

Abstract
Despite a rising demand for anti-obesity therapeutics, few effective pharmacological options are clinically available that target the synthesis and accumulation of body fat. Moderate inhibition of mammalian glycerol-3-phosphate acyltransferase (GPAT) with 2-(alkanesulfonamido)benzoic acids has recently been described in vitro, accompanied by promising weight loss in vivo. In silico docking studies with 2-(octanesulfonamido)benzoic acid modeled into the active site of squash GPAT revealed an unoccupied volume lined with hydrophobic residues proximal to C-4 and C-5 of the benzoic acid ring. In an effort to produce more potent GPAT inhibitors, a series of 4- and 5-substituted analogs were designed, synthesized, and evaluated for inhibitory activity. In general, compounds containing hydrophobic substituents at the 4- and 5-positions, such as biphenyl and alkylphenyl hydrocarbons, exhibited an improved inhibitory activity against GPAT in vitro. The most active compound, 4-([1,1'-biphenyl]-4-carbonyl)-2-(octanesulfonamido)benzoic acid, demonstrated an IC50 of 8.5 µM and represents the best GPAT inhibitor discovered to date. Conversely, further substitution with hydroxyl or fluoro groups, led to a 3-fold decrease in activity. These results are consistent with the presence of a hydrophobic pocket and may support the binding model as a potential tool for developing more potent inhibitors.
Introduction
Obesity, defined as having a Body Mass Index (BMI) of 30 or higher, has been implicated in multiple diseases, including cardiovascular disease1, type-2 diabetes2, hypertension, nonalcoholic fatty liver disease3, infertility4, and certain types of cancer.5 According to the World Health Organization, it affects approximately 500 million adults and is the fifth leading risk of deaths globally, accounting for at least 2.8 million deaths a year. Despite an increase in obesity and obesity-related diseases, few effective pharmacological therapies currently exist to combat the physiological source of obesity, the accumulation of triacylglycerol (TAG). One approach involves exploiting the mechanisms of the lipid metabolic pathway, either by decreasing the rate of fat synthesis, as shown with the small molecule fatty acid synthase (FAS) inhibitors C756 and cerulenin7, or increasing the rate of fatty acid oxidation.8 GPAT catalyzes the transesterification of fatty acyl-CoAs with the primary hydroxyl group of glycerol-3-phosphate (G3P), the first committed and rate-limiting step in the biosynthesis of TAG.9 In mammals, mitochondrial GPAT (mtGPAT) is an integral membrane protein that co-localizes in the mitochondrion with carnitine-palmitoyl transferase I (CPT1), the rate-limiting enzyme in β-oxidation. These enzymes are co-regulated and compete for fatty acyl-CoA substrates.10 Inhibition of GPAT by a small molecule inhibitor has the potential to reduce de novo synthesis of TAG and to divert the flux of fatty acyl-CoAs to β-oxidation mediated by CPT1, thus making GPAT an attractive therapeutic target against obesity.
Recently, it has been demonstrated that o-(alkanesulfonamido)benzoic acids such as 1a–b (Figure 1) possess moderate inhibitory activity against mammalian GPAT in vitro.11 The 8-carbon derivative 1c has also been synthesized and exhibits an inhibitory activity comparable to 1b.12 The benzoic acid moiety of these compounds, which is deprotonated at physiological pH, is believed to mimic the anionic phosphate group of the native substrate. The relatively acidic aryl sulfonamide hydrogen was proposed to interact with the catalytic histidine responsible for deprotonating the primary hydroxyl group of G3P necessary for acylation. The long alkyl chain of the sulfonamide was designed to bind the lipophilic palmitoyl-CoA binding site.
Figure 1.

Structure of o-(alkanesulfonamido)benzoic acid GPAT inhibitors (1a–c) and proposed analogs
While structural information regarding mammalian GPAT is limited, X-ray crystal structures of squash GPAT revealed the conserved catalytic HXXXXD motif present in all GPATs as well as a putative G3P binding site comprised of conserved cationic residues Lys193, Arg235, and Arg237.13a–b A complex network of relatively hydrophobic pockets and tunnels emanating from the active site was also observed. Protein homology-based modelling of multiple human transferases, including AGPAT1, AGPAT2, and AGPAT9, with the squash GPAT crystal structure showed that, despite low sequence homology, the human transferases are similar in predicted secondary structure and hydrophobic residue distribution.14 These findings suggest that, although the overall architectures of the acyltransferase homologs differ, structural similarity in the region of the active site may be good. An in silico docking simulation of 1c with squash GPAT, as shown in Figure 2, displayed the inhibitor bound at the putative G3P binding pocket with the carboxylate anion positioned at the phosphate binding site and the long alkyl chain residing in the putative palmitoyl-CoA binding site described by Turnbull et al.13a Furthermore, the docking model showed a second hydrophobic tunnel, described previously by Tamada et al.,13b extending away from the G3P binding site and presenting from C-4 and C-5 of the benzoic acid ring of the docked inhibitor. This volume was lined by hydrophobic and aromatic residues including Phe98, Gly99, Tyr102, Ile103, Ala143, Leu176, Pro179, and Phe180.
Figure 2.

A) Compound 1c (yellow) docked into the active site of squash GPAT using the CDOCKER protocol with Accelrys Discovery Studio (version 2.1). B) Compound 14b manually docked into the binding site obtained for compound 1c.
In an attempt to improve upon the inhibitory activity of 1c, a series of potential inhibitors was designed and synthesized with various aryl substituents either directly-bound to the 4- or 5-positions of 1c or coupled via one of several linkers (keto-, vinyl-, or ethyl-). The aryl substituents were chosen to promote π-stacking with the aromatic residues in the potential hydrophobic channel as well as for synthetic accessibility. The linkers were chosen to examine the effects of pharmacophore separation and conformational rigidity versus flexibility. The 8-carbon chain of 1c was chosen for its ready commercial availability and, within experimental error, essentially unchanged GPAT inhibitory activity compared to the nonyl side chain reported previously.11 The candidates were tested for GPAT inhibition in vitro to develop a structure-activity relationship and assess the validity of the docking model as a tool to direct future medicinal chemistry efforts.
Results and Discussion
Synthesis of 4- and 5-substituted Analogs
An efficient, divergent route was devised to access the 4- and 5-substituted derivatives of 1c. Sulfonamide coupling of 4- and 5-bromoanthranilates 2 and 3, respectively, with octanesulfonyl chloride generated sulfonamides 4 and 5. Suzuki-Miyaura coupling reactions with various aryl boronic acids were then performed either under argon to afford coupling products 6a–l and 7a–l or under an atmosphere of carbon monoxide to afford keto-linker precursors 8a–b and 9a–d. Alternatively, coupling of 4 and 5 with various trans-arylvinyl boronic acids under an argon atmosphere provided vinyl-linker precursors 10a–b and 11a–b. Each of the benzoates was then hydrolyzed, either under Gassman conditions15 or in aqueous 1 M NaOH, to afford analogs 12a–l, 13a–l, 14a–b, 15a–d, 16a–b and 17a–b. Alternatively, hydrogenation of 10a–b and 11a–b prior to hydrolysis afforded ethyl-linked analogs 18a–b and 19a–b.
Inhibition Data
The inhibitory data for the 4-aryl-substituted analogs are presented in Table 1. Several of these compounds exhibited marked improvement over 1c. In general, hydrophobic substituents such as p-n-butylphenyl (12d), biphenyl (12e,f), and methoxyphenyl (12g,h) exhibited a moderate increase in inhibitory activity of up to 2-fold. Chlorophenyl analogs (12a–c) also demonstrated markedly more potent inhibition, particularly the meta-chloro analog which also was about 2-fold more active than 1c. Conversely, hydroxyl- and fluoro-containing analogs (12i–l) caused a modest decrease in inhibitory activity. The para-hydroxyl analog, for instance, showed a nearly 3-fold decline in inhibition.
Table 1.
GPAT Inhibition by 4-Substituted Derivativesa
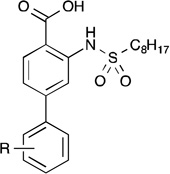 | ||
|---|---|---|
| Compound | R | IC50 (µM) ± SD |
| 12a | o-Cl | 26.9 ± 4.1 |
| 12b | m-Cl | 13.8 ± 2.5 |
| 12c | p-Cl | 18.1 ± 3.5 |
| 12d | p-n-C4H9 | 15.7 ± 1.6 |
| 12e | o-C6H5 | 17.1 ± 0.8 |
| 12f | p-C6H5 | 13.5 ± 3.2 |
| 12g | o-OCH3 | 16.1 ± 0.5 |
| 12h | p-OCH3 | 13.3 ± 1.1 |
| 12i | o-F | 47.2 ± 2.0 |
| 12j | m-F | 32.6 ± 3.6 |
| 12k | o-OH | 63.1 ± 5.5 |
| 12l | p-OH | 64.8 ± 3.8 |
A description of assay conditions can be found in the Supplemental Information.
The inhibitory data for the 5-aryl analogs is presented in Table 2. Generally, the structure-activity relationship of this class mirrors that of the 4-aryl class. For instance, several of the compounds showed notable improvement over 1c, particularly those containing chlorophenyl and hydrophobic substituents. Specifically, both the meta-chloro (13b) and para-n-butyl (13d) analogs effected a 2.9-fold increase in inhibitory activity. Also similar to the 4-aryl class, hydroxyphenyl and fluorophenyl groups were not well tolerated, decreasing inhibition by nearly 3-fold.
Table 2.
GPAT Inhibition by 5-Substituted Derivativesa
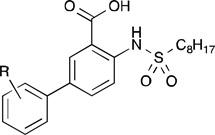 | ||
|---|---|---|
| Compound | R | IC50 (µM) ± SD |
| 13a | o-Cl | 14.4 ± 1.3 |
| 13b | m-Cl | 8.9 ± 0.9 |
| 13c | p-Cl | 9.2 ± 0.7 |
| 13d | p-n-C4H9 | 9.0 ± 0.4 |
| 13e | o-C6H5 | 10.1 ± 0.7 |
| 13f | p-C6H5 | 29.1 ± 6.3 |
| 13g | o-OCH3 | 27.6 ± 2.0 |
| 13h | p-OCH3 | 19.5 ± 5.5 |
| 13i | o-F | 34.6 ± 2.0 |
| 13j | m-F | 37.6 ± 9.7 |
| 13k | o-OH | 61.7 ± 13.8 |
| 13l | p-OH | 47.3 ± 6.4 |
A description of assay conditions can be found in the Supplemental Information.
The data for the 4- and 5-aryl analogs are consistent with the presence of a hydrophobic binding pocket extending from C-4 and C-5 of the benzoic acid of 1. Both steric and electronic factors may be affecting binding in this pocket. Sterically, the pocket is large enough around the 4-position of 1c to accommodate the relatively voluminous and rigid ortho- and para-biphenyl analogs 12e and 12f. The region near the 5-position, however, appears more constrained in size. Consequently, the para-biphenyl analog 13f shows a large decrease in activity, possibly due to steric interactions with residues on the edge of the binding pocket. The ortho-biphenyl analog 13e, however, may orient the terminal phenyl substituent away from the edge and back into the binding pocket resulting in a 2.5-fold increase in activity. Van der Waals and π-stacking interactions seem to be favored within the pocket as phenyl- alkylphenyl-, and alkoxyphenyl substituents showed an increase in activity. Conversely, the more polarized fluorine-containing analogs demonstrated moderately diminished activity possibly due to binding at a site intolerant of the inhibitor’s high charge density or disruption of key π-π interactions within the hydrophobic channel.
While the 3-fold increase in observed activity for the 4-aryl and 5-aryl derivatives was promising, we worried that the kinked nature of the hydrophobic tunnel observed in the docking model was preventing the analogs from extending fully into it. Therefore, we next sought to incorporate various linker regions on a subset of the more promising hydrophobic leads. Keto, vinyl, and ethyl linkers were chosen to examine the effect of separating the substituents from the pharmacophore and to explore conformational rigidity versus flexibility in extending the substituents into the hydrophobic tunnel. Inhibition data for the 4- and 5-substituted analogs are shown in Table 3 and Table 4, respectively. In general, the analogs possessing a keto linker were slightly to moderately less potent than the corresponding structures without a linker. For example, 5-keto-substituted para-n-butyl analog 15a exhibited a 3.5-fold decrease in inhibition compared to compound 13d. The para-biphenyl analogs 14b and 15b, however, were notable exceptions. Compound 14b showed a 2-fold more potent inhibition compared to the linker-free analog 13f. While the rigid linear carbon skeleton of 13f may sterically interfere with residues on the face of the hydrophobic pocket, the ketone linker in 15b could potentially direct the large biphenyl moiety back towards the cavity. Compound 14b, with an IC50 of 8.5 µM, was 3-fold more potent than 1c and, to our knowledge, represents the most active GPAT inhibitor discovered to date.
Table 3.
4-Substituted Inhibitors with Linkersa
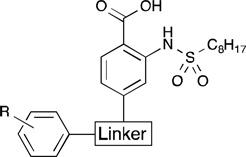 | |||
|---|---|---|---|
| Compound | Linker | R | IC50 (µM) |
| 14a | p-n-C4H9 | 15.8±0.8 | |
| 14b | p-C6H5 | 8.5±2.1 | |
| 16a | -H | 38.0±9.9 | |
| 16b | p-OCH3 | 38.7±6.3 | |
| 18a | -H | 37.5±4.8 | |
| 18b | p-OCH3 | 43.0±9.3 | |
A description of assay conditions can be found in the Supplemental Information.
Table 4.
5-Substituted Inhibitors with Linkersa
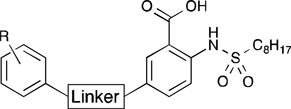 | |||
|---|---|---|---|
| Compound | Linker | R | IC50 (µM) |
| 15a | p-n-C4H9 | 31.7±3.2 | |
| 15b | p-C6H5 | 13.0±0.6 | |
| 15c | o-OCH3 | 59.2±3.8 | |
| 15d | p-OCH3 | 44.3±9.2 | |
| 17a | -H | 16.3±5.3 | |
| 17b | p-OCH3 | 37.1±8.3 | |
| 19a | -H | 37.5±4.8 | |
| 19b | p-OCH3 | 26.3±2.4 | |
A description of assay conditions can be found in the Supplemental Information.
The rigid vinyl and conformationally-flexible ethyl linkers were generally not well tolerated. For example, 16b, a vinyl-linked para-methoxyphenyl analog, produced an activity of 37.5 µM compared to 13.3 µM for the linker-free derivative 12h. No significant benefit was apparent from the incorporation of the conformationally-flexible ethyl linkers. The same para-methoxyphenyl substituent with a linker region in compound 18b demonstrated an IC50 of 43.0 µM or 3.2-fold lower activity than the corresponding compound 12h.
Conclusions
Several 4- and 5-substituted analogs of GPAT inhibitor (octanesulfonamido)benzoic acid were synthesized and analyzed for in vitro inhibitory activity. In general, hydrophobic substituents led to an increase in inhibitory activity while more polar and hydrogen-bonding substituents showed a decrease in inhibitory activity, consistent with the presence of a hydrophobic pocket identified by in silico studies. Taking advantage of the hydrophobic pocket, p-biphenylketone-substituted 14b improved the inhibitory activity 3-fold to 8.5 µM, representing the most potent GPAT inhibitor to date. Future studies will aim to determine whether further extension of aryl and alkyl substituents into this channel confers additional inhibitory activity and if the observed increases in vitro correlate with in vivo TAG biosynthesis and overall fat metabolism in keeping with earlier studies.16
Supplementary Material
Scheme 1.

Reagents and Conditions: a) ClSO2C8H17, NEt3, CH2Cl2, 62–76%; b) Pd(PPh3)4, Ar-B(OH)2, Na2CO3, PhMe/MeOH, 90 °C, 45–80%; c) Pd(PPh3)4, Ar-B(OH)2, K2CO3, dioxane, 90 °C, CO (1 atm), 28–46%; d) Pd(PPh3)4, (E)-Ar-(CH)2B(OH)2, K2CO3, PhCH3, 105 °C, 58–83%; e) Pd/C, H2 (1 atm), MeOH, 80–91%; f) KOtBu, H2O, Et2O, 0 °C-r.t., 71–94%; g) 1 M NaOH, THF, 40 °C, 73–92%.
Acknowledgements
We would like to thank Dr. I. P. Mortimer for performing HRMS analyses. This work was supported by the NIH 1R43DK65423 to FASgen, Inc. Under a license agreement between FASgen, Inc. and The Johns Hopkins University, C.A.T. is entitled to share royalty received by the University on sales of products described in this article. C.A.T. owns FASgen, Inc. stock, which is subject to certain restrictions under university policy.
Footnotes
The Johns Hopkins University, in accordance with its conflict of interest policies, is managing the terms of this arrangement.
Electronic Supplementary Information (ESI) available: Experimental details including synthetic procedures and characterization data for all novel compounds is available online free of charge. See DOI: 10.1039/b000000x/
Notes and references
- 1.a) Poirier P, Giles TD, Bray GA, et al. Arterioscler. Thromb. Vasc. Biol. 2006;26:968–976. doi: 10.1161/01.ATV.0000216787.85457.f3. [DOI] [PubMed] [Google Scholar]; b) Bastien M, Poirier P, Lemieux I, Després J-P. Prog. Cardiovasc. Dis. 2014;56:369–381. doi: 10.1016/j.pcad.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 2.Haslam DW, James WP. Obesity. Lancet. 2005;366:1197–1209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- 3.a) Hall JE. Hypertension. 2003;41:625–633. doi: 10.1161/01.HYP.0000052314.95497.78. [DOI] [PubMed] [Google Scholar]; b) Adams ST, Salhab M, Hussain ZI, Miller GV, Leveson SH. Blood Pressure. 2013;22:131–137. doi: 10.3109/08037051.2012.749570. [DOI] [PubMed] [Google Scholar]
- 4.Hammoud AO, Gibson M, Peterson CM, Meikle AW, Carrell DT. Fertil. Steril. 2008;90:897–904. doi: 10.1016/j.fertnstert.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 5.a) Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. N. Engl. J. Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]; b) Pantasri T, Norman RJ. Gynecol. Endocrinol. 2014;30:90–94. doi: 10.3109/09513590.2013.850660. [DOI] [PubMed] [Google Scholar]
- 6.a) Loftus TM, Jaworski DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Science. 2000;288:2379–2381. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]; b) Gilbert CA, Slingerland JM. Annu. Rev. Med. 2013;64:45–57. doi: 10.1146/annurev-med-121211-091527. [DOI] [PubMed] [Google Scholar]
- 7.Makimura H, Mizuno TM, Yang XJ, Silverstein J, Beasley J, Mobbs CV. Diabetes. 2001;50:733–739. doi: 10.2337/diabetes.50.4.733. [DOI] [PubMed] [Google Scholar]
- 8.Thupari JN, Landree LE, Ronnett GV, Kuhajda FP. Proc. Natl. Acad. Sci. U.S.A. 2002;99:9498–9502. doi: 10.1073/pnas.132128899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coleman RA, Lewin TM, Muoio DM. Annu. Rev. Nutr. 1998;18:331–351. [Google Scholar]
- 10.Muoio DM, Seefeld K, Witters LA, Coleman RA. Biochem. J. 1999;338(Pt 3):783–791. [PMC free article] [PubMed] [Google Scholar]
- 11.Wydysh EA, Medghalchi SM, Vadlamudi A, Townsend CA. J. Med. Chem. 2009;52:3317–3327. doi: 10.1021/jm900251a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.See Supplemental Information for the synthetic protocol to compound 1c
- 13.a) Turnbull AP, Rafferty JB, Sedelnikova SE, Slabas AR, Schierer TP, Kroon JT, Simon JW, Fawcett T, Nishida I, Murata N, Rice DW. Structure. 2001;9:347–353. doi: 10.1016/s0969-2126(01)00595-0. [DOI] [PubMed] [Google Scholar]; b) Tamada T, Feese MD, Ferri SR, Kato Y, Yajima R, Toguri T, Kuroki R. Acta Crystallogr D Biol Crystallogr. 2003;60:13–21. doi: 10.1107/s0907444903020778. [DOI] [PubMed] [Google Scholar]
- 14.Agarwal AK, Sukumaran S, Bartz R, Barnes RI, Garg A. Journal of Endocrinology. 2007;193:445–457. doi: 10.1677/JOE-07-0027. [DOI] [PubMed] [Google Scholar]
- 15.Gassman PG, Schenk WN. J. Org. Chem. 1977;42:918–920. [Google Scholar]
- 16.Kuhajda FP, Aja S, Tu Y, Han WF, Medghalchi SM, Meskini El R, Landree LE, Peterson JM, Daniels K, Wong K, Wydysh EA, Townsend CA, Ronnett GV. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301:R116–R130. doi: 10.1152/ajpregu.00147.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.