneration, it is also possible that a deficiency of cholesterol in distal axons contributes to the neurological phenotype.
Fig. 2.

Cholesterol sequestration in LEs/Ls of Npc1-deficient neurons. Primary hippocampal neurons from E17 Npc1−/− (A–C) and Npc+/+ (D–F) mice were cultured for 14 days in serum-free medium and then stained for cholesterol (filipin) (A, D) and with antibodies directed against lysosome-associated membrane protein-1, LAMP1 (B, E); merge (C, F). Scale bars indicate 20 µm.
The NPC1 protein is present in recycling endosomes of presynaptic nerve terminals, and the morphology of synaptic vesicles isolated from brains of Npc1−/− mice is aberrant (117). Moreover, defects in synaptic transmission have been observed in several brain areas and in experimental models of NPC1 deficiency. Synaptic membranes are relatively rich in cholesterol, which is required for synaptic transmission at many levels. For example, the activity of pre- and postsynaptic receptors is influenced by membrane cholesterol content (reviewed in Refs. 118, 119). In addition, the formation of presynaptic SNARE complexes, the interaction between synaptobrevin and synaptophysin, the fusion of synaptic vesicles with presynaptic membranes, and the sorting of synaptic vesicles during endocytosis all depend on cholesterol (120–124). Thus, extraction of cholesterol from neurons, by brief incubation with cyclodextrin, impaired exocytosis and endocytosis of synaptic vesicles (114, 125–128). Moreover, defects in exocytosis and mobilization of synaptic vesicles, as well as increased spontaneous neurotransmission, were observed in cultured hippocampal neurons from Npc1−/− mice (114, 126, 129). The inhibitory GABAergic synapses were more severely affected than glutamatergic synapses, indicating an imbalance between inhibitory and excitatory neurotransmission (129). Studies in hippocampal slices also revealed hyperexcitability of the neuronal network in NPC1-deficient brains (114). Furthermore, NPC1 deficiency alters long-term synaptic plasticity in several brain regions (129–132). The presynaptic defects in NPC1-deficient neurons are independent of the presence of glial cells (133).
The mechanism by which defective export of cholesterol from LEs/Ls leads to neuropathology is poorly understood. Thus, an important question is, in which cell type(s) of the brain is NPC deficiency responsible for the neurological deficits? The three major types of cells in the brain are neurons, astrocytes, and microglia, all of which sequester cholesterol in LEs/Ls in response to NPC deficiency (113, 115, 134, 135). Clearly, inflammation is widespread in NPC-deficient brains. Microglia are resident immune cells of the CNS and proliferate and become activated in response to changes in their environment. Microglia participate in phagocytosis of injured/dying cells (136) and can generate a protective inflammatory response (137, 138). On the other hand, chronic activation of microglia can induce neuroinflammation and neuronal death in several neurodegenerative disorders (reviewed in Ref. 139). Microglial proliferation and activation are rampant in specific brain regions of NPC disease patients and Npc1−/− mice, prior to onset of neurological symptoms (50, 52, 135). Thus, the microglial activation that occurs in NPC disease could either be a protective mechanism induced in response to neuronal death or could contribute directly to neuronal death. In mouse brains, NPC1 deficiency increased expression of mRNAs encoding proinflammatory cytokines, such as tumor necrosis factor (105, 108, 140). Moreover, treatment of Npc1−/− mice with a liver X receptor agonist reduced microglial activation and neuroinflammation and prolonged survival of the mice (140). However, this agonist alters the expression of genes involved in multiple pathways, and one cannot conclude that the decrease in neuroinflammation was directly linked to the increased life span. The majority of recent data indicate that inflammation in NPC-deficient brains is primarily a response to the neurodegeneration. For example, when NPC1 was eliminated only in neurons of mice, neuropathology developed typical of that in mice in which NPC1 was eliminated globally (141). Moreover, expression of NPC1 only in neurons of Npc1−/− mice prevented the neurodegeneration (77). In addition, in microglia-neuron cocultures, the presence of activated Npc1−/− microglia did not cause neuron death (135). Consistent with the idea that inflammation is secondary to neuron death, deletion of the macrophage inflammatory gene Mip1a/Ccl3 in Npc1−/− mice did not prevent neurodegeneration (142), nor did deletion of the complement system in Npc1−/− mice reduce neuronal death or prolong survival (143). In combination, these experiments imply that microglial activation in NPC-deficient brains is primarily a response to neuronal death, rather than a direct cause of the neurodegeneration.
The proliferation and activation of astrocytes are also widespread in NPC1-deficient brains (50). However, this astrogliosis does not appear to be the primary cause of the neurodegeneration. For example, in a hybrid mouse model, Npc1+/+ neurons were not killed by surrounding Npc1−/− astrocytes/microglia (144). Furthermore, genetic deletion of NPC1 in astrocytes alone did not cause the neuropathology exhibited by Npc1−/− mice (141). Nor did Tet-induced expression of NPC1 in astrocytes alone of Npc1−/− mice slow disease progression, except for a small delay in weight loss (77). In contrast, one study reported that cholesterol storage was decreased, and survival of Npc1−/− mice was increased, by expression of NPC1 in astrocytes under control of the glial fibrillary acidic protein promoter. However, NPC1 expression in other cell types was not completely excluded (145). On the other hand, neuron-specific deletion of NPC1 caused activation of astrocytes and microglia in the murine brain (141, 143). Consequently, the astrogliosis and inflammation in NPC-deficient brains appear to be largely secondary to NPC deficiency in neurons.
Overall, these observations in NPC-deficient cells of the brain strongly imply that the absence of NPC1 in neurons alone is sufficient to cause the neuropathology. Consistent with this conclusion, NPC1 expression only in neurons of Npc1−/− mice prevented the neurodegeneration (77, 146). More specifically, expression of NPC1 in cerebellar Purkinje neurons (the neurons most affected in NPC disease), but not in neurons of other forebrain areas, of Npc1−/− mice markedly extended life span and lessened the ataxia (77). In addition, elimination of NPC1 specifically in Purkinje neurons caused ataxia and loss of Purkinje cells in an anterior-to-posterior gradient, but, surprisingly, these mice did not exhibit weight loss or premature death (76). In another study, NPC1 was eliminated from neurons in nearly all brain areas, although NPC1 was still expressed in Purkinje cells, yet the mice exhibited the same neurodegenerative phenotype as that of NPC1-null mice (141). Thus, although lack of NPC1 in Purkinje cells contributes to Purkinje cell death and the neuropathology in NPC disease, lack of NPC1 in other types of neurons is clearly also detrimental. Overall, these observations demonstrate that NPC deficiency in neurons, rather than in astrocytes or microglia, is the primary cause of neuropathology in NPC disease.
CYCLODEXTRIN AS A THERAPY FOR NPC DISEASE
Potential therapies for NPC disease have focused on reduction of lipid storage in LEs/Ls. An important consideration when testing therapies for NPC disease patients is how disease progression can be quantitatively assessed. Thus, validation of biomarkers of NPC disease progression is an important goal (147–149). Because gangliosides accumulate in LEs/Ls of NPC-deficient cells, miglustat, which efficiently crosses the blood-brain barrier, has been used for treatment of NPC patients. Miglustat inhibits glucosylceramide synthase, a key enzyme of glycosphingolipid synthesis (reviewed in Ref. 150). In the feline model of NPC disease, miglustat decreased GM2 ganglioside accumulation and slightly delayed development of neurological symptoms (151). Similarly, in Npc1−/− mice, miglustat reduced ganglioside storage in LEs/Ls. In addition, miglustat modestly extended life span of (87) and produced some neurological improvements in NPC patients (152–156). However, the mechanism underlying these clinical improvements is not clear because reduction of gangliosides in LEs/Ls of NPC-deficient mice does not appear to be beneficial (93–95).
Several other treatments for NPC disease are currently being investigated (reviewed in Ref. 157). One of the most promising is inhibition of histone deacetylases (HDACs). HDACs appear to play important roles in neuronal survival (158, 159). HDAC inhibitors have already been approved for clinical use in humans for treatment of certain cancers. A clinically approved HDAC inhibitor decreased HDAC expression in NPC1-deficient cells and reduced cholesterol accumulation in LEs/Ls (160, 161). Interestingly, however, treatment of NPC2-deficient human fibroblasts with an HDAC inhibitor did not reduce cholesterol storage in LEs/Ls (161). The expression of several HDAC genes is increased by NPC deficiency in human fibroblasts (160, 161). Because HDAC inhibitors increase the amount of NPC1 protein (162), these inhibitors might be useful in NPC patients in which residual NPC1 activity remains or where partially active NPC1 protein is mislocalized (163). The antiapoptotic agent imatinib has also been suggested as an agent that might prevent neurodegeneration in NPC disease by inhibiting the c-Abl pathway, the activity of which is increased in Npc1−/− Purkinje cells (164). Imatinib treatment of Npc1−/− mice partially prevented Purkinje cell death and modestly improved neurological parameters, but only slightly prolonged life (164).
A very exciting approach toward treatment of NPC disease is based on the discovery that the cholesterol-binding agent CYCLO markedly delays neurodegeneration and increases life span of Npc1−/− mice [(95, 98, 99); reviewed in Ref. 165]. These studies were prompted by a report that in 7-day-old Npc1−/− mice injection of the neurosteroid allopregnanolone, dissolved in CYCLO, delayed the neurodegeneration and approximately doubled life span (166). Subsequent experiments revealed, however, that injection of allopregnanolone alone, without CYCLO, into 7-day-old Npc1−/− mice did not produce the beneficial effects (98). On the other hand, a single intraperitoneal injection of CYCLO (4,000 mg/kg body weight), the carrier of the allopregnanolone (166), did reduce the neurodegeneration and increased life span by ∼50% (95, 98). Thus, the reported beneficial effects of allopregnanolone in Npc1−/− mice (166) appear to be attributable to CYCLO. Additional benefits are gained by repeated administrations of CYCLO. For example, subcutaneous injection of CYCLO (4,000 mg/kg) into Npc1−/− mice once per week for 7 weeks reduced cholesterol accumulation in most tissues, markedly improved neurodegeneration and liver function, and approximately doubled life span (60), although the pulmonary disease was resistant to CYCLO (60). When CYCLO (4,000 mg/kg) was subcutaneously injected into Npc1−/− mice on alternate days, starting at either day 7 or day 21 after birth, the neuronal storage of cholesterol and gangliosides was reduced, neurodegeneration was delayed, and life span was prolonged (99). Importantly, injection of CYCLO into NPC2-deficient mice was similarly beneficial (99, 167). In careful “balance” studies on quantification of the cholesterol excreted into bile by Npc1−/− mice, CYCLO reduced cholesterol storage in almost all tissues and promoted the excretion of cholesterol-derived bile acids (100). Importantly, although CYCLO injection mobilized LE/L cholesterol and reduced inflammatory parameters in 49-day-old Npc1−/− mice (Fig. 3), their life span was not increased, probably because extensive tissue damage had already occurred (100). Moreover, CYCLO is cleared from the body 6 times more rapidly in mature mice than in 7-day-old Npc1−/− pups, which could further contribute to the lower efficacy of CYCLO in older mice (100).
Fig. 3.

CYCLO prevents Purkinje cells loss in Npc1−/− mice. Npc1−/− (B, C, E, F, H, I) and Npc+/+ (A, D, G) mice (7 days old) were given a single intraperitoneal injection of CYCLO (4,000 mg/kg body weight). At 49 days of age, sections of the cerebellum were stained with hematoxylin and eosin (A–C), for calbindin (Purkinje cell marker) (D–F), and for glial fibrillary acidic protein (marker of astrocyte activation) (G–I). Scale bars indicate 100 µm. Arrows indicate loss of Purkinje cells in a band-like pattern (indicated by brackets) in untreated Npc1−/− mice; *, necrotic Purkinje cells. Reproduced from Ref. 98 with permission.
The studies described previously on the remarkable effects of subcutaneous injection of CYCLO into 7-day-old mice (Fig. 3) (95, 98, 99) imply that at least some CYCLO crosses the blood-brain barrier and enters the CNS. Nevertheless, a recent study concluded that the amount of CYCLO that crosses the blood-brain barrier from plasma in mature mice is very small, and that the neurological benefits of peripherally injected CYCLO are the result of CYCLO binding to the cerebral vascular endothelium (168). Other studies, however, have indicated that a small percentage of CYCLO in the peripheral circulation does cross the blood-brain barrier (169); it is also possible that in 7-day-old, but not older, mice the blood-brain barrier is incompletely closed and is partially permeable to CYCLO. In light of the poor permeability of the blood-brain barrier to CYCLO, CYCLO was injected directly into the CNS of Npc1−/− mice. The CYCLO diffused throughout all areas of the brain, and the benefits lasted more than a week (170). CYCLO was, therefore, infused continuously by intracerebroventricular delivery (35 mg/kg/day) into brains of Npc1−/− mice for 4 weeks, starting at 3 weeks of age. When this treatment was combined with weekly subcutaneous injections of CYCLO (4,000 mg/kg), the histology of all regions of the brain was normal and neurodegeneration was eliminated (170). Importantly, in Npc1+/+ mice, cholesterol homeostasis was not altered when CYCLO was delivered peripherally or intracerebroventricularly (170). The ED50 for the therapeutic effect of CYCLO is 0.5 mg/kg (100, 170). The concentration of CYCLO required to mobilize LE/L cholesterol in NPC1-deficient cells in either peripheral tissues or in the CNS of Npc1−/− mice is <1 mM, probably ∼0.1 mM (100, 115, 170). No toxic effects of these low doses of CYCLO were evident in Npc1−/− mice, and CYCLO has been approved by the Federal Drug Administration (USA) as a drug delivery vehicle in humans. These exciting studies suggest that the simultaneous delivery of CYCLO by subcutaneous injection into the periphery and by direct infusion into the CNS might represent a valid, if cumbersome, therapeutic approach for treatment of NPC patients. Indeed, a clinical trial in which CYCLO is being delivered intrathecally via osmotic minipumps to a few NPC patients was initiated at the National Institutes of Health (USA) in 2013; the outcome is anxiously awaited.
The effectiveness of CYCLO was also evaluated in the feline NPC model. As in NPC-deficient mice, CYCLO (4,000 mg/kg) markedly attenuated the neurodegeneration and liver disease in the cats (79). However, a dose-dependent hearing defect developed in the CYCLO-treated cats suggests that CYCLO damages the peripheral auditory pathway (79). This unexpected side effect was further investigated by administration of a single dose of CYCLO subcutaneously (8,000 mg/kg), or intrathecally (120 mg), to Npc1+/+ cats. The cats exhibited a significant increase in hearing threshold. However, the mechanism by which CYCLO caused this effect has not been established (79). Consequently, it will be important to determine whether CYCLO causes a hearing defect in NPC patients.
MECHANISM BY WHICH CYCLO IMPROVES THE NPC PHENOTYPE
Cyclodextrins, such as CYCLO, are cyclic oligosaccharides consisting of multiple glycopyranose units (reviewed in Ref. 97) and have been used for delivery of hydrophobic drugs to humans. The cyclodextrins are water soluble but contain a hydrophobic pocket. Cyclodextrins typically exist as hexamers (α-cyclodextrins), heptamers (β-cyclodextrins), or octamers (γ-cyclodextrins), which contain hydrophobic cavities of different sizes. The β-cyclodextrins have the highest affinity for cholesterol and are the most efficient in extracting cholesterol from cells. In contrast, α-cyclodextrins are the most effective at extracting phospholipids from cells: the hydrophobic cavity of α-cyclodextrins appears to be too small to accommodate cholesterol. The binding pocket of γ-cyclodextrins is less hydrophobic than that of β-cyclodextrins. The water solubility of β-cyclodextrins can be increased by modification of the hydrophilic groups with moieties such as 2-hydroxypropyl groups. The methyl- and 2-hydroxypropyl derivatives of β-cyclodextrin are most commonly used experimentally to deplete cells of cholesterol. For example, exposure of cells to 5–10 mM β-cyclodextrins for >2 h removes 80–90% of cellular cholesterol (171). In addition, complexation of β-cyclodextrin with cholesterol is used to deliver cholesterol to cells (172).
Accumulating evidence indicates that the primary mechanism by which CYCLO mobilizes cholesterol from LEs/Ls of NPC-deficient cells depends on bulk-phase endocytosis (173) or clathrin-mediated endocytosis (174) of CYCLO into LEs/Ls. Time course experiments have shown that cholesterol removal from LEs/Ls requires the internalization of CYCLO into the cells, rather than extraction of cholesterol from the plasma membrane (173). For example, CYCLO was covalently linked to fluorescent dextran, and trafficking of the fluorescent conjugate was visualized in NPC-deficient cells. Cholesterol was efficiently removed from LEs/Ls and was CYCLO localized to intracellular organelles (173). In addition, LE/L cholesterol was mobilized in Npc1−/− fibroblasts even by incubation with CYCLO that was preloaded with cholesterol (173). Several studies have shown that cholesterol released from LEs/Ls of NPC-deficient cells by CYCLO reaches the cytosolic compartment and is accessible to the ER. An indication of increased ER cholesterol content is that production of cholesteryl esters (the storage form of cholesterol) via the ER localized enzyme ACAT is stimulated. Indeed, cholesterol esterification by ACAT was increased by up to 14-fold in various Npc1−/− mice tissues and cells after CYCLO treatment (98, 115, 170, 173, 175), demonstrating that the liberated cholesterol must have been accessible to ACAT on the ER. In cultured cerebellar neurons, astrocytes, and microglia from Npc1−/− mice, the sequestered cholesterol was mobilized to the ER by low concentrations (0.1–1.0 mM) of CYCLO (115). Another indicator of increased cholesterol availability at the ER is decreased expression/processing of SREBP2, a key transcription factor that regulates cholesterol synthesis and uptake (33). CYCLO decreased expression of mRNAs encoding the LDL receptor and 3-hydroxy-3-methylglutaryl-CoA reductase in Npc1−/− mice and, correspondingly, reduced the rate of cholesterol synthesis in the brain and peripheral tissues (98, 100, 170). These observations clearly demonstrate that in Npc1−/− cells CYCLO releases cholesterol stored in LEs/Ls so that the cholesterol is mobilized to the ER.
In order to determine which cell types in the brain are responsive to CYCLO, and to investigate further the mechanism by which CYCLO mobilizes LE/L cholesterol in NPC-deficient brains, primary cultures of Npc1−/− mouse neurons and astrocytes were exposed to 0.1 to 10 mM CYCLO for 24 h. Not surprisingly, because 5–10 mM CYCLO rapidly depletes cholesterol from the plasma membrane, 10 mM CYCLO was profoundly toxic to neurons and markedly altered astrocyte morphology (115). In contrast, 0.1 and 1.0 mM CYCLO were not obviously toxic to these cells and essentially abolished cholesterol accumulation in LEs/Ls of Npc1−/− neurons, astrocytes, and microglia. Importantly, however, 0.1 mM CYCLO and 1.0 mM CYCLO exerted opposite effects on cholesterol homeostasis. Consistent with the hypothesis that CYCLO releases cholesterol from LEs/Ls to the ER, 0.1 mM CYCLO reduced cholesterol synthesis in Npc1−/− astrocytes and neurons and profoundly enhanced cholesterol esterification in astrocytes (115). On the other hand, CYCLO did not stimulate cholesterol esterification in cerebellar neurons from Npc1−/− mice, probably because ACAT activity and cholesteryl ester content are very low in neurons compared with other cells (115). In marked contrast to 0.1 mM CYCLO, 1.0 mM CYCLO increased cholesterol synthesis and expression of genes involved in cholesterol uptake and synthesis in these cells, while dramatically decreasing cholesterol esterification, suggesting that CYCLO had depleted cholesterol even from the ER. These data strongly support the idea that 0.1 mM CYCLO releases cholesterol trapped in LEs/Ls of astrocytes and neurons, thereby increasing the ER content of cholesterol. In contrast, higher concentrations of CYCLO (1.0 mM and greater) appear to extract cholesterol from the plasma membrane and eventually deplete cholesterol from the ER in addition to removing cholesterol from LEs/Ls. These studies indicate that the concentration of CYCLO that would be beneficial in brains of NPC patients is ∼0.1 mM, consistent with the finding that in brains of Npc1−/− mice in which CYCLO reversed the cholesterol transport defect, the concentration of CYCLO was ∼0.1 mM (170). Thus, in brains, and probably also other tissues, concentrations of CYCLO >1 mM would be expected to deplete cellular cholesterol and impair neuron survival (115). On the other hand, in Npc1+/+ mice and cells that do not sequester cholesterol in LEs/Ls, CYCLO does not alter cholesterol homeostasis (115, 176). Even CYCLO that is preloaded with cholesterol can mobilize cholesterol from LEs/Ls without depleting cholesterol from the plasma membrane, suggesting that cholesterol-loaded CYCLO might be less toxic to the brain than CYCLO not loaded with cholesterol (173).
A model for the action of CYCLO that is consistent with the previous data is that CYCLO is taken up by cells via endocytosis and enters LEs/Ls. Cholesterol transfer within the LEs/Ls is normally mediated by the tandem action of NPC1 and NPC2 in a process that requires a physical association between NPC1 and NPC2 (Fig. 1) (27). However, in the presence of CYCLO, cholesterol stored in LEs/Ls is transported to and across the LE/L limiting membrane, independent of either NPC1 or NPC2, and is subsequently transported to the ER (by an undefined mechanism). Because CYCLO reduces cholesterol storage in LEs/Ls of either NPC1- or NPC2-deficient cells (99, 173), CYCLO can bypass the function of both proteins. In vitro studies have shown that CYCLO markedly accelerates cholesterol transfer between membranes via a collisional mechanism involving direct interaction between CYCLO and membranes (177). The mechanism by which CYCLO releases cholesterol from LEs/Ls was further investigated using two types of β-cyclodextrin (167, 176). Previous studies had shown that CYCLO, at concentrations of >1 mM, removes cholesterol from membranes into the aqueous medium by formation of a 2:1 CYCLO-cholesterol molar complex. In contrast, sulfobutylether-β-cyclodextrin forms a 1:1 complex with cholesterol that cannot solubilize cholesterol from membranes. At lower concentrations (<1 mM), however, both CYCLO and sulfobutylether-β-cyclodextrin can shuttle cholesterol between membranes (171). To determine whether CYCLO removed cholesterol from LEs/Ls of NPC-deficient cells by solubilization or by a shuttle mechanism, Npc1−/− mice were injected with CYCLO or sulfobutylether-β-cyclodextrin (167, 176). With both types of cyclodextrin, the mobilized cholesterol did not rapidly (at least within 12 h) enter the plasma, either in lipoproteins or in cholesterol-CYCLO complexes, nor was the released cholesterol excreted into urine (176). Furthermore, both cyclodextrins reduced expression of SREBP-responsive genes to a similar extent. Thus, the transfer of stored cholesterol out of LEs/Ls of Npc1−/− cells does not depend on the ability of CYCLO to solubilize cholesterol into an intracellular or extracellular aqueous milieu. More likely, CYCLO shuttles cholesterol between lumenal vesicles within LEs/Ls and the LE/L limiting membrane, so that the cholesterol reaches the exit site on the limiting membranes of LEs/Ls in the absence of NPC1 and NPC2 (176). In an attempt to increase the efficiency of cholesterol clearance from LEs/Ls, supramolecular complexes of cyclodextrin (polyrotaxanes) were designed in which cyclodextrin was threaded onto a biocompatible, linear polymer that was capped by bulky end groups that were amenable to enzymatic cleavage (178, 179). These complexes are endocytosed by cells, and cyclodextrin is released by enzymatic cleavage of the end caps so that high concentrations of cyclodextrin can be delivered to LEs/Ls, while cyclodextrin’s activity on the plasma membrane is avoided. Thus, treatment of NPC2-deficient fibroblasts with CYCLO-polyrotaxane complexes efficiently mobilized cholesterol from LEs/Ls (178, 179). Whether these complexes will also be effective in the CNS remains to be determined.
Several other neurodegenerative disorders, such as Alzheimer’s disease and Huntington’s disease, are also associated with impaired cholesterol homeostasis in the brain (reviewed in Ref. 180). Alzheimer’s disease and NPC disease have some similarities, including alterations in lysosomal function (39, 181), increased deposition of amyloid plaques (182–184), and the presence of neurofibrillary tangles in humans (185) [but not in Npc1−/− mice (106)]. NPC1 expression is higher in the hippocampus and frontal cortex of Alzheimer’s patients than in controls (186). Moreover, genetic overexpression of amyloid precursor protein in Npc1−/− mice decreased their life span, whereas CYCLO prolonged survival (187). CYCLO reduced cholesterol sequestration in brains of mouse models of Alzheimer’s disease and lowered levels of amyloid by reducing β-cleavage of amyloid precursor protein and increasing expression of genes involved in clearance of β-amyloid plaques (188). These observations suggest that CYCLO might be a useful therapeutic agent for treatment of Alzheimer’s disease and, perhaps also, of other neurodegenerative disorders in which cholesterol homoestasis is disrupted.
CONCLUSIONS
Identification of the defective genes in NPC disease led to elucidation of the mechanism by which NPC1 and NPC2 proteins mediate the egress of cholesterol from LEs/Ls. Currently, no effective treatment is available for NPC disease patients, although the remarkable discovery that treatment of Npc1−/− and Npc2−/− mice with the cholesterol-sequestering agent CYCLO improves the neurodegeneration and prolongs life are very encouraging. Clearly, however, because of the low permeability of the blood-brain barrier to CYCLO, administration of CYCLO to the CNS of NPC patients is problematic. Thus, a reasonable protocol might be to administer CYCLO simultaneously to the periphery and also to the brain of NPC patients. Such an approach might treat both the liver disease and the neurodegeneration. An unresolved issue, however, is that CYCLO is ineffective against lung disease in NPC-deficient mice. The most effective, nontoxic concentration of CYCLO in the brain must also be established in NPC patients. Another important requirement for clinical studies with CYCLO in NPC patients is that a reliable and convenient method must be developed for assessment of clinical progression of the disease. The observation that CYCLO treatment of NPC-deficient cats caused a hearing defect raises some concerns about this side effect in humans treated with CYCLO. Although CYCLO is a very promising agent for preventing the neurodegeneration in NPC disease, it is unlikely that CYCLO or any of the other proposed treatments would be able to replace neurons that had already been lost. Thus, one of the greatest challenges for treatment for NPC patients is obtaining an early diagnosis of the disease.
Footnotes
Abbreviations:
- CYCLO
- 2-hydroxypropyl-β-cyclodextrin
- ER
- endoplasmic reticulum
- HDAC
- histone deacetylase
- LE/L
- late endosome/lysosome
- NPC
- Niemann-Pick type C
- SREBP
- sterol response element-binding protein
REFERENCES
- 1.Ledesma M. D., Prinetti A., Sonnino S., Schuchman E. H. 2011. Brain pathology in Niemann Pick disease type A: insights from the acid sphingomyelinase knockout mice. J. Neurochem. 116: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanier M. T., Millat G. 2003. Niemann-Pick disease type C. Clin. Genet. 64: 269–281. [DOI] [PubMed] [Google Scholar]
- 3.Vanier M. T. 2010. Niemann-Pick disease type C. Orphanet J. Rare Dis. 5: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown M. S., Goldstein J. L. 1976. Receptor-mediated control of cholesterol metabolism. Science. 191: 150–154. [DOI] [PubMed] [Google Scholar]
- 5.Sokol J., Blanchette-Mackie J., Kruth H. S., Dwyer N. K., Amende L. M., Butler J. D., Robinson E., Patel S., Brady R. O., Comly M. E., et al. 1988. Type C Niemann-Pick disease. Lysosomal accumulation and defective intracellular mobilization of low density lipoprotein cholesterol. J. Biol. Chem. 263: 3411–3417. [PubMed] [Google Scholar]
- 6.Pentchev P. G., Comly M. E., Kruth H. S., Vanier M. T., Wenger D. A., Patel S., Brady R. O. 1985. A defect in cholesterol esterification in Niemann-Pick disease (type C) patients. Proc. Natl. Acad. Sci. USA. 82: 8247–8251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liscum L., Ruggiero R. M., Faust J. R. 1989. The intracellular transport of low density-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J. Cell Biol. 108: 1625–1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wojtanik K. M., Liscum L. 2003. The transport of low density lipoprotein-derived cholesterol to the plasma membrane is defective in NPC1 cells. J. Biol. Chem. 278: 14850–14856. [DOI] [PubMed] [Google Scholar]
- 9.Kristiana I., Yang H., Brown A. J. 2008. Different kinetics of cholesterol delivery to components of the cholesterol homeostatic machinery: implications for cholesterol trafficking to the endoplasmic reticulum. Biochim. Biophys. Acta. 1781: 724–730. [DOI] [PubMed] [Google Scholar]
- 10.Cruz J. C., Chang T-Y. 2000. Fate of endogenously synthesized cholesterol in Niemann Pick type C1 cells. J. Biol. Chem. 275: 41309–41316. [DOI] [PubMed] [Google Scholar]
- 11.Carstea E. D., Polymeropoulos M. H., Parker C. C., Detera-Wadleigh S. D., O’Neill R. R., Patterson M. C., Goldin E., Xiao H., Straub R. E., Vanier M. T., et al. 1993. Linkage of Niemann-Pick disease type C to human chromosome 18. Proc. Natl. Acad. Sci. USA. 90: 2002–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carstea E. D., Morris J. A., Coleman K. G., Loftus S. K., Zhang D., Cummings C., Gu J., Rosenfeld M. A., Pavan W. J., Krizman D. B., et al. 1997. Niemann-Pick C1 disease gene: homology to mediators of cholesterol homeostasis. Science. 277: 228–231. [DOI] [PubMed] [Google Scholar]
- 13.Sleat D. E., Donnelly R. J., Lackland H., Liu C-G., Sohar I., Pullarkat R. K., Lobel P. 1997. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 277: 1802–1805. [DOI] [PubMed] [Google Scholar]
- 14.Naureckiene S., Sleat D. E., Lackland H., Fensom A., Vanier M. T., Wattiaux R., Jadot M., Lobel P. 2000. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 290: 2298–2301. [DOI] [PubMed] [Google Scholar]
- 15.Okamura N., Kiuchi S., Tamba M., Kashima T., Hiramoto S., Baba T., Dacheux F., Dacheux J. L., Sugita Y., Jin Y. Z. 1999. A porcine homolog of the major secretory protein of human epididymis, HE1, specifically binds cholesterol. Biochim. Biophys. Acta. 1438: 377–387. [DOI] [PubMed] [Google Scholar]
- 16.Cheruku S. R., Xu Z., Dutia R., Lobel P., Storch J. 2006. Mechanism of cholesterol transfer from the Niemann-Pick type C2 protein to model membranes supports a role in lysosomal cholesterol transport. J. Biol. Chem. 281: 31594–31604. [DOI] [PubMed] [Google Scholar]
- 17.Xu S., Benoff B., Liou H. L., Lobel P., Stock A. M. 2007. Structural basis of sterol binding by NPC2, a lysosomal protein deficient in Niemann-Pick type C2 disease. J. Biol. Chem. 282: 23525–23531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon H. J., Abi-Mosleh L., Wang M. L., Deisenhofer J., Goldstein J. L., Brown M. S., Infante R. E. 2009. Structure of N-terminal domain of NPC1 reveals distinct subdomains for binding and transfer of cholesterol. Cell. 137: 1213–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., Goldstein J. L. 2010. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12: 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Infante R. E., Abi-Mosleh L., Radhakrishnan A., Dale J. D., Brown M. S., Goldstein J. L. 2008. Purified NPC1 protein. I. Binding of cholesterol and oxysterols to a 1278-amino acid membrane protein. J. Biol. Chem. 283: 1052–1063. [DOI] [PubMed] [Google Scholar]
- 21.Infante R. E., Radhakrishnan A., Abi-Mosleh L., Kinch L. N., Wang M. L., Grishin N. V., Goldstein J. L., Brown M. S. 2008. Purified NPC1 protein: II. Localization of sterol binding to a 240-amino acid soluble luminal loop. J. Biol. Chem. 283: 1064–1075. [DOI] [PubMed] [Google Scholar]
- 22.Liu R., Lu P., Chu J. W. K., Sharom F. J. 2009. Characterization of fluorescent sterol binding to purified human NPC1. J. Biol. Chem. 284: 1840–1852. [DOI] [PubMed] [Google Scholar]
- 23.Xu Z., Farver W., Kodukula S., Storch J. 2008. Regulation of sterol transport between membranes and NPC2. Biochemistry. 47: 11134–11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chevallier J., Chamoun Z., Jiang G., Prestwich G., Sakai N., Matile S., Parton R. G., Gruenberg J. 2008. Lysobisphosphatidic acid controls endosomal cholesterol levels. J. Biol. Chem. 283: 27871–27880. [DOI] [PubMed] [Google Scholar]
- 25.Vanier M. T., Duthel S., Rodriguiez-Lafrasse C., Pentchev P. G., Carstea E. D. 1996. Genetic heterogeneity in Niemann-Pick C disease: a study using somatic cell hybridization and linkage analysis. Am. J. Hum. Genet. 58: 118–125. [PMC free article] [PubMed] [Google Scholar]
- 26.Sleat D. E., Wiseman J. A., El-Banna M., Price S. M., Verot L., Shen M. M., Tint G. S., Vanier M. T., Walkley S. U., Lobel P. 2004. Genetic evidence for nonredundant functional cooperativity between NPC1 and NPC2 in lipid transport. Proc. Natl. Acad. Sci. USA. 101: 5886–5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Deffieu M. S., Pfeffer S. R. 2011. Niemann-Pick type C 1 function requires lumenal domain residues that mediate cholesterol-dependent NPC2 binding. Proc. Natl. Acad. Sci. USA. 108: 18932–18936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choudhury A., Dominguez M., Puri V., Sharma D. K., Narita K., Wheatley C. L., Marks D. L., Pagano R. E. 2002. Rab proteins mediate Golgi transport of caveola-internalized glycosphingolipids and correct lipid trafficking in Niemann-Pick C cells. J. Clin. Invest. 109: 1541–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choudhury A., Sharma D. K., Marks D. L., Pagano R. E. 2004. Elevated endosomal cholesterol levels in Niemann-Pick cells inhibit rab4 and perturb membrane recycling. Mol. Biol. Cell. 15: 4500–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Du X., Kumar J., Ferguson C., Schulz T. A., Ong Y. S., Hong W., Prinz W. A., Parton R. G., Brown A. J., Yang H. 2011. A role for oxysterol-binding protein-related protein 5 in endosomal cholesterol trafficking. J. Cell Biol. 192: 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du X., Kazim A. S., Dawes I. W., Brown A. J., Yang H. 2013. The AAA ATPase VPS4/SKD1 regulates endosomal cholesterol trafficking independently of ESCRT-III. Traffic. 14: 107–119. [DOI] [PubMed] [Google Scholar]
- 32.Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. 2008. Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8: 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brown M. S., Goldstein J. L. 1997. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 89: 331–340. [DOI] [PubMed] [Google Scholar]
- 34.Liscum L., Faust J. R. 1987. Low density lipoprotein (LDL)-mediated suppression of cholesterol synthesis and LDL uptake is defective in Niemann-Pick type C fibroblasts. J. Biol. Chem. 262: 17002–17008. [PubMed] [Google Scholar]
- 35.Lloyd-Evans E., Morgan A. J., He X., Smith D. A., Elliot-Smith E., Sillence D. J., Churchill G. C., Schuchman E. H., Galione A., Platt F. M. 2008. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 14: 1247–1255. [DOI] [PubMed] [Google Scholar]
- 36.Fu R., Yanjanin N. M., Bianconi S., Pavan W. J., Porter F. D. 2010. Oxidative stress in Niemann-Pick disease, type C. Mol. Genet. Metab. 101: 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vázquez M. C., Balboa E., Alvarez A. R., Zanlungo S. 2012. Oxidative stress: a pathogenic mechanism for Niemann-Pick type C disease. Oxid. Med. Cell. Longev. 2012: 205713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldman S. D., Krise J. P. 2010. Niemann-Pick C1 functions independently of Niemann-Pick C2 in the initial stage of retrograde transport of membrane-impermeable lysosomal cargo. J. Biol. Chem. 285: 4983–4994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amritraj A., Peake K., Kodam A., Salio C., Merighi A., Vance J. E., Kar S. 2009. Increased activity and altered subcellular distribution of lysosomal enzymes determine neuronal vulnerability in Niemann-Pick type C1-deficient mice. Am. J. Pathol. 175: 2540–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charman M., Kennedy B. E., Osborne N., Karten B. 2010. MLN64 mediates egress of cholesterol from endosomes to mitochondria in the absence of functional Niemann-Pick Type C1 protein. J. Lipid Res. 51: 1023–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu W., Gong J. S., Ko M., Garver W. S., Yanagisawa K., Michikawa M. 2005. Altered cholesterol metabolism in Niemann-Pick type C1 mouse brains affects mitochondrial function. J. Biol. Chem. 280: 11731–11739. [DOI] [PubMed] [Google Scholar]
- 42.Fernández A., Llacuna L., Fernández-Checa J. C., Colell A. 2009. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J. Neurosci. 29: 6394–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ordonez M. P., Roberts E. A., Kidwell C. U., Yuan S. H., Plaisted W. C., Goldstein L. S. 2012. Disruption and therapeutic rescue of autophagy in a human neuronal model of Niemann Pick type C1. Hum. Mol. Genet. 21: 2651–2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kennedy B. E., Charman M., Karten B. 2012. Niemann-Pick Type C2 protein contributes to the transport of endosomal cholesterol to mitochondria without interacting with NPC1. J. Lipid Res. 53: 2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pentchev P. G., Vanier M. T., Suzuki K., Patterson M. C. 1995. Niemann-Pick disease type C: a cellular cholesterol lipidosis. In The metabolic and molecular bases of inherited disease. C. R. Scriver, A. L. Beaudet, W. S. Sly, and D. Valle, editors. 7th edition. McGraw-Hill, New York. 2625–2639. [Google Scholar]
- 46.Vanier M. T. 1999. Lipid changes in Niemann-Pick disease type C: personal experience and review of the literature. Neurochem. Res. 24: 481–489. [DOI] [PubMed] [Google Scholar]
- 47.Yamada A., Saji M., Ukita Y., Shinoda Y., Taniguchi M., Higaki K., Ninomiya H., Ohno K. 2001. Progressive neuronal loss in the ventral posterior lateral and medial nuclei of thalamus in Niemann-Pick disease type C mouse brain. Brain Dev. 23: 288–297. [DOI] [PubMed] [Google Scholar]
- 48.Sarna J., Miranda S. R., Schuchman E. H., Hawkes R. 2001. Patterned cerebellar Purkinje cell death in a transgenic mouse model of Niemann Pick type A/B disease. Eur. J. Neurosci. 13: 1873–1880. [DOI] [PubMed] [Google Scholar]
- 49.Zervas M., Dobrenis K., Walkley S. U. 2001. Neurons in Niemann-Pick disease type C accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 60: 49–64. [DOI] [PubMed] [Google Scholar]
- 50.Baudry M., Yao Y., Simmons D., Liu J., Bi X. 2003. Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 184: 887–903. [DOI] [PubMed] [Google Scholar]
- 51.Walkley S. U., Suzuki K. 2004. Consequences of NPC1 and NPC2 loss of function in mammalian neurons. Biochim. Biophys. Acta. 1685: 48–62. [DOI] [PubMed] [Google Scholar]
- 52.Pressey S. N., Smith D. A., Wong A. M., Platt F. M., Cooper J. D. 2012. Early glial activation, synaptic changes and axonal pathology in the thalamocortical system of Niemann-Pick type C1 mice. Neurobiol. Dis. 45: 1086–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beltroy E. P., Richardson J. A., Horton J. D., Turley S. D., Dietschy J. M. 2005. Cholesterol accumulation and liver cell death in mice with Niemann-Pick type C disease. Hepatology. 42: 886–893. [DOI] [PubMed] [Google Scholar]
- 54.Kulinski A., Vance J. E. 2007. Lipid homeostasis and lipoprotein secretion in Niemann-Pick C-deficient hepatocytes. J. Biol. Chem. 282: 1627–1637. [DOI] [PubMed] [Google Scholar]
- 55.Rimkunas V. M., Graham M. J., Crooke R. M., Liscum L. 2009. TNF-{alpha} plays a role in hepatocyte apoptosis in Niemann-Pick type C liver disease. J. Lipid Res. 50: 327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sayre N. L., Rimkunas V. M., Graham M. J., Crooke R. M., Liscum L. 2010. Recovery from liver disease in a Niemann-Pick type C mouse model. J. Lipid Res. 51: 2372–2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pittman R. C., Steinberg D. 1984. Sites and mechanisms of uptake and degradation of high density and low density lipoproteins. J. Lipid Res. 25: 1577–1585. [PubMed] [Google Scholar]
- 58.Xie C., Turley S. D., Dietschy J. M. 1999. Cholesterol accumulation in tissues of the Niemann-Pick type C mouse is determined by the rate of lipoprotein-cholesterol uptake through the coated-pit pathway in each organ. Proc. Natl. Acad. Sci. USA. 96: 11992–11997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Osono Y., Woollett L. A., Herz J., Dietschy J. M. 1995. Role of the low density lipoprotein receptor in the flux of cholesterol through the plasma and across the tissues of the mouse. J. Clin. Invest. 95: 1124–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramirez C. M., Liu B., Taylor A. M., Repa J. J., Burns D. K., Weinberg A. G., Turley S. D., Dietschy J. M. 2010. Weekly cyclodextrin administration normalizes cholesterol metabolism in nearly every organ of the Niemann-Pick type C1 mouse and markedly prolongs life. Pediatr. Res. 68: 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roszell B. R., Tao J. Q., Yu K. J., Huang S., Bates S. R. 2012. Characterization of the Niemann-Pick C pathway in alveolar type II cells and lamellar bodies of the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 302: L919–L932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Roszell B. R., Tao J. Q., Yu K. J., Gao L., Huang S., Ning Y., Feinstein S. I., Vite C. H., Bates S. R. 2013. Pulmonary abnormalities in animal models due to Niemann-Pick type C1 (NPC1) or C2 (NPC2) disease. PLoS ONE. 8: e67084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ramirez C. M., Lopez A. M., Le L. Q., Posey K. S., Weinberg A. G., Turley S. D. 2014. Ontogenic changes in lung cholesterol metabolism, lipid content, and histology in mice with Niemann-Pick type C disease. Biochim. Biophys. Acta. 1841: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Claudepierre T., Paques M., Simonutti M., Buard I., Sahel J., Maue R. A., Picaud S., Pfrieger F. W. 2010. Lack of Niemann-Pick type C1 induces age-related degeneration in the mouse retina. Mol. Cell. Neurosci. 43: 164–176. [DOI] [PubMed] [Google Scholar]
- 65.Cologna S. M., Jiang X. S., Backlund P. S., Cluzeau C. V., Dail M. K., Yanjanin N. M., Siebel S., Toth C. L., Jun H. S., Wassif C. A., et al. 2012. Quantitative proteomic analysis of Niemann-Pick disease, type C1 cerebellum identifies protein biomarkers and provides pathological insight. PLoS ONE. 7: e47845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kennedy B. E., Leblanc V. G., Mailman T. M., Fice D., Burton I., Karakach T. K., Karten B. 2013. Pre-symptomatic activation of antioxidant responses and alterations in glucose and pyruvate metabolism in Niemann-Pick type C1-deficient murine brain. PLoS ONE. 8: e82685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porter F. D., Scherrer D. E., Lanier M. H., Langmade S. J., Molugu V., Gale S. E., Olzeski D., Sidhu R., Dietzen D. J., Fu R., et al. 2010. Cholesterol oxidation products are sensitive and specific blood-based biomarkers for Niemann-Pick C1 disease. Sci. Transl. Med. 2: 56ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cadigan K. M., Spillane D. M., Chang T-Y. 1990. Isolation and characterization of Chinese hamster ovary cell mutants defective in intracellular low density lipoprotein cholesterol trafficking. J. Cell Biol. 110: 295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chang T. Y., Reid P. C., Sugii S., Ohgami N., Cruz J. C., Chang C. C. 2005. Niemann-Pick type C disease and intracellular cholesterol trafficking. J. Biol. Chem. 280: 20917–20920. [DOI] [PubMed] [Google Scholar]
- 70.Sparrow S. M., Carter J. M., Ridgway N. D., Cook H. W., Byers D. M. 1999. U18666A inhibits intracellular cholesterol transport and neurotransmitter release in human neuroblastoma cells. Neurochem. Res. 24: 69–77. [DOI] [PubMed] [Google Scholar]
- 71.Cenedella R. J. 2009. Cholesterol synthesis inhibitor U18666A and the role of sterol metabolism and trafficking in numerous pathophysiological processes. Lipids. 44: 477–487. [DOI] [PubMed] [Google Scholar]
- 72.Loftus S. K., Morris J. A., Carstea E. D., Gu J. Z., Cummings C., Brown A., Ellison J., Ohno K., Rosenfeld M. A., Tagle D. A., et al. 1997. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 277: 232–235. [DOI] [PubMed] [Google Scholar]
- 73.Maue R. A., Burgess R. W., Wang B., Wooley C. M., Seburn K. L., Vanier M. T., Rogers M. A., Chang C. C., Chang T. Y., Harris B. T., et al. 2012. A novel mouse model of Niemann-Pick type C disease carrying a D1005G-Npc1 mutation comparable to commonly observed human mutations. Hum. Mol. Genet. 21: 730–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rimkunas V. M., Graham M. J., Crooke R. M., Liscum L. 2008. In vivo antisense oligonucleotide reduction of NPC1 expression as a novel mouse model for Niemann Pick type C- associated liver disease. Hepatology. 47: 1504–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nielsen G. K., Dagnaes-Hansen F., Holm I. E., Meaney S., Symula D., Andersen N. T., Heegaard C. W. 2011. Protein replacement therapy partially corrects the cholesterol-storage phenotype in a mouse model of Niemann-Pick type C2 disease. PLoS ONE. 6: e27287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Elrick M. J., Pacheco C. D., Yu T., Dadgar N., Shakkottai V. G., Ware C., Paulson H. L., Lieberman A. P. 2010. Conditional Niemann-Pick C mice demonstrate cell autonomous Purkinje cell neurodegeneration. Hum. Mol. Genet. 19: 837–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lopez M. E., Klein A. D., Dimbil U. J., Scott M. P. 2011. Anatomically defined neuron-based rescue of neurodegenerative niemann-pick type C disorder. J. Neurosci. 31: 4367–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vite C. H., Ding W., Bryan C., O’Donnell P., Cullen K., Aleman D., Haskins M. E., Van Winkle T. 2008. Clinical, electrophysiological, and serum biochemical measures of progressive neurological and hepatic dysfunction in feline Niemann-Pick type C disease. Pediatr. Res. 64: 544–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ward S., O’Donnell P., Fernandez S., Vite C. H. 2010. 2-hydroxypropyl-beta-cyclodextrin raises hearing threshold in normal cats and in cats with Niemann-Pick type C disease. Pediatr. Res. 68: 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sturley S. L., Patterson M. C., Pentchev P. 2009. Unraveling the sterol-trafficking defect in Niemann-Pick C disease. Proc. Natl. Acad. Sci. USA. 106: 2093–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huang X., Suyama K., Buchanan J., Zhu A. J., Scott M. P. 2005. A Drosophila model of the Niemann-Pick type C lysosome storage disease: dnpc1a is required for molting and sterol homeostasis. Development. 132: 5115–5124. [DOI] [PubMed] [Google Scholar]
- 82.Phillips S. E., Woodruff E. A., III, Liang P., Patten M., Broadie K. 2008. Neuronal loss of Drosophila NPC1a causes cholesterol aggregation and age-progressive neurodegeneration. J. Neurosci. 28: 6569–6582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Swaroop M., Thorne N., Rao M. S., Austin C. P., McKew J. C., Zheng W. 2012. Evaluation of cholesterol reduction activity of methyl-beta-cyclodextrin using differentiated human neurons and astrocytes. J. Biomol. Screen. 17: 1243–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bergamin N., Dardis A., Beltrami A., Cesselli D., Rigo S., Zampieri S., Domenis R., Bembi B., Beltrami C. A. 2013. A human neuronal model of Niemann Pick C disease developed from stem cells isolated from patient’s skin. Orphanet J. Rare Dis. 8: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Trilck M., Hubner R., Seibler P., Klein C., Rolfs A., Frech M. J. 2013. Niemann-Pick type C1 patient-specific induced pluripotent stem cells display disease specific hallmarks. Orphanet J. Rare Dis. 8: 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Devlin C., Pipalia N. H., Liao X., Schuchman E. H., Maxfield F. R., Tabas I. 2010. Improvement in lipid and protein trafficking in Niemann-Pick C1 cells by correction of a secondary enzyme defect. Traffic. 11: 601–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zervas M., Somers K. L., Thrall M. A., Walkley S. U. 2001. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr. Biol. 11: 1283–1287. [DOI] [PubMed] [Google Scholar]
- 88.te Vruchte D., Lloyd-Evans E., Veldman R. J., Neville D. C., Dwek R. A., Platt F. M., van Blitterswijk W. J., Sillence D. J. 2004. Accumulation of glycosphingolipids in Niemann-Pick C disease disrupts endosomal transport. J. Biol. Chem. 279: 26167–26175. [DOI] [PubMed] [Google Scholar]
- 89.Zhou S., Davidson C., McGlynn R., Stephney G., Dobrenis K., Vanier M. T., Walkley S. U. 2011. Endosomal/lysosomal processing of gangliosides affects neuronal cholesterol sequestration in Niemann-Pick disease type C. Am. J. Pathol. 179: 890–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kobayashi T., Beuchat M. H., Lindsay M., Frias S., Palmiter R. D., Sakuraba H., Parton R. G., Gruenberg J. 1999. Late endosomal membranes rich in lysobisphosphatidic acid regulate cholesterol transport. Nat. Cell Biol. 1: 113–118. [DOI] [PubMed] [Google Scholar]
- 91.Liou H. L., Dixit S. S., Xu S., Tint G. S., Stock A. M., Lobel P. 2006. NPC2, the protein deficient in Niemann-Pick C2 disease, consists of multiple glycoforms that bind a variety of sterols. J. Biol. Chem. 281: 36710–36723. [DOI] [PubMed] [Google Scholar]
- 92.Xie X., Brown M. S., Shelton J. M., Richardson J. A., Goldstein J. L., Liang G. 2011. Amino acid substitution in NPC1 that abolishes cholesterol binding reproduces phenotype of complete NPC1 deficiency in mice. Proc. Natl. Acad. Sci. USA. 108: 15330–15335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Liu Y., Wu Y-P., Wada R., Neufeld E. B., Mullin K. A., Howard A. C., Pentchev P. G., Vanier M. T., Suzuki K., Proia R. L. 2000. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum. Mol. Genet. 9: 1087–1092. [DOI] [PubMed] [Google Scholar]
- 94.Li H., Turley S. D., Liu B., Repa J. J., Dietschy J. M. 2008. GM2/GD2 and GM3 gangliosides have no effect on cellular cholesterol pools or turnover in normal or NPC1 mice. J. Lipid Res. 49: 1816–1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu B., Li H., Repa J. J., Turley S. D., Dietschy J. M. 2008. Genetic variations and treatments that affect the lifespan of the NPC1 mouse. J. Lipid Res. 49: 663–669. [DOI] [PubMed] [Google Scholar]
- 96.Reagan J. W., Hubbert M. L., Shelness G. S. 2000. Posttranslational regulation of acid sphingomyelinase in Niemann-Pick type C1 fibroblasts and free cholesterol-enriched Chinese hamster ovary cells. J. Biol. Chem. 275: 38104–38110. [DOI] [PubMed] [Google Scholar]
- 97.Zidovetzki R., Levitan I. 2007. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim. Biophys. Acta. 1768: 1311–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., Dietschy J. M. 2009. Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. USA. 106: 2377–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Davidson C. D., Ali N. F., Micsenyi M. C., Stephney G., Renault S., Dobrenis K., Ory D. S., Vanier M. T., Walkley S. U. 2009. Chronic cyclodextrin treatment of murine Niemann-Pick C disease ameliorates neuronal cholesterol and glycosphingolipid storage and disease progression. PLoS ONE. 4: e6951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu B., Ramirez C. M., Miller A. M., Repa J. J., Turley S. D., Dietschy J. M. 2010. Cyclodextrin overcomes the transport defect in nearly every organ of NPC1 mice leading to excretion of sequestered cholesterol as bile acid. J. Lipid Res. 51: 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dietschy J. M., Turley S. D. 2001. Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 12: 105–112. [DOI] [PubMed] [Google Scholar]
- 102.Dietschy J. M., Turley S. D. 2004. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45: 1375–1397. [DOI] [PubMed] [Google Scholar]
- 103.Higashi Y., Murayama S., Pentchev P. G., Suzuki K. 1993. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 85: 175–184. [DOI] [PubMed] [Google Scholar]
- 104.Sarna J. R., Larouche M., Marzban H., Sillitoe R. V., Rancourt D. E., Hawkes R. 2003. Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 456: 279–291. [DOI] [PubMed] [Google Scholar]
- 105.Li H., Repa J. J., Valasek M. A., Beltroy E. P., Turley S. D., German D. C., Dietschy J. M. 2005. Molecular, anatomical, and biochemical events associated with neurodegeneration in mice with Niemann-Pick type C disease. J. Neuropathol. Exp. Neurol. 64: 323–333. [DOI] [PubMed] [Google Scholar]
- 106.German D. C., Quintero E. M., Liang C. L., Ng B., Punia S., Xie C., Dietschy J. M. 2001. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J. Comp. Neurol. 433: 415–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ong W. Y., Kumar U., Switzer R. C., Sidhu A., Suresh G., Hu C. Y., Patel S. C. 2001. Neurodegeneration in Niemann-Pick type C disease mice. Exp. Brain Res. 141: 218–231. [DOI] [PubMed] [Google Scholar]
- 108.Wu Y. P., Mizukami H., Matsuda J., Saito Y., Proia R. L., Suzuki K. 2005. Apoptosis accompanied by up-regulation of TNF-alpha death pathway genes in the brain of Niemann-Pick type C disease. Mol. Genet. Metab. 84: 9–17. [DOI] [PubMed] [Google Scholar]
- 109.Luan Z., Saito Y., Miyata H., Ohama E., Ninomiya H., Ohno K. 2008. Brainstem neuropathology in a mouse model of Niemann-Pick disease type C. J. Neurol. Sci. 268: 108–116. [DOI] [PubMed] [Google Scholar]
- 110.Xie C., Burns D. K., Turley S. D., Dietschy J. M. 2000. Cholesterol is sequestered in the brains of mice with Niemann-Pick type C disease but turnover is increased. J. Neuropathol. Exp. Neurol. 59: 1106–1117. [DOI] [PubMed] [Google Scholar]
- 111.Takikita S., Fukuda T., Mohri I., Yagi T., Suzuki K. 2004. Perturbed myelination process of premyelinating oligodendrocyte in Niemann-Pick type C mouse. J. Neuropathol. Exp. Neurol. 63: 660–673. [DOI] [PubMed] [Google Scholar]
- 112.Yu T., Lieberman A. P. 2013. Npc1 acting in neurons and glia is essential for the formation and maintenance of CNS myelin. PLoS Genet. 9: e1003462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Karten B., Vance D. E., Campenot R. B., Vance J. E. 2002. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 83: 1154–1163. [DOI] [PubMed] [Google Scholar]
- 114.Hawes C. M., Wiemer H., Krueger S. R., Karten B. 2010. Pre-synaptic defects of NPC1-deficient hippocampal neurons are not directly related to plasma membrane cholesterol. J. Neurochem. 114: 311–322. [DOI] [PubMed] [Google Scholar]
- 115.Peake K. B., Vance J. E. 2012. Normalization of cholesterol homeostasis by 2-hydroxypropyl-beta-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 287: 9290–9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Karten B., Vance D. E., Campenot R. B., Vance J. E. 2003. Trafficking of cholesterol from cell bodies to distal axons in Niemann Pick C1-deficient neurons. J. Biol. Chem. 278: 4168–4175. [DOI] [PubMed] [Google Scholar]
- 117.Karten B., Campenot R. B., Vance D. E., Vance J. E. 2006. The Niemann-Pick C1 protein in recycling endosomes of presynaptic nerve terminals. J. Lipid Res. 47: 504–514. [DOI] [PubMed] [Google Scholar]
- 118.Zonta B., Minichiello L. 2013. Synaptic membrane rafts: traffic lights for local neurotrophin signaling? Front. Synaptic Neurosci. 5: 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sebastião A. M., Colino-Oliveira M., Assaife-Lopes N., Dias R. B., Ribeiro J. A. 2013. Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology. 64: 97–107. [DOI] [PubMed] [Google Scholar]
- 120.Mitter D., Reisinger C., Hinz B., Hollmann S., Yelamanchili S. V., Treiber-Held S., Ohm T. G., Herrmann A., Ahnert-Hilger G. 2003. The synaptophysin/synaptobrevin interaction critically depends on the cholesterol content. J. Neurochem. 84: 35–42. [DOI] [PubMed] [Google Scholar]
- 121.Cho W. J., Jeremic A., Jin H., Ren G., Jena B. P. 2007. Neuronal fusion pore assembly requires membrane cholesterol. Cell Biol. Int. 31: 1301–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Churchward M. A., Coorssen J. R. 2009. Cholesterol, regulated exocytosis and the physiological fusion machine. Biochem. J. 423: 1–14. [DOI] [PubMed] [Google Scholar]
- 123.Chang J., Kim S. A., Lu X., Su Z., Kim S. K., Shin Y. K. 2009. Fusion step-specific influence of cholesterol on SNARE-mediated membrane fusion. Biophys. J. 96: 1839–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dason J. S., Smith A. J., Marin L., Charlton M. P. 2014. Cholesterol and F-actin are required for clustering of recycling synaptic vesicle proteins in the presynaptic plasma membrane. J. Physiol. 592: 621–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Zamir O., Charlton M. P. 2006. Cholesterol and synaptic transmitter release at crayfish neuromuscular junctions. J. Physiol. 571: 83–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wasser C. R., Ertunc M., Liu X., Kavalali E. T. 2007. Cholesterol-dependent balance between evoked and spontaneous synaptic vesicle recycling. J. Physiol. 579: 413–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Linetti A., Fratangeli A., Taverna E., Valnegri P., Francolini M., Cappello V., Matteoli M., Passafaro M., Rosa P. 2010. Cholesterol reduction impairs exocytosis of synaptic vesicles. J. Cell Sci. 123: 595–605. [DOI] [PubMed] [Google Scholar]
- 128.Dason J. S., Smith A. J., Marin L., Charlton M. P. 2010. Vesicular sterols are essential for synaptic vesicle cycling. J. Neurosci. 30: 15856–15865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Xu S., Zhou S., Xia D., Xia J., Chen G., Duan S., Luo J. 2010. Defects of synaptic vesicle turnover at excitatory and inhibitory synapses in Niemann-Pick C1-deficient neurons. Neuroscience. 167: 608–620. [DOI] [PubMed] [Google Scholar]
- 130.Zhou S. Y., Xu S. J., Yan Y. G., Yu H. M., Ling S. C., Luo J. H. 2011. Decreased purinergic inhibition of synaptic activity in a mouse model of Niemann-Pick disease type C. Hippocampus. 21: 212–219. [DOI] [PubMed] [Google Scholar]
- 131.Sun C. L., Su L. D., Li Q., Wang X. X., Shen Y. 2011. Cerebellar long-term depression is deficient in Niemann-Pick type C disease mice. Cerebellum. 10: 88–95. [DOI] [PubMed] [Google Scholar]
- 132.Avchalumov Y., Kirschstein T., Lukas J., Luo J., Wree A., Rolfs A., Kohling R. 2012. Increased excitability and compromised long-term potentiation in the neocortex of NPC1(−/−) mice. Brain Res. 1444: 20–26. [DOI] [PubMed] [Google Scholar]
- 133.Xu S., Chen X., Wei X., Liu G., Wang Q. 2011. Presynaptic impairment in Niemann-Pick C1-deficient neurons: not dependent on presence of glial cells. Neurosci. Lett. 496: 54–59. [DOI] [PubMed] [Google Scholar]
- 134.Karten B., Hayashi H., Francis G. A., Campenot R. B., Vance D. E., Vance J. E. 2005. Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem. J. 387: 779–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Peake K. B., Campenot R. B., Vance D. E., Vance J. E. 2011. Activation and function of primary microglia from Niemann-Pick type C-deficient mice. Biochim. Biophys. Acta. 1812: 1121–1129. [DOI] [PubMed] [Google Scholar]
- 136.Giulian D., Chen J., Ingeman J. E., George J. K., Noponen M. 1989. The role of mononuclear phagocytes in wound healing after traumatic injury to adult mammalian brain. J. Neurosci. 9: 4416–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Buttini M., Appel K., Sauter A., Gebicke-Haerter P. J., Boddeke H. W. 1996. Expression of tumor necrosis factor alpha after focal cerebral ischaemia in the rat. Neuroscience. 71: 1–16. [DOI] [PubMed] [Google Scholar]
- 138.Nakamura Y., Si Q. S., Kataoka K. 1999. Lipopolysaccharide-induced microglial activation in culture: temporal profiles of morphological change and release of cytokines and nitric oxide. Neurosci. Res. 35: 95–100. [DOI] [PubMed] [Google Scholar]
- 139.Dheen S. T., Kaur C., Ling E. A. 2007. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 14: 1189–1197. [DOI] [PubMed] [Google Scholar]
- 140.Repa J. J., Li H., Frank-Cannon T. C., Valasek M. A., Turley S. D., Tansey M. G., Dietschy J. M. 2007. Liver X receptor activation enhances cholesterol loss from the brain, decreases neuroinflammation, and increases survival of the NPC1 mouse. J. Neurosci. 27: 14470–14480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Yu T., Shakkottai V. G., Chung C., Lieberman A. P. 2011. Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 20: 4440–4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lopez M. E., Klein A. D., Hong J., Dimbil U. J., Scott M. P. 2012. Neuronal and epithelial cell rescue resolves chronic systemic inflammation in the lipid storage disorder Niemann-Pick C. Hum. Mol. Genet. 21: 2946–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lopez M. E., Klein A. D., Scott M. P. 2012. Complement is dispensable for neurodegeneration in Niemann-Pick disease type C. J. Neuroinflammation. 9: 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ko D. C., Milenkovic L., Beier S. M., Manuel H., Buchanan J., Scott M. P. 2005. Cell-autonomous death of cerebellar Purkinje neurons with autophagy in Niemann-Pick type C disease. PLoS Genet. 1: 81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhang M., Strnatka D., Donohue C., Hallows J. L., Vincent I., Erickson R. P. 2008. Astrocyte-only Npc1 reduces neuronal cholesterol and triples life span of Npc1(−/−) mice. J. Neurosci. Res. 86: 2848–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Loftus S. K., Erickson R. P., Walkley S. U., Bryant M. A., Incao A., Heidenreich R. A., Pavan W. J. 2002. Rescue of neurodegeneration in Niemann-Pick C mice by a prion-promoter-driven Npc1 cDNA transgene. Hum. Mol. Genet. 11: 3107–3114. [DOI] [PubMed] [Google Scholar]
- 147.Jiang X., Sidhu R., Porter F. D., Yanjanin N. M., Speak A. O., te Vruchte D. T., Platt F. M., Fujiwara H., Scherrer D. E., Zhang J., et al. 2011. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 52: 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cluzeau C. V., Watkins-Chow D. E., Fu R., Borate B., Yanjanin N., Dail M. K., Davidson C. D., Walkley S. U., Ory D. S., Wassif C. A., et al. 2012. Microarray expression analysis and identification of serum biomarkers for Niemann-Pick disease, type C1. Hum. Mol. Genet. 21: 3632–3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fan M., Sidhu R., Fujiwara H., Tortelli B., Zhang J., Davidson C., Walkley S. U., Bagel J. H., Vite C., Yanjanin N. M., et al. 2013. Identification of Niemann-Pick C1 disease biomarkers through sphingolipid profiling. J. Lipid Res. 54: 2800–2814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lyseng-Williamson K. A. 2014. Miglustat: a review of its use in Niemann-Pick disease type C. Drugs. 74: 61–74. [DOI] [PubMed] [Google Scholar]
- 151.Stein V. M., Crooks A., Ding W., Prociuk M., O’Donnell P., Bryan C., Sikora T., Dingemanse J., Vanier M. T., Walkley S. U., et al. 2012. Miglustat improves Purkinje cell survival and alters microglial phenotype in feline Niemann-Pick disease type C. J. Neuropathol. Exp. Neurol. 71: 434–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Patterson M. C., Vecchio D., Prady H., Abel L., Wraith J. E. 2007. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. Lancet Neurol. 6: 765–772. [DOI] [PubMed] [Google Scholar]
- 153.Galanaud D., Tourbah A., Lehéricy S., Leveque N., Heron B., Billette de Villemeur T., Guffon N., Feillet F., Baumann N., Vanier M. T., et al. 2008. 24 month-treatment with miglustat of three patients with Niemann-Pick disease type C: follow up using brain spectroscopy. Mol. Genet. Metab. 96: 55–58. [DOI] [PubMed] [Google Scholar]
- 154.Wraith J. E., Vecchio D., Jacklin E., Abel L., Chadha-Boreham H., Luzy C., Giorgino R., Patterson M. C. 2010. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: long-term data from a clinical trial. Mol. Genet. Metab. 99: 351–357. [DOI] [PubMed] [Google Scholar]
- 155.Ginocchio V. M., D’Amico A., Bertini E., Ceravolo F., Dardis A., Verrigni D., Bembi B., Dionisi-Vici C., Deodato F. 2013. Efficacy of miglustat in Niemann-Pick C disease: a single centre experience. Mol. Genet. Metab. 110: 329–335. [DOI] [PubMed] [Google Scholar]
- 156.Chien Y. H., Peng S. F., Yang C. C., Lee N. C., Tsai L. K., Huang A. C., Su S. C., Tseng C. C., Hwu W. L. 2013. Long-term efficacy of miglustat in paediatric patients with Niemann-Pick disease type C. J. Inherit. Metab. Dis. 36: 129–137. [DOI] [PubMed] [Google Scholar]
- 157.Patterson M. C., Hendriksz C. J., Walterfang M., Sedel F., Vanier M. T., Wijburg F.NP-C Guidelines Working Group. 2012. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol. Genet. Metab. 106: 330–344. [DOI] [PubMed] [Google Scholar]
- 158.Bardai F. H., Price V., Zaayman M., Wang L., D’Mello S. R. 2012. Histone deacetylase-1 (HDAC1) is a molecular switch between neuronal survival and death. J. Biol. Chem. 287: 35444–35453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Nunes M. J., Moutinho M., Gama M. J., Rodrigues C. M., Rodrigues E. 2013. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS ONE. 8: e53394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Munkacsi A. B., Chen F. W., Brinkman M. A., Higaki K., Gutierrez G. D., Chaudhari J., Layer J. V., Tong A., Bard M., Boone C., et al. 2011. An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human Niemann-Pick type C disease. J. Biol. Chem. 286: 23842–23851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pipalia N. H., Cosner C. C., Huang A., Chatterjee A., Bourbon P., Farley N., Helquist P., Wiest O., Maxfield F. R. 2011. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in Niemann-Pick type C1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA. 108: 5620–5625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Helquist P., Maxfield F. R., Wiech N. L., Wiest O. 2013. Treatment of Niemann–pick type C disease by histone deacetylase inhibitors. Neurotherapeutics. 10: 688–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gelsthorpe M. E., Baumann N., Millard E., Gale S. E., Langmade S. J., Schaffer J. E., Ory D. S. 2008. Niemann-Pick type C1 I1061T mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 283: 8229–8236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Alvarez A. R., Klein A., Castro J., Cancino G. I., Amigo J., Mosqueira M., Vargas L. M., Yevenes L. F., Bronfman F. C., Zanlungo S. 2008. Imatinib therapy blocks cerebellar apoptosis and improves neurological symptoms in a mouse model of Niemann-Pick type C disease. FASEB J. 22: 3617–3627. [DOI] [PubMed] [Google Scholar]
- 165.Vance J. E., Peake K. B. 2011. Function of the Niemann-Pick type C proteins and their bypass by cyclodextrin. Curr. Opin. Lipidol. 22: 204–209. [DOI] [PubMed] [Google Scholar]
- 166.Griffin L. D., Gong W., Verot L., Mellon S. H. 2004. Niemann-Pick type C disease involves disrupted neurosteroidogenesis and responds to allopregnanolone. Nat. Med. 10: 704–711. [DOI] [PubMed] [Google Scholar]
- 167.Ramirez C. M., Liu B., Aqul A., Taylor A. M., Repa J. J., Turley S. D., Dietschy J. M. 2011. Quantitative role of LAL, NPC2, and NPC1 in lysosomal cholesterol processing defined by genetic and pharmacological manipulations. J. Lipid Res. 52: 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pontikis C. C., Davidson C. D., Walkley S. U., Platt F. M., Begley D. J. 2013. Cyclodextrin alleviates neuronal storage of cholesterol in Niemann-Pick C disease without evidence of detectable blood-brain barrier permeability. J. Inherit. Metab. Dis. 36: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Monnaert V., Tilloy S., Bricout H., Fenart L., Cecchelli R., Monflier E. 2004. Behavior of alpha-, beta-, and gamma-cyclodextrins and their derivatives on an in vitro model of blood-brain barrier. J. Pharmacol. Exp. Ther. 310: 745–751. [DOI] [PubMed] [Google Scholar]
- 170.Aqul A., Liu B., Ramirez C. M., Pieper A. A., Estill S. J., Burns D. K., Repa J. J., Turley S. D., Dietschy J. M. 2011. Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31: 9404–9413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Yancey P. G., Rodrigueza W. V., Kilsdonk E. P., Stoudt G. W., Johnson W. J., Phillips M. C., Rothblat G. H. 1996. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J. Biol. Chem. 271: 16026–16034. [DOI] [PubMed] [Google Scholar]
- 172.Levitan I., Christian A. E., Tulenko T. N., Rothblat G. H. 2000. Membrane cholesterol content modulates activation of volume-regulated anion current in bovine endothelial cells. J. Gen. Physiol. 115: 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rosenbaum A. I., Zhang G., Warren J. D., Maxfield F. R. 2010. Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. USA. 107: 5477–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Plazzo A. P., Hofer C. T., Jicsinszky L., Fenyvesi E., Szente L., Schiller J., Herrmann A., Muller P. 2012. Uptake of a fluorescent methyl-beta-cyclodextrin via clathrin-dependent endocytosis. Chem. Phys. Lipids. 165: 505–511. [DOI] [PubMed] [Google Scholar]
- 175.Abi-Mosleh L., Infante R. E., Radhakrishnan A., Goldstein J. L., Brown M. S. 2009. Cyclodextrin overcomes deficient lysosome-to-endoplasmic reticulum transport of cholesterol in Niemann-Pick type C cells. Proc. Natl. Acad. Sci. USA. 106: 19316–19321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Taylor A. M., Liu B., Mari Y., Liu B., Repa J. J. 2012. Cyclodextrin mediates rapid changes in lipid balance in Npc1−/− mice without carrying cholesterol through the bloodstream. J. Lipid Res. 53: 2331–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.McCauliff L. A., Xu Z., Storch J. 2011. Sterol transfer between cyclodextrin and membranes: similar but not identical mechanism to NPC2-mediated cholesterol transfer. Biochemistry. 50: 7341–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Collins C. J., McCauliff L. A., Hyun S. H., Zhang Z., Paul L. N., Kulkarni A., Zick K., Wirth M., Storch J., Thompson D. H. 2013. Synthesis, characterization, and evaluation of pluronic-based beta-cyclodextrin polyrotaxanes for mobilization of accumulated cholesterol from Niemann-Pick type C fibroblasts. Biochemistry. 52: 3242–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Mondjinou Y. A., McCauliff L. A., Kulkarni A., Paul L., Hyun S. H., Zhang Z., Wu Z., Wirth M., Storch J., Thompson D. H. 2013. Synthesis of 2-hydroxypropyl-beta-cyclodextrin/pluronic-based polyrotaxanes via heterogeneous reaction as potential Niemann-Pick type C therapeutics. Biomacromolecules. 14: 4189–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Vance J. E., Tasseva G. 2013. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta. 1831: 543–554. [DOI] [PubMed] [Google Scholar]
- 181.Amritraj A., Wang Y., Revett T. J., Vergote D., Westaway D., Kar S. 2013. Role of cathepsin D in U18666A-induced neuronal cell death: potential implication in Niemann-Pick type C disease pathogenesis. J. Biol. Chem. 288: 3136–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Yamazaki T., Chang T-Y., Haass C., Ihara Y. 2001. Accumulation and aggregation of amyloid {beta}-protein in late endosomes of Niemann-Pick type C cells. J. Biol. Chem. 276: 4454–4460. [DOI] [PubMed] [Google Scholar]
- 183.Chua C. C., Lim M. L., Wong B. S. 2010. Altered apolipoprotein E glycosylation is associated with Abeta(42) accumulation in an animal model of Niemann-Pick Type C disease. J. Neurochem. 112: 1619–1626. [DOI] [PubMed] [Google Scholar]
- 184.Malnar M., Kosicek M., Lisica A., Posavec M., Krolo A., Njavro J., Omerbasic D., Tahirovic S., Hecimovic S. 2012. Cholesterol-depletion corrects APP and BACE1 misstrafficking in NPC1-deficient cells. Biochim. Biophys. Acta. 1822: 1270–1283. [DOI] [PubMed] [Google Scholar]
- 185.Vanier M. T., Suzuki K. 1998. Recent advances in elucidating Niemann-Pick C disease. Brain Pathol. 8: 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Kågedal K., Kim W. S., Appelqvist H., Chan S., Cheng D., Agholme L., Barnham K., McCann H., Halliday G., Garner B. 2010. Increased expression of the lysosomal cholesterol transporter NPC1 in Alzheimer’s disease. Biochim. Biophys. Acta. 1801: 831–838. [DOI] [PubMed] [Google Scholar]
- 187.Maulik M., Ghoshal B., Kim J., Wang Y., Yang J., Westaway D., Kar S. 2012. Mutant human APP exacerbates pathology in a mouse model of NPC and its reversal by a beta-cyclodextrin. Hum. Mol. Genet. 21: 4857–4875. [DOI] [PubMed] [Google Scholar]
- 188.Yao J., Ho D., Calingasan N. Y., Pipalia N. H., Lin M. T., Beal M. F. 2012. Neuroprotection by cyclodextrin in cell and mouse models of Alzheimer disease. J. Exp. Med. 209: 2501–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]