

Abstract
The pregnane X receptor (PXR) was isolated as a xenosensor regulating xenobiotic responses. In this study, we show that PXR plays an endobiotic role by impacting lipid homeostasis. Expression of an activated PXR in the livers of transgenic mice resulted in an increased hepatic deposit of triglycerides. This PXR-mediated lipid accumulation was independent of the activation of the lipogenic transcriptional factor SREBP-1c (sterol regulatory element-binding protein 1c) and its primary lipogenic target enzymes, including fatty-acid synthase (FAS) and acetyl-CoA carboxylase 1 (ACC-1). Instead, the lipid accumulation in transgenic mice was associated with an increased expression of the free fatty acid transporter CD36 and several accessory lipogenic enzymes, such as stearoyl-CoA desaturase-1 (SCD-1) and long chain free fatty acid elongase. Studies using transgenic and knock-out mice showed that PXR is both necessary and sufficient for Cd36 activation. Promoter analyses revealed a DR-3-type of PXR-response element in the mouse Cd36 promoter, establishing Cd36 as a direct transcriptional target of PXR. The hepatic lipid accumulation and Cd36 induction were also seen in the hPXR “humanized” mice treated with the hPXR agonist rifampicin. The activation of PXR was also associated with an inhibition of pro-β-oxidative genes, such as peroxisome proliferator-activated receptor α (PPARα) and thiolase, and an up-regulation of PPARγ, a positive regulator of CD36. The cross-regulation of CD36 by PXR and PPARγ suggests that this fatty acid transporter may function as a common target of orphan nuclear receptors in their regulation of lipid homeostasis.
Lipid homeostasis is accomplished by complex physiological mechanisms. Disruptions of lipid formation and catabolism, including those controlled by the liver, have been implicated in various cardiovascular and metabolic diseases, such as atherosclerosis, obesity, and diabetes.
Hepatic lipid homeostasis is tightly maintained by balanced lipid formation (lipogenesis), catabolism (β-oxidation), and secretion. Lipogenic enzymes, including FAS,5 ACC-1, SCD-1, and FAE, are highly expressed in the liver. CD36, a free fatty acid transporter, is also important for lipogenesis because it is responsible for the high affinity uptake of fatty acids (1–3).
β-Oxidation is an important step of fatty acid catabolism. The enzymes responsible for β-oxidation include the orphan receptor PPARα and its target genes, such as the acyl-CoA oxidase, bifunctional enzymes, and 3-ketoacyl-CoA thiolase (thiolase) (4, 5).
Very low density lipoprotein (VLDL) plays an important role in hepatic lipid secretion. Cholesteryl ester is a major component of VLDL, and its formation from cholesterol is catalyzed by the acyl-coenzyme A:cholesterol acyltransferase (ACAT) (6). Once formed, cholesteryl esters are secreted into the bloodstream as part of the VLDL. A defect in the assembly of VLDL has been proposed to be responsible for the induction of fatty liver (7). ACAT2 is the major cholesterol-esterifying enzyme in mouse enterocytes and hepatocytes. An ACAT2 deficiency has been shown to lead to a significant decrease in the percentage of cholesteryl ester in plasma VLDL (8).
Several orphan nuclear receptors have been implicated in lipid homeostasis, consistent with the notion that orphan receptors are essential regulators in the expression of metabolic gene products (9, 10). Liver X receptors (LXRs), including the α and β isoforms, have been shown to promote lipogenesis (11–13). LXR activation increases plasma triglyceride levels through the transcriptional activation of SREBP-1c, a transcriptional factor known to regulate the expression of a battery of lipogenic enzymes that include SCD-1, ACC, and FAS (14–17). Genetic and pharmacological studies have revealed distinct roles for PPARs in lipid metabolism. PPARα enhances the metabolic usage of fatty acids by inducing enzymes involved in β-oxidation (4, 18). Indeed, PPARα-selective agonists, such as fibrates, have been widely used to treat hyper-lipidemia by increasing fatty acid oxidation and improving plasma lipid profiles. PPARγ serves as an essential regulator of adipocyte differentiation and promotes lipid storage in mature adipocytes (19–25). CD36, a receptor for the oxidized low density lipoprotein and long chain fatty acids, has been shown to be a PPARγ target gene (26). PPARδ is also involved in fat burning and obesity (27). PPARδ agonists were shown to lower plasma triglyceride levels in obese monkey models (28). More recently, expression of an activated form of PPARδ in the adipose tissues of transgenic mice was shown to activate fat metabolism and produce lean mice that are resistant to obesity induced either genetically or by a high fat diet (27). In contrast, PPARδ null mice are prone to high fat-induced obesity (27).
PXR was originally isolated as a xenobiotic receptor that regulates the expression of the drug-metabolizing cytochrome P450 3A (CYP3A) isozymes (29–33). Accumulating evidence has pointed to a role for PXR as a master regulator of mammalian xenobiotic response by controlling the expression of phase I and phase II enzymes, as well as drug transporters. This regulation is achieved through the binding of PXR to its response elements found within the promoters of target genes (34–36). Because many of the PXR target enzymes and transporters are responsible for the biotransformation and elimination of endogenous and exogenous chemicals, PXR-mediated gene regulation has also been implicated in the homeostasis of both xenobiotics, such as drugs and toxins, and endobiotics, such as bile acids, bilirubin, and steroid hormones (34, 37–40). PXR is highly expressed in the liver, a major organ for lipogenesis, fatty acid β-oxidation, and lipid secretion. Whether or not PXR-mediated gene regulation plays a role in lipid homeostasis is not known.
In this study, we show that PXR plays an important role in lipid homeostasis by activating genes that facilitate lipogenesis and suppressing the β-oxidative pathways. CD36 has been shown to be regulated by PPARγ (26). The PXR-mediated transcriptional regulation of Cd36 establishes this free fatty acid transporter as a shared target of orphan nuclear receptors in their regulation of lipid homeostasis.
MATERIALS AND METHODS
Animals and Treatment
The Alb-VP-hPXR transgenic (33), PXR null (33), and FABP-VP-hPXR transgenic mice (41) have been described previously. The FABP-hPXR transgenic mice were created by W. Xie at the Salk Institute Transgenic Core Facility when he was working in the laboratory of Dr. Ronald M. Evans. The cloning of the FABP promoter has been described previously (41). The wild type hPXR cDNA was used as we described previously (33). The transgene in the mice is estimated to be five copies based on Southern blot analysis. The FABP-hPXR transgene was subsequently bred onto the PXR-null background, and the resultant “humanized” mice have a mixed background of CB6F1 and C57BL/6J. Mice were housed in a pathogen-free animal facility under a standard 12-h light/dark cycle with free access to water and food. Age-matched 8–10 week-old mice were used for all experiments. For the RIF treatment, 8 week-old female mice received a daily gavage of RIF (10 mg/kg) for 5 weeks before being sacrificed. The use of mice in this study has complied with all relevant federal guidelines and institutional policies.
Measurement of Circulating and Tissue Lipid Levels
To measure circulating lipid levels, mice were fasted for 16 h prior to sacrificing and blood collection. Blood samples were collected into Vacutainer hematology tubes containing 3% EDTA (BD Biosciences). The plasma levels of triglycerides and cholesterol were measured using assay kits from Stanbio Laboratory (Boerne, TX). To measure liver lipid content, tissues were homogenized in 2 ml of buffer that contains 18 mM Tris, pH 7.5, 300 mM mannitol, 50 mM EGTA, and 0.1 mM phenylmethysulfonyl fluoride. 400 μl of homogenate was mixed with 4 ml of chloroform/methanol (2:1) and incubated overnight at room temperature with occasional shaking. 800 μl of H2O was then added, vortexed, and centrifuged for 5 min at 3,000 × g, and the lower lipid phase was collected and dried under nitrogen gas (42). The lipid pellets were then dissolved in a mixture of 60 μl of tert-butyl alcohol and 40 μl of Triton X-114/methanol (2:1) mixture. Triglyceride and cholesterol levels were then measured using the Stanbio assay kits.
Northern Blot Analysis and Real Time PCR
Total RNA was prepared from liver tissue using TRIzol reagent (Invitrogen). Northern blot analysis using radiolabeled cDNA probes was performed as described previously (33, 43, 44). The membranes were stripped and re-probed with Gapdh cDNA probe for loading control. Real time PCR using pre-designed Assay-On-Demand TaqMan reagents was performed with the ABI 7300 Real Time PCR System. The ABI assay codes are as follows: Pparα, Mm00440939m1; thiolase, Mm00624282 m1; Cyp51, Mm00490968_m1; and Dgat-1, Mm00515643_m1. The sequences of the CYBR-green probes are available upon request.
Plasmid Constructs and Transfection Assays
The 5′-regulatory sequences (−2116 bp to +53 bp) of the mouse Cd36 gene were cloned by PCR using the following pair of oligonucleotides: forward primer, 5′-CGGGGTACCATACATAAAAAGCAACCCAACTC-3′; reverse primer, 5′-TCCCCCGGGCTGTGAAGAAGAAAAAGTCCTC-3′. The design of oligonucleotides was based on sequences deposited in Gen-BankTM (accession number NM_007643). The mCd36 gene-containing bacterial artificial chromosome clone (clone identification number, RP23-226P20) from the Children’s Hospital Oakland Research Institute, BacPAK Resource Center (Oakland, CA), was used as the PCR template. The hCD36 promoter (−1961 bp to +57 bp) was cloned by PCR using the human placenta genomic DNA as the template and the following pair of oligonucleotides: forward primer, 5′-CGGGGTAC-CTCAAAATATGGTGGGTGCATAG-3′; reverse primer, 5′-TCC-CCCGGGTCTGGGTGATGGGAAAAATC-3′. The PCR-amplified sequences were cloned into the pGL3 vector (Promega). Site-directed mutagenesis was performed by the PCR overextension method and confirmed by DNA sequencing (33, 45). To generate the synthetic thymidine kinase (tk) reporter genes, three copies of the wild type or mutant DR-3 element were synthesized and inserted into the tk-Luc vector. Transient transfections were performed on HepG2 cells seeded into 48-well tissue culture plates using the polyethyleneimine polymer transfection agent as described previously (46). For each of the three wells, the plasmid-polyethyleneimine complexes were formed by incubating 0.4 μg of expression vector for each nuclear receptor and 0.8 μg of reporter gene and 0.5 μg of β-galactosidase plasmid with 10 μl of polyethyleneimine at room temperature for 10 min in a total volume of 300 μl of serum-free minimum Eagle’s medium. The complexes were then diluted with 300 μl of serum-free minimum Eagle’s medium and applied at 200 μl per well. After 12 h of incubation, the transfection medium was replaced with minimum Eagle’s medium that contained 10% fetal bovine serum laced with the appropriate solvent or drugs. Cells were lysed 24 h later and assayed for luciferase and β-galactosidase activities.
Electrophoretic Mobility Shift Assay (EMSA)
The receptor proteins were prepared using the TNT in vitro transcription and translation system (Promega). The binding reactions were as described previously (40, 43, 45). Protein-DNA complexes were resolved by electrophoresis through 8% polyacrylamide gel in 0.5×TBE at 4 °C for 3–4 h. The probe sequences are shown in the figures. 50× unlabeled competitor DNA was used for all of the competitions.
Chromatin Immunoprecipitation (ChIP) Assay
Three-week-old wild type female mice were pretreated with an intraperitoneal injection of Me2SO or PCN (40 mg/kg) for 30 min before being transfected with the pCMX-HA-mPXR or pCMX-HA control plasmid by a hydrodynamic and liver-specific gene delivery method (47). Five micrograms of plasmid were used for each mouse. The mice were sacrificed 8 h after transfection, and the liver tissues were harvested for ChIP assay. 50 mg of each tissue was minced and added with formaldehyde to a final concentration of 1%. After rocking at room temperature for 15 min, glycine was added to a final concentration of 0.125 M to stop the cross-linking. The tissues were collected by centrifuge and were homogenized by a Dounce homogenizer in cold phosphate-buffered saline. The resultant cell pellets were then resuspended in lysis buffer (5 mM PIPES, pH 8.0, 85 mM KCl, 0.5% Nonidet P-40) supplemented with proteinase inhibitor mixture. Nuclei pellets were collected by centrifugation at 5,000 rpm for 5 min at 4 °C and resuspended in 100 μl of nuclei lysis buffer (50 mM Tris-Cl, pH 8.1, 10 mM EDTA, 1% SDS) supplemented with proteinase inhibitor mixture. The ChIP procedures followed the Upstate protocol (catalog number 17-295). Nuclei were sonicated and precleared with protein A-agarose/salmon sperm DNA before immunoprecipitation with 2 μg of HA antibody (catalog number sc-805, Santa Cruz Biotechnology) or no antibody overnight at 4 °C. The precipitated complexes were collected with protein A-agarose/salmon sperm DNA and then washed and eluted in elution buffer. DNA in the precipitated samples was reverse cross-linked at 65 °C for 4 h, and the DNA was recovered by phenol/chloroform extraction and ethanol precipitation. PCR was carried out with mCd36-specific primers encompassing the DR-3 site. Cyp3a11-specific primers that encompass the PXR-responsive DR-3 site was used as the positive control. Gapdh was used as the loading control. The PCR conditions are as follows: 95 °C for 30 s; 55 °C for 30 s; and 72 °C for 40 s for 28 cycles. The primers used are as follows: mCd36/DR-3, 5′-CCCCTTCTATACTTTGTTTTCCATT-3′ and 5′-CTGA-AAGTCTTCAGGTTCATGCTA-3′; mCyp3A11/DR-3, 5′-ATGGGTA-GACCGTGACAAC-3′ and 5-GATCAAGCCAGTCGATGGATC-3′; and Gapdh, 5′-TACTAGCGGTTTTACGGGCG-3′ and 5′-TCGA-ACAGGAGGAGCAGAGAGCGA-3′.
Human and Mouse Primary Hepatocyte Preparation and Drug Treatment
Human livers were obtained through the liver tissue procurement and distribution system, and hepatocytes were isolated by three-step collagenase perfusion (48, 49). Mouse hepatocytes were also prepared by collagenase perfusion. Cells were plated on gelatin-coated T25 flask and maintained in hepatocyte maintenance medium (Cambrex BioScience, Walkersville, MA) supplemented with dexamethasone (0.1 mM), insulin (0.1 mM), gentamicin (50 μg/ml), and amphotericin (50 ng/ml) and incubated overnight. Cells were treated with appropriate drugs for 48 h prior to RNA harvest and Northern blot analysis.
RESULTS
Hepatic Lipid Accumulation in Transgenic Mice Expressing the Activated PXR
We have reported previously the creation of the Alb-VP-hPXR transgenic mice that express the activated hPXR (VP-hPXR) in the liver (33). Of note, although hPXR was used to create the transgenic mice, to our knowledge there is no species specificity in the PXR downstream target genes. Histological examination of adult wild type (Fig. 1A) and Alb-VP-hPXR transgenic mice (Fig. 1B) revealed remarkable differences. There was marked microvesicular steatosis that was most pronounced in zone 3 of the liver acinus (around the central veins). Microvesicular steatosis is characterized by the presence of many small fat droplets within the affected cell, and the nucleus of the involved hepatocyte typically remains centrally located. The lipid nature of the steatosis was confirmed by Oil-Red O staining (compare Fig. 1, C to D). The Alb-VP-hPXR mice also had hepatomegaly (Fig. 1E). By 2.5 months, the transgenic mice had an increase of 79% in liver weight when measured as a percentage of total body weight (Fig. 1G), whereas the size of the kidney remained unchanged (Fig. 1, F and G). The transgenic mice also had smaller body sizes and lower body weights throughout the post-natal development. Fig. 1H shows the growth curve of transgenic males as compared with age- and litter size-matched wild type males. The growth retardation of VP-hPXR mice was most apparent at 4–5 weeks of age, with a decrease of about 20% in body weight as compared with the wild type mice. This percentage decreased to about 10% by 8–9 weeks of age and persisted thereafter. A similar pattern of growth retardation was also observed in female transgenic mice (data not shown). The mechanism for this growth retardation is currently unknown.
FIGURE 1. Hepatic lipid accumulation in the Alb-VP-hPXR transgenic mice expressing the activated PXR.

A and B, liver paraffin sections of the wild type (A) and transgenic (B) mice were stained with hematoxylin and eosin. C and D, liver frozen sections of the wild type (C) and transgenic (D) mice were stained with Oil-Red O. CV, central vein. Magnification for all histology panels is ×200. E and F, enlarged liver (E) but not kidney (F) in the transgenic mice. G, the liver and kidney weights measured as percentages of the total body weight. H, growth retardation in the transgenic mice. Male transgenic (TG) mice and their wild type littermates were weaned and weighed at day 22 after birth and weighed every 3 days thereafter. The results are presented as the averages and S.D.
The liver tissue and circulating levels of lipids in the Alb-VP-hPXR transgenic mice were measured in order to gain insight into the effect of PXR on lipid homeostasis. The triglyceride content in the livers of the transgenic mice was nearly 20 times that in the wild type mice (Fig. 2A). In contrast, no significant changes in the hepatic cholesterol levels were seen in the transgenic mice (Fig. 2B). Despite the hepatic triglyceride accumulation, the circulating levels of both triglycerides (Fig. 2C) and cholesterol (Fig. 2D) were unaffected in the transgenic mice. The fasting levels of circulating free fatty acids were modestly lower in transgenic mice than those of the wild type mice, but the difference was not statistically significant (Fig. 2E).
FIGURE 2. Hepatic and circulating lipid levels in the Alb-VP-hPXR transgenic mice.

A–D, the liver (A and B) and plasma (C and D) levels of triglyceride (A and C) and cholesterol (B and D) from the wild type and transgenic (TG) mice were measured. E, the plasma levels of free fatty acids in the wild type and transgenic mice. Results represent the averages and S.D. from the indicated number of mice per group. FFA, free fatty acid. *, p < 0.01. Results are derived from males and they are repeatable in females (data not shown).
Effects of PXR Activation on the Expression of Genes That Impact Lipid Metabolism
The hepatic steatosis prompted us to examine the effect of PXR activation on the expression of a battery of genes whose products are known to affect lipid homeostasis. The hepatic lipid homeostasis is maintained by balanced lipid formation (lipogenesis), catabolism (β-oxidation), and secretion. Northern blot analyses showed that the expression of SREBP-1c, a major regulator in the de novo hepatic lipid biosynthesis, was unchanged (Fig. 3A), suggesting that the lipid accumulation in the transgenic mice is SREBP-independent. Consistent with the lack of SREBP-1c activation, the mRNA levels of Fas and Acc-1, two important SREBP-1c target lipogenic enzymes, were unaffected. Interestingly, the expression of SCD-1 and FAE, two downstream lipogenic enzymes that have also been shown to be regulated by SREBP-1c (50), was increased in the transgenic mice (Fig. 3A). Northern blot analyses also revealed an increased expression of CD36, a fatty acid translocase that is responsible for the high affinity uptake of fatty acids. PPARγ, an orphan nuclear receptor known to play a role in the positive regulation of Cd36, was also induced (Fig. 3A). Cyp3a11, a known PXR target gene, is induced as expected (Fig. 3A).
FIGURE 3. Effect of PXR activation on the expression of genes that impact lipid metabolism.

A, Northern blot analyses on the hepatic expression of genes that are involved in lipogenesis in the wild type and Alb-VP-hPXR transgenic mice. CYP3A11 was included as the positive control for PXR activation. B, the hepatic but not intestinal expression of CD36 was induced in the FABP-VP-hPXR transgenic (TG) mice. C, real time PCR analyses on the hepatic expression of PPARα, thiolase, diglycerol acyltransferase, and CYP51. D, Northern blot analyses on the hepatic expression of genes that are involved in gluconeogenesis. E, the hepatic and intestinal expression of ACAT2 was unaffected in the FABP-VP-hPXR transgenic mice. Lanes in A and D represent pooled samples from six mice. Lanes in B represent individual mice. *, p < 0.01.
We have recently created transgenic mice with targeted expression of VP-hPXR in both the liver and intestine (41). By using this transgenic model, we found that the PXR effect on the CD36 expression is tissue-specific, as hepatic, but not intestinal, expression of CD36 was induced in the transgenic mice (Fig. 3B). CYP3A11, in contrast, was induced in both tissues as expected, suggesting that the tissue specificity of the transgenic effect is gene-specific.
The expression of genes implicated in β-oxidation was profiled by real time PCR. As shown in Fig. 3C, the expression of PPARα and thiolase was down-regulated by the transgene. The down-regulation of PPARα and thiolase suggests that the inhibition of β-oxidation may have contributed to the lipid accumulation phenotype.
Real time PCR also showed that the expression of diglycerol acyltransferase, which catalyzes the final step in triglyceride biosynthesis (51), and sterol 14-demethylase P450 (CYP51), a crucial enzyme in the cholesterol biosynthesis pathway (52), was unchanged in the transgenic mice (Fig. 3C). The lack of CYP51 induction is consistent with the normal tissue and circulating levels of cholesterol (Fig. 2, B and D).
The expression of enzymes responsible for gluconeogenesis was evaluated by Northern blot analysis. Among the three major gluconeogenic enzymes, the expression of the phosphoenolpyruvate carboxykinase and glucose-6-phosphatase was reduced, but the mRNA level of fructose 1,6-bisphosphate 1 was unchanged (Fig. 3D). The implication of phosphoenolpyruvate carboxykinase and glucose-6-phosphatase down-regulation in the lipid phenotypic exhibition is currently unknown.
Defects in lipid secretion have been shown to contribute to fatty liver formation. The liver- and intestine-specific ACAT2 is known to promote lipid secretion by catalyzing cholesterol esterification and the subsequent formation of VLDL, the major lipoprotein that facilitates hepatic lipid secretion (53). We found that the expression of ACAT2 was not affected by the transgene in either the liver or the intestine (Fig. 3E).
PXR Is Necessary for the Pharmacological Regulation of CD36, SCD-1, FAE, and PPARγ in Hepatocytes
To examine the effects of a pharmacological activation of PXR on the regulation of lipogenic enzymes, primary hepatocytes were prepared from the wild type and PXR null mice. The cells were mock-treated or treated with the PXR ligand PCN and evaluated for gene expression by Northern blot analysis. Consistent with the observations in transgenic mice, PCN treatment of the wild type hepatocytes induced the mRNA expression of CD36, SCD-1, FAE, and PPARγ (Fig. 4A). This induction was abolished in hepatocytes derived from the PXR null mice (Fig. 4B). The Northern blot results were confirmed by real time PCR analysis on hepatocytes prepared from independent groups of mice (Fig. 4C). Interestingly, the real time PCR results showed that the basal expression of CD36, SCD-1, FAE, and PPARγ was increased in the vehicle-treated PXR null mice (Fig. 4C).
FIGURE 4. PXR is necessary for the pharmacological regulation of Cd36, Scd-1, Fae, and Pparγin the hepatocytes.
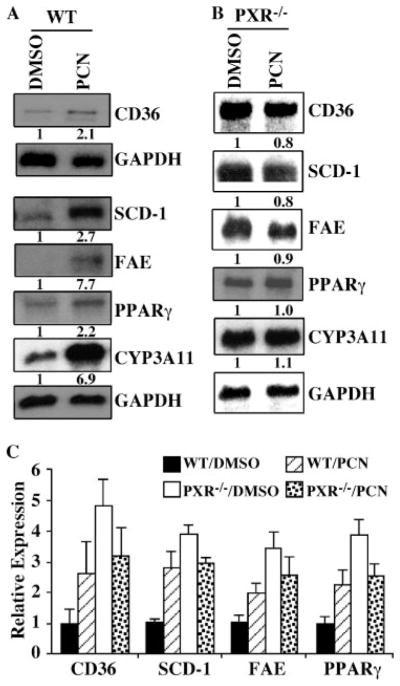
A and B, Northern blot analyses on RNA derived from the vehicle- or PCN-treated hepatocytes from the wild type (A) or PXR null (B) mice. Note the RNA samples in (A and B) are derived from a single mouse, and the Northern blotting and hybridization were performed independently for these two genotypes. The fold inductions versus the respective Me2SO (DMSO) controls are labeled. C, simultaneous real time PCR analysis on RNA derived from the wild type (n = 4) and PXR null (n = 4) hepatocytes. The concentration of ligand is 10 μM.
Cd36 Is a Direct Transcriptional Target of PXR
Having demonstrated that PXR is necessary and sufficient for mCd36 regulation in the hepatocytes, we proceeded to determine whether or not mCd36 is a direct transcriptional target of PXR. Inspection of the mCd36 promoter revealed a DR-3-type (direct repeat spaced by three nucleotides) of nuclear receptor-response element (AGGTCAtaaAGTGCA) (Fig. 5A), similar to the PXR-response element found in the rat Cyp3a23 gene (29, 30). EMSA showed that both hPXR/RXR and VP-hPXR/RXR heterodimers can bind to this DR-3 (Fig. 5B). The binding of mCd36/DR-3 by hPXR and VP-hPXR was specific, as evidenced by the efficient competition of binding by excess unlabeled wild type Cd36/DR-3 or Cyp3a23/DR-3 but not by the mutant-binding site with the mCd36/DR-3 disrupted (Fig. 5B). The binding of mPXR/RXR heterodimers to the mCd36/DR-3 is shown in Fig. 5C. ChIP assay was used to demonstrate the recruitment of mPXR onto the mCd36 promoter. In this experiment, the HA-tagged mPXR or the HA vector control plasmid was transfected into the livers of the wild type mice by a hydrodynamic gene delivery method (47). Mice were treated with Me2SO or PCN (40 mg/kg) for 8 h before sacrificing. ChIP assay was performed with the use of an anti-HA antibody. As shown in Fig. 5D, treatment with PCN resulted in the recruitment of HA-mPXR onto the mCd36 promoter. ChIP on the Cyp3a11 promoter was included as the positive control.
FIGURE 5. Cd36 is a direct transcriptional target of PXR.

A, the partial DNA sequences of the mouse Cd36 gene promoter. The DR-3 element is capitalized and labeled. The mutant variant is shown with the mutated nucleotides (nt) underlined. B, hPXR/RXR or VP-hPXR/RXR heterodimers bound to mCd36/DR-3 as revealed by EMSA. Arrows indicate specific shift bands. C, binding of the mPXR/RXR heterodimers to mCd36/DR-3 as revealed by EMSA. The Cyp3a11/DR-3 binding was included as the positive control. D, ChIP analysis to show the recruitment of mPXR onto the mCD36 promoter. HA-tagged mPXR or the HA vector control was used to transfect wild type mouse livers by a hydrodynamic gene delivery method. Mice were treated with Me2SO (DMSO) or PCN for 8 h before sacrificing. ChIP assay was performed with the use of an anti-HA antibody (Ab). Each lane represents one mouse. E, hPXR and VP-hPXR activate the synthetic thymidine kinase (tk) report containing the wild type but not the mutant mCd36/DR-3 element in transient transfections and luciferase reporter gene assays. The averages of the reporter alone transfections were arbitrarily set as 1 (data not shown). F, mPXR activates the natural mCd36 promoter (−2116 bp to +53 bp), and hPXR activates the hCD36 promoter (−1961 bp to +57 bp) reporter genes in a ligand-dependent manner. Results shown in E and F are fold induction over the reporter alone transfections (set as 1) and represent the averages and S.E. from triplicate assays. The concentration of ligand is 10 μM.
Transient transfections and reporter gene assays were used to determine whether mCd36/DR-3 is necessary and sufficient in mediating PXR transactivation. For this purpose, heterologous tk-luciferase reporter genes that contain three copies of the wild type or mutant mCd36/DR-3 element were generated and tested for PXR transactivation in human hepatoma HepG2 cells. As shown in Fig. 5E, the synthetic tk reporter gene was activated by hPXR in the presence of its agonist RIF and by VP-PXR in the absence of an agonist. This activation was abolished when the DR-3 element was mutated (Fig. 5E). A similar pattern of ligand-dependent activation of mPXR activation was seen when a reporter gene that contains the natural promoter of mCd36 (−2116 to +53 bp) was used (Fig. 5F). The DR-3 element was also necessary in the context of this natural promoter, as the disruption of DR-3 abolished the mPXR activation (Fig. 5F). Interestingly, the hCD36 promoter (−1961 bp to +57 bp) was also transactivated by hPXR in a ligand-dependent manner (Fig. 5F), although our promoter analysis failed to identify a conserved DR-3 site in the hCD36 promoter (data not shown).
Hepatic Lipid Accumulation and CD36 Induction in Rifampicin-treated hPXR “Humanized” Mice
The PXR ligand effect on hepatic lipogenesis and CD36 regulation was confirmed in the newly created humanized mice in which the FABP-hPXR transgene was introduced into the PXR null background. Compared with the previously described Alb-hPXR humanized mice (33), the FABP-hPXR transgene targets the expression of hPXR to both the liver and intestine (Fig. 6A), under the control of the rat Fabp promoter (41). In this experiment, the FABP-hPXR humanized mice were mock-treated or treated with the hPXR agonist RIF (10 mg/kg) for 5 weeks before sacrificing and analysis. As shown in Fig. 6B, liver sections from the RIF-treated mice showed more Oil-Red O staining compared with their vehicle-treated counterparts. Similar to the lipid accumulation that we have observed in the Alb-VP-hPXR mice, the liver accumulation of triglycerides (Fig. 6C), but not cholesterol (Fig. 6D), was observed in the RIF-treated mice. The triglyceride accumulation was significant, although the magnitude of increase was less than what was observed in the Alb-VP-hPXR mice. RIF treatment also resulted in a modest but nonsignificant decrease in the circulating levels of free fatty acid (Fig. 6E). Consistent with the lipid accumulation phenotype, the RIF treatment resulted in an increased hepatic expression of CD36, SCD-1, FAE, and PPARγ but not SREBP-1c (Fig. 6F). These results are consistent with an anecdotal clinical report that the liver steatosis in tuberculosis patients appeared to be associated with the RIF treatment (54).
FIGURE 6. Hepatic lipid accumulation and Cd36 induction in rifampicin-treated hPXR humanized mice.

A, creation of the FABP-hPXR transgenic mice. The FABP-hPXR transgene construct is depicted, and the expression of the transgene in the liver and intestinal tracts was confirmed by Northern blot analysis. B, Oil-Red O staining of liver sections of the humanized mice that have been treated with the vehicle or RIF for 5 weeks. C–E, the liver levels of triglyceride (C) and cholesterol (D) and plasma levels of free fatty acids (E ) from the vehicle- and RIF-treated female humanized mice. F, real time PCR analysis on liver RNA derived from the vehicle- and RIF-treated mice. C–F, n = 3 for the vehicle group and n = 5 for the RIF group. Results represent the averages and S.D. RIF, rifampicin; FFA, free fatty acid. *, p < 0.05. Results are derived from females and they are repeatable in males (data not shown).
DISCUSSION
In this study, we show that the genetic (using the VP-hPXR trans-gene) or pharmacological (using the PXR agonist) activation of PXR caused hepatic steatosis, which is characterized by a marked accumulation of hepatic triglycerides. This PXR-mediated lipid accumulation likely resulted from a combined effect of increased hepatic free fatty acid uptake, lipogenesis, and suppression of β-oxidation.
The PXR-mediated lipogenesis is independent of SREBP-1c, which is distinct from that mediated by LXR (16, 17). Although the expression of SREBP-1c and its target gene FAS and ACC-1 was unaffected, SCD-1 and FAE were induced in transgenic mice and in PCN-treated hepatocytes in a PXR-dependent manner. It remains to be determined whether SCD-1 and FAE are direct transcriptional targets of PXR. Nevertheless, the lack of SREBP-1c activation in the transgenic mice suggests that PXR can mediate the SREBP-1c-independent SCD-1 and FAE activation, which may have contributed to the steatosis phenotype.
The induction of mCd36 in the PXR transgenic mice is intriguing. CD36 is a receptor for oxidized low density lipoprotein and long chain fatty acids. It is abundantly expressed in peripheral tissues and is involved in the high affinity uptake of fatty acids (1, 2, 55, 56). Our promoter analysis results have provided evidence to support mCd36 as a direct transcriptional target of PXR. Genetic and pharmacological activation of PXR was sufficient to induce mCd36 expression. The ligand-inducible expression of mCd36 was abolished in the absence of PXR. The activation of the hCD36 promoter by hPXR suggests that this regulatory pathway may be conserved in the humans, although a PXR-response element in the hCD36 promoter has yet to be identified. In addition to the role of PXR, the induction of PPARγ may have also contributed to the CD36 activation because Cd36 has been shown to be a PPARγ target gene (26). The shared regulation of CD36 by PXR and PPARγ suggests that CD36 may be the common target of orphan nuclear receptors in their regulation of the lipid metabolism.
The increased expression of PPARγ may have also contributed to the hepatic lipid accumulation in the transgenic mice. Overexpression of PPARγ1 in the PPARα null mice has been reported to induce the expression of adipocyte-specific genes and lipogenesis-related genes (24). The mechanism of PPARγ up-regulation in the VP-hPXR transgenic mice remains to be determined. A 2.7-kb Pparγ1 promoter (19) failed to be activated by either PXR or VP-PXR in transient transfections and reporter gene assays (data not shown). The role of PPARγ in PXR-mediated hepatic lipogenesis can be evaluated with the use of PPARγ small interfering RNA or the liver-specific PPARγ null mice (25). Nevertheless, the induction of PPARγ in the VP-hPXR mice suggests a potential cross-talk between these two receptors in mediating hepatic steatosis.
The implications of the transgene-induced down-regulation of glu-coneogenic phosphoenolpyruvate carboxykinase and glucose-6-phospha-tase enzyme expression in the lipid phenotypes have yet to be determined. However, these observations are consistent with a recent report that activation of constitutive androstane receptor and PXR down-regulated the transcriptional activity of FOXO1, a positive regulator of the gluconeogenic enzymes (57).
We recognize that many of the phenotypes, including CD36 regulation and triglyceride accumulation, are more dramatic in the VP-hPXR transgenic mice compared with the ligand-treated wild type hPXR transgenic mice. We consider the use of the VP-fusion receptor transgene to have unique advantages over drug treatment, because we know that treatments with receptor pan-agonists, such as bile acids, may affect multiple receptors depending upon the tissue context (37, 38). Moreover, several lines of evidence suggest that ligand treatment may have additional transcriptional consequences independent of the presence of endogenous xenobiotic receptor. Bypassing the requirement of ligand treatment, the VP fusion of receptors provides a unique strategy not only to study the biological consequences of receptor activation but also to identify target genes. The utility and practicality of this strategy have been proven in our previous creation and characterization of the VP-hPXR and VP-CAR transgenic mice (33, 39, 40, 45). Even though the VP fusion receptor represents a unique tool to genetically dissect the gene regulation by PXR, we recognize that the level of PXR expression and/or activity in the VP-PXR mice may be substantially higher than the endogenous PXR activity in response to endogenous ligands in normal physiology. However, the limitation of this genetic model does not exclude the potential that pharmacological modulation of PXR activity may affect lipogenesis. The significance of the wild type receptor is supported by our use of the wild type PXR receptor in demonstrating the CD36 regulation as well as the PXR ligand effect on lipogenesis.
In summary, we have revealed an endobiotic role of PXR in lipid homeostasis, adding another level of complexity for the function of this orphan receptor. It remains to be determined whether this PXR-mediated and SREBP-independent lipogenesis plays a role in lipid-associated metabolite diseases, such as obesity and diabetes.
Acknowledgments
We thank Drs. Richard Lin and Harleen Ahuja for their help in the initial phase of this study.
Footnotes
This work was supported in part by National Institutes of Health Grants ES012479 and CA107011.
The abbreviations used are: FAS, fatty-acid synthase; ACAT, acyl-coenzyme A:cholesterol acyltransferase; ACC-1, acetyl-CoA-carboxylase 1; CYP, cytochrome P450; EMSA, electrophoretic mobility shift assay; FAE, long chain free fatty acid elongase; LXR, liver X receptor; PCN, pregnenolone-16α-carbonitrile; PPAR, peroxisome proliferator-activated receptor; PXR, pregnane X receptor; RXR, retinoic acid X receptor; SCD-1, stearoyl-CoA desaturase-1; SREBP, sterol regulatory element-binding protein; VLDL, very low density lipoprotein; FABP, fatty acid binding protein; HA, hemagglutinin; ChIP, chromatin immunoprecipitation; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; PIPES, 1,4-piperazinediethanesulfonic acid; h, human; RIF, rifampicin; h, human; m, mouse; VP, viral protein.
References
- 1.Abumrad NA, El-Maghrabi MR, Amri EZ, Lopez E, Grimaldi PA. J Biol Chem. 1993;268:17665–17668. [PubMed] [Google Scholar]
- 2.Abumrad N, Harmon C, Ibrahimi A. J Lipid Res. 1998;39:2309–2318. [PubMed] [Google Scholar]
- 3.Bonen A, Campbell SE, Benton CR, Chabowski A, Coort SL, Han XX, Koonen DP, Glatz JF, Luiken JJ. Proc Nutr Soc. 2004;63:245–249. doi: 10.1079/PNS2004331. [DOI] [PubMed] [Google Scholar]
- 4.Berger J, Moller DE. Annu Rev Med. 2002;53:409–435. doi: 10.1146/annurev.med.53.082901.104018. [DOI] [PubMed] [Google Scholar]
- 5.Barish GD, Evans RM. Trends Endocrinol Metab. 2004;15:158–165. doi: 10.1016/j.tem.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 6.Chang TY, Chang CCY, Cheng D. Annu Rev Biochem. 1997;66:613–638. doi: 10.1146/annurev.biochem.66.1.613. [DOI] [PubMed] [Google Scholar]
- 7.Nagayoshi A, Matsuki N, Saito H, Tsukamoto K, Kaneko K, Wakashima M, Kinoshita M, Yamanaka M, Teramoto T. J Biochem (Tokyo) 1995;117:787–793. doi: 10.1093/oxfordjournals.jbchem.a124777. [DOI] [PubMed] [Google Scholar]
- 8.Lee RG, Shah R, Sawyer JK, Hamilton RL, Parks JS, Rudel LL. J Lipid Res. 2005;46:1205–1212. doi: 10.1194/jlr.M500018-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 10.Evans RM, Barish GD, Wan YX. Nat Med. 2004;10:1–7. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- 11.Repa JJ, Mangelsdorf DJ. Nat Med. 2002;8:1243–1248. doi: 10.1038/nm1102-1243. [DOI] [PubMed] [Google Scholar]
- 12.Tontonoz P, Mangelsdorf DJ. Mol Endocrinol. 2003;17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 13.Barish GD, Evans RM. Cell. 2004;119:149–151. doi: 10.1016/j.cell.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 14.Kim JB, Spiegelman BM. Genes Dev. 1996;10:1096–1107. doi: 10.1101/gad.10.9.1096. [DOI] [PubMed] [Google Scholar]
- 15.Shimano H, Yahagi N, Amemiya-Kudo M, Hasty AH, Osuga J, Tamura Y, Shionoiri F, Iizuka Y, Ohashi K, Harada K, Gotoda T, Ishibashi S, Yamada N. J Biol Chem. 1999;274:35832–35839. doi: 10.1074/jbc.274.50.35832. [DOI] [PubMed] [Google Scholar]
- 16.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, Li L, Schwendner S, Wang S, Thoolen M, Mangelsdorf DJ, Lustig KD, Shan B. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. J Clin Investig. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y, Qi C, Korenberg JR, Chen XN, Noya D, Rao MS, Reddy JK. Proc Natl Acad Sci U S A. 1995;92:7921–7925. doi: 10.1073/pnas.92.17.7921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM. Mol Cell. 1999;4:611–617. doi: 10.1016/s1097-2765(00)80211-7. [DOI] [PubMed] [Google Scholar]
- 21.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, Evans RM. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 22.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, Eto K, Tsubamoto Y, Okuno A, Murakami K, Sekihara H, Hasegawa G, Naito M, Toyoshima Y, Tanaka S, Shiota K, Kitamura T, Fujita T, Ezaki O, Aizawa S, Kadowaki T. Mol Cell. 1999;4:597–609. doi: 10.1016/s1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 23.Rosen ED, Spiegelman BM. J Biol Chem. 2001;276:37731–37734. doi: 10.1074/jbc.R100034200. [DOI] [PubMed] [Google Scholar]
- 24.Yu S, Matsusue K, Kashireddy P, Cao WQ, Yeldandi V, Yeldandi AV, Rao MS, Gonzalez FJ, Reddy JK. J Biol Chem. 2003;278:498–505. doi: 10.1074/jbc.M210062200. [DOI] [PubMed] [Google Scholar]
- 25.Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- 26.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 27.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 28.Oliver WR, Jr, Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM. Proc Natl Acad Sci U S A. 2001;98:5306–5311. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blumberg B, Sabbagh W, Juguilon H, Bolado J, Jr, Ong ES, Evans RM. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kliewer SA, Moore JT, Wade L, Staudinger JL, Jones MA, McKee DD, Oliver BM, Willson TM, Zetterstrom RH, Perlmann T, Lehmann J. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 31.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Backman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Proc Natl Acad Sci U S A. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. J Clin Investig. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xie W, Barwick JL, Downes M, Blumberg B, Simon CM, Nelson MC, Neuschwander-Tetri BA, Brunt EM, Guzelian PS, Evans RM. Nature. 2000;406:435–439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 34.Xie W, Uppal H, Saini SPS, Mu Y, Little JM, Radominska-Pandya A, Zemaitis MA. Drug Discov Today. 2004;9:442–449. doi: 10.1016/S1359-6446(04)03061-2. [DOI] [PubMed] [Google Scholar]
- 35.Sonoda J, Rosenfeld JM, Xu L, Evans RM, Xie W. Curr Drug Metab. 2003;4:59–72. doi: 10.2174/1389200033336739. [DOI] [PubMed] [Google Scholar]
- 36.Zhou J, Zhang J, Xie W. Curr Drug Metab. 2005;6:289–298. doi: 10.2174/1389200054633853. [DOI] [PubMed] [Google Scholar]
- 37.Xie W, Radominska-Pandya A, Shi Y, Simon CM, Nelson MC, Ong ES, Waxman DJ, Evans RM. Proc Natl Acad Sci U S A. 2001;98:3375–3380. doi: 10.1073/pnas.051014398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie W, Yeuh MF, Radominska-Pandya A, Saini SPS, Negishi Y, Bottroff BS, Cabrera GY, Tukey RH, Evans RM. Proc Natl Acad Sci U S A. 2003;100:4150–4155. doi: 10.1073/pnas.0438010100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saini SP, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W. Mol Pharmacol. 2004;65:292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- 41.Gong H, Singh SV, Singh SP, Mu Y, Lee JH, Saini SPS, Toma D, Kagan VE, Day BW, Zimniak P, Xie W. Mol Endocrinol. 2006;20:279–290. doi: 10.1210/me.2005-0205. [DOI] [PubMed] [Google Scholar]
- 42.Zhao AZ, Huan JN, Gupta S, Pal R, Sahu A. Nat Neurosci. 2002;5:727–728. doi: 10.1038/nn885. [DOI] [PubMed] [Google Scholar]
- 43.Saini SP, Mu Y, Gong H, Toma D, Uppal H, Ren S, Li S, Poloyac SM, Xie W. Hepatology. 2005;41:497–505. doi: 10.1002/hep.20570. [DOI] [PubMed] [Google Scholar]
- 44.Uppal H, Toma D, Saini SP, Ren S, Jones TJ, Xie W. Hepatology. 2005;41:168–176. doi: 10.1002/hep.20512. [DOI] [PubMed] [Google Scholar]
- 45.Xie W, Barwick JL, Simon CM, Pierce A, Safe S, Blumberg B, Guzelian PS, Evans RM. Genes Dev. 2000;14:3014–3023. doi: 10.1101/gad.846800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mu Y, Stephenson CRJ, Kendall C, Saini SPS, Toma D, Cai H, Strom S, Day BW, Wipf P, Xie W. Mol Pharmacol. 2005;68:403–413. doi: 10.1124/mol.105.013292. [DOI] [PubMed] [Google Scholar]
- 47.Liu F, Song YK, Liu D. Gene Ther. 1999;6:1258–1266. doi: 10.1038/sj.gt.3300947. [DOI] [PubMed] [Google Scholar]
- 48.Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Chacko J, Trump BF. In Vitro (Rockville) 1981;17:913–925. doi: 10.1007/BF02618288. [DOI] [PubMed] [Google Scholar]
- 49.Strom SC, Chowdhury JR, Fox IJ. Semin Liver Dis. 1999;19:39–48. doi: 10.1055/s-2007-1007096. [DOI] [PubMed] [Google Scholar]
- 50.Matsuzaka T, Shimano H, Yahagi N, Yoshikawa T, Amemiya-Kudo M, Hasty AH, Okazaki H, Tamura Y, Iizuka Y, Ohashi K, Osuga J, Takahashi A, Yato S, Sone H, Ishibashi S, Yamada N. J Lipid Res. 2002;43:911–920. [PubMed] [Google Scholar]
- 51.Owen MR, Corstorphine CC, Zammit VA. Biochem J. 1997;323:17–21. doi: 10.1042/bj3230017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lamb DC, Kelly DE, Kelly SL. FEBS Lett. 1998;425:263–265. doi: 10.1016/s0014-5793(98)00247-6. [DOI] [PubMed] [Google Scholar]
- 53.Buhman KK, Accad M, Novak S, Choi RS, Wong JS, Hamilton RL, Turley S, Farese RV., Jr Nat Med. 2000;6:1341–1347. doi: 10.1038/82153. [DOI] [PubMed] [Google Scholar]
- 54.Morere P, Nouvet G, Stain JP, Paillot B, Metayer J, Hemet J. Sem Hop. 1975;51:2095–2102. [PubMed] [Google Scholar]
- 55.Endemann G, Stanton LW, Madden KS, Bryant CM, White RT, Protter AA. J Biol Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 56.Van Nieuwenhoven FA, Verstijnen CP, Abumrad NA, Willemsen PH, Van Eys GJ, Van der Vusse GJ, Glatz JF. Biochem Biophys Res Commun. 1995;207:747–752. doi: 10.1006/bbrc.1995.1250. [DOI] [PubMed] [Google Scholar]
- 57.Kodama S, Koike C, Negishi M, Yamamoto Y. Mol Cell Biol. 2004;24:7931–7940. doi: 10.1128/MCB.24.18.7931-7940.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]