

Abstract
Mutations in the Huntington locus (htt) have devastating consequences. Gain-of-poly-Q repeats in Htt protein causes Huntington's disease (HD), while htt-/- mutants display early embryonic lethality. Despite its importance, the function of Htt remains elusive. To address this, we compared more than 3,700 compounds in three syngeneic mouse embryonic stem cell (mESC) lines: htt-/-, extended poly-Q (Htt-Q140/7), and wildtype mESCs (Htt-Q7/7) using untargeted metabolite profiling. While Htt-Q140/7 cells, did not show major differences in cellular bioenergetics, we find extensive metabolic aberrations in htt-/- mESCs, including: (i) complete failure of ATP production despite preservation of the mitochondrial membrane potential; (ii) near-maximal glycolysis, with little or no glycolytic reserve; (iii) marked ketogenesis; (iv) depletion of intracellular NTPs; (v) accelerated purine biosynthesis and salvage; and (vi) loss of mitochondrial structural integrity. Together, our findings reveal that Htt is necessary for mitochondrial structure and function from the earliest stages of embryogenesis, providing a molecular explanation for htt-/- early embryonic lethality.
Keywords: Huntington's Disease, embryonic stem cells, metabolomics, metabolism, untargeted metabolite profiling, LC-MS/MS, mitochondria, mitochondrial bioenergetics, mitochondrial respiration, oxygen consumption, glycolysis, AMP kinase
Introduction
The Huntington locus (htt) in the mammalian genome encodes a large protein called Huntingtin (Htt). Htt protein is maternally expressed from the first cell stage in the fertilized egg and subsequently is ubiquitously found in all cells of the embryo and adult. Mutations affecting the locus have dramatic consequences in both mouse and humans. Homozygote htt-/- embryos undergo developmental arrest, fail to undergo proper gastrulation, lack a proper node, display a shortened primitive streak, and exhibit an impaired patterning of embryonic germ layers - early embryonic lethality occurs by e7.0-7.5 [1], [2].
In contrast to the lethality of htt-/- embryos, mutation of a single htt allele that expands the poly-Q (CAG) repeat at the N-terminal domain of Htt protein results in the dominantly inherited Huntington's Disease (HD) [1], [3]. HD is a devastating neurodegenerative disease typified by a progressive movement disorder, cognitive decline, and psychological impairment due to the death of medium spiny neurons in the striatum [4], and other areas of the brain [5]. Some aspects of the disease can be recapitulated in rodent models by genetically increasing the length of the poly-Q repeat. As a platform to investigate Htt loss- and gain-of-function effects in the same genetic background, experiments were performed in syngeneic knock-in mouse embryonic stem cells (mESC) containing one copy of a humanized exon 1 (with an extended polyglutamine tract and adjacent proline-rich region; Htt-Q140/Q7), as well as knockout (htt-/-) [6], [7], in comparison with mESC expressing the mouse htt gene ( Htt-Q7/7). Importantly, studies of htt-/- mESC afford an opportunity to assess Htt protein function because viability is maintained as pluripotent cells, despite the lethality that invariably ensues in htt-/- mice during embryogenesis.
The embryonic functions of the Htt protein remain essentially unknown. Notwithstanding, Htt has been implicated in diverse cell processes in multiple investigated cell types. These include, but are not limited to: trafficking of growth factor complexes [8], [9], transcriptional regulation of a large variety of genes [10]–[12], mitotic spindle orientation [13], cell adhesion [14], endocytosis and vesicular transport [15]–[18], neuronal survival and neurogenesis [4], [19], [20]. Additionally, Htt protein is present in mitochondria of rat [21], mouse, and human somatic cells[22] and therefore, may be important for mitochondrial activities. In human lymphoblasts, the ATP/ADP ratio, a key measure of cellular energy reserves has been correlated with the length of the poly-Q extension in Htt, even when this length is in the normal range [23]. This apparent correlation raises the possibility that energy metabolism modulation may be a normal cellular role of Htt, not just a function acquired by the extension of the poly-Q region. Although Htt plays a crucial role in both embryonic development and disease initiation, the precise molecular and cellular functions of Htt in early embryonic cells and in the brain remain unknown [2]. In particular, the connection between Htt and metabolism during early development is essentially unexplored to date.
In this study, we used global untargeted metabolite profiling to compare the small molecule metabolome (50 – 1,000 Da) of mutant htt in mESC, both gain- and loss-of-function, in comparison with the metabolome of wild-type mESCs. Our findings demonstrate that htt mutation results in profound dysregulation of key metabolic pathways during pluripotency. These studies reveal that mouse htt-/- ESCs exhibit severe bioenergetic and metabolic defects, including an essentially complete failure of mitochondrial ATP generation, associated with defects in mitochondrial structure. Conversely, mutant mESCs with poly-Q (Q140/7) do not exhibit apparent mitochondrial structural defects, but do show metabolic alterations, including a higher respiratory capacity with wild type levels of nucleotide mono-, di- and tri-phosphates. Our findings provide a molecular explanation for the early embryonic lethality phenotype of the htt-/-mouse, and reveal a previously unrecognized but essential role for Htt in supporting mitochondrial bioenergetics, metabolism and structural integrity during early embryogenesis.
Materials and Methods
Cell culture
Htt-/-, Htt-Q7/7, and Htt-Q140/7 mES cells were provided by Dr. Marcy E. Macdonald (Harvard University) and propagated on 0.1% gelatin-coated plates, under serum- and feeder-free culture conditions in medium containing a 1:1 mix of DMEM-F12 (Invitrogen Cat# 12634) and Neurobasal medium (Invitrogen Cat# 21103), supplemented with N2 (Invitrogen Cat# 17502), B27 supplement without Vitamin A (Invitrogen Cat# 12587), 2 mM GlutaMAX (Invitrogen Cat# 35050), 25 μg/ml of BSA (Invitrogen Cat# 15260), 10 μg/ml insulin (Sigma-Aldrich Cat# 19278), O.1mM β-mercaptoethanol (Invitrogen Cat# 21985), 10 ng/ml LIF (Millipore Cat# ESG1107), 0.5μM PD0325901 (Axon Med Chem, Cat# Axon 1408) and 3μM CHIR99021 (EMD Millipore, Cat# 361559). Cell culture medium was changed daily and the cells were passaged by trypsinization every three days.
Metabolite extraction for metabolomic analysis
Cells were grown in 6-well plates to 70% confluency Culture medium and cells were collected separately for analysis by untargeted metabolite profiling. For media analysis, medium was centrifuged at 1500 rpm for 10 min at 4°C to remove dead cells and debris; supernatants were collected and frozen at -80°C until the day of analysis. For analysis, media metabolites were directly extracted in 1:200 vol:vol 70% acetonitrile and aqueous 0.2% ammonium hydroxide and 3 μl of this extract was subjected to untargeted metabolite profiling by LC/MS. For analysis of cellular metabolites, cells were quickly washed twice with ice-cold PBS, followed by metabolic quenching and metabolite extraction using -70°C 80:20 methanol:water (LC-MS grade methanol, Fisher Scientific). Cold-quenched cells were scrape-harvested using a teflon cell scraper and transferred with 80% cold MeOH to 2.0ml Tissuelyzer tubes (Qiagen). The Cell-MeOH mixtures were incubated on dry ice for 10 min then subjected to bead-beating for 45 sec using a Tissuelyser cell disrupter (Qiagen). Extracts were centrifuged for 5 min at 5,000 rpm to pellet insoluble material and supernatants were tranfered to clean tubes. This extraction was repeated two additional times and all three supernatants were pooled, dried in a speed-vac (Savant) and stored at -80°C until analysis. For normalization of sample analyses, post-extracted cell pellets were solubilized in 200 μl 0.2M aqueous NaOH at 95°C for 20 min and the pellet protein was quantified using the BioRad DC assay. On the day of metabolite analysis, dried cell extracts were reconstituted in 70% acetonitrile with 0.2% ammonium hydroxide at a relative protein concentration of 8 μg/μl and 3 μl of this reconsitued extract was injected ifor LC/MS-based untargted metabolite profiling.
Untargeted metabolite profiling
Both cell and culture medium extracts were analyzed by LC/MS essentially as described previously [24], using a platform comprised of an Agilent Model 1200 liquid chromatography system coupled to an Agilent Model 6230 time-of-flight MS analyzer. Metabolite separation was performed by using aqueous neutral phase gradient chromatography on a Diamond Hydride column (Microsolv) and mobile phases as follows: A: 50% isopropanol, containing 0.025% acetic acid, and B: 90% acetonitrile containing 5mM ammonium acetate. Raw data were analyzed using Agilent MassHunter Qual software, and Mass Profiler Professional software. Briefly, Qual performs untargeted molecular feature extraction to generate compounds/metabolites based on the elution profile at identical mass and retention times within a specifed mass accuracy (5 ppm). Aligned molecular features detected in all biological replicates from at least one cell group (i.e., htt-/-, Htt-Q140/7 mutant and Htt-Q7/7 wildtype) were directly applied for statistical analysis across treatment groups by Mass Profiler Professional. The Bonferroni family-wise-error-rate correction was applied for multiple testing correction of p-values (corrected for p < 0.05).
Differentially-expressed Metabolite identification
Differentially-expressed metabolites in htt mutant mESC, with fold-changes greater than 2.0 (p<0.05), compared to htt wildtype mESC, were searched against an in-house annotated METLIN Personal Metabolite Database (Agilent Technologies), based on accurate monoisotopic neutral masses (<5 ppm). A molecular formula generator (MFG) algorithm in MPP was used to generate and score empirical molecular formulae based on a weighted consideration of monoisotopic mass accuracy, isotope abundance ratios, and spacing between isotope peaks. A putative compound ID was tentatively assigned when METLIN and MFG scores concurred for a given candidate molecule. Tentatively assigned compound identities were assigned and verified based on a match of LC retention times and/or MS/MS fragmentation spectra to that of pure molecule standards in our progressively growing database.
Mitochondrial oxygen consumption and glycolysis-derived lactate flux
Glycolysis and oxidative phosphorylation flux analysis was performed using kits from Seahorse Biosciences, in accord with the manufacturer's recommendations. For these assays, cells were seeded on XF96 cell culture microplates (Seahorse Biosciences Cat# 101085-004) and grown to 70% confluency prior to analysis. On the day of assay, culture media were changed to XF Assay medium (Seahorse Biosciences Cat# 102365-100), supplemented with 5 g/L of glucose, 2 mM glutamine, and 2 mM pyruvate for oxidative phosphorylation assay and supplemented with only 2 mM pyruvate for assay of glycolysis rates. Prior to assay, plates were transferred into a non-CO2 incubator at 37° sand kept for 1 h. To quantify oxidative phosphorylation rates, we first measured initial (basal) oxygen consumption rates, followed by a series of changes in oxygen consumption when cells were sequentially treated with 2.5 μM oligomycin, 0.75 μM FCCP, and finally a compbination of 1 μM rotenone and 1 μM antimycin. Following each addition, oxygen levels in the culture medium were monitored at 20 s intervals and the overall oxygen consumption rate (OCR) was calculated. For the glycolysis stress test, after collecting baseline acidification rate (lactate release) data, cells were sequentially treated with 10 mM glucose, 1 μM oligomycin, and 100 mM 2-deoxy-glucose and evoked changes in acidification rate were quantified after each addition. For ths purpose, changes in culture medium pH was monitored every 20 s and used to calculate the overall extracellular acidification rate (ECAR).
Measurement of Mitochondrial Abundance
Mitotracker Red dye (Invitrogen, Cat# M7512) was used for the measurement of mitochondrial abundance. Cells were trypsinized and 105 cells were treated with Mitotracker Red dye (50nM) in mESC medium at 37°C for 30 minutes. After incubation, the cells were spun down, washed with medium, resuspended in PBS containing 0.5% bovine serum albumin and then analyzed for fluorescence by flow cytometry. Flow cytometry analysis was performed using a BD LSRII instrument at Rockefeller University Flow Cytometry core facility.
Western Blot Analysis
Whole cell lysate was harvested using trypsin / EDTA and lysed in RIPA buffer containing protease inhibitors (Roche, 11-836-170-001) and PMSF (1mM). Lysate was centrifuged at >13,000g for 10 minutes at 4C. Protein concentrations of lysate were measured by Bradford assay (Bio-Rad) and 60ug total protein was loaded. Westerns were performed using the Novex NuPage SDS-PAGE blot system. Blots were probed with anti-cytochrome c (Millipore, #MAB1800), anti- Cytochrome b-c1 subunit RISP (Millipore, #ABC322), anti-VDAC (Millipore, #MABN504), or anti-Actin (Cell Signaling, #3700).
Measurement of Mitochondrial Membrane Potential
A mitoprobe JC-1 assay kit (Invitrogen, Cat# M34152) was used for measurement of mitochondrial membrane potential according to manufacturer's instructions. Briefly, the cells were trypsinized, 105 cells were resuspended in mESC medium, treated with 2 μM JC-1 at 37°C for 30 minutes, washed and resuspended for fluorescence analysis by flow cytometry. TMRE (Invitrogen T-699) and MitoTracker DeepRed (Invitrogen M22426) co-staining, Cells were trypisinized, washed twice with growth medium and incubated in medium containing TMRE (200 nM) and/or MitoTracker DeepRed (200 nM) for 30 minutes at 37°C for 30 minutes. After incubation, cells were washed twice with media, resuspended in PBS and analyzed using a BD FACSArray instrument.
Electron microscopy
Cells were seeded on 6 well plates and grown to 90% confluence. The plates were processed according to previously published protocols [25] and imaged at the Weill Cornell Medical College EM core facility. Mitochondria were counted blind using >200 mitochondria from EM images. ‘Good’ mitochondria were regular in shape, had defined outer membrane and well developed cristae. ‘Poor’ mitochondria were those determined to have an irregular shape, compromised outer membrane, abnormal cristae, and /or a ‘vesicular’ morphology.
Results
Comparative metabolite profiling of htt-/- and Htt-Q140/7 in embryonic stem cells
To broadly discover metabolic consequences in both htt-/- and Htt-Q140/7 mutant mouse stem cells, we performed untargeted liquid chromatography/mass spectrometry (LC/MS)-based metabolite profiling and compared to findings with wildtype Htt-Q7/7 stem cells. This analysis contrasted metabolite profiles in three syngeneic cell lines that differ only with regard to their htt genotypes when cultured under pluripotency conditions (see Materials and Methods). Profiling data were acquired using aqueous neutral phase LC and both negative-ion and positive-ion MS detection for broad coverage of potential changes in hydrophilic metabolite levels that contribute to intermediary metabolism. Together, this metabolomic analysis surveyed 3758 molecular features of 50 – 1,000 Da mass/charge (m/z), considering only molecules that were detected in all biological replicates from at least one genetic group. Each genetic group comprised 9-10 replicate mESC cultures (Figure S1). Notably, 1384 species were quantified as negative ions (Figure S1A) and 2374 species were detected as positive ions (Figure S2A). Principal component analysis (PCA) and unsupervised hierarchical cluster analysis (HCA) revealed within-group clustering and between-group separation of metabolites in samples prepared from each of the three different cell lines (Figure S1B-C and S2B-C). Considering the 1384 species observed as negative ions, levels of 60 metabolites in htt-/- and 164 species in Htt-Q140/7 were significantly altered when compared to Htt-Q7/7 (p <0.05 and fold-change ≥ 2.0; Figure S1D). Among these, 30 differences from Htt-Q7/7, overlapped between both htt-/- and Htt-Q140/7 mutants (Figure S1D, Table S1). A significant and marked effect of htt mutants on metabolite expression levels was similarly observed with positive ion detection, where 62 species in htt-/- and 192 species in Htt-Q140/7 differed significantly from Htt-Q7/7 (p <0.05 and fold-change ≥ 2.0; Figure S2D). Together, these findings establish that both htt-/- and Htt-Q140 mutations profoundly perturb metabolism in pluripotent embryonic stem cells.
htt-/- mESCs display accelerated glycolysis and ketogenesis
Despite the relative inefficiency of aerobic glycolysis for ATP generation, as compared to ATP generation by mitochondrial respiration, aerobic glycolysis is the predominant bioenergetic phenotype for rapidly proliferating cells (e.g., cancer cells) and is also typical of pluripotent embryonic stem cells [26]. A hallmark feature of aerobic glycolysis is increased glucose uptake and accelerated metabolism to pyruvate, followed by NADH-mediated reduction to lactate and extracellular export. To assess whether htt mutations are associated with an altered rate of glycolysis, we compared relative levels of glucose, lactate, and pyruvate, both intracellularly and in 24h cell culture medium. Interestingly, htt-/- mutants displayed significantly increased levels of lactate, both intracellularly and extracellularly, compared to Htt-Q7 and Htt-Q140 (Figure 1). Conversely, lactate release into the cell culture medium was relatively diminished in Htt-Q140/7 mutant cells. In accord with a relatively accelerated rate of aerobic glycolysis in htt-/- cells, the observed increase in lactate release was associated with a reciprocal decrease in intracellular and culture medium pyruvate levels (Figure 1). Additionally, htt-/- cells exhibited a 15-20% decrease in intracellular glucose and increased glucose uptake from the culture medium, when compared with normal Htt-Q7/7 and mutant Htt-Q140/7 cells, consistent with enhanced glucose consumption for aerobic glycolysis (Figure 1). These findings demonstrate that htt-/- mutation in mESC is associated with a metabolic switch toward increased aerobic glycolysis.
Figure 1.
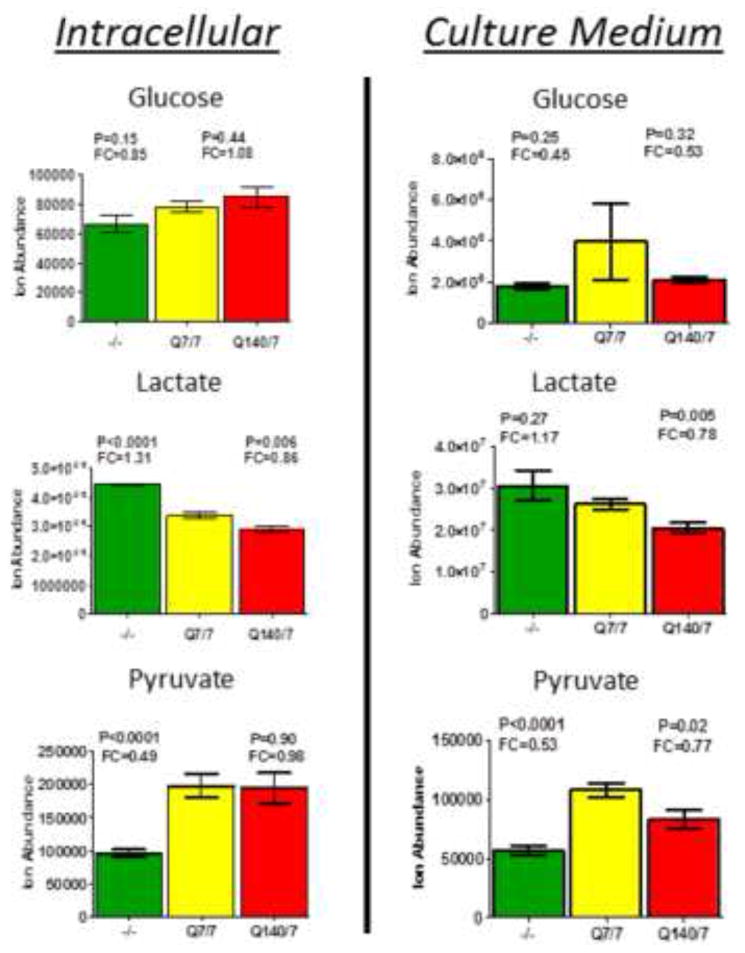
Measurement of relative levels of glucose, lactate and pyruvate in cells and cell culture medium indicates an increased rate of glycolysis in htt-/- mESCs, but no significant effect of Htt-Q140/7 mutation. Bars depict relative ion abundances measured by LC-MS for the indicated metabolites, measured both in cells (left panels) and in culture media (right panels). Observed fold-changes and p-values are quantified for htt-/- mESC (green bars) and Htt-Q140/7 mutant mESC (red bars), in comparison with wildtype Htt-Q7/7 mESC (yellow bars). Error bars are SEM.
Targeted metabolite profiling of data described in Figures S1 and S2, confirmed by chromatographic retention time and MS/MS fragmentation of standards, provided for a broad assessment of the effects of htt mutations on metabolic pathway intermediates, including those of glycolysis and the Krebs cycle (Figure S3). Notably, whereas htt-/- mutants showed a significant decrease in levels of glycolytic hexose intermediates (glucose-6-phosphate and fructose 1,6-diphosphate), all glycolytic triose intermediates except pyruvate increased in abundance. This finding is consistent with accelerated use of glucose for glycolysis, resulting in build-up of triose intermediates, increased pyruvate reduction to lactate and efflux of lactate into the culture medium. Increased conversion of pyruvate to lactate, in lieu of reductive decarboxylation by pyruvate decarboxylase to acetyl CoA for subsequent entry into the Krebs cycle, is in accord with a more than 50% decrease in acetyl CoA levels observed in htt-/- mESCs (Figure S3B). In contrast, acetyl CoA levels were increased by almost 2-fold in Htt-Q140/7 cells (Figure S3B). It is notable that β-oxidation of lipids provides an additional source of acetyl CoA for entry into the Krebs cycle, beyond that provided by glycolysis. However, insufficient catabolic consumption of acetyl CoA by the Krebs cycle can promote acetyl CoA condensation reactions that result in accumulation of ketone body products, including acetoacetic acid and β-hydroxybutyric acid. We observed that htt-/- cells exhibit a significant accumulation of these ketone bodies intracellularly and release ketones and hydroxyacids into the cell culture medium (Figure S4). It is notable that although Htt-Q140/7 mESC did not exhibit altered levels of nucleotide mono-, di-, and triphosphate levels relative to wild-type Htt-Q7/7 mESC, other metabolite levels were significantly altered – this included changes in nucleotide bases, lipid profiles, carnitines, sulfur metabolites and some amino acids (Figure S5). Taken together, these findings demonstrate that metabolism in both htt-/- and Htt-Q140/7 mESCs is profoundly aberrant; in the case of htt-/- mESC this perturbation is characterized by an increased rate of aerobic glycolysis and failure to efficiently oxidize acetyl CoA by the Krebs cycle.
htt-/- mESCs are depleted of intracellular nucleotide triphosphates
Our finding of a ketogenic metabolism in htt-/- mESCs suggests impaired NADH production for support of ATP generation by aerobic respiration in mitochondria. Since glycolysis is far less bio-energetically efficient than aerobic respiration (2 vs. 36 mols ATP/mole glucose generated), we wondered whether htt-/- mutation may be associated with a relative bioenergetic deficit, reflected in decreased ATP/AMP and ATP/ADP ratios. To test this possibility, untargeted negative ion profiling data were assessed for relative levels of adenine nucleotide tri-, di-, and mono-phosphates in htt-/- relative to Htt-Q7/7 and Htt-Q140/7 cells (Figure 2). Notably, htt-/-cells exhibited a 50% decrease in ATP levels, concomitant with 2-fold increases in both ADP and AMP levels, compared to both Htt-Q7/7 and Htt-Q140/7. This relative energetic collapse observed in htt-/- cells for adenine nucleotides extended to other nucleotides of the guanine, cytosine, thymidine, and uridine mono-, di-, and tri-phosphate series. Indeed, levels of GTP, CTP, TTP and UTP were all decreased significantly in htt-/- compared to Htt-Q7/7 cells, concomitant with reciprocal increases in cognate mono- and diphosphate levels (with the sole exception of GDP, which remained unchanged in htt-/- cells; Figure 2). In sharp contrast to the profound nucleotide triphosphate deficit and monophosphate/diphosphate increases in htt-/- mESC, nucleotide levels in Htt-Q140/7 were indistinguishable from levels in Htt-Q7/7 cells. Dissipation of the cellular reservoir of ATP and other nucleotide triphosphates in htt-/- cells demonstrates that Htt protein activity is critical for the maintenance of high energy phospho-metabolites.
Figure 2.
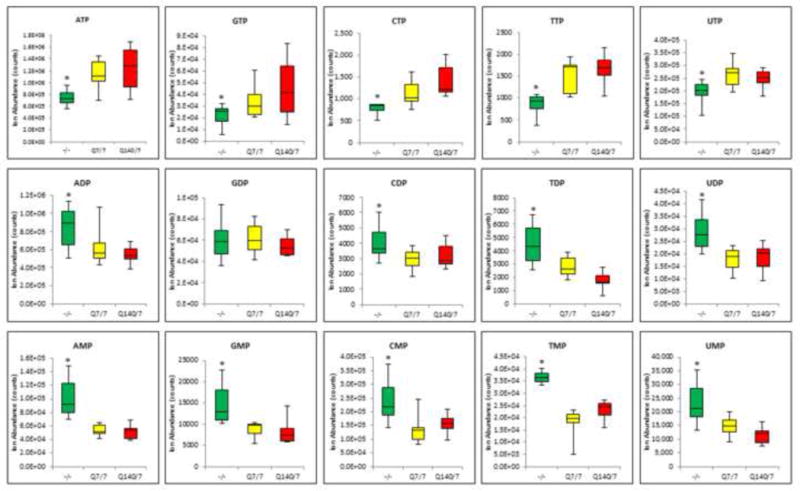
High-energy nucleotide triphosphate levels are significantly decreased in htt-/- mESCs and nucleotide di- and mono-phosphate levels are significantly increased, as compared with Htt-Q7/7 control mESC. In contrast, Htt-Q140/7 mutation does not alter the levels of nucleotide Tri-, di- or mono-phosphates. Results are mean values +/- SD (n = 9-10). * Denotes a significant difference with p<0.05.
htt-/- mutation accelerates purine biosynthetic and salvage pathways
Interestingly, examination of the 30 molecules that are differentially expressed between htt-/-and Htt-Q7/7 (Figure S1D, Table S1) led to the identification of several molecules that are uniquely expressed in htt-/- cells. One such unique molecule (RT=10.97 min, Mass=295.0618 Da) was observed to have a formula consistent with C8H14N3O7P and was molecularly identified as 5-aminoimidazole ribonucleotide (AIR, Figure 3). AIR is a pivotal intermediate in purine nucleotide biosynthesis, which involves 10 enzymes for the ultimate conversion of phosphoribosylpyrophosphate to the first purine metabolite, inosine monophosphate. Examination of the relative abundance of all other intermediates involved in this pathway revealed that in addition to AIR, levels of 5 other purine biosynthetic intermediates were also markedly increased in htt-/- versus Htt-Q7/7 cells. Indeed, similar to AIR, 3 out of 5 intermediates were only detected in htt-/- cells – these include phosphor-ribosyl-formyl-glycineamidine (FGAM), formamidoimidazole-4-carboxamide ribotide (FAICAR), and 5′-phosphoribosyl-4-(N-succinocarboxamide)-5-aminoimidazole SAICAR). The 2 other differentially-expressed purine pathway intermediates were 5-aminoimidazole-4-carboxamide ribotide (AICAR, increased 7.1-fold) and the final pathway product, inosine monophosphate (IMP, increased 3.3-fold). In addition to purine biosynthesis intermediates, all detected purine salvage pathway metabolites and products were significantly increased in htt-/- cells, except for ATP and GTP, which significantly decreased (Figure 2). Together, these results reveal a substantial acceleration of purine synthesis and turnover in htt-/- mESCs, suggesting the operation of a biochemical regulatory mechanism that increases purine biosynthesis, potentially as compensation for the relative paucity of ATP, GTP and other nucleotide triphosphates in htt-/-mESC.
Figure 3.
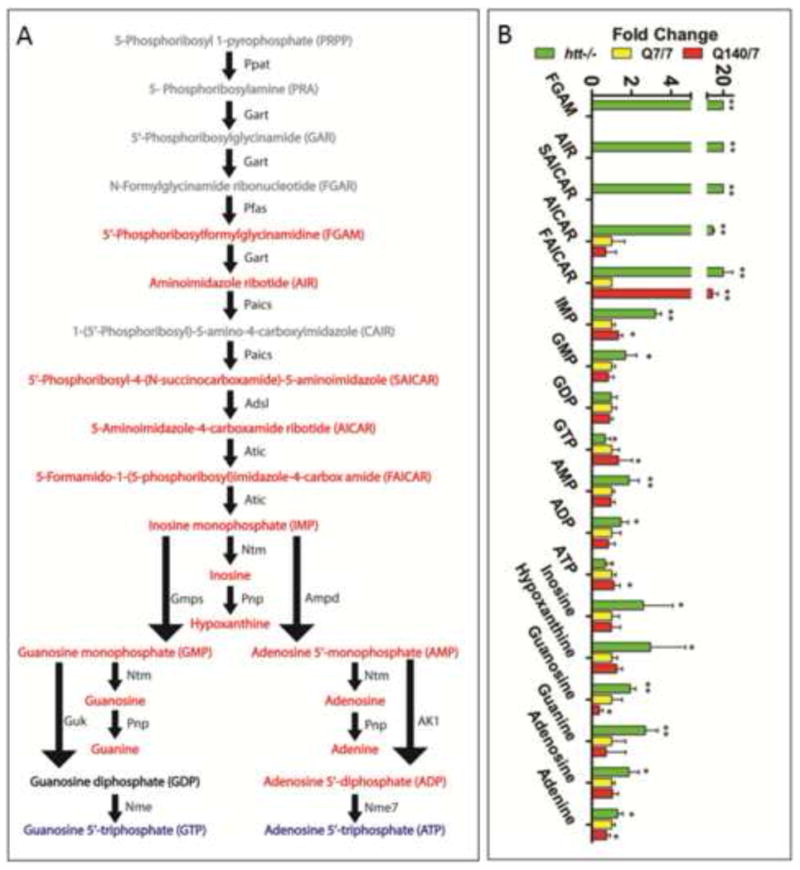
There is a marked intracellular accumulation of metabolites of de novo purine biosynthesis and purine salvage pathways in htt-/- compared with Htt-Q7/7 wildtype and Htt-Q140/7 mutant mESCs. The colors of fonts denote metabolites in htt-/- mESC with abundances that are significantly increased (red), decreased (blue), unchanged (black) and undetected (grey), in comparison with Htt-Q7/7 mESC. For each observed change, the measured p-values and fold-changes are given. * Denotes a significant difference with p<0.05. The nomenclature for labeling of metabolic enzymes (names given adjacent to pathway arrows) is from the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway map. Abbreviations: PPAT, Phosphoribosyl Pyrophosphate Amidotransferase; GART, Phosphoribosylglycinamide Formyltransferase; PFAS, Phosphoribosylformylglycinamidine Synthase; PAICS, Phosphoribosylaminoimidazole Carboxylase; ADSL, Adenylosuccinate Lyase; ATIC, 5-Aminoimidazole-4-Carboxamide Ribonucleotide Formyltransferase; GMPS, Guanine Monphosphate Synthetase; GUK, Guanylate Kinase; NME, Nucleoside Diphosphate Kinase; 5′-NT, 5′-Nucleotidase; PNP, Purine Nucleoside Phosphorylase; AMPD, Adenosine Monophosphate Deaminase; AK1, Adenylate Kinase 1; NME7, NME/NM23 Family Member 7.
Htt protein activity is necessary for mitochondrial ATP synthesis
AMP-activated protein kinase (AMPK) serves as a master switch that regulates cellular bioenergetics, including an activation of glucose uptake and accelerated glycolysis [27]. Since AMPK is allosterically activated in cells in response to an elevated ratio of AMP/ATP as well as by direct binding of the AMP analog AICAR [28], both of which were increased in htt-/- cells, we considered whether htt-/- cells favored glycolysis over mitochondrial respiration as an ATP source. To directly test this possibility, both mitochondrial and glycolysis respiration rates were measured in all three mESC lines using an extracellular flux analyzer. Results showed that htt-/-cells consume 70% less oxygen compared to wildtype Htt-Q7/7 cells. In contrast, Htt-Q140/7 mESC displayed a relative 50% increase in oxygen consumption. Because oxygen consumption by htt-/- mESC was not decreased by the selective mitochondrial ATP synthase inhibitor, oligomycin, it is apparent that htt-/- mESC mitochondria do not contribute to ATP synthesis. This is in contrast to a marked degree of oligomycin-inhibited mitochondrial ATP synthesis in both Htt-Q7/7 and Htt-Q140/7 mESC.
To investigate whether htt-/- impairs electron transfer through the mitochondrial respiratory complexes, we measured the maximal respiratory capacity of htt-/- mESC for support of oxidative phosphorylation. Toward this end, we studied the effect of the mitochondrial uncoupling agent, carbonylcyanide-4-(trifluorometoxy)-phenylhydrazine (FCCP) on cellular oxygen consumption. Treatment of htt-/- cells with FCCP elicited a very modest increase in oxygen consumption, indicative of a minimal mitochondrial respiratory capacity (Figure 4). In contrast, Htt-Q140/7 mESC exhibited an apparently accelerated rate of mitochondrial ATP synthesis, compared to Q7/7 mESC, and additionally exhibited a greater mitochondrial respiratory reserve (Figure 4A). Together, these results suggest htt gene expression is essential for mitochondrial ATP synthesis and respiratory chain function and Htt mutations can alter mitochondrial bioenergetic capacities.
Figure 4.
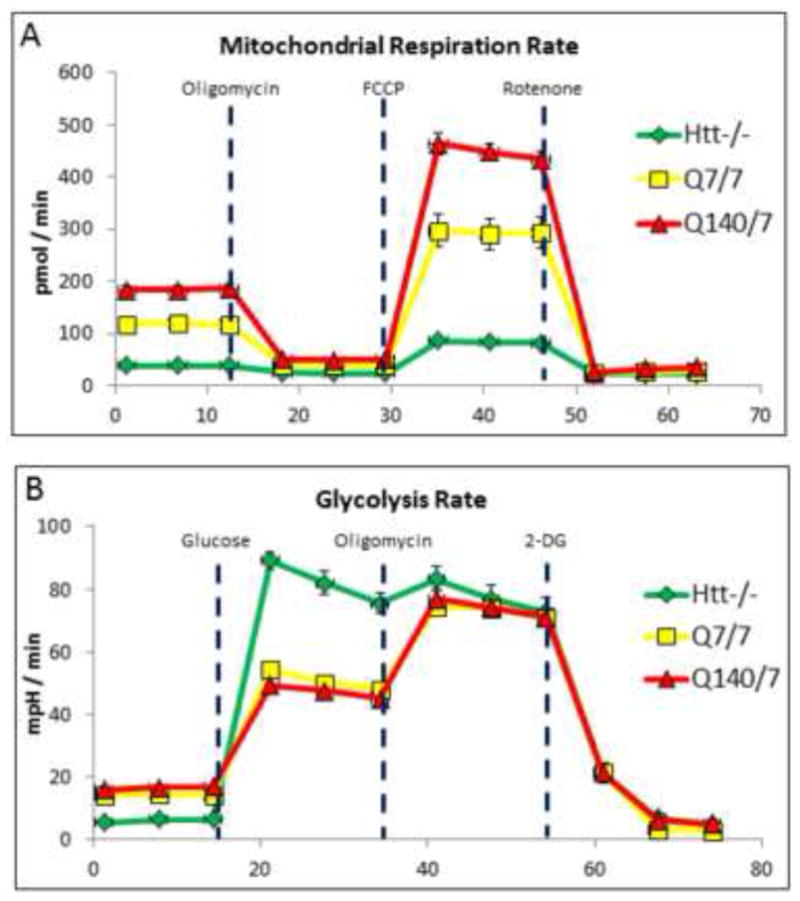
Influence of htt-/- and Htt-Q140/7 mutations on mitochondrial and glycolytic fluxes in mESCs. (A) Mitochondrial ATP synthesis rate, based on oligomycin-inhibited oxygen consumption rate (OCR). At the indicated time, oligomycin was added to reveal the extent to which oxygen is being consumed for ATP synthesis. FCCP, an uncoupler of mitochondrial respiration was subsequently added to define the maximal capacity of cells for mitochondrial respiration. Finally, the combination of rotenone and antimycin was added to elucidate total mitochondrial oxygen consumption. Results demonstrated that ATP synthesis and synthetic capacity are dramatically impaired in htt-/- mESC, while ATP production rate and capacity are relatively increased in Htt-Q140/7 cells. (B) Glycolysis rate, based on glucose-dependent extracellular acidification rate (ECAR). Initially, cells are in glucose-free medium, followed by addition of glucose at the indicated time. Oligomycin is subsequently added to block mitochondrial respiration and reveal the glycolytic reserve capacity (i.e., maximal rate of glycolysis). Finally, an inhibitor of glycolysis, 2-deoxyglucose (2-DG), is added to ascertain the extent to which OCR derives from glycolysis. These results demonstrate that htt-/- mESC conduct glycolysis at a near-maximal rate for energy production. In both panels, points depict mean values for 8 replicate determinations +/- SEM.
To test if htt-/- cells alter their rate of glycolysis-derived ATP in compensation for an apparent lack of mitochondrial ATP generation, extracellular acidification rate (ECAR) was quantified to assess glycolytic flux. After equilibrating mESC cell lines with glucose-free culture medium, 10 mM glucose was added to activate glycolysis and ECAR was quantified. While the glycolytic rate of Htt-Q7/7 and Htt-Q140/7 mESCs were essentially indistinguishable, htt-/- cells displayed an ≈2-fold greater rate of glycolysis (Figure 4B). Upon inhibition of oxidative phosphorylation with oligomycin, mESC lines responded with the anticipated increase in glycolysis, offsetting the elicited inhibition of mitochondria-derived ATP generation and revealing the cells maximal capacity for glycolysis. Whereas ECAR measurements demonstrated large and essentially identical glycolytic reserve capacities for both Htt-Q7/7 and Htt-Q140/7, htt-/- mESCs exhibited no detectable glycolysis reserve capacity whatsoever (i.e., cells were already operating at a maximum glycolytic rate prior to oligomycin treatment). A final addition of 2-deoxyglucose to all cell lines resulted in a rapid and near-complete decrease in ECAR, confirming that the measurements were indeed due to glycolysis-derived lactate release . Taken together, these studies indicate that htt-/- mESCs are incapable of mitochondrial ATP synthesis and instead continually generate ATP at near maximal capacity by glycolysis, without any apparent functional reserve.
Electron microscopy shows severe structural problems in htt-/- mitochondria
Studies were performed in order to determine whether the failure of mitochondrial production of ATP in htt-/- mESCs is associated with conspicuous alterations in mitochondrial abundance, structural integrity, or functional capacity. In one line of investigation, electron microscopy (EM) was employed to compare structural features of mitochondria in the three mESC lines. Interestingly, EM images revealed that many mitochondria in htt-/- mESC are structurally abnormal exhibiting a relative paucity of cristae compared to Htt-Q7/7 and Htt-Q140/7 lines (Figure 5A-G). Additionally, mitochondria in htt-/- mESC exhibited fragmentation, suggestive of initial stages of autophagy (as indicated by the loss of inner membrane structure) [29] (Figure 5B). These findings reveal that mitochondria are structurally aberrant in the absence of Htt protein.
Figure 5.
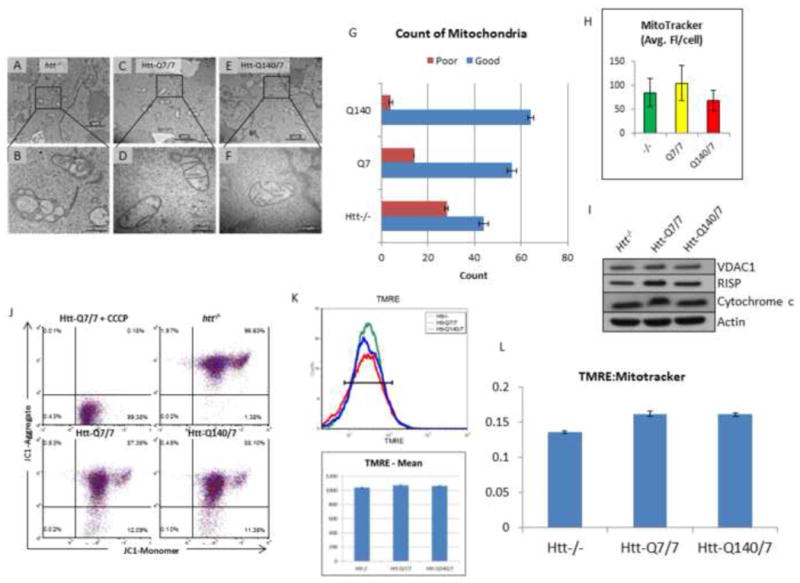
Mitochondrial EM structure is profoundly aberrant in htt-/-, relative to Htt-Q7/7 mESCs, whereas neither mitochondrial abundance nor membrane potential is significantly affected. (A-F) EM images at 15000X (top panels), and 60000X (bottom panels) comparing htt-/-(A-B), Htt-Q7/7 (C-D), and Htt-Q140/7 (E-F) mESC. Results reveal profound structural aberrations in htt-/- mESC, indicative of loss of inner membrane cristae. Scale bars are shown at lower right. (G) Counts from EM images of healthy (Good) and unhealthy (Poor) mitochondria show htt-/- mESC have more poor quality mitochondria. Mitochondria were counted in a blind manner. Error bars are standard deviation. (H) Relative mitochondrial abundance in htt-/- and Htt-Q140/7 mESCs, compared with wildtype Htt-Q7/7 mESCs, expressed on a per cell basis. Selective staining with MitoTracker red fluorescent dye, followed by FACS analysis was used to assess relative mitochondria abundance. Fluorescence values are given as mean values +/- SD. Notably, MitoTracker fluorescence was indistinguishable for all three htt mESC lines tested. (I) Equivalent mitochondrial abundance is shown by western blot analysis of whole cell lysate of htt-/-, Htt-Q7/7, and Htt-Q140/7 for the micochondrial protein VDAC1, Cytochrome b-c1 subunit RISP, and Cytochrome c. Equal amounts of protein were loaded and Actin were used controls. (J) Mitochondria from htt-/- mESC are polarized like mitochondria from Htt-Q7/7 and Htt-Q140/7 mESCs. FACS analysis assessed mitochondria potential after staining htt-/-, Htt-Q140/7 and Htt-Q7/7 mESCs with the mitochondrial voltage-sensitive fluorescent dye, JC-1. Negative control with the mitochondrial uncoupling agent CCCP depicts fluorescence spectral changes that occur when the mitochondrial potential was eliminated. Notably, JC-1 fluorescence results reveal polarization of mitochondria in all 3 htt lines. (K) Mitochondria from htt-/- mESC are polarized like mitochondria from Htt-Q7/7 and Htt-Q140/7 mESCs. FACS analysis assessed mitochondria potential after staining htt-/-, Htt-Q140/7 and Htt-Q7/7 mESCs with the mitochondrial voltage-sensitive fluorescent dye, TMRE. (L) The ratio of polarization to the number of mitochondria is roughly equivalent between htt-/-, Htt-Q7/7 and Htt-Q140/7. Intensity levels of TMRE were divided by the intensity levels of MitoTracker.
We next assessed whether relative mitochondrial abundances differed in htt-/-, Htt-Q140/7, and Htt-Q7/7 mESC. Toward this end, mitochondrial abundance was compared on a per cell basis using FACS analysis, based on mitotracker dye. Results demonstrated similar average fluorescence levels in all three cell lines (Figure 5H), indicating similar mitochondrial abundances and that loss of mitochondria from htt-/- mESCs does not explain their relatively low levels of total ATP and the apparent inability of mitochondria from these cells to produce ATP. We next took whole cell lysate from all three lines and probed for relative expression levels of the mitochondria specific proteins VDAC1, RISP (cyctochrome b-c1 subunit 5), and cytochrome c. Results demonstrated that levels of these proteins across all three lines were indistinguishable (Figure 5I).
Based on the mitochondrial structure perturbation in htt-/- cells, we next investigated the functional capacity of htt-/- cells to maintain their normal membrane potential. Toward this end, we used the fluorescent dyes JC1 and TMRE as relative indicators of inner mitochondrial membrane potential. The greater the membrane potential the more JC1 and TMRE aggregation within mitochondria, and this is revealed by distinct fluorescence emission signals. In JC1 experiments, a mitochondrial uncoupling agent (CCCP) was used as a control, for quantification of fluorescence emission upon complete dissipation of the electrical potential across the mitochondrial inner membrane. As expected, treatment of cells with CCCP, resulted in a complete loss of the JC-1 aggregate fluorescence signal (Figure 5J,). In TMRE experiments (Figure 5K), we additionally utilized mitotracker dye measure the abundance of mitochondria in relation to their membrane potential (Figure 5L). Surprisingly, results of these studies revealed that the mitochondrial membrane potential was completely preserved in htt-/- cells, despite their apparently complete loss of ATP-generating capacity (Figure 4A), and conspicuous structural deficits (Figure 5B). In fact, the mitochondrial membrane potential was similar in all three lines (Figure 5J-L). Paradoxically, we noted that the Htt-/- actually exhibit fewer depolarized mitochondria compared to either Htt-Q7/7 or Htt-Q140/7 (Figure S6). These results demonstrate that failure of ATP production in htt-/- cells is not associated with a diminished abundance or relative loss of inner mitochondrial membrane polarization, but is associated with a loss of structural integrity.
Discussion
The early embryonic lethality of htt-/- mice reveals an essential role for Htt in mammalian development. However, despite decades of investigation focused on elucidating the basis for neurotoxicity of mutant Htt protein, the fundamental role of wildtype Htt remains unknown. In the present study we sought to define the broad impact of htt-/- and Htt-Q140/7 mutants on cell metabolism in pluripotent mESCs, a model of early development. Toward this end, we performed an untargeted metabolite profiling study in attempt to uncover differences in mESC in three syngeneic lines. Our findings demonstrate that extensive metabolic aberrations occur in both htt-/- and Htt-Q140/7, compared with Htt-Q7/7 mESCs. Most notable among these aberrations was a profound energy deficit in htt-/- cells, indicated by decreases in all nucleotide tri-phosphate (NTP) levels, along with reciprocal increases in nucleotide di- and mono-phosphate levels. In Htt-Q140/7 mESC, on the other hand, NTP levels were not markedly impacted although other energy metabolites are altered (Figure S5). This is consistent with findings that patients with HD do not show a drastically altered ATP concentration [30]. It is thought that downstream modifiers influence the role mutant Htt plays in energy metabolism in the cell. Mitochondrial and glycolytic flux measurements in htt-/- mESCs showed that NTP deficits were in conjunction with an essentially complete failure of ATP production via oxidative phosphorylation and compensatory acceleration of glycolysis-derived ATP production. Our findings strongly suggest that glycolysis may operate at a maximal rate in htt-/- mESC, leaving no glycolytic reserve to accommodate increases in cellular energy demands. Electron microscopic analysis of htt-/- mESCs further revealed severe defects in mitochondrial structural integrity. Taken together, these findings demonstrate that Htt protein is necessary for both mitochondrial structure and function from the earliest stages of embryogenesis.
It is notable that htt-/- embryos are smaller compared to their wild type littermates and die by E7.5 with increased apoptosis [31]. Defects in both the transfer of nutrition from the visceral endoderm to the embryo [32] and a general aberration in forming and patterning of the primitive streak have been suggested as the basis for embryonic death [2]. In view of the present metabolomic findings, we propose that disruption of oxidative phosphorylation is, in fact, the key driver of early developmental death of htt-/- embryos. Notably, htt-/- pluripotent mESCs are viable and exhibit a rate of proliferation that is indistinguishable from syngeneic wildtype Htt-Q7/7 cells. The viability of mESCs and pre-E7.5 embryos can be explained by the predominant bioenergetic reliance on glycolysis, allowing for survival despite of the failed oxidative phosphorylation. With further cell development and differentiation beyond E7.5, however, mitochondrial-derived ATP becomes progressively greater [33]–[36], offering a likely explanation for the lethality phenotype of htt-/- mutant mouse.
In contrast with our finding that NTPs are preserved at their wild type levels in Htt-Q140/7 mESCs, a recent investigation of htt mutant mESCs observed a progressive decrease in ATP/ADP ratios with increasing poly-Q lengths (studied to a maximal length of Q111/7) [3]. Moreover, this study did not observe the decrease in ATP/ADP that we found in htt-/- mESCs. A likely explanation for our seemingly discrepant findings regards culture conditions and cell growth details, including days in culture, cell density at harvest, frequency of media changes and culture media composition. Notably, the ability or inability of glycolysis alone to support all ATP needs of the cell will predictably be a function of whether the cells are expanding in rapid growth phase or quiescent, whether the media is relatively rich or poor, how frequently the cells are supplemented with fresh media and in what quantity. In this regard, we recognize that the above-mentioned study grew mESC in serum-supplemented medium, while we cultured cells in defined medium. One possibility is that undefined growth factors in serum compensate for the loss of mitochondrial ATP generation by htt-/- mESCs and thereby cloak the phenotype we observed herein. Clearly, direct metabolite profiling of htt-/- embryos will ultimately shed light to this apparent contrast between the two studies. Unfortunately, however, the number of mouse embryos required to perform such a study is prohibitive with current technologies. We are currently using CRISPR technology to generate htt-/- mutants in Xenopus taking advantage of the fact that early embryogenesis (from the fertilized egg onward) can be observed in a large number (hundreds) of embryos to overcome this limitation. Direct imaging and measurement of metabolomic profiles during very early stages of Xenopus embryonic development will inevitably provide further insights into the functions Htt in energy metabolism and in generation of respiration competent mitochondria, in vivo.
Surprisingly, and despite significant structural aberration, we observed no significant decrease in the abundance of mitochondria in htt-/- or Htt-Q140/7 mESCs. It is also remarkable that mitochondria from htt-/- mESC maintain their inner membrane potential despite their apparent inability to contribute to ATP generation. This unusual observation can be explained by one of the two scenarios that we model in Figure 6. In the first scenario, “normal” mitochondrial potential in htt-/- mESCs arises non-autonomously, at the expense of extra-mitochondrial ATP (Figure 6A). In this case, membrane potential might be maintained through reversed directionality of the mitochondrial ATP synthase complex (complex V in Figure 6), a phenomenon previously reported to occur in some settings [37]. Thus, glycolysis-derived ATP can be consumed for generation of a proton gradient (and ADP) in the inner mitochondrial membrane by a reversed directionality of the ATP synthase complex – this would establish a membrane potential and protect against cytotoxicity owing to mitochondria-dependent apoptosis. If operational, a reversal of ATP synthase directionality would require that glycolysis produces not only enough ATP to meet cellular demands for biochemical, mobility and transport processes, but would need to additionally provide ATP for preservation of the mitochondrial potential and hence, cell viability. Such a critical glycolytic demand for ATP would predictably cause cells to exist in a fragile state, susceptible to death by even modest transient increases in cell demands for ATP. In the second scenario, Htt may act in a mitochondria autonomous manner for coupling of the mitochondrial proton gradient to ATP generation (Figure 6B). If so, the absence of Htt would limit dissipation of the mitochondrial membrane potential and thereby promote the viability of cells despite an apparent failure in mitochondrial ATP synthesis. From the two scenarios we currently favor the first. This is due to our observation that tagged-Htt when overexpressed in human embryonic stem cells (hESCs) fails to localize to mitochondria, thus suggesting a non-autonomous effect (data not shown). Regardless of the mechanism however, this aspect of Htt function on mitochondria membrane potential was unexpected and previously unnoticed. The construction of syngeneic htt-/-, and gain-of poly-Q mutants, currently in progress in hESCs, and their comparison with wildtype cells, will allow performing the same analysis directly in humans. The comparison of the three human lines will identify metabolomic differences, and comparison of the human and mouse htt-mutants data sets will allow for a clear distinction between evolutionarily conserved versus specie-specific differences between human and mouse.
Figure 6.
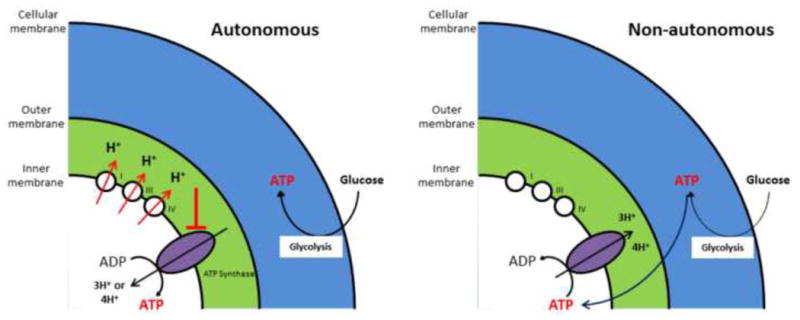
Schematic depicting two alternative models that may explain the existence of a mitochondrial inner membrane potential in htt-/- mESC, despite a lack of mitochondrial ATP generation by these cells. The two models are distinguished as arising via either a mitochondria autonomous mechanism, wherein the respiratory complexes generate a proton gradient, but ATP synthase is unable to effectively use this proton gradient for ATP production (left panel), or arising via a mitochondria non-autonomous mechanism, wherein glycolysis-generated ATP serves to drive mitochondrial ATP synthase in reverse, resulting in a proton gradient at the expense of glycolysis-derived ATP (right panel). Details of these models are considered in the text (Adapted from Takeda et al, 2004).
We show that Htt-Q140/7 mutant that models HD, do not display a defect in mitochondrial ATP synthesis in embryonic cells. In mature neurons however, HD clearly leads to mitochondrial ATP deficit and cell death. Therefore, an interesting and unresolved question is: at what point of development does the initial damages from gain-of poly-Q mutation that ultimately leads to HD, becomes cytotoxic? Providing the answer to this question at the cellular and embryological level will provide important clues on the progression of HD, and inevitably have a strong impact in the rationale design of HD treatments.
Finally, we note that mitochondrial biology is emerging as a key focus point in understanding the cellular and molecular basis of other neurodegenerative diseases. These include: Alzheimer's disease (AD), Parkinson's disease (PD) and amyotrophic lateral sclerosis (ALS) [38], [39]. In some instances, mitochondrial function is hampered due to aberrant protein-protein interactions that are disease specific. For example, amyloid precursor protein (APP) has only been observed associated with mitochondrial protein import channels TOMM40 and TIMM23 in AD brains, not in controls [40], [41]. This interaction blocks transfer of proteins into the mitochondria and perturbs energy metabolism [41]. Similarly, certain mutants of SOD1 cause inherited ALS by associating with mitochondria and changing mitochondrial respiratory rate [42], [43]. Mitochondrial dysfunction can also be caused by the loss of normal protein function. For example, recessive mutations of PINK1, a mitochondrial kinase, have been shown to cause early-onset PD [44]. PINKl's interactions suggest that this is due to disruption of mitochondrial trafficking [45]. While the impact of a gain-of-poly-Q mutant Htt protein on mitochondria has been previously noticed in mature neuronal cells [46], [47], we currently have a very little understanding of how the loss or impairment of normal Htt activity contributes to HD pathology [48]. Our study demonstrates that Htt protein is necessary for maintenance of mitochondrial structure and function, and thus links HD to a panel of neurodegenerative conditions that are either caused by, or arise in conjunction with aberrant mitochondria. Understanding the molecular bases that connect Htt to mitochondrial function will provide novel insights into mitochondrial biogenesis and may translate into unanticipated strategies for therapeutic intervention in HD patients.
Supplementary Material
Highlights.
Loss (htt-/-) and mutant Huntingtin (Htt-Q140/7) aberrantly affects metabolism in mESC
nucleotide phosphate levels are altered in htt-/- mESCs
metabolite levels were significantly altered in Htt-Q140/7
The htt-/- mESC display accelerated glycolysis and a lack of oxidative phosphorylation
Mitochondria are physically altered in htt-/- mESC
Acknowledgments
We thank the Cure Huntington Disease Inc. (CHDI), and Dr. Scott Zeitlin for providing all three mESC lines used in this study, as wells as Lee Cohen-Gould for expert electron microscopy. We thank the members of the Gross and Brivanlou laboratories for constructive criticism of the manuscript. Funds from The Rockefeller University, and CHDI (CEN5401544) to AHB, as well as NIH (P01 HD067244, R37 HL87062 and RR027305) to SSG were used to support this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Duyao MP, Auerbach AB, Ryan A, Persichetti F, Barnes GT, McNeil SM, Ge P, Vonsattel JP, Gusella JF, Joyner AL. Inactivation of the mouse Huntington's disease gene homolog Hdh. Science. 1995 Jul;269(no. 5222):407–410. doi: 10.1126/science.7618107. [DOI] [PubMed] [Google Scholar]
- 2.Woda JM, Calzonetti T, Hilditch-Maguire P, Duyao MP, Conlon RA, MacDonald ME. Inactivation of the Huntington's disease gene (Hdh) impairs anterior streak formation and early patterning of the mouse embryo. BMCDev Biol. 2005;5:17. doi: 10.1186/1471-213X-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jacobsen JC, Gregory GC, Woda JM, Thompson MN, Coser KR, Murthy V, Kohane IS, Gusella JF, Seong IS, MacDonald ME, Shioda T, Lee JM. HD CAG-correlated gene expression changes support a simple dominant gain of function. Hum Mol Genet. 2011 Jul;20(no. 14):2846–2860. doi: 10.1093/hmg/ddr195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988 Aug;85(no. 15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabrizi SJ, Scahill RI, Durr A, Roos RA, Leavitt BR, Jones R, Landwehrmeyer GB, Fox NC, Johnson H, Hicks SL, Kennard C, Craufurd D, Frost C, Langbehn DR, Reilmann R, Stout JC, TRACK-HD Investigators Biological and clinical changes in premanifest and early stage Huntington's disease in the TRACK-HD study: the 12-month longitudinal analysis. Lancet Neurol. 2011 Jan;10(no. 1):31–42. doi: 10.1016/S1474-4422(10)70276-3. [DOI] [PubMed] [Google Scholar]
- 6.Zheng S, Ghitani N, Blackburn JS, Liu JP, Zeitlin SO. A series of N-terminal epitope tagged Hdh knock-in alleles expressing normal and mutant huntingtin: their application to understanding the effect of increasing the length of normal Huntingtin's polyglutamine stretch on CAG140 mouse model pathogenesis. Mol Brain. 2012;5:28. doi: 10.1186/1756-6606-5-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nasir J, Floresco SB, O'Kusky JR, Diewert VM, Richman JM, Zeisler J, Borowski A, Marth JD, Phillips AG, Hayden MR. Targeted disruption of the Huntington's disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 1995 Jun;81(no. 5):811–823. doi: 10.1016/0092-8674(95)90542-1. [DOI] [PubMed] [Google Scholar]
- 8.del Toro D, Canals JM, Ginés S, Kojima M, Egea G, Alberch J. Mutant huntingtin impairs the post-Golgi trafficking of brain-derived neurotrophic factor but not its Val66Met polymorphism. J Neurosci Off J Soc Neurosci. 2006 Dec;26(no. 49):12748–12757. doi: 10.1523/JNEUROSCI.3873-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liot G, Zala D, Pla P, Mottet G, Piel M, Saudou F. Mutant Huntingtin alters retrograde transport of TrkB receptors in striatal dendrites. J Neurosci Off J Soc Neurosci. 2013 Apr;33(no. 15):6298–6309. doi: 10.1523/JNEUROSCI.2033-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cha JHJ. Transcriptional signatures in Huntington's disease. Prog Neurobiol. 2007 Nov;83(no. 4):228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Seong IS, Woda JM, Song JJ, Lloret A, Abeyrathne PD, Woo CJ, Gregory G, Lee JM, Wheeler VC, Walz T, Kingston RE, Gusella JF, Conlon RA, Macdonald ME. Huntingtin facilitates polycomb repressive complex 2. Hum Mol Genet. 2010;19:573–583. doi: 10.1093/hmg/ddp524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001 Jul;293(no. 5529):493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- 13.Godin JD, Colombo K, Molina-Calavita M, Keryer G, Zala D, Charrin BC, Dietrich P, Volvert ML, Guillemot F, Dragatsis I, Bellaiche Y, Saudou F, Nguyen L, Humbert S. Huntingtin is required for mitotic spindle orientation and mammalian neurogenesis. Neuron. 2010;67:392–406. doi: 10.1016/j.neuron.2010.06.027. [DOI] [PubMed] [Google Scholar]
- 14.Lo Sardo V, Zuccato C, Gaudenzi G, Vitali B, Ramos C, Tartari M, Myre MA, Walker JA, Pistocchi A, Conti L, Valenza M, Drung B, Schmidt B, Gusella J, Zeitlin S, Cotelli F, Cattaneo E. An evolutionary recent neuroepithelial cell adhesion function of huntingtin implicates ADAM10-Ncadherin. Nat Neurosci. 2012 Ma;15(no. 5):713–721. doi: 10.1038/nn.3080. [DOI] [PubMed] [Google Scholar]
- 15.Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, Hayden MR. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem. 2001 Oct;276(no. 42):39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- 16.Pardo R, Molina-Calavita M, Poizat G, Keryer G, Humbert S, Saudou F. pARIS-htt: an optimised expression platform to study huntingtin reveals functional domains required for vesicular trafficking. Mol Brain. 2010;3:17. doi: 10.1186/1756-6606-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qin ZH, Wang Y, Sapp E, Cuiffo B, Wanker E, Hayden MR, Kegel KB, Aronin N, DiFiglia M. Huntingtin bodies sequester vesicle-associated proteins by a polyproline-dependent interaction. J Neurosci. 2004;24:269–281. doi: 10.1523/JNEUROSCI.1409-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith R, Bacos K, Fedele V, Soulet D, Walz HA, Obermiiller S, Lindqvist A, Bjorkqvist M, Klein P, Onnerfjord P, Brundin P, Mulder H, Li JY. Mutant huntingtin interacts with {beta}-tubulin and disrupts vesicular transport and insulin secretion. Hum Mol Genet. 2009 Oct;18(no. 20):3942–3954. doi: 10.1093/hmg/ddp336. [DOI] [PubMed] [Google Scholar]
- 19.Reiner A, Dragatsis I, Zeitlin S, Goldowitz D. Wild-type huntingtin plays a role in brain development and neuronal survival. Mol Neurobiol. 2003 Dec;28(no. 3):259–276. doi: 10.1385/MN:28:3:259. [DOI] [PubMed] [Google Scholar]
- 20.White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat Genet. 1997 Dec;17(no. 4):404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]
- 21.Gutekunst CA, Li SH, Yi H, Ferrante RJ, Li XJ, Hersch SM. The cellular and subcellular localization of huntingtin-associated protein 1 (HAP1): comparison with huntingtin in rat and human. J Neurosci Off J Soc Neurosci. 1998 Oct;18(no. 19):7674–7686. doi: 10.1523/JNEUROSCI.18-19-07674.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choo YS, Johnson GVW, MacDonald M, Detloff PJ, Lesort M. Mutant huntingtin directly increases susceptibility of mitochondria to the calcium-induced permeability transition and cytochrome c release. Hum Mol Genet. 2004 Jul;13(no. 14):1407–1420. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 23.Seong IS, Ivanova E, Lee JM, Choo YS, Fossale E, Anderson M, Gusella JF, Laramie JM, Myers RH, Lesort M, MacDonald ME. HD CAG repeat implicates a dominant property of huntingtin in mitochondrial energy metabolism. Hum Mol Genet. 2005 Oct;14(no. 19):2871–2880. doi: 10.1093/hmg/ddi319. [DOI] [PubMed] [Google Scholar]
- 24.Chen Q, Park HC, Goligorsky MS, Chander P, Fischer SM, Gross SS. Untargeted plasma metabolite profiling reveals the broad systemic consequences of xanthine oxidoreductase inactivation in mice. PloS One. 2012;7(no. 6):e37149. doi: 10.1371/journal.pone.0037149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cohen-Gould L. Handling Cell Culture Monolayers for Transmission Electron Microscopy. Microsc Today. 2013;21(no. 03):36–39. [Google Scholar]
- 26.Folmes CDL, Dzeja PP, Nelson TJ, Terzic A. Metabolic plasticity in stem cell homeostasis and differentiation. Cell Stem Cell. 2012 Nov;11(no. 5):596–606. doi: 10.1016/j.stem.2012.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan HX, Xiong Y, Guan KL. Nutrient sensing, metabolism, and cell growth control. Mol Cell. 2013 Feb;49(no. 3):379–387. doi: 10.1016/j.molcel.2013.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Merrill GF, Kurth EJ, Hardie DG, Winder WW. AICA riboside increases AMP-activated protein kinase, fatty acid oxidation, and glucose uptake in rat muscle. Am J Physiol. 1997 Dec;273(no. 6 Pt 1):E1107–1112. doi: 10.1152/ajpendo.1997.273.6.E1107. [DOI] [PubMed] [Google Scholar]
- 29.Kubli DA, Gustafsson ÅB. Mitochondria and Mitophagy The Yin and Yang of Cell Death Control. Circ Res. 2012 Oct;111(no. 9):1208–1221. doi: 10.1161/CIRCRESAHA.112.265819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arning L, Haghikia A, Taherzadeh-Fard E, Saft C, Andrich J, Pula B, Höxtermann S, Wieczorek S, Akkad DA, Perrech M, Gold R, Epplen JT, Chan A. Mitochondrial haplogroup H correlates with ATP levels and age at onset in Huntington disease. J Mol Med. 2010 Apr;88(no. 4):431–436. doi: 10.1007/s00109-010-0589-2. [DOI] [PubMed] [Google Scholar]
- 31.Zeitlin S, Liu JP, Chapman DL, Papaioannou VE, Efstratiadis A. Increased apoptosis and early embryonic lethality in mice nullizygous for the Huntington's disease gene homologue. Nat Genet. 1995 Oct;11(no. 2):155–163. doi: 10.1038/ng1095-155. [DOI] [PubMed] [Google Scholar]
- 32.Dragatsis I, Efstratiadis A, Zeitlin S. Mouse mutant embryos lacking huntingtin are rescued from lethality by wild-type extraembryonic tissues. Dev Camb Engl. 1998 Apr;125(no. 8):1529–1539. doi: 10.1242/dev.125.8.1529. [DOI] [PubMed] [Google Scholar]
- 33.Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A. Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells. Nat Clin Pract Cardiovasc Med. 2007 Feb;4(Suppl 1):S60–67. doi: 10.1038/ncpcardio0766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vempati UD, Torraco A, Moraes CT. Mouse models of oxidative phosphorylation dysfunction and disease. Methods San Diego Calif. 2008 Dec;46(no. 4):241–247. doi: 10.1016/j.ymeth.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilding M, Coppola G, Dale B, Matteo LD. Mitochondria and human preimplantation embryo development. Reproduction. 2009 Apr;137(no. 4):619–624. doi: 10.1530/REP-08-0444. [DOI] [PubMed] [Google Scholar]
- 36.Baker C, Ebert S. Development of aerobic metabolism in utero: requirement for mitochondrial function during embryonic and foetal periods. OA Biotechnol. Apr 1;2(no. 2):16. [Google Scholar]
- 37.Garedew A, Henderson S0, Moncada S. Activated macrophages utilize glycolytic ATP to maintain mitochondrial membrane potential and prevent apoptotic cell death. Cell Death Differ. 2010 Oct;17(no. 10):1540–1550. doi: 10.1038/cdd.2010.27. [DOI] [PubMed] [Google Scholar]
- 38.Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci. 2012;107:355–415. doi: 10.1016/B978-0-12-385883-2.00005-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012 Nov;322(no. 1–2):254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 40.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer's disease brain is associated with mitochondrial dysfunction. J Neurosci Off J Soc Neurosci. 2006 Aug;26(no. 35):9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bettayeb K, Oumata N, Zhang Y, Luo W, Bustos V, Galons H, Greengard P, Meijer L, Flajolet M. Small-molecule inducers of Aβ-42 peptide production share a common mechanism of action. FASEB J Off Publ Fed Am Soc Exp Biol. 2012 Dec;26(no. 12):5115–5123. doi: 10.1096/fj.12-212985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ferri A, Cozzolino M, Crosio C, Nencini M, Casciati A, Gralla EB, Rotilio G, Valentine JS, Carrì MT. Familial ALS-superoxide dismutases associate with mitochondria and shift their redox potentials. Proc Natl Acad Sci U S A. 2006 Sep;103(no. 37):13860–13865. doi: 10.1073/pnas.0605814103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faes L, Callewaert G. Mitochondrial dysfunction in familial amyotrophic lateral sclerosis. J Bioenerg Biomembr. 2011 Dec;43(no. 6):587–592. doi: 10.1007/s10863-011-9393-0. [DOI] [PubMed] [Google Scholar]
- 44.Hatano Y, Li Y, Sato K, Asakawa S, Yamamura Y, Tomiyama H, Yoshino H, Asahina M, Kobayashi S, Hassin-Baer S, Lu CS, Ng AR, Rosales RL, Shimizu N, Toda T, Mizuno Y, Hattori N. Novel PINK1 mutations in early-onset parkinsonism. Ann Neurol. 2004 Sep;56(no. 3):424–427. doi: 10.1002/ana.20251. [DOI] [PubMed] [Google Scholar]
- 45.Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry (Mosc) 2009 Mar;48(no. 9):2045–2052. doi: 10.1021/bi8019178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bossy-Wetzel E, Petrilli A, Knott AB. Mutant huntingtin and mitochondrial dysfunction. Trends Neurosci. 2008 Dec;31(no. 12):609–616. doi: 10.1016/j.tins.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Browne SE. Mitochondria and Huntington's disease pathogenesis: insight from genetic and chemical models. Ann N Y Acad Sci. 2008 Dec;1147:358–382. doi: 10.1196/annals.1427.018. [DOI] [PubMed] [Google Scholar]
- 48.Mochel F, Haller RG. Energy deficit in Huntington disease: why it matters. J Clin Invest. 2011 Feb;121(no. 2):493–499. doi: 10.1172/JCI45691. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.