

Abstract
The SNP-SNP interactome has rarely been explored in the context of neuroimaging genetics mainly due to the complexity of conducting ∼1011 pairwise statistical tests. However, recent advances in machine learning, specifically the iterative sure independence screening (SIS) method, have enabled the analysis of datasets where the number of predictors is much larger than the number of observations. Using an implementation of the SIS algorithm (called EPISIS), we used exhaustive search of the genome-wide, SNP-SNP interactome to identify and prioritize SNPs for interaction analysis. We identified a significant SNP pair, rs1345203 and rs1213205, associated with temporal lobe volume. We further examined the full-brain, voxelwise effects of the interaction in the ADNI dataset and separately in an independent dataset of healthy twins (QTIM). We found that each additional loading in the epistatic effect was associated with ∼5% greater brain regional brain volume (a protective effect) in both the ADNI and QTIM samples.
Keywords: epistasis, interaction, genome, sure independence, tensor-based morphometry
1 Introduction
Traditional univariate methods can test the association of common genetic variants with complex quantitative traits, but they only consider the marginal effect of a single locus and potentially miss variance explained by synergistic or interacting effects of pairs or sets of SNPs1 [1]. For many complex traits, the similarity of family members drops faster than would be expected as relatedness decreases [2]. This implies that there are non-additive (epistatic) interactions involved in the etiology of many complex traits. Statistical interactions have been demonstrated to be plausible representations of the complex interactions of genes in biological pathways [3-4].
Some prior studies have examined second-order interactive effects of SNPs on brain structure [5-7]. However, none of these studies has considered genome-wide genotype data; the closest conceptually related study tested for SNP effects on diffusion imaging measures, and aggregated all SNPs with correlated effects into a network [8]. The concept here is different, and aims to assess gene pairs that influence each other's effects on the brain. Prior studies tested interaction effects only for a limited number of popular candidate genes. Any approach based on pre-selecting a pair of genes will overlook a vast search space of potential interactions among SNPs in the genome that have no obvious prior connection. Also, a large main effect is not necessary to be able to detect significant second-order interactions [9]. Given this, prior hypotheses focusing on SNPs with large individual effects may also overlook large second-order effects. Importantly, power estimates for detecting interactive effects are comparable to those for single SNP tests [1]. In simulation studies, the inclusion of interaction terms can boost the power to detect main effects, at least for certain genetic tests [10]. Here we examined the genome-wide, SNP-SNP interactome2 to test genetic associations with a quantitative biomarker of Alzheimer's disease (temporal lobe volume) in the public Alzheimer's Disease Neuroimaging Initiative (ADNI) dataset. We further examine the whole-brain effects of interaction pairs in statistical parametric maps generated with tensor-based morphometry (TBM); we also replicate our tests in an independent, non-overlapping dataset of young healthy twins from the Queensland Twin Imaging (QTIM) study [11].
2 Methods
2.1 Imaging Parameters and Study Information
We downloaded the full baseline set of 818 high-resolution, T1-weighted structural MRI brain scans from the Alzheimer's Disease Neuroimaging Initiative (ADNI). ADNI is a multi-site, longitudinal study of patients with Alzheimer's disease (AD), mild cognitive impairment (MCI) and healthy elderly controls (HC). Subjects were scanned with a standardized protocol to maximize consistency across sites. We used the baseline 1.5 Tesla MRI scans, i.e., the T1-weighted 3D MP-RAGE scans, with TR /TE = 2400/1000 ms, flip angle = 8°, slice thickness = 1.2 mm, and a final voxel resolution = 0.9375 × 0.9375 × 1.2 mm3. Raw MRI scans were pre-processed to remove signal inhomogeneity, non-brain tissue, and affine registered to the MNI template (using 9 parameters).
Additionally, we obtained 753 high-resolution, T1-weighted structural MRI brain scans from the Queensland Twin Imaging (QTIM) study. QTIM is a longitudinal neuroimaging and genetic study of young, healthy twins and their family members. All structural MRI scans were acquired on a single 4-Tesla scanner (Bruker Medspec): T1-weighted images, inversion recovery rapid gradient echo sequence, TR /TE = 1500/3.35 ms, flip angle = 8°, slice thickness = 0.9 mm, 256 × 256 acquisition matrix, with a final voxel resolution = 0.9375 × 0.9375 × 0.9 mm3. Raw MRI scans were pre-processed to remove signal inhomogeneity, non-brain tissue, and affine registered to the ICBM template (using 9 parameters).
2.2 Genotype Pre-processing and Study Demographics
Genome-wide genotyping data were available for the full set of ADNI subjects. We performed standard quality control procedures to ascertain the largest homogenous genetic sub-population in the dataset, using multi-dimensional scaling (MDS) compared to a dataset of subjects of known genetic identity (HapMap III; http://hapmap.ncbi.nlm.nih.gov/). The largest subset contained 737 subjects from the CEU population (Caucasians). We therefore removed the remaining 81 subjects from our analysis to limit the effects of genetic stratification on our statistical analyses [12]. Additionally, we applied filter rules to the genotype data to remove rare SNPs (minor allele frequency < 0.01), violations of Hardy-Weinberg Equilibrium (HWE p < 5.7×10-7), and poor call rate (<95%). Data were further “phased” to impute any missing individual genotypes after filtering using the MaCH program [13] following the ENIGMA imputation protocol [14]. After filtering and phasing, 534,033 SNPs remained.
All QTIM subjects were ascertained for genetic similarity, so no subjects were removed before analysis. All 753 subjects in the QTIM dataset clustered with the CEU population, in the MDS analysis. The same genotype filter rules from the ADNI dataset were applied to the QTIM sample's genetic data. After filtering and phasing, 521,232 SNPs remained.
After all rounds of genotype pre-processing, the ADNI sample contained 737 subjects (mean age±sd: 75.5±6.8 yrs; 436 males) comprised of 173 patients diagnosed with Alzheimer's disease, 358 subjects with mild cognitive impairment, and 206 healthy elderly controls. The QTIM sample contained 753 subjects (mean age±sd: 23.1±3.0 yrs; 286 males) and consisted of 110 monozygotic twin pairs, 147 dizygotic twin pairs, 3 dizygotic twin trios, 143 singletons, and 87 siblings from 438 families.
2.3 Tensor-Based Morphometric Differences in the Full Brain
We calculated information on regional brain morphometry using an elastic, nonlinear registration algorithm (3DMI) [15] applied to the entire brain. Voxelwise volumetric differences were stored, using the Jacobian value of the deformation matrix obtained by nonlinearly registering a subject's scan to a study-specific minimum deformation template (MDT). Scans from the ADNI and QTIM datasets were processed and analyzed separately (using separate study templates). The MDT for the ADNI sample is a nonlinear average of 40 age-and-sex matched healthy elderly controls [16]. The MDT for the QTIM is a nonlinear average of 32 age- and sex- matched, unrelated subjects [17]. Nonlinear registration with 3DMI yields a 110 × 110 × 110 voxel statistical parametric map, where the Jacobian value at each voxel represents the expansion required to match the same voxel in the study-specific MDT.
2.4 Genome-Wide, Gene-Gene Interaction Testing
The EPISIS software is an implementation of the machine-learning algorithm called sure independence screening (SIS) developed by Fan and Lv [18]. The SIS algorithm is a correlation learning method that can be applied to ultra-high dimensional datasets where the number of predictors p is much greater than the number of observations n. Despite the development of robust methods for cases where p>n (e.g., the Dantzig selector of Candes and Tao [19]) the properties of the selector fail when p≫n. Fan and Lv [18] developed the SIS algorithm to reduce the ultra-high dimension of p to a moderately-sized subset, while guaranteeing that the subset still explains the maximum amount of variance explained by the full set of predictors.
We conducted an exhaustive search of association tests of genome-wide SNP-SNP interactions with temporal lobe volume (computed by integrating the Jacobian over an temporal lobe ROI on the MDT) [20] in the ADNI dataset using the EPISIS software. EPISIS utilizes the massively parallel processing available in GPGPU (General-purpose computing on graphics processing units) framework to test p(p-1)/2 SNP-SNP interactions in the ADNI dataset in a feasible timeframe. We used the SIS algorithm with cell-wise dummy coding (CDC) [21] to reduce the full predictor space into a subset d of n/log(n) interaction terms [18]. After screening the full set of possible two-way SNP-SNP interactions, we applied ridge regression [22-23] to the subset of interaction terms (the multiplicative loading of each SNP-SNP pair) and selected significant SNP-SNP interaction terms using the extended Bayesian Information Criterion (EBIC) [24] with γ = 0.5. The choice of the parameter γ was chosen based on simulations [21]. The EPISIS software is implemented in CUDA and optimized for parallel processing across multiple NVIDIA GPU cards as detailed elsewhere [21]. A single exhaustive search of the genome-wide, SNP-SNP interactome with EPISIS was completed in 7 hours (using one NVIDIA Tesla C2050 GPU card).
2.5 Voxelwise Interaction Analysis and Replication
We tested the significant SNP-SNP interaction pair selected by ridge regression for association with voxelwise, regional volume differences (V) at each point, i, in the full brain. The association test at each voxel in the ADNI dataset followed the multiplicative interaction model in multiple linear regression:
| (1) |
Additionally, we used QTIM as an independent replication sample of the top SNP-SNP interaction pair identified by ridge regression after EPISIS. The voxelwise association tests assume the multiplicative interaction model, detailed previously. Due to the family design of the QTIM sample, we tested association using mixed-effects modeling as implemented in the R package kinship (version 1.3) in order to account for relatedness.
3 Results
After screening the full set of SNP-SNP interaction pairs for association with temporal lobe volume in the ADNI dataset, we obtained a subset d of SNP-SNP interaction pairs such that d = n/log(n). The subset is chosen by ranking the marginal correlation coefficients of each interaction pair and selecting the top d SNP-SNP pairs (correlation learning) [18], in this case d = 111 pairs. Next, we applied ridge regression to the pruned subset of SNP-SNP interaction pairs. Using the extended BIC (γ = 0.5) [21] to estimate significance in our ridge regression, we identified a significant interaction between rs1345203 and rs1213205. The distribution of alleles for each SNP and their interaction is given in Table 1.
Table 1.
The distribution of alleles for the significant SNPs and the number of subjects with each genotype by study. For rs1345203 the minor allele is G and the major allele is A in both studies. The minor allele is A and the major allele is G for rs1213205. The association testing assumes an additive model (each subject is assigned a value 0,1,2 based on the number of minor alleles they have at a given SNP). The interaction column gives the number of subjects in each category after multiplying together the counts of each of the alleles.
| Study | rs1345203 | rs1213205 | Interaction |
|---|---|---|---|
| ADNI (n=737) | G/G: 27 | A/A: 93 | 0 loadings: 612 |
| A/G: 223 | G/A: 297 | 1 loadings: 79 | |
| A/A: 487 | G/G: 347 | 2 loadings: 46 | |
|
| |||
| QTIM (n=753) | G/G: 5 | A/A: 78 | 0 loadings: 664 |
| A/G: 193 | G/A: 300 | 1 loadings: 70 | |
| A/A: 555 | G/G: 375 | 2 loadings: 19 | |
We further examined the significant SNP pair, rs1345203 and rs1213205, for whole-brain effects in the statistical parametric maps generated using tensor-based morphometry (TBM). In the ADNI dataset, we found broad effects bilaterally in the temporal and occipital lobes (Fig. 1) after correcting for multiple tests at a 5% false discovery rate (FDR) using the searchlight FDR method [25].
Fig. 1.
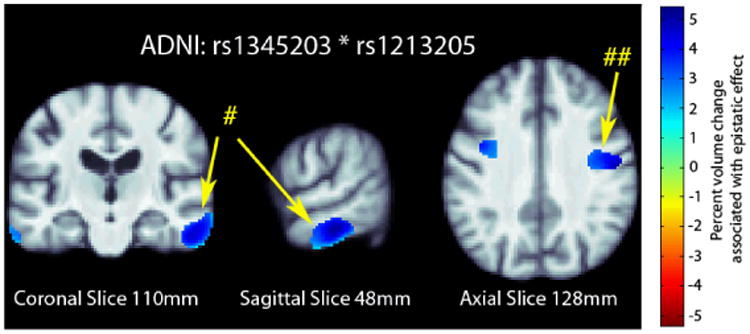
3D maps of percent tissue change for each additional genetic variant in the interaction in ADNI. Only significant regions are shown after correcting for multiple comparisons with searchlight FDR [25] at a 5% false discovery rate. Images follow radiological orientation. The origin is placed at the Posterior-Right-Inferior corner. Cooler colors over the tissue represent tissue expansion (larger regional brain volume) compared to an average template. There is a clear protective effect of the epistatic loadings bilaterally in the temporal (# in the figure) and occipital lobes (## in the figure): as the number of alleles a subject has increases, the amount of local brain tissue they have is also increased on average.
We examined the whole-brain effects of the SNP pair on voxelwise, regional brain volume in the statistical parametric maps in an independent dataset (QTIM). The distribution of alleles for each SNP and their interaction in the QTIM sample is given in Table 1. In the QTIM, we identified significant effects in the left temporal lobe and along the border of the left frontal and occipital lobes (Fig. 2) after correction for multiple tests at 5% false discovery rate (FDR) using the searchlight FDR method.
Fig. 2.
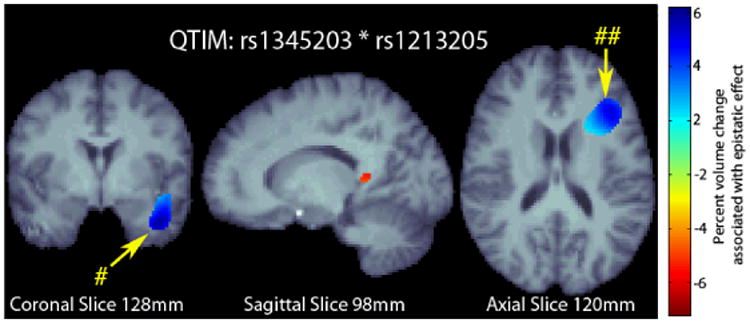
3D maps of percent tissue change for each additional genetic variant in the interaction in QTIM. Only significant regions are shown after correcting for multiple comparisons with searchlight FDR [25] at a 5% false discovery rate. Images follow radiological orientation. The origin is placed at the Posterior-Right-Inferior corner. Cooler colors over the tissue represent tissue expansion (larger regional brain volume) compared to an average template. There is a clear protective effect of the epistatic loadings in the left temporal (# in the figure) and along the boundary of the frontal and occipital lobes (## in the figure): as the number of alleles a subject has increases, the amount of local brain tissue they have is also increased on average.
4 Discussion
The genome is incredibly complex and statistical epistasis has been suggested as an appropriate model for the biological interactions among genes and protein products in related pathways [3-4]. Here we examined the multiplicative effect of SNP-SNP pairs on brain volume differences. Significant interaction terms explain additional variance in brain volume beyond what is already explained by the additive SNP terms. In our primary tests of associations with temporal lobe volume in the ADNI dataset, we screened 1011 possible SNP-SNP interaction pairs using the GPU acceleration implemented in the EPISIS software. The top 111 interaction pairs were selected after ranking the marginal effect of each SNP-SNP pair on temporal lobe volume, using an implementation of the sure independence screening (SIS) algorithm [18]. We used ridge regression and the extended BIC [24] to identify a significant interaction between rs1345203 and rs1213205. The functional relevance of the two SNPs is as yet unknown. However, data obtained from the ENCODE dataset (http://genome.ucsc.edu/) show that rs1345203 is located in a transcription factor gene (ELF1/CEBPB) that demonstrates regulatory influence on the DNA structure. The SNP rs1213205 is located in a region of hypersensitivity to cleavage by DNase regulatory elements. It is worth noting that the parameter choices made in the interaction analysis may influence the results, however, parameters were chosen based on the recommended values for EPISIS [21] and SIS [18]. Additional work is still required to identify precisely how these two SNPs might affect brain structure, and to further replicate their interaction. Specifically, we need to identify how changes at a given SNP are related to changes in activity in gene transcription or translation into protein products involved in similar biological pathways.
Footnotes
SNP (=single nucleotide polymorphism): a single-letter variant in the genome; these variations are common, even in healthy human populations, and their effects on brain measures can be assessed using association testing, at one SNP or up to a million genotyped SNPs.
Interactome: The study of interactions between genetic variants or sets of variants in terms of their effects on traits such as brain measures.
References
- 1.Marchini J, et al. Genome-wide strategies for detecting multiple loci that influence complex diseases. Nature Genetics. 2005;37(4):413–417. doi: 10.1038/ng1537. [DOI] [PubMed] [Google Scholar]
- 2.Wray NR, et al. Multi-locus models of genetic risk of disease. Genome Med. 2010;2(10) doi: 10.1186/gm131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore JH, Williams SM. Epistasis and its implications for personal genetics. American Journal of Human Genetics. 2009;85(3):309. doi: 10.1016/j.ajhg.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stich B, et al. Power to detect higher-order epistatic interactions in a metabolic pathway using a new mapping strategy. Genetics. 2007;176(1):563–570. doi: 10.1534/genetics.106.067033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pezawas L, et al. Evidence of biologic epistasis between BDNF and SLC6A4 and implications for depression. Molecular Psychiatry. 2008;13(7):709–716. doi: 10.1038/mp.2008.32. [DOI] [PubMed] [Google Scholar]
- 6.Tan HY, et al. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. PNAS. 2007;104(30):12536–12541. doi: 10.1073/pnas.0610125104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, et al. Evidence of Epistasis Between the Catechol-O-Methyltransferase and Aldehyde Dehydrogenase 3B1. Genes in Paranoid Schizophrenia Biological Psychiatry. 2009;65(12):1048–1054. doi: 10.1016/j.biopsych.2008.11.027. [DOI] [PubMed] [Google Scholar]
- 8.Chiang MC, et al. Gene network effects on brain microstructure and intellectual performance identified in 472 twins. Journal of Neuroscience. 2012;32(25):8732–8745. doi: 10.1523/JNEUROSCI.5993-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cordell HJ. Detecting gene–gene interactions that underlie human diseases. Nature Reviews Genetics. 2009;10(6):392–404. doi: 10.1038/nrg2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cordell HJ, et al. Statistical modeling of interlocus interactions in a complex disease: rejection of the multiplicative model of epistasis in type 1 diabetes. Genetics. 2001;158(1):357–367. doi: 10.1093/genetics/158.1.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Zubicaray GI, et al. Meeting the challenges of neuroimaging genetics. Brain Imaging Behavior. 2008;2:258–263. doi: 10.1007/s11682-008-9029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265(5181):2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 13.Abecasis GR, et al. MaCH: Using Sequence and Genotype Data to Estimate Haplotypes and Unobserved Genotypes. Genetic Epidemiology. 2010;34(8):816–834. doi: 10.1002/gepi.20533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.ENIGMA2 Genetics Support Team. ENIGMA2 1KGP cookbook (v3) [Online] [accessed 27 July 2012];The Enhancing Neuroimaging Genetics through Meta-Analysis (ENIGMA) consortium. [Google Scholar]
- 15.Leow A, et al. Inverse consistent mapping in 3D deformable image registration: its construction and statistical properties. Inf Process Med Imaging. 2005;19:493–503. doi: 10.1007/11505730_41. 2005. [DOI] [PubMed] [Google Scholar]
- 16.Hua X, et al. Unbiased tensor-based morphometry: Improved robustness and sample size estimates for Alzheimer's disease clinical trials. Neuroimage. 2012 doi: 10.1016/j.neuroimage.2012.10.086. EPUB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jahanshad N, et al. Brain structure in healthy adults is related to serum transferrin and the H63D polymorphism in the HFE gene. Proc Natl Acad Sci. 2012;109(14):E851–9. doi: 10.1073/pnas.1105543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fan J, Lv J. Sure independence screening for ultrahigh dimensional feature space. Journal of the Royal Statistical Society: Series B (Statistical Methodology) 2008;70(5):849–911. doi: 10.1111/j.1467-9868.2008.00674.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Candes E, Tao T. The Dantzig selector: Statistical estimation when p is much larger than n. The Annals of Statistics. 2007;35(6):2313–2351. [Google Scholar]
- 20.Stein JL, et al. Genome-wide analysis reveals novel genes influencing temporal lobe structure with relevance to neurodegeneration in Alzheimer's disease. Neuroimage. 2010;51(2):542–554. doi: 10.1016/j.neuroimage.2010.02.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ueki M, Tamiya G. Ultrahigh-dimensional variable selection method for whole-genome gene-gene interaction analysis. BMC Bioinformatics. 2012;13(1):72. doi: 10.1186/1471-2105-13-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoerl AE. Application of ridge analysis to regression problems. Chemical Engineering Progress. 1962;58:54–59. [Google Scholar]
- 23.Kohannim O, et al. Boosting power to detect genetic associations in imaging using multi-locus, genome-wide scans and ridge regression. Biomedical Imaging: From Nano to Macro IEEE. 2011 EPUB. [Google Scholar]
- 24.Chen J, Chen Z. Extended Bayesian information criteria for model selection with large model spaces. Biometrika. 2008;95(3):759–771. [Google Scholar]
- 25.Langers DR, et al. Enhanced signal detection in neuroimaging by means of regional control of the global false discovery rate. NeuroImage. 2007;38(1):43–56. doi: 10.1016/j.neuroimage.2007.07.031. [DOI] [PubMed] [Google Scholar]