

Abstract
Wheezing and asthma are significant clinical problems for infants and young children, particularly following premature birth. Recurrent wheezing in infants can progress to persistent asthma. As in adults, altered airway structure (remodeling) and function (increased bronchoconstriction) are also important in neonatal and pediatric airway diseases. Accumulating evidence suggests that airway disease in children is influenced by perinatal factors including perturbations in normal fetal lung development, postnatal interventions in the intensive care unit, and environmental and other insults in the neonatal period. Here, in addition to genetics, maternal health, environmental processes, innate immunity, and impaired lung development/function can all influence pathogenesis of airway disease in children. We summarize current understanding of how prenatal and postnatal factors can contribute to development of airway diseases in neonates and children. Understanding these mechanisms will help identify and develop novel therapies for childhood airway diseases.
Keywords: Infant, Child, Perinatal, Asthma, Wheezing, Remodeling, Bronchoconstriction
Introduction
Diseases such as bronchopulmonary dysplasia (BPD), recurrent wheezing and asthma are significant causes of pulmonary morbidity in infants and young children worldwide, and adversely impact childhood quality of life, education and physical activity [1]. Asthma is particularly prevalent and is characterized by airway obstruction and hyperreactivity resulting from inflammation, structural changes (airway remodeling) involving enhanced epithelial and smooth muscle cell proliferation, increased airway smooth muscle (ASM) cell contractility, and impaired relaxation [2,3]. Innate and adaptive immune responses facilitate chronic inflammation characteristic of asthma and regulate airway remodeling [4]. Severity of airway disease is highly variable and there are a variety of factors that contribute to asthma pathogenesis in children [3]. Similarly, the long-term sequelae of BPD involve wheezing and asthma as well as failure to thrive, resulting in significant morbidity. Such mechanistic complexity, due to the variety of factors, make it important to identify preventable or modifiable factors to develop targeted and effective therapies for infants at risk of developing lung diseases.
While a number of genetic and environmental factors can contribute to lung diseases at different time points in a child’s life, the perinatal period (5 months antepartum to 2 months postnatal) is a critical time window during which lung development and growth are substantially influenced. A number of maternal, fetal, neonatal, environmental, and iatrogenic factors influence the subsequent course of diseases in infancy and childhood (Table 1). Our goal is to highlight some of these important contributing perinatal factors, and their potential mechanisms of action that can result in lung diseases, particularly in the conducting airways. Furthermore, no single review can comprehensively integrate the myriad of likely extrinsic vs. innate factors, including the important influence of genetics. The intent here is to establish the link between perinatal events and subsequent progression of lung diseases, thus stimulating further bench and clinical investigations.
Table 1. Key Effects of Detrimental Prenatal and Postnatal Factors in Lung Development.
Data derived from multiple references. See text for details and individual references from bibliography.
| Model | Key Findings | Mechanisms | |
|---|---|---|---|
| Prenatal Factors | Intra-amniotic,-uterine, - peritoneal endotoxin |
|
|
| Intrauterine growth restriction |
|
|
|
| Maternal smoking |
|
|
|
| Maternal Diabetes |
|
|
|
| Pollution |
|
|
|
| Postnatal Factors | Respiratory Support |
|
|
| Respiratory Infection |
|
|
|
| Allergens |
|
|
|
| Pollution |
|
|
A particularly important contributory factor to neonatal/pediatric lung disease is preterm birth. While approximately 12% of infants in the United States are born prematurely [5], the healthcare and financial burden imposed by this small fraction is disproportionately large (approximately $25 billion/year in United States) [6]. Preterm infants are at higher risk of developing lung diseases including bronchopulmonary dysplasia, recurrent wheeze, and asthma compared to term infants [7]. This risk is further modulated by multiple maternal factors that uniquely affect preterm infants such as chorioamnionitis, systemic infections, and metabolic disease that are all linked to development of airway disease [8]. Maternal behaviors, including diet and smoking, as well as environmental factors such as pollution and tobacco smoke may also be important contributors and modulators [9].
Prematurity and subsequent pulmonary immaturity often require respiratory support in the neonatal intensive care unit (ICU) placing the neonate at high risk for ongoing respiratory problems due to effects of mechanical ventilation and supplemental oxygen. Early preterm infants (24–28 weeks) are at highest risk of developing BPD [10]. Preterm infants have immature alveolar structure resulting in reduced airway tethering, airway collapse, and/or increased airway resistance which increases the respiratory load on a highly compliant chest wall [11]. Thus, altered alveolarization and airway architecture can contribute to airway dysfunction and may explain why preterm infants with BPD develop airway reactivity. Although late preterm infants (born at 33–36 weeks gestation) are less likely to develop BPD, they are still at risk of developing recurrent wheezing and asthma [12,13]. Given these important considerations, this review attempts to highlight factors that not only contribute to prematurity but also influence subsequent lung development and diseases.
Beyond prematurity, many innate and extrinsic factors can influence the development of lung disease during the perinatal period. Genetic and innate immune responses are likely important in initial sensitization to environmental factors such as allergens, while the exacerbating effects of allergens, pollution, and tobacco smoke in asthma are recognized [14]. Additional factors such as deficits in growth and nutrition, recurrent infections (particularly respiratory viruses), and impaired lung function due to other causes may also play an important role [15]. These postnatal factors are also discussed in this review. While detailed discussion of genetics and innate immunity is beyond the scope of this review, the reader is referred to other reviews on these subjects [8,9,15].
Prenatal Factors
Chorioamnionitis
Chorioamnionitis is a common source of in utero inflammation defined as the presence of inflammation within fetal membranes. Clinically, chorioamnionitis is diagnosed by the presence of maternal fever greater than 38°C, maternal tachycardia, fetal tachycardia, uterine tenderness, purulent or malodorous amniotic fluid, and/or maternal leukocytosis [16]. Inflammation in this condition is a result of bacterial or viral infection of the amniotic fluid, fetal membranes, placenta, and/or uterus. Chorioamnionitis is a leading cause of preterm labor, and is highly associated with preterm birth [8]. Neonates born to mothers with chorioamnionitis are at increased risk of mortality and of developing significant lung morbidities including acute respiratory distress syndrome, BPD, and reactive airway disorders [8]. Recent studies indicate that chorioamnionitis is correlated with an increase in risk of preterm infants for developing asthma, even after correction for confounding factors [8,17,18].
An important, yet unanswered, question is regarding the relative importance of microbes that cause chorioamnionitis vs. microbial products vs. host (maternal)-derived inflammatory mediators that cross the placenta. These issues are difficult to resolve given that all three processes occur concurrently. Nonetheless, the fetal lung is a major target of the inflammatory mediators induced by in utero infection (Figure 1). Chorioamnionitis-induced pro-inflammatory mediators primarily reach the lung through fetal breathing and swallowing of amniotic fluid. Fetal inflammatory response, as measured by proteins identified in the blood of neonates exposed to chorioamnionitis, is characterized by an upregulation of cytokines, chemokines, adhesion molecules, matrix metalloproteinases (MMPs), and angiogenic factors.
Figure 1.
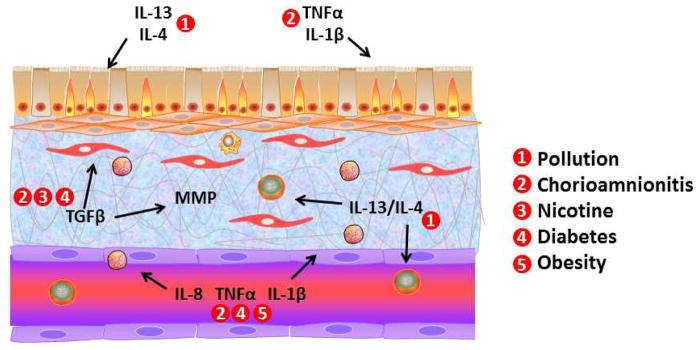
Prenatal factors and underlying mechanisms in lung development. Environmental insults such as pollution and environmental tobacco smoke exposure can have substantial influence on the inflammatory milieu and production of pro-fibrotic factors such as TGF-β and matrix metalloproteinases during critical periods of in utero lung development. Infectious and inflammatory insults, particularly chorioamnionitis can substantially lung development via cytokines such as TNFα and IL1β which target not only the developing airways but the pulmonary vasculature as well. Such factors can pre-dispose the newborn to premature birth and subsequent bronchopulmonary dysplasia, especially in the setting of supplemental oxygen and mechanical ventilation in the intensive care unit. Maternal factors such as diabetes and obesity can also influence in utero lung development, with a potential role of chronic inflammation that overlaps with other factors such as infections which are more likely to occur in mothers with these chronic diseases.
Animal models of intrauterine inflammation and infection suggest that chorioamnionitis substantially dysregulates lung development by arresting alveolarization and vascular development. For example, lipopolysaccharide (LPS) exposure in mice, with concomitant postnatal hyperoxia (simulating oxygen supplementation in the neonatal ICU of the premature newborn of a chorioamnionitis mother) arrests alveolarization, enhances fibrosis, and impairs pulmonary function, resulting in pathophysiology similar to human BPD [19]. Moreover, intra-amniotic injection of LPS in fetal mice during the saccular period of lung development causes dilation of airways and inhibition of airway branching resulting in altered lung structure [20]. This is a particularly devastating insult since airway branching is fixed by the time of birth, unlike the continued postnatal development of alveoli.
While the mechanisms underlying chorioamnionitis-induced lung injury are still under investigation, one important target may be fibroblast growth factors (FGF) which are vital for synchronized epithelial-mesenchymal branching morphogenesis and alveolarization [21]. In a study examining fetal rat lung explants, endotoxin exposure during the pseudoglandular period was found to alter lung morphogenesis with accompanying decreases in FGF9, FGF-10, and FGF-2R [20]. Another investigation suggested that nuclear factor-κB (NF-κB) activation is responsible for inhibiting the expression of FGF10 [22]. One study in mice found that chorioamnionitis increases angiogenesis during the saccular stage, which may be mediated in part by chemokines, ultimately contributing to altered vascularization, impaired gas exchange, and respiratory disease [23].
Given the known inflammatory response of both mother and fetus to chorioamnionitis, it is somewhat surprising that studies have found that chorioamnionitis may actually enhance lung maturation. The maturation response involves increases in surfactant, improved pulmonary compliance and gas exchange, and thinning of the lung mesenchyme. Surfactant serves an essential purpose of decreasing alveolar surface tension to reduce the work of breathing and prevent alveolar collapse at exhalation. A study in sheep showed that intra-amniotic injections of E. coli endotoxin administered prior to a preterm delivery cause increases in lung compliance, lung gas volumes, and alveolar phosphatidylcholine independent of cortisol [24]. Likewise, a prospective clinical study by Watterberg et al. first reported that chorioamnionitis decreases incidence of respiratory distress syndrome in preterm infants; however the incidence of chronic lung injury, noted by the manifestation of BPD is increased [25]. In preterm sheep, injection of bacterial LPS into the amniotic fluid results in increased surfactant production and lung growth [26].
The published clinical studies examining the effects of antenatal inflammation on neonatal respiratory outcomes differ considerably, and there is no clear consensus on how mechanistically intrauterine inflammation impacts neonatal pulmonary outcomes, especially given the opposing effects of increased inflammation and enhanced lung maturation. Furthermore, given the fact that many lung development mechanisms common to several different cell types are likely affected in antenatal inflammation, it is difficult to justify clinical interventions when there is lack of knowledge regarding the actual mechanisms by which inflammation affects the lung. From a mechanistic perspective, pro-inflammatory mediators that are released following exposure to antenatal inflammation likely contribute to lung maturation. E. coli LPS increases the number of alveolar type II cells in fetal mouse lungs, potentially explaining the increased surfactant production [27]. A study that exposed fetal sheep to intrauterine inflammation demonstrated higher prostaglandin (PG)E2 concentration [28]. PGE2 increases surfactant secretion in rat alveolar type 2 cells [29]. Thus, inflammatory mediators, such as LPS, can stimulate lung maturation through upregulation of the PGE2 signaling pathway resulting in increased surfactant production by alveolar type II cells.
While these animal data provide clues to enhanced surfactant production and improved lung maturation, mechanisms detrimental to the lung that result in chorioamnionitis leading to increased risk for postnatal lung diseases, particularly reactive airway disease, are largely unknown. Airway remodeling through altered cell proliferation and migration is a key feature of diseases such as asthma. In fetuses exposed to chorioamnionitis, there is increased apoptosis of distal airway epithelial cells as well as increased endothelial and smooth muscle cell proliferation [30]. Additionally, intra-amniotic LPS reduces expression of the plasma membrane protein caveolin-1 [31], which can have substantial implications for cell proliferation and intracellular signaling. For example, we have previously shown that human fetal airway smooth muscle cells express caveolin-1 [32] and exposure to LPS results in reduced caveolin-1 but increased cell proliferation, and altered extracellular matrix (ECM) production, which should lead to increased airway stiffness. In this regard, chorioamnionitis also increases expression of mediators that regulate alveolar and airway ECM. Elevated MMP-8 and MMP-9 levels have been reported in bronchoalveolar lavage fluid from premature infants exposed to chorioamnionitis [33]. Conversely, no significant differences in levels of tissue inhibitors of metalloproteinases (TIMP)-1 have been noted between preterm infants exposed to chorioamnionitis compared to preterm infants without chorioamnionitis exposure [33]. Intra-amniotic LPS increases phosphorylation of signaling proteins such as Smad2/3, Stat-1, and Stat-3 in the lung suggesting activation of the TGFβ signaling pathways which regulate ECM composition [31]. Overall, these data suggest that chorioamnionitis activates a number of pathways that can contribute to airway remodeling with functional consequences of stiffer, fibrotic airways and hyperreactivity resulting from greater smooth muscle mass.
While prevention, early recognition, and treatment of chorioamnionitis are obviously preferable, in the event of preterm labor acceleration of lung maturation is beneficial. In this regard, it is interesting that the commonly used corticosteroid, Betamethasone, increases lung maturation, but does not inhibit inflammation in the presence of LPS [34]. This raises the intriguing question: In the presence of chorioamnionitis, are steroids more effective in enhancing lung maturation? And could this influence the long-term consequences to lung function? Along a similar line, hyperoxia (80% O2) following exposure to intra-amniotic LPS actually results in enhanced postnatal alveolar and vascular development [35]. This has implications for exposure to supplemental oxygen in the neonatal ICU following premature birth under conditions of chorioamnionitis. However, the mechanisms of interactions between infection/inflammation and oxygen, and the long-term consequences of hyperoxia with inflammation on the airway or lung are not known.
Intrauterine Growth Restriction
Intrauterine growth restriction (IUGR) is a complication of pregnancy observed in both preterm and full-term births, with an incidence of 5–12% depending on the stringency of its definition. Given that IUGR also occurs in full-term babies, it becomes important to distinguish the effects of IUGR per se on lung development and subsequent disease from that resulting from prematurity alone. Fetal growth restriction has been linked to the inability of the placenta to meet the needs of the fetus for oxygen and metabolic substrates, and this environment may have an additional impact on the fetal lung. Studies have demonstrated an association between growth-restricted neonates and increased likelihood of developing cardiovascular disease, diabetes mellitus, metabolic syndrome, and asthma later in life [36]. Recent studies have identified an association between altered fetal growth and the development of wheezing at 3 years of age [37]. This association appears to be independent of gestational age at birth.
Processes related to IUGR limit fetal lung growth and maturation, making the lung more vulnerable to postnatal insults. Studies have shown that premature, small for gestational age infants are at higher risk for developing chronic lung disease compared with premature infants that are at appropriate weight for gestational age [38], again highlighting the distinction between IUGR and prematurity. In animal studies, mice with fetal growth restriction had decreased expression of mRNA for surfactant proteins [39]. In ewes, growth restricted offspring demonstrated decreased pulmonary vascularization and alveolarization [40]. A second study in ewes evaluated the impact of IUGR during late gestation on fetal pulmonary structure. Evaluation was performed near term as well as 8 weeks postnatally demonstrating decreased alveolar number and increased septal thickness, with these structural changes being more notable 8 weeks post-delivery [41]. These structural pulmonary changes could certainly cause impaired gas exchange and lung function. In fact, another study examining growth-restricted neonatal ewes identified a reduction in pulmonary diffusion capacity, hypoxemia, increases in metabolic rate, and ventilation relative to body weight in comparison to non-growth restricted ewes [42]. Further studies are needed to elucidate the mechanisms that contribute to IUGR-induced deficits in lung development and function.
Maternal Infection
Maternal infections during pregnancy can increase the risk of childhood asthma. Prior studies have shown that childhood asthma is observed at higher frequency in mothers who experienced respiratory tract infections, febrile infectious diseases, urinary tract infections, or vaginitis during pregnancy [43]. A Finnish prospective study observed a trend that a febrile infection early during pregnancy makes the risk of childhood asthma even higher [44].
The mechanisms by which maternal infections result in increased risk of childhood asthma are currently under investigation. One hypothesis is that allergic sensitization starts in utero and the asthma phenotype is programmed prior to birth such that postnatal challenges can result in asthma per se. Indeed, epidemiological effects of protective factors observed under the ‘hygiene hypothesis’ are thought to be due to the effects of endotoxin and other microbial exposure via the innate immune system. For example, the PARSIFAL study previously found that prenatal exposure to environmental factors in the farming sector is associated with increased expression of innate immune receptors such as the TLRs and with decreased incidence of atopic sensitization [45]. Failure of such tolerance mechanisms may occur in the setting of maternal infection, leading to enhanced sensitization of the fetus and newborn.
Interestingly, the mechanisms underlying the correlation between maternal infections and asthma in the progeny can be potentially gleaned from information on maternal periodontal disease. While obviously not a hugely prevalent problem, the association between maternal periodontitis, preterm birth, and adverse perinatal outcomes [46] may provide mechanistic insight. Studies have shown that chronic inflammation from periodontitis results in elevated systemic levels of cytokines such as TNF-α, IL-8 and IL-1β. These cytokines subsequently promote release of uterine stimulating factors such as PGE2 which impact placental function, onset of labor, contractions, and fetal outcomes [47]. Although no studies have specifically examined the correlation between maternal periodontal disease and development of neonatal lung disease, the observed increase in cytokines, in addition to the various inflammatory mediators associated with periodontitis could certainly have an impact on fetal lung development and postnatal pulmonary function. Other studies focusing on different etiologies of maternal inflammation have indicated that exposure to chronic inflammation in utero can impact fetal organ development. In utero exposures to cytokines such as TNF-α, IL-8, and IL-1β, have all been implicated in disruptions of fetal lung development. Furthermore, as with chorioamnionitis, it is important to consider the roles of microbes causing periodontal disease vs. their products such as LPS and/or maternal factors such as cytokines.
Maternal Smoking
Tobacco abuse continues to be a major prenatal problem with 10–12% of women admitting to tobacco use during pregnancy [48]. Moreover, approximately half of women smokers continue to do so early during pregnancy despite substantial evidence of the deleterious impact of in utero tobacco exposure on fetal development [48]. Additionally, maternal exposure to second-hand smoke due to paternal or grandparental smoking has been associated with increased risk of neonatal development of asthma and airway disease [49,50]. Mechanistically, nicotine in tobacco products results in elevated placental vascular resistance, leading to decreased uterine blood flow, increased carboxyhemoglobin, fetal hypoxia, and impaired fetal development [51]. Nicotine, fat soluble, and additive components of tobacco can cross the placenta, be detected in amniotic fluid, and have an adverse impact on fetal lung development [52]. Certainly, given the multitude of detrimental compounds present in cigarette smoke, several other mechanisms may also be at play, but have not been systematically elucidated.
Studies have shown that both maternal smoking during pregnancy and maternal exposure to tobacco results in abnormal lung function in the offspring. These abnormalities in pulmonary function can be detected in the neonatal period, continuing into childhood, and persisting throughout adult life [53]. In terms of asthma and respiratory morbidity, in utero exposure to tobacco smoke is known to impair lung function and increase airway hyperreactivity, potentially forming the underlying basis for the known association between maternal smoking during pregnancy and asthma. Indeed, maternal smoking increases neonatal Th2 cytokine responses to antigen (representing asthma pathophysiology), decreases production of the anti-inflammatory interferon gamma, and impairs TLR-mediated responses. These smoking related effects on lung structure, airway function and immune function are likely to have both short term and long term effects in the context of asthma.
Maternal smoking, before and during pregnancy, increases airway reactivity and thickening in pups exposed to house dust mite [54]. These findings are attributed at least in part to alterations in Wnt signaling, and lower expression of genes involved in the Wnt signaling pathway in newborn mouse offspring from mothers exposed to cigarette smoke during pregnancy [55]. The Wnt-β signaling pathway regulates expression of multiple genes that are critical for mediating lung development [55]. In rats, maternal smoking in pregnancy results in increased alveolar size and decreased number of alveoli in rat offspring [56]. In utero nicotine exposure has been shown to increase IL-13 and TGF-β in the neonatal lung (Figure 1) [57]. Additionally, in utero nicotine exposure increases responsiveness to methacholine and smooth muscle layer thickness and collagen III deposition around the airways in mouse offspring [58]. In utero nicotine exposure has been shown to increase procollagen type I and III mRNA expression in neonatal monkeys [59]. These data suggests that nicotine can lead to increased collagen deposition, resulting in increased airway wall dimension in the fetal lung, thereby influencing lung mechanics postnatally [59]. Furthermore, studies have demonstrated that in utero exposure to smoke alters alveolar-airway attachments, leading to abnormal airway function due to reduced forces that can counteract airway narrowing [60], akin to adult COPD. Overall, these data highlight the detrimental effects of cigarette smoke exposure in fetal and neonatal lung development, and only emphasize the need for greater efforts to curb or eliminate maternal smoking.
Maternal Diet
Studies suggest that development of asthma and wheezing is influenced by the maternal nutritional status and antenatal environmental conditions. Epidemiologic and immunologic studies also indicate that maternal diet may affect the development of neonatal allergic diseases [61]. There are a number of studies exploring the relationship between maternal nutritional status, specifically the presence or absence of particular nutrients and dietary supplements and childhood asthma.
The biologically active form of vitamin D, 1-α-hydroxyvitamin D3 (1α,25(OH)2D3), is a critical nutrient which regulates numerous physiologic functions including calcium metabolism, skeletal integrity, and immune responses [62]. Vitamin D signaling occurs predominantly through binding to the Vitamin D receptor, VDR. Serum levels of <20 ng/mL of the precursor to Vitamin D, 25(OH)D, corresponds to Vitamin D deficiency [63]. In clinical studies, low maternal Vitamin D levels have been associated with increased occurrence in wheezing in 5 year-old children [64]. Conversely, another study found that higher antepartum maternal consumption of Vitamin D was associated with a lower risk of recurrent wheeze in children at 3 years of age [65]. However, these results are not universal, with another study showing no significant associations between maternal late-pregnancy serum 25-hydroxyvitamin D levels and risk of asthma, or for transient or persistent wheeze at 1, 3, or 6 years of age [66]. Furthermore, there is one human study suggesting that vitamin D supplementation in the first year of life may actually increase asthma [67].
There has been considerable interest in understanding the mechanisms by which Vitamin D can influence airway structure and function, particularly in adult asthma. In the adult airway, proposed mechanisms include: ability of vitamin D to increase differentiation and recruitment of macrophages, enhance responsiveness to corticosteroids, and downregulate atopy [62]. In mouse models of asthma, notably higher expression of TNF-α in the lungs of vitamin D deficient mice have been noted, corresponding to a role for vitamin D deficiency in increasing airway inflammation [68]. Additionally, Vitamin D has also been shown to mediate airway smooth muscle cell proliferation and cytokine-induced chemokine expression [69,70]. There is currently a paucity of information on the potential mechanisms by which Vitamin D could influence the developing airway; however, it is hypothesized that mechanisms similar to the adult airway may be involved. For example, in ongoing studies, we have found that in human fetal airway smooth muscle cells, Vitamin D can reverse inflammation-induced changes in ECM deposition and agonist induced Ca2+ responses.
A number of studies have shown that omega-3 polyunsaturated fatty acids (PUFAs) have anti-inflammatory properties and function as immunomodulators [71]. There have been investigations examining the protective role of fish consumption in the development of asthma [72]. One study demonstrated decrease in persistent wheeze with increased fish intake in non-breastfed children [73]. A recent study suggests that increased omega-3 PUFA lung tissue concentration reduces ovalbumin (OVA)-induced airway inflammation and hyperresponsiveness [74]. In a murine model, maternal docosahexaenoic acid (DHA) supplementation enhances alveolarization and reduces inflammation in newborn pups exposed to hyperoxia [75]. These data overall suggest that maternal omega-3 PUFA supplementation may be useful in preventing the development of airway disease in infants.
Maternal Metabolic Syndrome
Metabolic syndrome in pregnant women encompasses a spectrum of conditions including obesity and insulin resistance which increases their risk of preterm delivery [76,77]. More than two-thirds of North American women of childbearing age are overweight (BMI 25–30) or obese (>=30) [76]. Metabolic syndrome results in elevated levels of pro-inflammatory mediators, such as IL-6 and C-reactive protein, in the circulation and placenta (Figure 1) [78,79]. Pregnant mothers with metabolic syndrome are at higher risk of having infants who develop wheezing and asthma, regardless of gestation at birth [80,81]. This leads to the question whether maternal metabolic syndrome has an impact on development of postnatal neonatal lung diseases. Here it is also important to distinguish between the topic of maternal issues of obesity vs. excessive weight gain of the child which can be influenced by several factors. Given the substantial rise in maternal obesity and excessive gestational weight gain, the prevention of pediatric obesity through prenatal interventions during pregnancy represents a novel area for future research [82]. However, this aspect is beyond the scope of this review.
Children of obese mothers have an increased risk of recurrent wheezing [83]. A large population based study of children born in United States cities from 1998–2000 followed after delivery showed that maternal obesity correlated with a 52% chance of asthma in the offspring by age 3 [84]. A Norwegian study identified a linear rise in the risk of wheezing at up to 18 months of age with increased maternal BMI [80]. Moreover, a Danish National Cohort Study found that maternal BMI greater than or equal to 35 was associated with severe asthma diagnosis by age 7 years [85]. With all of these studies, it is important to differentiate between obesity during pregnancy vs. continued post-partum obesity: another area that is not well studied.
There is also an association between maternal type 2 diabetes and development of neonatal airway disease [81]. Diabetes during pregnancy is classified by the White’s system which takes into account maternal age at onset of diabetes, duration of diabetes diagnosis, and the severity of the diabetes. The White’s classification distinguishes between gestational diabetes (type A) and pre-gestational diabetes. Diabetes in pregnancy can increase the risk of abnormal fetal organogenesis. One proposed mechanism by which diabetes induces embryonic anomalies is through increased oxidative stress [86]. In newborn rats exposed to intrauterine hyperglycemia, examination of their postnatal lung development revealed diminished alveolarization [87]. In vivo and in vitro experiments established that hyperglycemia and the resulting fetal hyperinsulinism resulted in delayed fetal lung maturation [88]. Moreover, exposure to intrauterine maternal hyperglycemia results in upregulation of genes coding for pro-collagen Iα and IIIα, as well as an upregulation of MMP-14, suggesting that intrauterine environmental factors apart from inflammatory conditions may also impact pulmonary development [89]. Although these studies suggest that obesity and diabetes influence neonatal airway disease, future clinical studies are needed to determine how these complications contribute to the development of airway disease. Furthermore, additional studies in animal models will help to identify mechanisms by which maternal obesity and diabetes alter lung development and airway reactivity.
Environment
Maternal exposure to deleterious environmental factors negatively impacts the developing fetus, potentially altering immune and respiratory system maturation, subsequently influencing the risk of acquiring chronic lung diseases postnatally [61]. Traffic or motor vehicle related air pollution is common especially in urban areas, and typically includes: carbon monoxide, particulate matter, nitrogen oxides, and ozone. Recent studies have revealed that ozone exposure in asthmatic individuals tends to increase the risk of airway injury and further enhance inflammation [90]. Although, there is concern that environmental factors such as air pollution may be associated with increasing prevalence of asthma the pathophysiologic processes involved in pollutant-mediated effects remain undefined.
Recent studies suggest that maternal exposures can influence development of airway disease in offspring. Maternal inhalation of diesel exhaust increases pro-inflammatory markers in fetal mouse lungs [91]. An animal study using OVA exposure showed that neonates of mothers exposed to titanium dioxide and diesel exhaust particles developed airway hyperresponsiveness and allergic inflammation [92]. In another murine model, maternal exposure to an urban particulate matter increased placental TNF-α, IL-6, and keratinocyte-derived cytokine levels [93]. Postnatal ozone exposure increased pro-inflammatory cytokine levels in whole lung compared with postnatal air exposed groups [93]. Interestingly, ozone exposure increased airway reactivity but had no effect on alveolar volume or surface density [93]. The mechanisms by which maternal exposure to air pollutants influences postnatal pulmonary structure and function are not well elucidated. A proposed explanation is that the organic components of diesel exhaust particles contain polycyclic aromatic hydrocarbons resulting in a greater production of Th2 cytokines which are known to mediate allergic and asthmatic disease (Figure 1) [93]. It is possible that maternal Th2 cytokines can potentially traverse the placental barrier and affect the developing fetus [94]. These data suggest that pregnancy exposure to either “inert” particles or known toxic substances such as diesel exhaust particles can exacerbate pulmonary inflammation and asthma in offspring.
Postnatal Factors
Respiratory Support
Immaturity of the lung and respiratory system accompanying premature birth frequently necessitates administration of supplemental oxygen (hyperoxia) with or without additional mechanical ventilation. Studies show that hyperoxia and ventilation are pro-inflammatory, and detrimental to alveolar development and airway structure and function. Prolonged respiratory support leads to alveolar simplification and impaired lung function in preterm infants [10]. Similar pathologies are observed in animal models of neonatal lung disease involving hyperoxia or mechanical ventilation and represent the “new BPD” in the post-surfactant era [95,96].
Airway dysfunction in preterm infants is due to changes in airway function and structure and over and above alveolar or vascular dysmorphogenesis. Preterm infants who developed BPD show persistent airflow obstruction [97] and increased bronchial hyperresponsiveness to methacholine [98]. Although late preterm infants do not develop severe deficits in alveolar structure/function they are still vulnerable to developing airway dysfunction [99]. Early studies showed that hyperoxia increases airway reactivity in rodents [100]. This phenotype attributed to increased thickness of the airway wall, a characteristic of remodeling. Ventilation of the neonatal lung further increases airway reactivity in newborn mice [101]. The combination of hyperoxia and ventilation can synergistically worsen airway function, although the extent of such interactions are likely to vary considerably depending on the level of prematurity, and intensity/duration of oxygen and ventilation.
The extracellular matrix (ECM) is a key component of both alveolar and airway structure. Elasticity and stiffness of the parenchyma is impacted by the composition of structural proteins, such as collagen and elastin, within the ECM. Transforming growth factor-β (TGF-β) is a key mediator of ECM composition and is highly expressed during lung development [102,103]. Dysregulation of TGF-β signaling results in abnormal alveolar development [104]. Preterm infants who develop BPD show altered TGF-β levels in bronchoalveolar lavage [105]. Histology from preterm infants with BPD demonstrates increased collagen expression and deposition in the parenchyma [106]. Expression of MMP-2 MMP-8, MMP-9, and TIMP-2 are all increased in preterm infants who developed BPD [107]. However, these changes may also be influenced by gestational age [108]. Overall, ECM remodeling in preterm infants mostly likely varies, but could contribute to altered alveolar and airway structure resulting in airway dysfunction.
Hyperoxia increases TGF-β (Figure 2) and bone morphogenetic protein (BMP) expression and signaling in newborn mice [102]. Additionally, hyperoxia increases expression of ECM proteins including collagen I, MMPs, and TIMP [96,102] as well as non-structural ECM proteins such as periostin [109]. These data suggest that hyperoxia-induced changes to ECM composition are consistent with the observed changes in lung structure of preterm infants.
Figure 2.
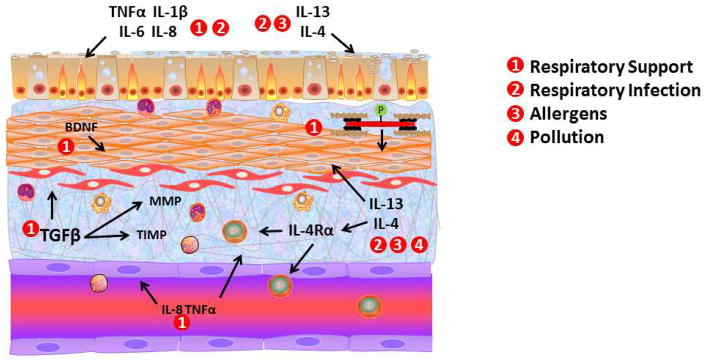
Postnatal factors and underlying mechanisms in lung development. Although prenatal factors already predispose the newborn lung to diseases such as BPD, asthma and wheezing, both environmental and iatrogenic factors in early postnatal life can be either exacerbating or causative insults. For example, in premature infants, supplemental oxygen with or without mechanical ventilation in the neonatal intensive care unit enhances the inflammatory and pro-fibrotic environment in both airways and pulmonary vasculature contributing to BPD and asthma. Isolated or superimposed respiratory infections (particular RSV) can also activate common inflammatory and pro-fibrotic mechanisms that contribute to wheezing and asthma later in life. What is less clear is the factors that sustain these detrimental changes following the neonatal period. Here, growth factors such as BDNF may be contributory. Certainly, ongoing environmental insults such as pollution, allergens and environmental tobacco smoke exposure particularly by parents can continue to have detrimental effects on postnatal lung development leading to chronic airway diseases.
The mechanisms by which hyperoxia mediates its effects on airway contraction, relaxation, and remodeling are still under investigation. Electric field stimulation induced tracheal relaxation is reduced by hyperoxia, and is attributable to lower levels of PGE2 and cAMP [110]. Newborn rats exposed to >95% O2 for 7 days have increased tracheal contractility in response to acetylcholine [111] while airway smooth muscle from newborn rats show increased contraction following 14 days of exposure to 60% O2 [112]. Expression of the neurotrophic factor, brain-derived neurotrophic factor (BDNF), and its receptor, tyrosine kinase B receptor (TrkB), is increased in newborn rats exposed to >95% O2 [113]. BDNF can increase airway smooth muscle cell proliferation and contractility [114,115], thus contributing to hyperoxia induced enhancement of airway tone (Figure 2). Separately, mechanical ventilation can also influence airway structure and function. For example, in fetal ovine lungs, mechanical ventilation increases collagen deposition and airway smooth muscle thickness [116]. Separately, mechanical ventilation increases contractility of tracheal rings isolated from newborn rats [117]. What is not known is whether parameters such as tidal volume and respiratory frequency, mechanical stretch on the developing airway, and superimposed hyperoxia influence the effects of mechanical ventilation.
Enhanced ASM proliferation is a key aspect of remodeling (Figure 2). ASM responds to cytokines and growth factors which may be enhanced by hyperoxia. Recent studies have suggested that fetal ASM cells proliferate in response to changes in redox balance [118]. Our recent study showed that moderate levels of hyperoxia (30–50% O2) increase proliferation of human fetal ASM, while higher oxygen concentrations induce apoptotic pathways [32], indicating that hyperoxia can detrimental to the developing airway regardless of concentration.
Inflammation is a typical response to hyperoxia or mechanical ventilation. Inflammation has been extensively studied in preterm infants, particularly those at risk of developing BPD. Preterm infants have elevated levels of pro-inflammatory mediators in tracheal aspirates and whole blood, suggesting systemic inflammatory responses [119]. Infants with BPD may also have increased mast cells and eosinophils [120,121] contributing to enhanced airway reactivity, as suggested by studies in the newborn rat lung [122,123]. However, not all studies show that inflammation contributes to airway dysfunction in preterm infants. Preschool children with prior BPD show hyperresponsiveness to methacholine but not adenosine monophosphate, which stimulates release of bronchoconstrictors by inflammatory cells [124]. Thus, wheezing in some former preterm infants with BPD may not be mediated by chronic inflammation. Additionally, late preterm infants who develop recurrent wheeze likely experience less inflammation than infants at risk of developing BPD. Thus the development of airway disease in preterm infants may be due to deficits in the parenchymal and airway structural network [124]. Further studies are needed to determine if airway dysfunction in this cohort is mediated by inflammation.
Children with airflow obstruction and severe asthma have increased oxidation in the airway [125]. Oxidative stress is a consequence of pro-inflammatory processes. The fraction of expired nitric oxide (FENO) is a marker of lung inflammation and increased in asthma. Recently, FENO has been identified as a potential marker for infants that will develop childhood asthma [126]. Monitoring the progression of airway inflammation may help with implementation of therapeutic strategies [127].
Lung Structure and Function
Clinical data show that preterm infants have impaired lung function, including airway obstruction [11,128]. Assessment of lung function and hyperresponsiveness may be useful tool to identify infants that are at risk of developing asthmatic symptoms later in life. Indeed, children with persistent asthmatic symptoms show lower lung function and airway hyperresponsiveness as infants [129,130]. Lower lung function in infants born at term is associated with development of wheezing between 1–3 years old [37], while one month old term infants with increased airway reactivity are at higher risk of developing airway disease and have lower forced expiratory volume (FEV1) and functional vital capacity (FVC) at age 6 [131]. Infants with lower lung function and airflow obstruction were more likely to develop asthma by adolescence [132,133]. These clinical data underline the need to understand the short-term and long-term influences of prematurity and other factors on the developing airway. Airway inflammation and remodeling are present in children with early wheeze and asthma [134,135]. Biopsies from children with asthma revealed thickening of the reticular basement membrane, epithelial injury, and inflammatory cells. Interestingly, these symptoms were found in children with atopy but without asthmatic symptoms [135]. Altered airway structure and inflammation correlates with lower FEV1 in children with difficult asthma [136]. In contrast to older children, infants with recurrent wheeze and reversible airflow obstruction do not present with reticular membrane thickening and increased eosinophils [137].
Postnatal Infection
Infants infected with rhinovirus or respiratory syncytial virus (RSV), are at an increased risk of developing asthma [138,139]. Rhinovirus, more than RSV, results in higher risk of developing asthma [139]. Viral infection alters immune responses and can interact with atopy to instigate asthmatic symptoms [138,140]. Although influenza A also presents in infants, it has not been linked to development of asthma. However, recent studies suggest that influenza be a factor for specific patient populations [141]. Preterm infants are at risk of hospitalization during infancy and acquiring viral infections, particularly RSV [142]. A recent trial suggests that infection with RSV during the first year of life, may contribute to development of recurrent wheezing in former preterm infants [143]. Thus former preterm infants with RSV infection could be particularly vulnerable to instigation of airway disease. Further investigation is needed to determine to what degree infections heighten the risk of developing recurrent wheeze and asthma.
Recent studies have investigated the impact of viral and bacterial infection on the developing lung and immune system. RSV infection increases airway inflammation in the developing lung of neonatal mice [144], which is exacerbated by OVA challenge and persists into adulthood [145]. The adverse effects of RSV on the airway may be mediated by Th2 responses (Figure 2) [146] since inhibition of Th2 responses reduces OVA-induced airway inflammation [147]. Bacterial pathogens also affect allergen-induced airway inflammation. In a murine model, Influenza A infection during the neonatal period exacerbates immune responses and airway hyperresponsiveness to house dust mite allergens [148]. Airway dysfunction persisted in these mice to adulthood [148]. A similar phenotype was reported in mice challenged with Chlamydophila pneumoniae as neonates and OVA as young adults [149]. These investigations suggest that infection during the neonatal period alters innate immune responses and can negatively impact airway inflammation and function. Modulation of Th2 responses in infants with pulmonary infection may reduce airway inflammation and susceptibility to developing airway disease. However, there is currently no information on the modulatory factors such as hyperoxia or mechanical ventilation that is often implemented upon re-admission to the NICU or the accompanying inflammation.
Allergens
Sensitization to allergens is a key component for pathogenesis of asthma and may develop early during fetal development and continues into infancy [14]. While innate immune responses as well as genetics play substantial roles in the progression towards asthma, exposures to indoor allergen such as house dust mite, cockroach, mold, and dander from animals early in life may induce sensitization, setting the stage for further disease, and increasing the risk of developing asthma [150]. Following sensitization, subsequent allergen exposures can trigger asthmatic symptoms [151]. Primary prevention studies show that reduced exposure to allergens early in life reduces sensitization and may influence the development of asthma [152,153]. Although these studies suggest that allergen exposure contributes to development of wheezing and asthma, it is unclear to what extent sensitization causes asthma [154].
From a pathophysiological perspective, allergen sensitization perpetuates chronic inflammation due, in part, to increased Th2-mediated responses that facilitate airway remodeling and hypercontractility (Figure 2) [155]. In adults, cytokines such as IL-4 and IL-13 stimulate changes in the airway that lead to an asthmatic phenotype including mucus secretion and ASM proliferation [2]. Children with asthmatic symptoms have inflammation that may be mediated by eosinophils [134] and T cells [135,136]. Data that provides insight into the mechanisms by which sensitization and subsequent allergen challenge could mediate airway inflammation and hyperresponsiveness infants, particularly preterm infants, remain limited. Future studies will be needed to elucidate early mechanisms that contribute to the pathophysiology of recurrent wheeze and asthma in young children.
Recent studies from animal models have begun to characterize the effects of allergen sensitization on the developing lung and adaptive immune response. Sensitization and challenge with allergens such as OVA or house dust mite induce robust inflammatory responses and airway hyperresponsiveness in newborn mice [156,157]. Following challenge, these mice develop increased eosinophil and T lymphocyte infiltration as well as airway remodeling that is characterized by increased collagen deposition and mucus production. Expression of Th2 cytokines, IL-4, IL-5, and IL-13, are increased in allergen challenged mice [156,157]. Allergen sensitization also alters ECM composition in the airway. Rhesus monkeys develop airway remodeling and increased collagen deposition following house dust mite sensitization [158].
Environmental Tobacco Smoke and Pollution
The effects of prenatal vs. postnatal environmental tobacco smoke (ETS) exposure are difficult to separate since mothers often smoke during and after pregnancy. In addition to the association between prenatal ETS exposure and wheezing and/or asthma in the offspring, evidence suggests a similar but less robust association in postnatal ETS exposure, with effects lasting into adolescence. Such effects of ETS (even that of in utero exposure) wane as the child reaches adulthood. Genetic polymorphisms for nicotine metabolizing genes such as glutathione S-transferase (GST) and pathways such as TGF-β and IL-1R may be important. Other immunological or epigenetic changes induced by ETS may also predispose to asthma. However, further systematic studies in these areas are required.
Postnatal exposure to pollutants has been attributed to development of airway disease in children. Children who live close to busy motorways show reduced lung function [159]. Young children who reside in high pollution areas have increased risk of developing asthma [160,161]. Exposure to diesel exhaust particles have been shown to instigate asthma in mice [162]. In 6 week old mice, inhalation of diesel exhaust particles has been shown to enhance allergic airway inflammation, hyperresponsiveness, and goblet cell hyperplasia [162]. Particulate matter induces epithelial-mesenchymal transition in airway epithelial cells in neonatal mice [163]. Persistent free radicals in the environment increase oxidation within the lung and increase airway responsiveness to methacholine [164]. Here, reduced antioxidant capacity may make neonatal animals more susceptible to particulate matter contributing to airway dysfunction [165,166]. Distal airway structure is also altered by particulate matter changing airway diameter and length [167].
Expert Commentary
The path of the fetus to a healthy extrauterine life is fraught with several obstacles, not all of which are due to genetic or physiological abnormalities. Maternal and societal behaviors and environmental insults clearly play a role, as reviewed above. Many of these factors interact substantially at different time points during lung development resulting in a variety of phenotypes. Furthermore, the contribution of the myriad of factors does not end with parturition but can frequently endanger the newborn, especially under conditions of prematurity. In this regard, the developing fetal and early postnatal lung is likely very susceptible to these insults for a number of reasons: 1) in utero where it is not required for oxygenation and ventilation, the lung is undergoing a period of very rapid growth and increasing complexity in terms of branching and alveolar genesis. Accordingly, factors that impair airway branching or induce alveolar and vascular dysmorphogenesis manifest their devastating effects postnatally when the baby (especially in prematurity) is born with the now-reduced adult number of airway branches and a simplified lung morphology leading to lifelong impairment of function; 2) in humans alveolar formation continues postnatally for a number of years. Accordingly, insults in the perinatal period can produce long-term effects in terms of lung structure and function; 3) even in the airways, exacerbated structural and functional changes induced by factors such as inflammation, infection or even oxygen can impair respiratory efforts, particularly in the setting of a premature respiratory control system and a highly compliant chest wall; 4) during early childhood the lung continues to remain one of the main initial exposure points for environmental and infectious factors, and can thus continue to suffer structural and functional changes that result in diseases such as asthma and wheezing.
Overall, these issues underline the importance of understanding the mechanisms by which various factors influence prenatal and postnatal lung development (Table 1), and the potential roles of timing and interactions between contributory factors. In this regard, a primary problem is premature birth. While identifying factors that induce premature birth is key to therapy, there is a considerable knowledge gap regarding controllable vs. non-controllable factors. In the prenatal period, factors such as chorioamnionitis and maternal infections may be preventable, but not entirely avoidable. Nonetheless, given the substantial detriment induced in the lung by these conditions, there is a clear need to identify at risk mothers with early antibiotic therapies. Similarly, early identification of IUGR and correction of factors such as placental insufficiency become essential. This only underlines the need for a well developed and implemented prenatal care plan. Within this realm is the need to allow for maximal in utero lung maturation prior to delivery, i.e. minimizing the extent of prematurity.
Compared to infectious causes, a number of factors that contribute to impaired lung development arise due to causes that are highly preventable, often by maternal efforts. Certainly tobacco abuse is one such factor that is the focus of much research as well as community outreach and education. While such campaigns have substantially reduced maternal smoking, the fact that more than 10% of women and 50% of smokers smoke during pregnancy, and also post-partum, suggests the need to better highlight the deleterious impact of both in utero and postnatal tobacco smoke exposure. Indeed, emerging studies in both animal models and human suggest that the combination of prematurity, hyperoxia with or without mechanical ventilation in the ICU, and subsequent exposure to tobacco smoke following discharge to home can be even more devastating than any of these factors individually. Similarly, a more nutritious, low-fat maternal diet with proper weight control and avoidance of gestational diabetes may be beneficial not only to maternal health but the general growth and well-being of the neonate and its developing lungs.
While prenatal factors can be controlled to a certain extent, premature birth does occur. Even with maternal corticosteroid therapy and the availability of surfactant, ICU management of the immature lung is difficult. Here, as reviewed above, factors such as supplemental oxygen and mechanical ventilation could potentially enhance the burden of premature birth placed on the developing lung. Furthermore, the issue of hyperoxia with or without mechanical ventilation is also relevant to term babies with respiratory insufficiency due to abnormal lung structure/function, infection or other causes. While it is unlikely that hyperoxia per se can be avoided, the limited data in both animal models and humans suggest that more oxygen is not necessarily good. Indeed, clinical protocols increasingly involve moderate levels of hyperoxia of limited duration, although even moderate hyperoxia may be detrimental to the developing lung as suggested by emerging studies. Nonetheless, one major “improvement” in the management of premature infants is the move away from endotracheal intubation and mechanical ventilation, replaced by equally effective nasal CPAP strategies. Whether the latter non-invasive approach also produces longer-term detrimental changes to the airway such as increased thickening or hyperreactivity, and impairs alveolar growth remains to be established. Along these lines, pharmacological interventions would be excellent adjuncts. However, the promise of inhaled nitric oxide has not been fulfilled, and new approaches are being sought. Here, phosphodiesterase inhibition as well as targeting of emerging signaling targets such as growth factors (BDNF, TGF-β, VEGF) are of interest. Assuming the survival of the premature infant and its discharge from the NICU, further detriment to the lung could be avoided through measures targeting cessation of maternal smoking, minimization of environmental tobacco smoke and pollutant exposures, and infections.
Five Year View
Both pre-clinical and clinical studies have identified a number of prenatal and postnatal factors that can have detrimental effects on the developing lung. Given that a number of such factors are controllable, clinical and research efforts should be geared towards understanding the mechanisms that underlie the effects of these factors on the developing lung (both short-term and long-term), and importantly public education of the importance of limiting such factors. From a clinical perspective, improved understanding of the effects of oxygen and ventilator strategies (including CPAP) will greatly enhance the adoption of intelligent approaches to balancing the need for sufficient oxygenation and ventilation against the harmful effects of these interventions. From a research standpoint, identification of novel signaling targets leading to altered airway structure and function will allow for pharmacological or other adjunct therapies to limit the development of asthma and other airway disorders as well as alveolar simplification and fibrosis. Overall, these complementary approaches will greatly assist in improving the pulmonary outcomes of premature infants and limit the short-term and long-term pulmonary morbidity in children.
Key Issues.
Development of lung disease in children and even in adults is influenced by both prenatal and postnatal factors
Prenatal factors such as infection and chorioamnionitis and maternal factors including smoking, obesity and diabetes can substantially impair normal fetal lung development. The mechanisms underlying changes to lung structure and function induced by these factors are under investigation, and should lead to novel therapies for limiting their detrimental effects
Postnatal factors that contribute to short-term and long-term pulmonary morbidity include treatments and procedures in the NICU necessitated by premature birth or perinatal respiratory problems. In this regard, maternal health, environmental processes, immune responses of the child, and impaired lung development and function can all influence the pathogenesis of lung disease in children. Understanding of the targets and effects of such factors on development of recurrent wheeze and asthma will help identify and develop novel therapies for children with airway diseases.
References
- 1.Asher MI, Montefort S, Bjorksten B, et al. Worldwide time trends in the prevalence of symptoms of asthma, allergic rhinoconjunctivitis, and eczema in childhood: ISAAC Phases One and Three repeat multicountry cross-sectional surveys. Lancet. 2006;368(9537):733–743. doi: 10.1016/S0140-6736(06)69283-0. [DOI] [PubMed] [Google Scholar]
- 2.Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18(5):716–725. doi: 10.1038/nm.2678. [DOI] [PubMed] [Google Scholar]
- 3.Bush A, Menzies-Gow A. Phenotypic differences between pediatric and adult asthma. Proc Am Thorac Soc. 2009;6(8):712–719. doi: 10.1513/pats.200906-046DP. [DOI] [PubMed] [Google Scholar]
- 4.Holgate ST. Innate and adaptive immune responses in asthma. Nat Med. 2012;18(5):673–683. doi: 10.1038/nm.2731. [DOI] [PubMed] [Google Scholar]
- 5.Reich ES. Pre-term births on the rise. Nature. 2012;485(7396):20. doi: 10.1038/485020a. [DOI] [PubMed] [Google Scholar]
- 6.Petrou S, Khan K. Economic costs associated with moderate and late preterm birth: primary and secondary evidence. Semin Fetal Neonatal Med. 2012;17(3):170–178. doi: 10.1016/j.siny.2012.02.001. [DOI] [PubMed] [Google Scholar]
- 7.Jaakkola JJ, Ahmed P, Ieromnimon A, et al. Preterm delivery and asthma: a systematic review and meta-analysis. J Allergy Clin Immunol. 2006;118(4):823–830. doi: 10.1016/j.jaci.2006.06.043. [DOI] [PubMed] [Google Scholar]
- 8.Kumar R, Yu Y, Story RE, et al. Prematurity, chorioamnionitis, and the development of recurrent wheezing: a prospective birth cohort study. J Allergy Clin Immunol. 2008;121(4):878–884. e876. doi: 10.1016/j.jaci.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Duijts L. Fetal and infant origins of asthma. Eur J Epidemiol. 2012;27(1):5–14. doi: 10.1007/s10654-012-9657-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10*.Jobe AH. The new bronchopulmonary dysplasia. Curr Opin Pediatr. 2011;23(2):167–172. doi: 10.1097/MOP.0b013e3283423e6b. Comprehensive review which addresses from a clinical perspective various factors that influence the development of bronchopulmomary dysplasia, while providing insights into preventive strategies. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Henschen M, Stocks J, Brookes I, Frey U. New aspects of airway mechanics in pre-term infants. Eur Respir J. 2006;27(5):913–920. doi: 10.1183/09031936.06.00036305. [DOI] [PubMed] [Google Scholar]
- 12.Martin RJ, Prakash YS, Hibbs AM. Why do former preterm infants wheeze? J Pediatr. 2013;162(3):443–444. doi: 10.1016/j.jpeds.2012.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13*.McEvoy C, Venigalla S, Schilling D, Clay N, Spitale P, Nguyen T. Respiratory function in healthy late preterm infants delivered at 33–36 weeks of gestation. J Pediatr. 2013;162(3):464–469. doi: 10.1016/j.jpeds.2012.09.042. Describes the effects of late term birth on lung function. Which suggests that this cohort is may be at risk of developing airway disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warner JO. The early life origins of asthma and related allergic disorders. Arch Dis Child. 2004;89(2):97–102. doi: 10.1136/adc.2002.013029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Turner S. Perinatal programming of childhood asthma: early fetal size, growth trajectory during infancy, and childhood asthma outcomes. Clin Dev Immunol. 2012;2012:962923. doi: 10.1155/2012/962923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fahey JO. Clinical management of intra-amniotic infection and chorioamnionitis: a review of the literature. J Midwifery Womens Health. 2008;53(3):227–235. doi: 10.1016/j.jmwh.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 17.Rusconi F, Galassi C, Forastiere F, et al. Maternal complications and procedures in pregnancy and at birth and wheezing phenotypes in children. Am J Respir Crit Care Med. 2007;175(1):16–21. doi: 10.1164/rccm.200512-1978OC. [DOI] [PubMed] [Google Scholar]
- 18.Getahun D, Strickland D, Zeiger RS, et al. Effect of chorioamnionitis on early childhood asthma. Arch Pediatr Adolesc Med. 2010;164(2):187–192. doi: 10.1001/archpediatrics.2009.238. [DOI] [PubMed] [Google Scholar]
- 19.Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol. 2010;108(5):1347–1356. doi: 10.1152/japplphysiol.01392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muratore CS, Luks FI, Zhou Y, Harty M, Reichner J, Tracy TF. Endotoxin alters early fetal lung morphogenesis. J Surg Res. 2009;155(2):225–230. doi: 10.1016/j.jss.2008.06.043. [DOI] [PubMed] [Google Scholar]
- 21.Weinstein M, Xu X, Ohyama K, Deng CX. FGFR-3 and FGFR-4 function cooperatively to direct alveogenesis in the murine lung. Development. 1998;125(18):3615–3623. doi: 10.1242/dev.125.18.3615. [DOI] [PubMed] [Google Scholar]
- 22.Benjamin JT, Carver BJ, Plosa EJ, et al. NF-kappaB activation limits airway branching through inhibition of Sp1-mediated fibroblast growth factor-10 expression. J Immunol. 2010;185(8):4896–4903. doi: 10.4049/jimmunol.1001857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Miller JD, Benjamin JT, Kelly DR, Frank DB, Prince LS. Chorioamnionitis stimulates angiogenesis in saccular stage fetal lungs via CC chemokines. Am J Physiol Lung Cell Mol Physiol. 2010;298(5):L637–645. doi: 10.1152/ajplung.00414.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jobe AH, Newnham JP, Willet KE, et al. Endotoxin-induced lung maturation in preterm lambs is not mediated by cortisol. Am J Respir Crit Care Med. 2000;162(5):1656–1661. doi: 10.1164/ajrccm.162.5.2003044. [DOI] [PubMed] [Google Scholar]
- 25*.Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics. 1996;97(2):210–215. Important clinical paper that introduces the notion that although chorioamnionitis can accelerate pulmonary maturation, it also increases lung inflammation, thereby contributing to subsequent lung injury. [PubMed] [Google Scholar]
- 26.Moss TJ, Newnham JP, Willett KE, Kramer BW, Jobe AH, Ikegami M. Early gestational intra-amniotic endotoxin: lung function, surfactant, and morphometry. Am J Respir Crit Care Med. 2002;165(6):805–811. doi: 10.1164/ajrccm.165.6.2108053. [DOI] [PubMed] [Google Scholar]
- 27.Prince LS, Okoh VO, Moninger TO, Matalon S. Lipopolysaccharide increases alveolar type II cell number in fetal mouse lungs through Toll-like receptor 4 and NF-kappaB. Am J Physiol Lung Cell Mol Physiol. 2004;287(5):L999–1006. doi: 10.1152/ajplung.00111.2004. [DOI] [PubMed] [Google Scholar]
- 28.Westover AJ, Hooper SB, Wallace MJ, Moss TJ. Prostaglandins mediate the fetal pulmonary response to intrauterine inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302(7):L664–678. doi: 10.1152/ajplung.00297.2011. [DOI] [PubMed] [Google Scholar]
- 29.Morsy MA, Isohama Y, Miyata T. Prostaglandin E(2) increases surfactant secretion via the EP(1) receptor in rat alveolar type II cells. Eur J Pharmacol. 2001;426(1–2):21–24. doi: 10.1016/s0014-2999(01)01211-0. [DOI] [PubMed] [Google Scholar]
- 30.May M, Marx A, Seidenspinner S, Speer CP. Apoptosis and proliferation in lungs of human fetuses exposed to chorioamnionitis. Histopathology. 2004;45(3):283–290. doi: 10.1111/j.1365-2559.2004.01936.x. [DOI] [PubMed] [Google Scholar]
- 31.Collins JJ, Kunzmann S, Kuypers E, et al. Antenatal glucocorticoids counteract LPS changes in TGF-beta pathway and caveolin-1 in ovine fetal lung. Am J Physiol Lung Cell Mol Physiol. 2013;304(6):L438–444. doi: 10.1152/ajplung.00251.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartman WR, Smelter DF, Sathish V, et al. Oxygen dose responsiveness of human fetal airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2012;303(8):L711–719. doi: 10.1152/ajplung.00037.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sweet DG, Curley AE, Chesshyre E, et al. The role of matrix metalloproteinases -9 and -2 in development of neonatal chronic lung disease. Acta Paediatr. 2004;93(6):791–796. doi: 10.1111/j.1651-2227.2004.tb03020.x. [DOI] [PubMed] [Google Scholar]
- 34.Kuypers E, Collins JJ, Kramer BW, et al. Intra-amniotic LPS and antenatal betamethasone: inflammation and maturation in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol. 2012;302(4):L380–389. doi: 10.1152/ajplung.00338.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tang JR, Seedorf GJ, Muehlethaler V, et al. Moderate postnatal hyperoxia accelerates lung growth and attenuates pulmonary hypertension in infant rats after exposure to intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol. 2010;299(6):L735–748. doi: 10.1152/ajplung.00153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamashiro KL, Moran TH. Perinatal environment and its influences on metabolic programming of offspring. Physiol Behav. 2010;100(5):560–566. doi: 10.1016/j.physbeh.2010.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pike KC, Crozier SR, Lucas JS, et al. Patterns of fetal and infant growth are related to atopy and wheezing disorders at age 3 years. Thorax. 2010;65(12):1099–1106. doi: 10.1136/thx.2010.134742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma P, McKay K, Rosenkrantz TS, Hussain N. Comparisons of mortality and pre-discharge respiratory outcomes in small-for-gestational-age and appropriate-for-gestational-age premature infants. BMC Pediatr. 2004;4:9. doi: 10.1186/1471-2431-4-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gortner L, Hilgendorff A, Bahner T, Ebsen M, Reiss I, Rudloff S. Hypoxia-induced intrauterine growth retardation: effects on pulmonary development and surfactant protein transcription. Biol Neonate. 2005;88(2):129–135. doi: 10.1159/000085895. [DOI] [PubMed] [Google Scholar]
- 40.Rozance PJ, Seedorf GJ, Brown A, et al. Intrauterine growth restriction decreases pulmonary alveolar and vessel growth and causes pulmonary artery endothelial cell dysfunction in vitro in fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2011;301(6):L860–871. doi: 10.1152/ajplung.00197.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maritz GS, Cock ML, Louey S, Joyce BJ, Albuquerque CA, Harding R. Effects of fetal growth restriction on lung development before and after birth: a morphometric analysis. Pediatr Pulmonol. 2001;32(3):201–210. doi: 10.1002/ppul.1109. [DOI] [PubMed] [Google Scholar]
- 42.Joyce BJ, Louey S, Davey MG, Cock ML, Hooper SB, Harding R. Compromised respiratory function in postnatal lambs after placental insufficiency and intrauterine growth restriction. Pediatr Res. 2001;50(5):641–649. doi: 10.1203/00006450-200111000-00018. [DOI] [PubMed] [Google Scholar]
- 43.Calvani M, Alessandri C, Sopo SM, et al. Infectious and uterus related complications during pregnancy and development of atopic and nonatopic asthma in children. Allergy. 2004;59(1):99–106. doi: 10.1046/j.1398-9995.2003.00338.x. [DOI] [PubMed] [Google Scholar]
- 44.Collier CH, Risnes K, Norwitz ER, Bracken MB, Illuzzi JL. Maternal Infection in Pregnancy and Risk of Asthma in Offspring. Matern Child Health J. 2013 doi: 10.1007/s10995-013-1220-2. [DOI] [PubMed] [Google Scholar]
- 45.Ege MJ, Mayer M, Normand AC, et al. Exposure to environmental microorganisms and childhood asthma. N Engl J Med. 2011;364(8):701–709. doi: 10.1056/NEJMoa1007302. [DOI] [PubMed] [Google Scholar]
- 46.Klebanoff M, Searle K. The role of inflammation in preterm birth--focus on periodontitis. Bjog. 2006;113 (Suppl 3):43–45. doi: 10.1111/j.1471-0528.2006.01121.x. [DOI] [PubMed] [Google Scholar]
- 47.Hasegawa K, Furuichi Y, Shimotsu A, et al. Associations between systemic status, periodontal status, serum cytokine levels, and delivery outcomes in pregnant women with a diagnosis of threatened premature labor. J Periodontol. 2003;74(12):1764–1770. doi: 10.1902/jop.2003.74.12.1764. [DOI] [PubMed] [Google Scholar]
- 48.Tong VT, Jones JR, Dietz PM, D’Angelo D, Bombard JM. Trends in smoking before, during, and after pregnancy - Pregnancy Risk Assessment Monitoring System (PRAMS), United States, 31 sites, 2000–2005. MMWR Surveill Summ. 2009;58(4):1–29. [PubMed] [Google Scholar]
- 49.Burke H, Leonardi-Bee J, Hashim A, et al. Prenatal and passive smoke exposure and incidence of asthma and wheeze: systematic review and meta-analysis. Pediatrics. 2012;129(4):735–744. doi: 10.1542/peds.2011-2196. [DOI] [PubMed] [Google Scholar]
- 50.Li YF, Langholz B, Salam MT, Gilliland FD. Maternal and grandmaternal smoking patterns are associated with early childhood asthma. Chest. 2005;127(4):1232–1241. doi: 10.1378/chest.127.4.1232. [DOI] [PubMed] [Google Scholar]
- 51.Herrmann M, King K, Weitzman M. Prenatal tobacco smoke and postnatal secondhand smoke exposure and child neurodevelopment. Curr Opin Pediatr. 2008;20(2):184–190. doi: 10.1097/MOP.0b013e3282f56165. [DOI] [PubMed] [Google Scholar]
- 52.Rehan VK, Wang Y, Sugano S, et al. In utero nicotine exposure alters fetal rat lung alveolar type II cell proliferation, differentiation, and metabolism. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L323–333. doi: 10.1152/ajplung.00071.2006. [DOI] [PubMed] [Google Scholar]
- 53.Landau LI. Tobacco smoke exposure and tracking of lung function into adult life. Paediatr Respir Rev. 2008;9(1):39–43. doi: 10.1016/j.prrv.2007.11.002. quiz 43–34. [DOI] [PubMed] [Google Scholar]
- 54.Blacquiere MJ, Timens W, Melgert BN, Geerlings M, Postma DS, Hylkema MN. Maternal smoking during pregnancy induces airway remodelling in mice offspring. Eur Respir J. 2009;33(5):1133–1140. doi: 10.1183/09031936.00129608. [DOI] [PubMed] [Google Scholar]
- 55.Blacquiere MJ, Timens W, van den Berg A, Geerlings M, Postma DS, Hylkema MN. Maternal smoking during pregnancy decreases Wnt signalling in neonatal mice. Thorax. 2010;65(6):553–554. doi: 10.1136/thx.2009.120154. [DOI] [PubMed] [Google Scholar]
- 56.Collins MH, Moessinger AC, Kleinerman J, et al. Fetal lung hypoplasia associated with maternal smoking: a morphometric analysis. Pediatr Res. 1985;19(4):408–412. doi: 10.1203/00006450-198519040-00018. [DOI] [PubMed] [Google Scholar]
- 57.Wongtrakool C, Grooms K, Ping XD, et al. In utero nicotine exposure promotes M2 activation in neonatal mouse alveolar macrophages. Pediatr Res. 2012;72(2):147–153. doi: 10.1038/pr.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wongtrakool C, Wang N, Hyde DM, Roman J, Spindel ER. Prenatal nicotine exposure alters lung function and airway geometry through alpha7 nicotinic receptors. Am J Respir Cell Mol Biol. 2012;46(5):695–702. doi: 10.1165/rcmb.2011-0028OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sekhon HS, Keller JA, Proskocil BJ, Martin EL, Spindel ER. Maternal nicotine exposure upregulates collagen gene expression in fetal monkey lung. Association with alpha7 nicotinic acetylcholine receptors. Am J Respir Cell Mol Biol. 2002;26(1):31–41. doi: 10.1165/ajrcmb.26.1.4170. [DOI] [PubMed] [Google Scholar]
- 60.Elliot JG, Carroll NG, James AL, Robinson PJ. Airway alveolar attachment points and exposure to cigarette smoke in utero. Am J Respir Crit Care Med. 2003;167(1):45–49. doi: 10.1164/rccm.2110005. [DOI] [PubMed] [Google Scholar]
- 61.Dietert RR. Maternal and childhood asthma: risk factors, interactions, and ramifications. Reprod Toxicol. 2011;32(2):198–204. doi: 10.1016/j.reprotox.2011.04.007. [DOI] [PubMed] [Google Scholar]
- 62.Paul G, Brehm JM, Alcorn JF, Holguin F, Aujla SJ, Celedon JC. Vitamin D and asthma. Am J Respir Crit Care Med. 2012;185(2):124–132. doi: 10.1164/rccm.201108-1502CI. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ross AC, Manson JE, Abrams SA, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96(1):53–58. doi: 10.1210/jc.2010-2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Devereux G, Litonjua AA, Turner SW, et al. Maternal vitamin D intake during pregnancy and early childhood wheezing. Am J Clin Nutr. 2007;85(3):853–859. doi: 10.1093/ajcn/85.3.853. [DOI] [PubMed] [Google Scholar]
- 65.Camargo CA, Jr, Rifas-Shiman SL, Litonjua AA, et al. Maternal intake of vitamin D during pregnancy and risk of recurrent wheeze in children at 3 y of age. Am J Clin Nutr. 2007;85(3):788–795. doi: 10.1093/ajcn/85.3.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pike KC, Inskip HM, Robinson S, et al. Maternal late-pregnancy serum 25-hydroxyvitamin D in relation to childhood wheeze and atopic outcomes. Thorax. 2012;67(11):950–956. doi: 10.1136/thoraxjnl-2012-201888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hypponen E, Sovio U, Wjst M, et al. Infant vitamin d supplementation and allergic conditions in adulthood: northern Finland birth cohort 1966. Ann N Y Acad Sci. 2004;1037:84–95. doi: 10.1196/annals.1337.013. [DOI] [PubMed] [Google Scholar]
- 68.Agrawal T, Gupta GK, Agrawal DK. Vitamin D deficiency decreases the expression of VDR and prohibitin in the lungs of mice with allergic airway inflammation. Exp Mol Pathol. 2012;93(1):74–81. doi: 10.1016/j.yexmp.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Banerjee A, Damera G, Bhandare R, et al. Vitamin D and glucocorticoids differentially modulate chemokine expression in human airway smooth muscle cells. Br J Pharmacol. 2008;155(1):84–92. doi: 10.1038/bjp.2008.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Damera G, Fogle HW, Lim P, et al. Vitamin D inhibits growth of human airway smooth muscle cells through growth factor-induced phosphorylation of retinoblastoma protein and checkpoint kinase 1. Br J Pharmacol. 2009;158(6):1429–1441. doi: 10.1111/j.1476-5381.2009.00428.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Prescott SL, Calder PC. N-3 polyunsaturated fatty acids and allergic disease. Curr Opin Clin Nutr Metab Care. 2004;7(2):123–129. doi: 10.1097/00075197-200403000-00004. [DOI] [PubMed] [Google Scholar]
- 72.Maslova E, Strom M, Oken E, et al. Fish intake during pregnancy and the risk of child asthma and allergic rhinitis - longitudinal evidence from the Danish National Birth Cohort. Br J Nutr. 2013:1–13. doi: 10.1017/S000711451300038X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Romieu I, Torrent M, Garcia-Esteban R, et al. Maternal fish intake during pregnancy and atopy and asthma in infancy. Clin Exp Allergy. 2007;37(4):518–525. doi: 10.1111/j.1365-2222.2007.02685.x. [DOI] [PubMed] [Google Scholar]
- 74.Bilal S, Haworth O, Wu L, Weylandt KH, Levy BD, Kang JX. Fat-1 transgenic mice with elevated omega-3 fatty acids are protected from allergic airway responses. Biochim Biophys Acta. 2011;1812(9):1164–1169. doi: 10.1016/j.bbadis.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rogers LK, Valentine CJ, Pennell M, et al. Maternal docosahexaenoic acid supplementation decreases lung inflammation in hyperoxia-exposed newborn mice. J Nutr. 2011;141(2):214–222. doi: 10.3945/jn.110.129882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yogev Y, Visser GH. Obesity, gestational diabetes and pregnancy outcome. Semin Fetal Neonatal Med. 2009;14(2):77–84. doi: 10.1016/j.siny.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 77.Cnattingius S, Villamor E, Johansson S, et al. Maternal obesity and risk of preterm delivery. Jama. 2013;309(22):2362–2370. doi: 10.1001/jama.2013.6295. [DOI] [PubMed] [Google Scholar]
- 78.Challier JC, Basu S, Bintein T, et al. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29(3):274–281. doi: 10.1016/j.placenta.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Basu S, Haghiac M, Surace P, et al. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring) 2011;19(3):476–482. doi: 10.1038/oby.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Haberg SE, Stigum H, London SJ, Nystad W, Nafstad P. Maternal obesity in pregnancy and respiratory health in early childhood. Paediatr Perinat Epidemiol. 2009;23(4):352–362. doi: 10.1111/j.1365-3016.2009.01034.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Azad MB, Becker AB, Kozyrskyj AL. Association of maternal diabetes and child asthma. Pediatr Pulmonol. 2012 doi: 10.1002/ppul.22668. [DOI] [PubMed] [Google Scholar]
- 82.Adamo KB, Ferraro ZM, Goldfield G, et al. The Maternal Obesity Management (MOM) Trial Protocol: A lifestyle intervention during pregnancy to minimize downstream obesity. Contemp Clin Trials. 2013;35(1):87–96. doi: 10.1016/j.cct.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 83.Kumar R, Story RE, Pongracic JA, et al. Maternal Pre-Pregnancy Obesity and Recurrent Wheezing in Early Childhood. Pediatr Allergy Immunol Pulmonol. 2010;23(3):183–190. doi: 10.1089/ped.2010.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Reichman NE, Nepomnyaschy L. Maternal pre-pregnancy obesity and diagnosis of asthma in offspring at age 3 years. Matern Child Health J. 2008;12(6):725–733. doi: 10.1007/s10995-007-0292-2. [DOI] [PubMed] [Google Scholar]
- 85.Harpsoe MC, Basit S, Bager P, et al. Maternal obesity, gestational weight gain, and risk of asthma and atopic disease in offspring: A study within the Danish National Birth Cohort. J Allergy Clin Immunol. 2013;131(4):1033–1040. doi: 10.1016/j.jaci.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 86.Ornoy A, Rand SB, Bischitz N. Hyperglycemia and hypoxia are interrelated in their teratogenic mechanism: studies on cultured rat embryos. Birth Defects Res B Dev Reprod Toxicol. 2010;89(2):106–115. doi: 10.1002/bdrb.20230. [DOI] [PubMed] [Google Scholar]
- 87.Koskinen A, Lukkarinen H, Moritz N, Aho H, Kaapa P, Soukka H. Fetal hyperglycemia alters lung structural development in neonatal rat. Pediatr Pulmonol. 2012;47(3):275–282. doi: 10.1002/ppul.21541. [DOI] [PubMed] [Google Scholar]
- 88.Bourbon JR, Farrell PM. Fetal lung development in the diabetic pregnancy. Pediatr Res. 1985;19(3):253–267. doi: 10.1203/00006450-198503000-00001. [DOI] [PubMed] [Google Scholar]
- 89.Koskinen A, Laiho A, Lukkarinen H, Kaapa P, Soukka H. Maternal hyperglycemia modifies extracellular matrix signaling pathways in neonatal rat lung. Neonatology. 2010;98(4):387–396. doi: 10.1159/000317010. [DOI] [PubMed] [Google Scholar]
- 90.Dye JA, Madden MC, Richards JH, Lehmann JR, Devlin RB, Costa DL. Ozone effects on airway responsiveness, lung injury, and inflammation. Comparative rat strain and in vivo/in vitro investigations. Inhal Toxicol. 1999;11(11):1015–1040. doi: 10.1080/089583799196664. [DOI] [PubMed] [Google Scholar]
- 91.Auten RL, Gilmour MI, Krantz QT, Potts EN, Mason SN, Foster WM. Maternal diesel inhalation increases airway hyperreactivity in ozone-exposed offspring. Am J Respir Cell Mol Biol. 2012;46(4):454–460. doi: 10.1165/rcmb.2011-0256OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 92.Fedulov AV, Leme A, Yang Z, et al. Pulmonary exposure to particles during pregnancy causes increased neonatal asthma susceptibility. Am J Respir Cell Mol Biol. 2008;38(1):57–67. doi: 10.1165/rcmb.2007-0124OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Auten RL, Potts EN, Mason SN, Fischer B, Huang Y, Foster WM. Maternal exposure to particulate matter increases postnatal ozone-induced airway hyperreactivity in juvenile mice. Am J Respir Crit Care Med. 2009;180(12):1218–1226. doi: 10.1164/rccm.200901-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 94.Lim RH, Kobzik L. Maternal transmission of asthma risk. Am J Reprod Immunol. 2009;61(1):1–10. doi: 10.1111/j.1600-0897.2008.00671.x. [DOI] [PubMed] [Google Scholar]
- 95.Hillman NH, Polglase GR, Pillow JJ, Saito M, Kallapur SG, Jobe AH. Inflammation and lung maturation from stretch injury in preterm fetal sheep. Am J Physiol Lung Cell Mol Physiol. 2011;300(2):L232–241. doi: 10.1152/ajplung.00294.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Velten M, Britt RD, Jr, Heyob KM, et al. Prenatal inflammation exacerbates hyperoxia-induced functional and structural changes in adult mice. Am J Physiol Regul Integr Comp Physiol. 2012;303(3):R279–290. doi: 10.1152/ajpregu.00029.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fakhoury KF, Sellers C, Smith EO, Rama JA, Fan LL. Serial measurements of lung function in a cohort of young children with bronchopulmonary dysplasia. Pediatrics. 2010;125(6):e1441–1447. doi: 10.1542/peds.2009-0668. [DOI] [PubMed] [Google Scholar]
- 98*.Halvorsen T, Skadberg BT, Eide GE, Roksund O, Aksnes L, Oymar K. Characteristics of asthma and airway hyper-responsiveness after premature birth. Pediatr Allergy Immunol. 2005;16(6):487–494. doi: 10.1111/j.1399-3038.2005.00314.x. Measurement of airway hyperresponsiveness in children who were former preterm infants with or without development of BPD. This study shows that preterm infants that do nto develop chronic lung diease can also develop airway hyperresponsiveness. [DOI] [PubMed] [Google Scholar]
- 99.Goyal NK, Fiks AG, Lorch SA. Association of late-preterm birth with asthma in young children: practice-based study. Pediatrics. 2011;128(4):e830–838. doi: 10.1542/peds.2011-0809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hershenson MB, Aghili S, Punjabi N, et al. Hyperoxia-induced airway hyperresponsiveness and remodeling in immature rats. Am J Physiol. 1992;262(3 Pt 1):L263–269. doi: 10.1152/ajplung.1992.262.3.L263. [DOI] [PubMed] [Google Scholar]
- 101.Iben SC, Haxhiu MA, Farver CF, Miller MJ, Martin RJ. Short-term mechanical ventilation increases airway reactivity in rat pups. Pediatr Res. 2006;60(2):136–140. doi: 10.1203/01.pdr.0000227447.55247.7d. [DOI] [PubMed] [Google Scholar]
- 102.Alejandre-Alcazar MA, Kwapiszewska G, Reiss I, et al. Hyperoxia modulates TGF-beta/BMP signaling in a mouse model of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol. 2007;292(2):L537–549. doi: 10.1152/ajplung.00050.2006. [DOI] [PubMed] [Google Scholar]
- 103.Alejandre-Alcazar MA, Michiels-Corsten M, Vicencio AG, et al. TGF-beta signaling is dynamically regulated during the alveolarization of rodent and human lungs. Dev Dyn. 2008;237(1):259–269. doi: 10.1002/dvdy.21403. [DOI] [PubMed] [Google Scholar]
- 104.Warburton D, Shi W, Xu B. TGF-beta-Smad3 signaling in emphysema and pulmonary fibrosis: an epigenetic aberration of normal development? Am J Physiol Lung Cell Mol Physiol. 2013;304(2):L83–85. doi: 10.1152/ajplung.00258.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Been JV, Debeer A, van Iwaarden JF, et al. Early alterations of growth factor patterns in bronchoalveolar lavage fluid from preterm infants developing bronchopulmonary dysplasia. Pediatr Res. 2010;67(1):83–89. doi: 10.1203/PDR.0b013e3181c13276. [DOI] [PubMed] [Google Scholar]
- 106.Kaarteenaho-Wiik R, Paakko P, Herva R, Risteli J, Soini Y. Type I and III collagen protein precursors and mRNA in the developing human lung. J Pathol. 2004;203(1):567–574. doi: 10.1002/path.1547. [DOI] [PubMed] [Google Scholar]
- 107.Cederqvist K, Sorsa T, Tervahartiala T, et al. Matrix metalloproteinases-2, -8, and -9 and TIMP-2 in tracheal aspirates from preterm infants with respiratory distress. Pediatrics. 2001;108(3):686–692. doi: 10.1542/peds.108.3.686. [DOI] [PubMed] [Google Scholar]
- 108.Sweet DG, McMahon KJ, Curley AE, O’Connor CM, Halliday HL. Type I collagenases in bronchoalveolar lavage fluid from preterm babies at risk of developing chronic lung disease. Arch Dis Child Fetal Neonatal Ed. 2001;84(3):F168–171. doi: 10.1136/fn.84.3.F168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bozyk PD, Bentley JK, Popova AP, et al. Neonatal periostin knockout mice are protected from hyperoxia-induced alveolar simplication. PLoS One. 2012;7(2):e31336. doi: 10.1371/journal.pone.0031336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mhanna MJ, Haxhiu MA, Jaber MA, et al. Hyperoxia impairs airway relaxation in immature rats via a cAMP-mediated mechanism. J Appl Physiol. 2004;96(5):1854–1860. doi: 10.1152/japplphysiol.01178.2002. [DOI] [PubMed] [Google Scholar]
- 111.Agani FH, Kuo NT, Chang CH, et al. Effect of hyperoxia on substance P expression and airway reactivity in the developing lung. Am J Physiol. 1997;273(1 Pt 1):L40–45. doi: 10.1152/ajplung.1997.273.1.L40. [DOI] [PubMed] [Google Scholar]
- 112.Belik J, Jankov RP, Pan J, Yi M, Chaudhry I, Tanswell AK. Chronic O2 exposure in the newborn rat results in decreased pulmonary arterial nitric oxide release and altered smooth muscle response to isoprostane. J Appl Physiol. 2004;96(2):725–730. doi: 10.1152/japplphysiol.00825.2003. [DOI] [PubMed] [Google Scholar]
- 113.Yao Q, Haxhiu MA, Zaidi SI, Liu S, Jafri A, Martin RJ. Hyperoxia enhances brain-derived neurotrophic factor and tyrosine kinase B receptor expression in peribronchial smooth muscle of neonatal rats. Am J Physiol Lung Cell Mol Physiol. 2005;289(2):L307–314. doi: 10.1152/ajplung.00030.2005. [DOI] [PubMed] [Google Scholar]
- 114.Abcejo AJ, Sathish V, Smelter DF, et al. Brain-derived neurotrophic factor enhances calcium regulatory mechanisms in human airway smooth muscle. PLoS One. 2012;7(8):e44343. doi: 10.1371/journal.pone.0044343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Aravamudan B, Thompson M, Pabelick C, Prakash YS. Brain-derived neurotrophic factor induces proliferation of human airway smooth muscle cells. J Cell Mol Med. 2012;16(4):812–823. doi: 10.1111/j.1582-4934.2011.01356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.O’Reilly M, Hooper SB, Allison BJ, et al. Persistent bronchiolar remodeling following brief ventilation of the very immature ovine lung. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L992–L1001. doi: 10.1152/ajplung.00099.2009. [DOI] [PubMed] [Google Scholar]
- 117.Fukunaga T, Davies P, Zhang L, Hashida Y, Motoyama EK. Prolonged high intermittent positive-pressure ventilation induces airway remodeling and reactivity in young rats. Am J Physiol. 1998;275(3 Pt 1):L567–573. doi: 10.1152/ajplung.1998.275.3.L567. [DOI] [PubMed] [Google Scholar]
- 118.Pandya HC, Snetkov VA, Twort CH, Ward JP, Hirst SJ. Oxygen regulates mitogen-stimulated proliferation of fetal human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. 2002;283(6):L1220–1230. doi: 10.1152/ajplung.00268.2001. [DOI] [PubMed] [Google Scholar]
- 119.Ambalavanan N, Carlo WA, D’Angio CT, et al. Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics. 2009;123(4):1132–1141. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brostrom EB, Katz-Salamon M, Lundahl J, Hallden G, Winbladh B. Eosinophil activation in preterm infants with lung disease. Acta Paediatr. 2007;96(1):23–28. doi: 10.1111/j.1651-2227.2006.00002.x. [DOI] [PubMed] [Google Scholar]
- 121.Bhattacharya S, Go D, Krenitsky DL, et al. Genome-wide transcriptional profiling reveals connective tissue mast cell accumulation in bronchopulmonary dysplasia. Am J Respir Crit Care Med. 2012;186(4):349–358. doi: 10.1164/rccm.201203-0406OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brock TG, Di Giulio C. Prolonged exposure to hyperoxia increases perivascular mast cells in rat lungs. J Histochem Cytochem. 2006;54(11):1239–1246. doi: 10.1369/jhc.6A7007.2006. [DOI] [PubMed] [Google Scholar]
- 123.Schultz ED, Potts EN, Mason SN, Foster WM, Auten RL. Mast cells mediate hyperoxia-induced airway hyper-reactivity in newborn rats. Pediatr Res. 2010;68(1):70–74. doi: 10.1203/PDR.0b013e3181e0cd97. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 124.Kim do K, Choi SH, Yu J, Yoo Y, Kim B, Koh YY. Bronchial responsiveness to methacholine and adenosine 5′-monophosphate in preschool children with bronchopulmonary dysplasia. Pediatr Pulmonol. 2006;41(6):538–543. doi: 10.1002/ppul.20402. [DOI] [PubMed] [Google Scholar]
- 125.Fitzpatrick AM, Teague WG, Holguin F, Yeh M, Brown LA. Airway glutathione homeostasis is altered in children with severe asthma: evidence for oxidant stress. J Allergy Clin Immunol. 2009;123(1):146–152. e148. doi: 10.1016/j.jaci.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Chawes BL, Buchvald F, Bischoff AL, et al. Elevated exhaled nitric oxide in high-risk neonates precedes transient early but not persistent wheeze. Am J Respir Crit Care Med. 2010;182(2):138–142. doi: 10.1164/rccm.200909-1377OC. [DOI] [PubMed] [Google Scholar]
- 127.Zacharasiewicz A, Wilson N, Lex C, et al. Clinical use of noninvasive measurements of airway inflammation in steroid reduction in children. Am J Respir Crit Care Med. 2005;171(10):1077–1082. doi: 10.1164/rccm.200409-1242OC. [DOI] [PubMed] [Google Scholar]
- 128.Filbrun AG, Popova AP, Linn MJ, McIntosh NA, Hershenson MB. Longitudinal measures of lung function in infants with bronchopulmonary dysplasia. Pediatr Pulmonol. 2011;46(4):369–375. doi: 10.1002/ppul.21378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kurukulaaratchy RJ, Matthews S, Holgate ST, Arshad SH. Predicting persistent disease among children who wheeze during early life. Eur Respir J. 2003;22(5):767–771. doi: 10.1183/09031936.03.00005903. [DOI] [PubMed] [Google Scholar]
- 130.Morgan WJ, Stern DA, Sherrill DL, et al. Outcome of asthma and wheezing in the first 6 years of life: follow-up through adolescence. Am J Respir Crit Care Med. 2005;172(10):1253–1258. doi: 10.1164/rccm.200504-525OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Palmer LJ, Rye PJ, Gibson NA, Burton PR, Landau LI, Lesouef PN. Airway responsiveness in early infancy predicts asthma, lung function, and respiratory symptoms by school age. Am J Respir Crit Care Med. 2001;163(1):37–42. doi: 10.1164/ajrccm.163.1.2005013. [DOI] [PubMed] [Google Scholar]
- 132.Bisgaard H, Jensen SM, Bonnelykke K. Interaction between asthma and lung function growth in early life. Am J Respir Crit Care Med. 2012;185(11):1183–1189. doi: 10.1164/rccm.201110-1922OC. [DOI] [PubMed] [Google Scholar]
- 133.Hovland V, Riiser A, Mowinckel P, Carlsen KH, Lodrup Carlsen KC. The significance of early recurrent wheeze for asthma outcomes in late childhood. Eur Respir J. 2013;41(4):838–845. doi: 10.1183/09031936.00071512. [DOI] [PubMed] [Google Scholar]
- 134.Saglani S, Payne DN, Zhu J, et al. Early detection of airway wall remodeling and eosinophilic inflammation in preschool wheezers. Am J Respir Crit Care Med. 2007;176(9):858–864. doi: 10.1164/rccm.200702-212OC. [DOI] [PubMed] [Google Scholar]
- 135.Turato G, Barbato A, Baraldo S, et al. Nonatopic children with multitrigger wheezing have airway pathology comparable to atopic asthma. Am J Respir Crit Care Med. 2008;178(5):476–482. doi: 10.1164/rccm.200712-1818OC. [DOI] [PubMed] [Google Scholar]
- 136.Payne DN, Qiu Y, Zhu J, et al. Airway inflammation in children with difficult asthma: relationships with airflow limitation and persistent symptoms. Thorax. 2004;59(10):862–869. doi: 10.1136/thx.2003.017244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137*.Saglani S, Malmstrom K, Pelkonen AS, et al. Airway remodeling and inflammation in symptomatic infants with reversible airflow obstruction. Am J Respir Crit Care Med. 2005;171(7):722–727. doi: 10.1164/rccm.200410-1404OC. Demonstrates that inflammation that is characteristic of adult asthma is not present in infants with reverible airflow obstruction. This study provide a pathological description of the airway from infants at risk of developing asthma. [DOI] [PubMed] [Google Scholar]
- 138.Kusel MM, de Klerk NH, Kebadze T, et al. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J Allergy Clin Immunol. 2007;119(5):1105–1110. doi: 10.1016/j.jaci.2006.12.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139*.Jackson DJ, Gangnon RE, Evans MD, et al. Wheezing rhinovirus illnesses in early life predict asthma development in high-risk children. Am J Respir Crit Care Med. 2008;178(7):667–672. doi: 10.1164/rccm.200802-309OC. Evaluates the effects of viral infection, most notably rhinovirus, on the risk of developing asthma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat Med. 2012;18(5):726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
- 141.Mandelcwajg A, Moulin F, Menager C, Rozenberg F, Lebon P, Gendrel D. Underestimation of influenza viral infection in childhood asthma exacerbations. J Pediatr. 2010;157(3):505–506. doi: 10.1016/j.jpeds.2010.04.067. [DOI] [PubMed] [Google Scholar]
- 142.Resch B, Pasnocht A, Gusenleitner W, Muller W. Rehospitalisations for respiratory disease and respiratory syncytial virus infection in preterm infants of 29–36 weeks gestational age. J Infect. 2005;50(5):397–403. doi: 10.1016/j.jinf.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 143.Blanken MO, Rovers MM, Molenaar JM, et al. Respiratory syncytial virus and recurrent wheeze in healthy preterm infants. N Engl J Med. 2013;368(19):1791–1799. doi: 10.1056/NEJMoa1211917. [DOI] [PubMed] [Google Scholar]
- 144.Becnel D, You D, Erskin J, Dimina DM, Cormier SA. A role for airway remodeling during respiratory syncytial virus infection. Respir Res. 2005;6:122. doi: 10.1186/1465-9921-6-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.You D, Becnel D, Wang K, Ripple M, Daly M, Cormier SA. Exposure of neonates to respiratory syncytial virus is critical in determining subsequent airway response in adults. Respir Res. 2006;7:107. doi: 10.1186/1465-9921-7-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ripple MJ, You D, Honnegowda S, et al. Immunomodulation with IL-4R alpha antisense oligonucleotide prevents respiratory syncytial virus-mediated pulmonary disease. J Immunol. 2010;185(8):4804–4811. doi: 10.4049/jimmunol.1000484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Siegle JS, Hansbro N, Dong C, Angkasekwinai P, Foster PS, Kumar RK. Blocking induction of T helper type 2 responses prevents development of disease in a model of childhood asthma. Clin Exp Immunol. 2011;165(1):19–28. doi: 10.1111/j.1365-2249.2011.04392.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Al-Garawi A, Fattouh R, Botelho F, et al. Influenza A facilitates sensitization to house dust mite in infant mice leading to an asthma phenotype in adulthood. Mucosal Immunol. 2011;4(6):682–694. doi: 10.1038/mi.2011.35. [DOI] [PubMed] [Google Scholar]
- 149.Horvat JC, Starkey MR, Kim RY, et al. Early-life chlamydial lung infection enhances allergic airways disease through age-dependent differences in immunopathology. J Allergy Clin Immunol. 2010;125(3):617–625. 625, e611–625, e616. doi: 10.1016/j.jaci.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 150.Illi S, von Mutius E, Lau S, Niggemann B, Gruber C, Wahn U. Perennial allergen sensitisation early in life and chronic asthma in children: a birth cohort study. Lancet. 2006;368(9537):763–770. doi: 10.1016/S0140-6736(06)69286-6. [DOI] [PubMed] [Google Scholar]
- 151.Salo PM, Arbes SJ, Jr, Crockett PW, Thorne PS, Cohn RD, Zeldin DC. Exposure to multiple indoor allergens in US homes and its relationship to asthma. J Allergy Clin Immunol. 2008;121(3):678–684. e672. doi: 10.1016/j.jaci.2007.12.1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arshad SH, Bateman B, Matthews SM. Primary prevention of asthma and atopy during childhood by allergen avoidance in infancy: a randomised controlled study. Thorax. 2003;58(6):489–493. doi: 10.1136/thorax.58.6.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mihrshahi S, Peat JK, Marks GB, et al. Eighteen-month outcomes of house dust mite avoidance and dietary fatty acid modification in the Childhood Asthma Prevention Study (CAPS) J Allergy Clin Immunol. 2003;111(1):162–168. doi: 10.1067/mai.2003.36. [DOI] [PubMed] [Google Scholar]
- 154.Arshad SH. Does exposure to indoor allergens contribute to the development of asthma and allergy? Curr Allergy Asthma Rep. 2010;10(1):49–55. doi: 10.1007/s11882-009-0082-6. [DOI] [PubMed] [Google Scholar]
- 155.Holt PG, Upham JW, Sly PD. Contemporaneous maturation of immunologic and respiratory functions during early childhood: implications for development of asthma prevention strategies. J Allergy Clin Immunol. 2005;116(1):16–24. doi: 10.1016/j.jaci.2005.04.017. quiz 25. [DOI] [PubMed] [Google Scholar]
- 156.Carnieli DS, Yoshioka E, Silva LF, et al. Inflammation and remodeling in infantile, juvenile, and adult allergic sensitized mice. Pediatr Pulmonol. 2011;46(7):650–665. doi: 10.1002/ppul.21436. [DOI] [PubMed] [Google Scholar]
- 157.Saglani S, Mathie SA, Gregory LG, Bell MJ, Bush A, Lloyd CM. Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am J Respir Cell Mol Biol. 2009;41(3):281–289. doi: 10.1165/rcmb.2008-0396OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Evans MJ, Fanucchi MV, Miller LA, Carlson MA, Nishio SJ, Hyde DM. Reduction of collagen VII anchoring fibrils in the airway basement membrane zone of infant rhesus monkeys exposed to house dust mite. Am J Physiol Lung Cell Mol Physiol. 2010;298(4):L543–547. doi: 10.1152/ajplung.00337.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brunekreef B, Janssen NA, de Hartog J, Harssema H, Knape M, van Vliet P. Air pollution from truck traffic and lung function in children living near motorways. Epidemiology. 1997;8(3):298–303. doi: 10.1097/00001648-199705000-00012. [DOI] [PubMed] [Google Scholar]
- 160.Carlsten C, Dybuncio A, Becker A, Chan-Yeung M, Brauer M. Traffic-related air pollution and incident asthma in a high-risk birth cohort. Occup Environ Med. 2011;68(4):291–295. doi: 10.1136/oem.2010.055152. [DOI] [PubMed] [Google Scholar]
- 161.Clark NA, Demers PA, Karr CJ, et al. Effect of early life exposure to air pollution on development of childhood asthma. Environ Health Perspect. 2010;118(2):284–290. doi: 10.1289/ehp.0900916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Miyabara Y, Ichinose T, Takano H, Lim HB, Sagai M. Effects of diesel exhaust on allergic airway inflammation in mice. J Allergy Clin Immunol. 1998;102(5):805–812. doi: 10.1016/s0091-6749(98)70021-1. [DOI] [PubMed] [Google Scholar]
- 163.Thevenot PT, Saravia J, Jin N, et al. Radical-containing ultrafine particulate matter initiates epithelial-to-mesenchymal transitions in airway epithelial cells. Am J Respir Cell Mol Biol. 2013;48(2):188–197. doi: 10.1165/rcmb.2012-0052OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Balakrishna S, Saravia J, Thevenot P, et al. Environmentally persistent free radicals induce airway hyperresponsiveness in neonatal rat lungs. Part Fibre Toxicol. 2011;8:11. doi: 10.1186/1743-8977-8-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Finkelstein JN, Johnston CJ. Enhanced sensitivity of the postnatal lung to environmental insults and oxidant stress. Pediatrics. 2004;113(4 Suppl):1092–1096. [PubMed] [Google Scholar]
- 166.Chan JK, Kodani SD, Charrier JG, et al. Age-specific effects on rat lung glutathione and antioxidant enzymes after inhaling ultrafine soot. Am J Respir Cell Mol Biol. 2013;48(1):114–124. doi: 10.1165/rcmb.2012-0108OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lee D, Wallis C, Wexler AS, et al. Small particles disrupt postnatal airway development. J Appl Physiol. 2010;109(4):1115–1124. doi: 10.1152/japplphysiol.00295.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]