

Abstract
The cellular prion protein (PrPC) is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein on the cell surface. Previous studies have demonstrated contradictory roles for PrPC in connection with the phagocytic ability of macrophages. In the present work, we investigated the function of PrPC in phagocytosis and cytokine expression in bone marrow-derived macrophages infected with Escherichia coli. E. coli infection induced an increase in the PRNP mRNA level. Knockout of PrPC promoted bacterial uptake; upregulated Rab5, Rab7, and Eea1 mRNA expression; and increased the recruitment of lysosomal-associated membrane protein-2 to phagosomes, suggesting enhanced microbicidal activity. Remarkably, knockout of PrPC suppressed the proliferation of internalized bacteria and increased the expression of cytokines such as interleukin-1β. Collectively, our data reveal an important role of PrPC as a negative regulator for phagocytosis, phagosome maturation, cytokine expression, and macrophage microbicidal activity.
Introduction
Phagocytosis of pathogens initiates the innate immune response [1], [2]. Macrophages rely heavily on phagocytosis and subsequent degradation of microbes to help clear the invading pathogens [3]. The initial stage of the elimination process is the internalization of particles into a plasma membrane-derived vacuole known as the phagosome. Nascent phagosomes lack the ability to kill pathogens and degrade ingested targets; these properties are acquired during the process of phagosome maturation [4]. After internalization, targets are delivered from phagosomes to lysosomes for degradation [5]. The low-molecular-weight GTPases Rab5 and Rab7, which govern the fusion of phagosomes with early and late endosomes, are associated with phagosome maturation [2], [6]. Additionally, early endosome antigen 1 (Eea1), a Rab5 effector, is present in very early phagosomes but disappears rapidly during maturation of the organelle [7]. Late phagosomes acquire markers such as lysosomal-associated membrane protein (LAMP)-1 and LAMP-2, which are required for acquisition of Rab7 and microbicidal properties [8]. These markers are important for phagosome fusion and maturation.
In addition to particle and pathogen internalization, activated macrophages initiate cytokine secretion, which is essential for host defense [2], [9]. Studies have demonstrated that failure to regulate cytokine secretion may induce a pathological state; indeed, excessive levels of tumor necrosis factor (TNF) and interleukin (IL)-6 lead to chronic inflammation [10], [11]. Therefore, regulatory control of phagocytosis is essential to limit damage to the host during pathogen clearance [12], and negative regulation of phagocytic activity may provide protection against pathological phagocytosis. Performing further studies on the specific signal transduction components that negatively regulate phagocytosis is essential.
Preliminary experiments have shown that cellular prion protein (PrPC) may play an important role in phagocytosis [13], [14]. PrPC is a glycosylphosphatidylinositol (GPI)-anchored glycoprotein encoded by a specific prion protein gene (PRNP) [15]. It is expressed mainly in the central nervous system (CNS) but is also expressed in other cell types, including macrophages [14], [16]. PrPC can be conformationally converted into PrPSC, which accumulates in the brain in prion disease [17]. Although a great deal has been learned about PrPSC and its role in prion propagation, the physiological functions of PrPC remain unclear. Numerous studies have suggested that PrPC may have protective functions, including providing protection against apoptotic and oxidative stress, facilitating cellular uptake or binding of copper ions, promoting transmembrane signaling, and participating in the formation and maintenance of synapses [18]. PrPC is also necessary for neuronal survival and maintenance of the myelin sheath around peripheral nerves [19], [20]. Additionally, although numerous reports have revealed a relationship between PrPC and the phagocytic ability of different cell lines following ingestion of various particles, the results are conflicting. Studies have supported that Rab7a interacts with PrPC and that endosomal compartments are involved in the trafficking and regulation of PrPC [21]; however, further studies are required to elucidate the specific signaling mechanisms mediating the important roles of PrPC in phagocytosis.
Therefore, in this study, we sought to investigate the role of PrPC during phagosome formation and maturation, and we hypothesized that PrPC may exert an important protective effect against internalized particles or pathogens.
Materials and Methods
Antibodies
The mouse monoclonal PrP antibody AH6 and rabbit anti-mouse β-actin antibody were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The rabbit polyclonal anti-LAMP2 antibody was obtained from ProteinTech Group (Chicago, USA). The secondary antibodies, horseradish peroxidase-conjugated Affinipure goat anti-rabbit IgG, horseradish peroxidase-conjugated Affinipure goat anti-mouse IgG and rhodamine-conjugated Affinipure goat anti-rabbit IgG were purchased from Zhongshan Golden Bridge Biological Technology (Beijing, China).
Enhanced green fluorescent protein (EGFP)-Escherichia coli preparation
The EGFP sequence from the pEGFP-N1 vector was cloned and inserted into the PET28a vector, yielding the recombinant plasmid PET28a-EGFP. This plasmid was transformed into DH5a competent cells for amplification (TransGen Biotech, Beijing, China). After incubation at 37°C for 10 h, the recombinant plasmid was isolated and transformed into BL21 (DE3) plyss competent cells (TransGen Biotech). When the OD600 of the culture reached 0.4–0.6, 0.5 mM isopropylthio-β-d-galactopyranoside (IPTG, ICN Pharmaceuticals, CA, USA) was added to induce expression of the EGFP protein. EGFP-E. coli was then plated on LB plates, and after 10 h, the number of colony-forming units (CFU) was determined and used for subsequent calculation of the EGFP-E. coli count per milliliter for other experiments.
Primary cell cultures
Bone marrow-derived macrophages (BMDMs) were derived from bone marrow cells extracted from the femurs and tibiae of 6- to 8-week-old female ZrchI type PRNP−/− mice [22] and wild-type C57BL/6 mice (Vital River Laboratory Animal Technology, Beijing, China). The mice were bred under strict specific pathogen-free conditions. All of the animal experiments were conducted in accordance with the guidelines of Beijing Municipality on the Review of Welfare and Ethics of Laboratory Animals approved by the Beijing Municipality Administration Office of Laboratory Animals (BAOLA). After euthanasia, the mice were immersed in 75% ethanol for 3 min. Then the femurs and tibiae were dissected using sterile scissors, and muscles connected to the bones were also removed. After three washes in sterile RPMI 1640 (Gibco, Grand Island, NY, USA), both epiphyses were removed using sterile scissors and forceps. The bones were flushed with a syringe filled with RPMI 1640 to extrude bone marrow into a 15 mL sterile polypropylene tube. The fresh bone marrow cells were cultured as previously described [23], with the following modifications. The cells were differentiated in a humidified incubator at 37°C with 5% CO2 in bone marrow differentiation medium, which was composed of RPMI 1640 supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco, Grand Island, NY, USA), 100 µg/mL streptomycin, 100 U/mL penicillin (Gibco), and 10 ng/mL macrophage colony-stimulating factor (M-CSF, Peprotech Asia, Rehovot, Israel). Seven days later, the cells were detached from the dishes, counted, reseeded, and cultivated in tissue culture plates overnight before any further experimental procedures. For regular culture, BMDMs were grown in RPMI 1640 medium containing 10% heat-inactivated FBS, 100 µg/mL streptomycin, 100 U/mL penicillin, and 2 ng/mL M-CSF.
Phagocytosis assays
Macrophages were plated on glass coverslips at a density of 2×105 cells/well in 24-well plates overnight. EGFP-E. coli organisms were washed in phosphate-buffered saline (PBS; Hyclone, Logan, UT, USA) twice and then resuspended at the appropriate concentration in RPMI 1640 without serum or antibiotics. Before infection, cells were washed twice in PBS to eliminate the effects of Fc and/or complement receptors. BMDMs were infected with EGFP-E. coli at a multiplicity of infection (MOI) of 10 and incubated at 37°C in the presence of 5% CO2. After 30 or 60 min, the macrophages were washed in cold PBS three times to stop the uptake of additional bacteria and remove extracellular bacteria. The cells were fixed with 4% paraformaldehyde (Solarbio Science & Technology Co., Beijing, China) and the nuclei were stained with DAPI staining solution (Beyotime Institute of Biotechnology, Shanghai, China). The phagocytic ability of the macrophages was examined by fluorescence microscopy. In each experiment, at least 300 macrophages from triplicate wells were counted. The results were expressed as the phagocytic index (PI), calculated using the following equation: PI = percentage of cells containing E. coli×mean number of E. coli organisms per cell.
Quantitative real-time polymerase chain reaction (qPCR)
An EASYspin Plus Cell/Tissue RNA Isolation Kit (Aidlab Biotechnologies Co., Beijing, China) was used to extract total RNA from adherent macrophages. Reverse transcription was performed using a RevertAid First-Strand cDNA Synthesis Kit (Fermentas, Vilnius, Lithuania) according to the manufacturer’s instructions. Based on previous studies [15], β-actin was selected as the internal control. qPCR was performed using an ABI Vii7 fluorescence detection system (Applied Biosystems, Foster City, CA, USA) with the primer pairs listed in Table 1, using an annealing temperature of 52°C. qPCR data were analyzed using the comparative CT method (2−ΔΔCT) [24]. All samples were analyzed in triplicate.
Table 1. Primers used for qPCR.
| Gene | Forward primer sequences (5′-3′) | Reverse primer sequences (5′-3′) |
| PRNP | GAGAACTTCACCGAGACC | GATGAGGAGGATGACAGG |
| Rab5a | CTGGTTCTTCGCTTTGTGA | ACTATGGCTGCTTGTGCTC |
| Rab7 | GGCTTCACAGGTTGGAC | GGCTTGGCTTGGAGATTG |
| Eea1 | GTGGCAGTCTAGTCAACG | CTTCGCCTTTAAGACACCTC |
| TNF-α | GCGGTGCCTATGTCTCAG | CACTTGGTGGTTTGCTACG |
| IL-1β | CAGGCTCCGAGATGAACAA | CCCAAGGCCACAGGTATTT |
| IL-6 | TTGCCTTCTTGGGACTGAT | TTGCCATTGCACAACTCTTT |
| β-actin | GATCATTGCTCCTCCTGAGC | AAAGGGTGTAAAACGCAGC |
Bacterial proliferation assay
BMDMs were plated in 24-well plates and infected with E. coli at a ratio of 1∶10 at 37°C for 1 h. The cells were then washed three times with PBS, and RPMI 1640 with 10 ng/mL gentamicin (Sigma-Aldrich, St. Louis, Missouri, USA) was added for 30 min (0-h time point) or for an additional 1 h and 2 h. The infected macrophages were then washed three times, lysed with a solution of 1% Triton X-100 (Amresco, Solon, Ohio, USA) in sterile water at 37°C for 20 min, and shaken vigorously. The lysates were diluted, plated on LB agar plates, and incubated for 10 h at 37°C. The colonies were counted and expressed as CFU. The proliferation index (i.e., the total number of divisions divided by the number of bacteria that underwent division) was also calculated. Infections were completed in triplicate.
Western blotting
After the cells were infected with E. coli for 1 h, the cell culture medium was discarded. The cells were then lysed immediately in ice-cold lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) containing protease and phosphatase inhibitors, and the total cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels, followed by transfer to 0.45-µm polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, USA). Nonspecific binding sites were blocked with 5% fat-free dried milk in Tris-buffered saline with Tween-20 (TBST) for 1 h at 37°C. The membranes were probed overnight at 4°C with the PrP antibody, β-actin antibody, and LAMP2 antibody. After the blots were washed with TBST three times, they were incubated with the respective secondary antibodies, horseradish peroxidase-conjugated Affinipure goat anti-mouse IgG and horseradish peroxidase-conjugated Affinipure goat anti-rabbit IgG (1∶5000), followed by enhanced chemiluminescence detection on an imaging system (Versadoc; Bio-Rad, Hercules, CA, USA).
Immunofluorescence and confocal microscopy
Macrophages were plated on glass coverslips at a density of 2×105 cells/well in 24-well plates and infected with EGFP-E. coli. One hour later, the cells were washed with PBS and fixed for 30 min with 4% paraformaldehyde. After fixation, cells were permeabilized with 0.3% Triton X-100. Cells were then blocked with 1% bovine serum albumin (BSA, Amresco, Solon, Ohio, USA) for 1 h at room temperature and incubated overnight with the rabbit polyclonal anti-LAMP2 antibody at 4°C. After washing in PBS, the rhodamine-conjugated Affinipure goat anti-rabbit IgG was added, and the cells were incubated for 1 h at 37°C in a humidified black box. The cells were then labeled with DAPI, mounted on slides, and observed under confocal microscopy. Negative controls were incubated without the primary antibody.
Statistical analyses
All assays were independently performed three times. The results have been expressed as mean ± standard deviation (SD). Statistical significance was assessed using unpaired two-tailed Student’s t-tests. SPSS software (Chicago, IL, USA) was used for the statistical analysis. Differences were considered significant when p<0.05.
Results
Infection of BMDMs with E. coli affected the mRNA expression of PRNP
BMDMs from wild-type C57BL mice were infected with EGFP-E. coli at an MOI of 10, and the mRNA expression of PRNP was examined. PRNP mRNA expression varied with time, and a significant increase in PRNP expression was observed at 30 and 45 min relative to the control (Figure 1).
Figure 1. Quantitative PCR analysis of the effects of E. coli infection on PRNP mRNA levels in macrophages collected from wild-type mice.
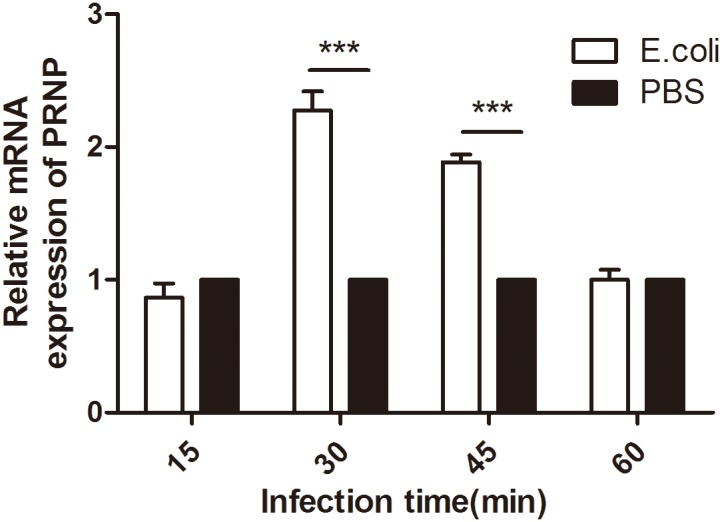
Murine bone marrow-derived macrophages were exposed to E. coli at an MOI of 10. Total RNA was collected at the indicated times, reverse transcribed into cDNA, and subjected to qPCR analysis. The expression of PRNP at each time point is expressed as the fold change relative to PRNP mRNA levels in control cells exposed to PBS only. Data are the mean ± SD of triplicate samples. **p≤0.01, ***p≤0.001.
Knockout of PRNP promoted phagocytosis
Next, we examined the expression of PrPC in BMDMs from PRNP−/− and wild-type mice. As expected, no PrPC expression was observed in PRNP−/− samples (Figure 2A). To determine the role of PrPC in phagocytic activity, BMDMs from PRNP−/− and wild-type mice were infected with EGFP-E. coli for 30 and 60 min, and the PI was then assessed as a measure of the phagocytic capacity. All of the EGFP-E. coli was within BMDMs (Figure 2B). The percentage of cells containing E. coli, the mean number of E. coli organisms per cell, and the PI were significantly higher in BMDMs from PRNP−/− mice compared to those from wild-type mice (Figure 2B, C, D, E). Furthermore, the CFU assay below gave the same result.
Figure 2. BMDMs from PRNP−/− mice exhibited greater phagocytic activity than those from wild-type (WT) mice.

Total proteins were collected from BMDMs and analyzed by western blotting with anti-PrP antibodies (AH6). The expected glycoforms of PrPC were observed in WT samples, whereas no bands were observed in PRNP−/− samples (A). Phagocytosis assays were performed using EGFP-E. coli (green). Cells were fixed and stained with DAPI (blue) to detect chromosomal DNA, and fluorescence microscopy was used to image the stained cells. The micrographs for 60 min are shown; the cell profile was observed through phase-contrast microscopy. (B). The percentage of cells containing E. coli (C), the mean count of E. coli per cell (D), and the phagocytic index (E) are shown. Data represent at least three independent experiments. **p≤0.01, ***p≤0.001.
Knockout of PRNP enhanced phagosome maturation
To further explore the role of PRNP in macrophage phagocytosis, we investigated the role of PRNP in phagosome maturation. Knockout of PRNP resulted in increased Rab5a, Rab7, and Eea1 mRNA levels and led to an increase in the recruitment of LAMP2 to phagosomes. Rab5a mRNA expression was significantly higher in PRNP−/− macrophages than in wild-type macrophages at 15, 30, and 45 min. Additionally, mRNA expression of Rab7 and Eea1 was significantly higher in PRNP−/− macrophages than in wild-type macrophages at 15, 30, 45, and 60 min (Figure 3A). Confocal immunofluorescence microscopy showed that recruitment of LAMP2 to phagosomes containing EGFP-E. coli increased in PRNP−/− macrophages following incubation with EGFP-E. coli for 1 h (Figure 3B). Additionally, densitometry analysis of western blotting data revealed that LAMP2 expression in PRNP−/− macrophages was almost twice that in wild-type macrophages (Figure 3C).
Figure 3. Knockout of PRNP regulated phagosome maturation.

(A) qPCR analysis of the effects of PrPC on the expression of Rab5a, Rab7, and Eea1. Macrophages from PRNP−/− and wild-type (WT) mice were incubated with E. coli at 10 MOI for the indicated times. Total mRNA was isolated and reverse transcribed. The expression levels of the three markers were analyzed at each time point and are expressed as the fold change relative to the mRNA level in control cells exposed to PBS only. (B) Murine BMDMs were infected with EGFP-E. coli (green) at 10 MOI. Confocal fluorescence microscopy was used to image LAMP2 staining (red) in PRNP−/− and WT macrophages at 1 h. Recruitment was quantified (right panel). (C) Western blot analysis of LAMP2 expression after infection for 1 h. Data represent at least three independent experiments. *p≤0.05, **p≤0.01, ***p≤0.001.
Knockout of PRNP suppressed the proliferation of internalized bacteria
Given that knockout of PRNP affected phagocytosis and phagosome maturation, we attempted to elucidate the role of PRNP in killing intracellular pathogens and in cytokine secretion. Macrophages were allowed to ingest E. coli for 1 h at 37°C and were then treated with gentamicin for various durations. The survival of E. coli engulfed by macrophages was analyzed by CFU enumeration. An increase in the number of CFUs was observed from 0 to 2 h. At the 0- and 1-h time points, the number of CFUs from PRNP−/− macrophages was significantly higher than that from wild-type macrophages, whereas the opposite effect was observed at the 2-h time point (Figure 4A). However, the proliferation index of PRNP−/− macrophages was much lower than that of wild-type macrophages (Figure 4B).
Figure 4. Survival of internalized E. coli in macrophages.
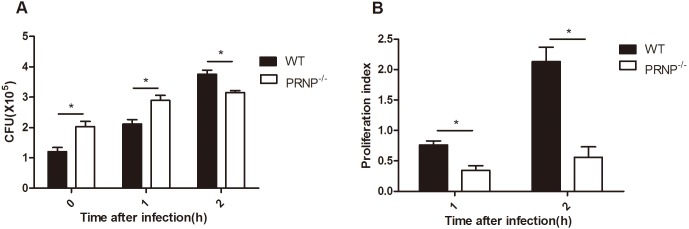
(A) PRNP−/− and wild-type (WT) macrophages were incubated with E. coli (MOI of 10) for 1 h. After washing, cells were incubated with RPMI-1640 containing gentamicin for 30 min (0-h time point) or for an additional 1–2 h. After each time point, cells were washed and lysed. Live bacteria in the lysates were counted after inoculation on LB agar plates. (B) The proliferation index at the 1-h and 2-h time points is shown. Data are representative of at least three independent experiments. *p≤0.05, **p≤0.01, ***p≤0.001.
Knockout of PRNP increased the expression of pro-inflammatory cytokines
We investigated whether knockout of PrPC affected the expression of pro-inflammatory cytokines. Knockout of PRNP dramatically increased the expression of IL-1β, IL-6, and TNF-α during the entire experimental procedure, as revealed by qPCR (Figure 5A, B, and C). The variation tendency in cytokine expression was similar among PRNP−/− and wild-type macrophages. However, the amplitude of variation of PRNP−/− macrophages was higher than that of wild-type macrophages.
Figure 5. mRNA expression of pro-inflammatory cytokines.

(A, B, and C) Total RNA was collected, reverse transcribed to cDNA, and analyzed by qPCR to assay the expression of IL-1β, IL-6, and TNF-α. The expression levels of these three cytokines were analyzed at each time point and are expressed as the fold change relative to the mRNA level in control cells exposed to PBS only. Data are representative of at least three independent experiments. *p≤0.05, **p≤0.01, ***p≤0.001.
Discussion
Macrophages act as the first line of protection in the innate immune response and play an important role in phagocytosis, antigen presentation, and inflammatory cytokine production. The classical activation of macrophages corresponds to the first phase, also known as the killing phase, of the innate immune response to acute stimuli and is characterized by the induction of a specific gene profile and the subsequent production of multiple cytoactive factors such as TNF-α, NO, and IL-1 that protect against tissue invaders [25]–[29]. A recent study performed in our laboratory showed that, in the long term, PrPC may actively participate in the regulation of microglia during the activation process [30]. However, the role of PrPC in the killing phase of macrophages has not been reported yet. Macrophages play an important role in facilitating the spread of prion infections from the periphery to the central nervous system [31], as prion protein normally is expressed on the surface of macrophages. To explore the role of PrPC in macrophage phagocytosis, microbicidal activity, and activation, we chose EGFP-E. coli as a representative of general pathogenic microbe for infection of BMDMs. We found that E. coli infection altered the mRNA expression of PRNP. It is possible that upregulation of PRNP expression interferes with BMDM activation, suggesting a possible role of PrPC in the host immune response. This observation is consistent with the effects of Mycobacterium bovis infection in BV2 microglia but differs from the findings of studies reporting exposure of microglia to interferon (IFN)-γ, IL-4, or IL-10 [15], [30]. This discrepancy may arise from the nature of bacterial infections, which are different from and more complex as a model than cytokine stimulation [15].
Because of the diversity among the bacteria, particles, cells, and methods used for the different experiments, reports on the relationship between PrPC and phagocytic ability are controversial. Moreover, distinct rates of phagocytosis cannot be attributed to random variations as a result of mixed genetic backgrounds [14]. In a study on M. bovis infection, PRNP silencing did not affect the number of viable bacilli in infected microglia [15]. Additionally, PrPC deficiency has been found to prevent swimming internalization of Brucella abortus into macrophages [32]. More efficient phagocytosis of zymosan particles was observed in ZrchI Prnp−/− mice than in Prnp+/+ mice [14]. Rikn Prnp−/− cells showed lower phagocytic activity than Prnp+/+ cells following ingestion of fluorescent beads [13], [33]. However, our results showed that PrPC exerted a negative regulatory function in phagocytosis during E. coli infection, which is consistent with a previously reported in vivo assay [14].
Phagosome maturation into the phagolysosome is the innate immune defense mechanism of macrophages. In the absence of PrPC, the mRNA expression of Rab5, Rab7, and Eea1 in BMDMs increased after infection with E. coli. Furthermore, increased recruitment of LAMP2 to phagosomes was observed, indicating that PrPC played a negative regulatory role in phagosome maturation. These observations prompted us to hypothesize that PRNP−/− macrophages formed phagosomes with an enhanced capacity to kill intracellular E. coli. Bacterial proliferation is the product of both replication and killing in the population as a whole [34]. Our data show that the number of CFUs increased in both wild-type and knockout macrophages, which may be explained by the observation that E. coli replicates inside macrophages and that the number of bacteria was higher than the clearance capacity of macrophages. However, the bacterial proliferation index observed in PRNP−/− macrophages was much lower than that in wild-type macrophages. Therefore, PRNP−/− macrophages were more efficient at bacterial clearance than wild-type macrophages, which indicates that PrPC plays a negative regulatory role in phagosome maturation. To our knowledge, this is the first evidence that PrPC has a negative effect on the defense function of macrophages in the killing phase.
Several lines of evidence indicate that NF-κB activation is critical for the induction of iNOS and the upregulation of inflammatory cytokines such as IL-1β, IL-6, and TNF-α [35]–[37]. Our previous study showed that macrophages exposed to neurotoxic prion peptides were activated through the activation of NF-κB [38]. In this study, we examined the effect of E. coli infection on the expression of the pro-inflammatory cytokines IL-1β, IL-6, and TNF-α in PRNP-knockout macrophages. Knockout of PRNP increased the expression of the pro-inflammatory cytokines, which would likely recruit more macrophages to inflammation sites and activate naïve macrophages to release ROS and active NO to kill the intracellular pathogens. Our CFU results for intracellular E. coli confirmed this effect. Moreover, recent studies have shown that recycling of endosomes is required for TNF-α trafficking. TNF-α is trafficked from the Golgi to recycling endosomes, which are delivered to the cell surface by the action of vesicle-associated membrane protein (VAMP) 3 [9]. In addition, IL-6 and IL-12 specifically induce the expression of Rab5 and Rab7 via the activation of extracellular signal-regulated kinase and p38 mitogen-activated protein kinase (MAPK), respectively, thereby modulating intracellular trafficking. Furthermore, TNF-α and IFN-γ significantly modify phagosome maturation [39]–[41]. These data suggest that cytokines can also modulate membrane trafficking, suggesting that PrPC plays a role in this process.
A study by Aguzzi’s group provided a new perspective [42]. The theoretical basis of that study was that CD47 is a ligand for the extracellular region of signal regulatory protein alpha (SIRPα) and that the binding of CD47 on apoptotic cells to SIRPα on macrophages transmits a “don’t eat me” signal that protects the targets from phagocytosis [42]–[45]. CD47 is known to be a transmembrane protein that is ubiquitously expressed in host cells, but not in E. coli [43]. Therefore, our findings can be attributed mainly to PrPC and not SIRPα. Accumulating evidence and the present study suggest that many other molecules play important roles in phagocytosis and cytokine secretion through different signaling pathways; such molecules include the soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), synaptotagmins (syts), Toll-like receptors (TLRs), Src family kinases, and cAMP [5], [9], [46]–[50]. Thus, although many studies have investigated the roles of PrPC, further studies are needed to clarify the relationships between PrPC and these molecules.
In summary, our results revealed that in the acute phase of macrophage activation after infection by E. coli, PrPC actively participates in the regulation of the process by protecting against excess inflammation through negative regulation of phagocytosis, phagosome maturation, cytokine expression, and microbicidal activity, which provides new insights into the physiological functions of PrPC in macrophages. Further studies are necessary to determine how PrPC regulates vesicular trafficking associated with phagocytosis and cytokine secretion.
Acknowledgments
We thank Dr. Charles Weissmann at the Scripps Research Institute for supplying the PRNP−/− knockout mice.
Funding Statement
This work was supported by the National “Twelfth Five-Year” Plan for Science & Technology Support (Project No. 2012AA101302), MoST-RCUK international cooperation project (Project No. 2013DFG32500), National Natural Science Foundation of China (Project No. 31172293, and No. 31272532), Funding of State Key Lab of Agrobiotechnology (Project No. 2012SKLAB06-14), and CAU Foreign Experts Major Projects (Project No: 2012z018). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Metchnikoff E. (1905) Immunity in infective diseases. Cambridge University Press, Cambridge.
- 2. Aderem A, Underhill DM (1999) Mechanisms of phagocytosis in macrophages. Annual review of immunology 17: 593–623. [DOI] [PubMed] [Google Scholar]
- 3. Sansonetti P (2001) Phagocytosis of bacterial pathogens: implications in the host response. Seminars in immunology 13: 381–390. [DOI] [PubMed] [Google Scholar]
- 4. Vieira O, Botelho R, Grinstein S (2002) Phagosome maturation: aging gracefully. Biochem J 366: 689–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blander JM, Medzhitov R (2004) Regulation of phagosome maturation by signals from toll-like receptors. Science 304: 1014–1018. [DOI] [PubMed] [Google Scholar]
- 6. Scott C, Botelho R, Grinstein S (2003) Phagosome maturation: a few bugs in the system. The journal of membrane biology 193: 137–152. [DOI] [PubMed] [Google Scholar]
- 7. Duclos S, Desjardins M (2000) Subversion of a young phagosome: the survival strategies of intracellular pathogens. Cellular microbiology 2: 365–377. [DOI] [PubMed] [Google Scholar]
- 8. Huynh KK, Eskelinen E-L, Scott CC, Malevanets A, Saftig P, et al. (2007) LAMP proteins are required for fusion of lysosomes with phagosomes. The EMBO journal 26: 313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murray RZ, Kay JG, Sangermani DG, Stow JL (2005) A role for the phagosome in cytokine secretion. Science 310: 1492–1495. [DOI] [PubMed] [Google Scholar]
- 10. Nishimoto N, Kishimoto T (2004) Inhibition of IL-6 for the treatment of inflammatory diseases. Current opinion in pharmacology 4: 386–391. [DOI] [PubMed] [Google Scholar]
- 11.Beutler B (1999) The role of tumor necrosis factor in health and disease. The journal of rheumatology. Supplement 57: 16–21. [PubMed]
- 12. Gresham HD, Dale BM, Potter JW, Chang PW, Vines CM, et al. (2000) Negative regulation of phagocytosis in murine macrophages by the Src kinase family member, Fgr. The journal of experimental medicine 191: 515–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Uraki R, Sakudo A, Ando S, Kitani H, Onodera T (2010) Enhancement of phagocytotic activity by prion protein in PrP-deficient macrophage cells. International journal of molecular medicine 26: 527–532. [DOI] [PubMed] [Google Scholar]
- 14. de Almeida CJ, Chiarini LB, da Silva JP, e Silva PM, Martins MA, et al. (2005) The cellular prion protein modulates phagocytosis and inflammatory response. Journal of leukocyte biology 77: 238–246. [DOI] [PubMed] [Google Scholar]
- 15. Ding T, Zhou X, Kouadir M, Shi F, Yang Y, et al. (2013) Cellular prion protein participates in the regulation of inflammatory response and apoptosis in BV2 microglia during infection with Mycobacterium bovis. Journal of molecular neuroscience 51: 1–9. [DOI] [PubMed] [Google Scholar]
- 16. Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, et al. (2008) Physiology of the prion protein. Physiological reviews 88: 673–728. [DOI] [PubMed] [Google Scholar]
- 17. Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, et al. (1993) Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences 90: 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westergard L, Christensen HM, Harris DA (2007) The cellular prion protein (PrPC): Its physiological function and role in disease. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease 1772: 629–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, et al. (2002) Cellular prion protein transduces neuroprotective signals. The EMBO journal 21: 3317–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, et al. (2010) Axonal prion protein is required for peripheral myelin maintenance. Nature neuroscience 13: 310–318. [DOI] [PubMed] [Google Scholar]
- 21. Zafar S, von Ahsen N, Oellerich M, Zerr I, Schulz-Schaeffer WJ, et al. (2011) Proteomics approach to identify the interacting partners of cellular prion protein and characterization of Rab7a interaction in neuronal cells. Journal of proteome research 10: 3123–3135. [DOI] [PubMed] [Google Scholar]
- 22. Rossi D, Cozzio A, Flechsig E, Klein MA, Rülicke T, et al. (2001) Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. The EMBO journal 20: 694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marim FM, Silveira TN, Lima Jr DS, Zamboni DS (2010) A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PLoS One 5: e15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wong ML, Medrano JF (2005) Real-time PCR for mRNA quantitation. Biotechniques 39: 75–85. [DOI] [PubMed] [Google Scholar]
- 25. Adams DO, Hamilton TA (1984) The cell biology of macrophage activation. Annual review of immunology 2: 283–318. [DOI] [PubMed] [Google Scholar]
- 26. Hume DA, Ross IL, Himes SR, Sasmono RT, Wells CA, et al. (2002) The mononuclear phagocyte system revisited. Journal of leukocyte biology 72: 621–627. [PubMed] [Google Scholar]
- 27. Nguyen MD, Julien J-P, Rivest S (2002) Innate immunity: the missing link in neuroprotection and neurodegeneration? Nature Reviews Neuroscience 3: 216–227. [DOI] [PubMed] [Google Scholar]
- 28. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318. [DOI] [PubMed] [Google Scholar]
- 29. Colton CA (2009) Heterogeneity of microglial activation in the innate immune response in the brain. Journal of neuroimmune pharmacology 4: 399–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi F, Yang L, Kouadir M, Yang Y, Ding T, et al. (2013) Prion protein participates in the regulation of classical and alternative activation of BV2 microglia. Journal of neurochemistry 124: 168–174. [DOI] [PubMed] [Google Scholar]
- 31. Zhou X, Xu G, Zhao D (2008) In vitro effect of prion peptide PrP 106–126 on mouse macrophages: Possible role of macrophages in transport and proliferation for prion protein. Microbial pathogenesis 44: 129–134. [DOI] [PubMed] [Google Scholar]
- 32. Watarai M, Kim S, Erdenebaatar J, Makino S, Horiuchi M, et al. (2003) Cellular prion protein promotes Brucella infection into macrophages. The journal of experimental medicine 198: 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nitta K, Sakudo A, Masuyama J, Xue G, Sugiura K, et al. (2009) Role of cellular prion proteins in the function of macrophages and dendritic cells. Protein and peptide letters 16: 239–246. [DOI] [PubMed] [Google Scholar]
- 34. Helaine S, Thompson JA, Watson KG, Liu M, Boyle C, et al. (2010) Dynamics of intracellular bacterial replication at the single cell level. Proceedings of the National Academy of Sciences of the United States of America 107: 3746–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baeuerle PA, Henkel T (1994) Function and activation of NF-kappaB in the immune system. Annual review of immunology 12: 141–179. [DOI] [PubMed] [Google Scholar]
- 36. Tak PP, Firestein GS (2001) NF-κB: a key role in inflammatory diseases. Journal of clinical investigation 107: 7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pahl HL (1999) Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18: 6853–6866. [DOI] [PubMed] [Google Scholar]
- 38. Lu Y, Liu A, Zhou X, Kouadir M, Zhao W, et al. (2012) Prion peptide PrP106–126 induces inducible nitric oxide synthase and proinflammatory cytokine gene expression through the activation of NF-κB in macrophage cells. DNA and cell biology 31: 833–838. [DOI] [PubMed] [Google Scholar]
- 39. Saito F, Kuwata H, Oiki E, Koike M, Uchiyama Y, et al. (2008) Inefficient phagosome maturation in infant macrophages. Biochemical and biophysical research communications 375: 113–118. [DOI] [PubMed] [Google Scholar]
- 40. Bhattacharya M, Ojha N, Solanki S, Mukhopadhyay CK, Madan R, et al. (2006) IL-6 and IL-12 specifically regulate the expression of Rab5 and Rab7 via distinct signaling pathways. The EMBO journal 25: 2878–2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. MacMicking JD, Taylor GA, McKinney JD (2003) Immune control of tuberculosis by IFN-gamma-inducible LRG-47. Science 302: 654–659. [DOI] [PubMed] [Google Scholar]
- 42. Nuvolone M, Kana V, Hutter G, Sakata D, Mortin-Toth SM, et al. (2013) SIRPα polymorphisms, but not the prion protein, control phagocytosis of apoptotic cells. The journal of experimental medicine 210: 2539–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Matozaki T, Murata Y, Okazawa H, Ohnishi H (2009) Functions and molecular mechanisms of the CD47–SIRPα signalling pathway. Trends in cell biology 19: 72–80. [DOI] [PubMed] [Google Scholar]
- 44. Kinchen JM, Ravichandran KS (2008) Phagocytic signaling: you can touch, but you can’t eat. Current biology 18: R521–R524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Barclay AN (2009) Signal regulatory protein alpha (SIRPα)/CD47 interaction and function. Current opinion in immunology 21: 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Braun V, Fraisier V, Raposo G, Hurbain I, Sibarita J-B, et al. (2004) TI-VAMP/VAMP7 is required for optimal phagocytosis of opsonised particles in macrophages. The EMBO journal 23: 4166–4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Doyle SE, O’Connell RM, Miranda GA, Vaidya SA, Chow EK, et al. (2004) Toll-like receptors induce a phagocytic gene program through p38. The journal of experimental medicine 199: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vinet AF, Fukuda M, Descoteaux A (2008) The exocytosis regulator synaptotagmin V controls phagocytosis in macrophages. The journal of immunology 181: 5289–5295. [DOI] [PubMed] [Google Scholar]
- 49. Duque GA, Fukuda M, Descoteaux A (2013) Synaptotagmin XI regulates phagocytosis and cytokine secretion in macrophages. The journal of immunology 190: 1737–1745. [DOI] [PubMed] [Google Scholar]
- 50. Rossi AG, McCutcheon JC, Roy N, Chilvers ER, Haslett C, et al. (1998) Regulation of macrophage phagocytosis of apoptotic cells by cAMP. The journal of immunology 160: 3562–3568. [PubMed] [Google Scholar]