

Abstract
The RAS/RAF mitogen activated protein kinase pathway (MAPK) is highly active in many tumor types including the majority of high-grade gliomas and expression of activated RAS or RAF in neural progenitor cells combined with either AKT activation or Ink4a/Arf loss leads to the development of high-grade gliomas in vivo. This strongly suggests that this pathway is necessary for glioma formation and maintenance. To further define the role of this pathway in the development of high-grade gliomas, we used the established RCAS/TVA glioma mouse model to test the ability of activated MAPK/extracellular signal-regulated kinase (ERK) kinase (MEK), a RAF effector, to induce tumors in vivo in the context of activated AKT or Ink4a/Arf loss. While expression of activated MEK alone in neural progenitor cells is not sufficient for tumorigenesis, the combination of activated MEK and AKT or MEK with Ink4a/Arf loss is transforming. The data reveal that activation of the classical RAS/MAPK pathway, which is mediated through MEK, leads to the development of high-grade gliomas in vivo and suggest that MEK may be a relevant target for glioma therapy. To test this, we treated both mouse and human glioma cells with the MEK inhibitor PD0325901. While this treatment induced apoptosis in a significant percentage of the cells, the effect was enhanced by combined treatment with the PI3K/mTOR inhibitor NVP-BEZ235. Our results demonstrate that combined inhibition of MEK and PI3K/mTOR is a rational strategy for the treatment of high-grade gliomas and may be an effective adjuvant therapy for this disease.
Keywords: MEK, AKT, Ink4a/Arf, glioma, mouse model, targeted therapy
Introduction
The current standard of care for patients with high-grade gliomas includes surgical resection followed by adjuvant radiotherapy and/or temozolomide chemotherapy. This treatment regimen increases the median survival of patients with high-grade gliomas from 12.1 to 14.6 months and was also found to significantly increase two year survival to 27.2 percent compared with 10.9 percent for radiotherapy alone (Stupp et al., 2009); however, more effective treatment strategies are badly needed because the majority of patients experience tumor recurrence and ultimately succumb to the disease (Stupp et al., 2009). Research in glioma therapy has recently shifted towards targeting the specific molecular alterations that underlie the pathogenesis of these cancers. Mutations or other genetic alterations in oncogenes or tumor suppressor genes often cause deregulation of signal transduction pathways but determining which of these can be effectively targeted remains to be defined.
Alterations in receptor tyrosine kinase (RTK) growth factor receptors (EGFR, PDGFR, MET and ERBB2), which result in constitutive downstream signaling of the classical mitogen activated protein kinase (MAPK) (e.g., RAS/RAF/MEK/ERK) and phosphatidylinositol 3-kinase (PI3K)/AKT pathways, are found in almost all World Health Organization (WHO) grade II, III, and IV astrocytomas. The MAPK pathway is also activated in the majority of WHO grade I tumors as a result of BRAF alterations (Jones et al., 2008). Together these data suggest that MAPK signaling is important for glioma development. Activation of the this pathway leads to cell cycle progression through Cyclin D1 activation (Furnari et al., 2007) and inhibition of apoptosis by regulation of BCL-2 family member gene expression and activity (Balmanno and Cook, 2009).
The majority of gliomas also have homozygous deletions that functionally inactivate the CDKN2A (cyclin-dependent kinase inhibitor 2A) locus (Ichimura et al., 1996). This locus encodes two independent protein products, p16INK4a and p14ARF [reviewed in (Chin et al., 1998; Roussel, 1999)]. p16INK4a is a specific inhibitor of Cyclin D/CDK4 or CDK6 complexes (Serrano et al., 1993). By inhibiting the kinase activity of CDK4 and CDK6, p16INK4a blocks RB phosphorylation and prevents progression from G1 to S in the cell cycle (Chin et al., 1998). p14ARF (p19Arf in mice) (Quelle et al., 1995), is a protein whose physiological role involves stabilization of TP53 levels (Chin et al., 1998) and cell cycle arrest in G1 and G2 in response to oncogenic stimuli (Kamijo et al., 1997; Zhang et al., 1998). Disruption of the TP53 and RB pathways also occurs in gliomas through direct mutation or deletion of TP53 and RB or amplification of HDM2 or CDK4, respectively (Parsons et al., 2008).
Astrocytic gliomas typically contain inactivating mutations in the AKT signaling repressor phosphatase and tensin homolog deleted on chromosome ten (PTEN). PTEN is a phosphatase that functions to inhibit the PI3K/AKT/mTOR pathway (Jiang et al., 1999). In the absence of PTEN, levels of active AKT are elevated (Holland et al., 2000). This signaling leads to progression through the cell cycle, increased proliferation, and inhibition of apoptosis. AKT activation has also been documented in gliomas as a result of AKT amplification and increased PI3K activity due to mutations within either the regulatory subunit PIK3RI or the catalytic subunit PIK3CA (TCGA, 2008). AKT activation has been shown to mimic loss of Pten in the context of glioma development in mice (Hu et al., 2005).
We and others have successfully used a somatic cell gene delivery method based on the RCAS/TVA system to evaluate the role(s) of specific genes in glioma initiation, progression, and maintenance. Expression of KRAS, CRAF, or BRAF in neural progenitor cells in combination with constitutive AKT activation, Pten loss, or Ink4a/Arf loss leads to the development of high grade gliomas in vivo (Holland et al., 2000; Hu et al., 2005; Lyustikman et al., 2008; Robinson et al., 2010; Uhrbom et al., 2002). While it has been widely accepted that MEK1 and MEK2 are downstream effectors of the RAF proteins, several studies have suggested that mammalian RAF proteins might have other substrates. These include but are not limited to NFκB, IκB, CDC25C, RB, BCL-2, and BAD (Wellbrock et al., 2004). In addition, CRAF has also been demonstrated to be an effector of BRAF (Wan et al., 2004). In this study we further delineated the downstream effectors of RAS and RAF in glioma development for the purpose of identifying additional therapeutic targets. Expression of a constituently active MEK1 mutant (MEK-GF) (Mansour et al., 1996) in combination with Ink4a/Arf loss or constitutive AKT activation led to the development of high grade gliomas in this model. While treatment with the MEK inhibitor PD0325901 induced apoptosis in the majority of human glioma cells tested, this effect was enhanced by combined treatment with the PI3K/mTOR inhibitor NVP-BEZ235. Our results demonstrate that combined inhibition of MEK and PI3K/mTOR is a rational strategy for the treatment of high-grade gliomas and may be an effective adjuvant therapy for this disease.
Results
Activated MEK induces anchorage-independent growth of astrocytes in vitro when combined with Ink4a/Arf loss or Akt activation
N-TVA/Ink4a/Arflox/lox primary astrocytes (Robinson et al., 2010) were used to evaluate the expression and transforming potential of RCASBP(A)MEK-GF alone or in combination with RCASBP(A)AKT or RCASBP(A)CRE. For comparison, the effect of CRAFΔ22W, BRAFV600E, and KRASG12D expression was evaluated concurrently. Western blot analysis of cell lysates confirmed gene expression in infected astrocytes (Supplementary Figure 1a) and demonstrated protein function though assessment of phoshporylated Erk 1/2 (P-Erk) and phoshporylated Akt (P-Akt) (Supplementary Figure 1b). N-TVA/Ink4a/Arflox/lox astrocytes infected with viruses containing CRAFΔ22W, BRAFV600E, MEK-GF, or KRASG12D in combination with CRE or AKT expression formed numerous colonies in soft agar demonstrating the ability of the mutants to induce anchorage-independent growth in immortalized astrocytes, a characteristic feature of transformation. In contrast, N-TVA/Ink4a/Arflox/lox astrocytes infected with any of the viruses alone did not result in significant growth in soft agar (Supplementary Figure 1b and data not shown).
Activated MEK induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation
Following intracranial infection of newborn N-TVA/Ink4a/Arflox/lox mice with different combinations of viral producing cells, survival was assessed for 12 weeks. All mice infected with viruses containing MEK-GF (14/14) or CRE (14/14) alone survived during the 12 week experimental time frame (Figure 1). A higher percentage of N-TVA/Ink4a/Arflox/lox mice infected with viruses containing MEK-GF and CRE survived during the experimental period (44%; 8/18) compared with mice infected with MEK-GF and AKT (31%; 8/26); however, this difference was not statistically significant (P = 0.758) (Figure 1). Brain tissue from all injected mice was analyzed histologically. While none of the mice infected with viruses containing MEK-GF or CRE alone developed tumors, gliomas were detected in 50% of mice injected with viruses containing either MEK-GF and CRE or MEK-GF and AKT. Immunostaining for the HA epitope tag confirmed expression of virally delivered AKT in MEK-GF + AKT induced tumors (Supplementary Figure 2a). PCR analysis on DNA extracted from the tumor samples confirmed the presence of MEK-GF in all tumors (Supplementary Figure 2b) and all tumors also expressed nestin, which drives TVA expression in these mice (Supplementary Figure 2c).
Figure 1. Kaplan-Meier survival curve.

N-TVA;Ink4a/Arflox/lox mice were injected with the indicated viruses at birth. Censored survival data were analyzed using a log-rank test of the Kaplan-Meier estimate of survival. No tumors were detected during the experimental period (12 weeks) in mice injected with MEK-GF (n = 14) or CRE (n = 14) alone and it has previously been shown that no tumors form in mice injected with AKT alone (Holland et al., 2000). There was no significant difference in survival between mice injected with viruses containing MEK-GF + AKT (n = 26) (small dashed line) or MEK + CRE (n = 18) (solid black line); P = 0.758; however, a significant difference in survival was observed between mice injected with MEK-GF + AKT or MEK + CRE when compared with mice injected with either virus alone; P = 0.002.
Gliomas induced by activated MEK in the context of Ink4a/Arf-deficiency or Akt expression displayed a range of morphologies
MEK-GF + CRE and MEK-GF + AKT induced tumors showed variable histologies but no specific feature distinguished either cohort (Figure 2). All tumors showed a multifocal appearance in the brain, mostly originating from the ventricular surfaces, forming solid masses protruding into the ventricle or expansively growing into the brain parenchyma (Figure 2a). All tumors showed a variable degree of infiltration into the CNS tissue, resulting in a diffuse overgrowth of underlying structures. A subset of tumors showed a biphasic phenotype, where an additional component of elongated, cytoplasm-rich tumor cells with a glial phenotype was encountered. Further features that were variably present were vascular endothelial proliferation (VEP) and geographic and/or palisading necrosis, both present in a subset of these tumors (Figure 2b). VEP or microvascular proliferation (MVP) is a feature of glial tumors that indicates malignant progression. This feature can be found in anaplastic oligodendrogliomas (AO), oligoastrocytomas (OA) and in GBM. Most tumors showed a uniform appearance of the tumor cell morphology with small, round monomorphic tumor cells, frequent mitotic activity and variable extent of clear cell cytoplasm, resembling anaplastic oligodendrogliomas (Figure 2c–f). Immunophenotyping of these tumors showed variable expression of the glial marker GFAP and markers of glial phenotype that are also seen in human gliomas, such as MAP-2 (not shown), Olig-2, and doublecortin, but they were negative for the neuronal marker synaptophysin, which would be expected to be positive in primitive neuroectodermal tumors (Figure 3). In conclusion, all tumors showed a glial phenotype with variable morphological resemblance to anaplastic astrocytomas, anaplastic oligodendrogliomas and glioblastomas (Figure 2). No consistent correlation between the genotype (e.g., CRE/AKT) and the presence of a specific cellular or architectural feature, necrosis, vascular endothelial proliferation, or infiltration was seen. The immunophenotype, although showing some variability, confirmed the range of differentiation across both genotypes.
Figure 2. Representative tumors induced by expression of MEK-GF and CRE or by MEK-GF and AKT.

a, b, c: Low power magnification of tumors induced by MEK-GF and CRE or by MEK-GF and AKT. All tumors show a glial phenotype but with variable morphology. a. MEK-CRE induced tumor showing diffuse growth with infiltration of the hippocampus (arrow pointing to a cluster of cells infiltrating perpendicularly to the dentate gyrus). The tumor grows diffusely, in monomorphic sheets of tumor cells but does not show necrosis or vessels with endothelial proliferation. b. Tumor induced by expression of MEK-GF + AKT, but with palisading necrosis (bright areas, arrows) which are a typical feature of GBM. c. A similar morphology, but absence of necrosis in a tumor induced by expression of MEK-GF + AKT. d, e, f: detail of these tumors showing a small-cell glial phenotype similar to (d) human anaplastic oligoastrocytomas, e, details of a palisading necrosis of neoplastic glial cells adjacent to a palisading necrosis and f, glial tumor with microvascular proliferation. Scale bar represents 600 μm for a–c and 80 μm for d–f.
Figure 3. Immunoprofiling of glial tumors.

All tumors show similar immunoprofiles, with slight variation in the expression of individual markers. (a–c) GFAP; (d–f) Olig2; (g–i) doublecortin, (j–l) synaptophysin; (m–o) CD15 (LeX). The MEK-GF + CRE induced tumor shows a loss of GFAP expression in a subset of cells, a feature that can be seen in anaplastic gliomas, such as oligoastrocytomas (OA) or even anaplastic astrocytomas (AA). There is expression of Olig-2 and doublecortin, but no expression of synaptophysin in the tumor tissue and very little CD15 expression. The MEK-GF + AKT induced gliomas show variable expression of Olig2 and doublecortin. The tumor cells are negative for synaptophysin and CD15. Representative images are shown. The scale bar represents 160 μm.
Glioma cells exhibit variable sensitivity to MEK and PI3K/mTOR inhibition
Using a genetic approach we previously demonstrated that tumors induced with KRAS are reliant on continued KRAS signaling for tumor maintenance in the context of activated AKT (Holmen and Williams, 2005). In this study, we used a pharmacological approach to evaluate the effect of MEK inhibition alone and in combination with PI3K/mTOR inhibition on glioma cell viability. The minimal concentrations required for complete inhibition of ERK phosphorylation with the MEK inhibitor PD0325901 and AKT phosphorylation with the dual PI3K/mTOR inhibitor NVP-BEZ235 were initially determined by single agent titration ranging from 100 nmol/L to 5 μmol/L in four human glioma cell lines and N-TVA/Ink4a/Arflox/lox astrocytes infected with viruses containing MEK-GF and CRE. Treatment with 0.2 μmol/L of the MEK inhibitor PD0325901 was sufficient for complete blockade of MEK signaling and 0.2 μmol/L of NVP-BEZ235 was sufficient for inhibition of AKT signaling in the mouse cells (Supplementary Figure 3). Treatment with 1 μmol/L of the MEK inhibitor PD0325901 was sufficient for complete blockade of MEK signaling and 1 μmol/L of NVP-BEZ235 was sufficient for inhibition of AKT signaling in the human cells (Supplementary Figure 3). To determine the effect of MEK inhibition on cell viability, N-TVA/Ink4a/Arflox/lox astrocytes infected with either RCASBP(A)MEK-GF, CRAFΔ22W, BRAFV600E or KRASG12D in combination with RCASBP(A)AKT or RCASBP(A)CRE were exposed to 0.5 μmol/L concentrations of PD0325901. After 48 hours, apoptosis was quantified using flow cytometry. Following inhibition of MEK signaling, no significant apoptosis was seen in N-TVA/Ink4a/Arflox/lox mouse astrocytes (M.A.) or in immortalized but non-transformed N-TVA/Ink4a/Arflox/lox astrocytes infected only with a virus containing CRE (Figure 4). However, treatment with PD0325901 led to significant apoptosis in N-TVA/Ink4a/Arflox/lox astrocytes infected with viruses containing CRE in combination with MEK-GF, BRAFV600E, or CRAFΔ22W. The ability of PD0325901 to induce apoptosis was reduced in N-TVA/Ink4a/Arflox/lox astrocytes infected with viruses containing KRASG12D or active AKT (Figure 4). To determine the effect of combined inhibition of MEK and AKT signaling, the same cell lines were treated with 0.5 μmol/L NVP-BEZ235, a dual PI3K/mTOR inhibitor, alone or in combination with PD0325901. Interestingly, treatment with NVP-BEZ235 exhibited significant single agent activity with ~30% cell death in N-TVA/Ink4a/Arflox/lox astrocytes infected only with a virus containing CRE. N-TVA/Ink4a/Arflox/lox astrocytes transformed with KRASG12D and CRE or CRAFΔ22W and AKT were the only other cells that were significantly more sensitive to treatment with NVP-BEZ235 than PD0325901 (Figure 4). The combination of PD0325901 and NVP-BEZ235 significantly increased apoptosis in all of the cell lines tested with the exception of KRASG12D/CRE and CRAFΔ22W/AKT. The enhanced activity was most prominent in N-TVA/Ink4a/Arflox/lox astrocytes infected with viruses containing KRAS and AKT. Treatment of these cells with PD0325901 or NVP-BEZ235 alone resulted in less than 5% apoptosis whereas inhibition of both pathways resulted in >30% apoptosis (Figure 4). Importantly, treatment with PD0325901 and NVP-BEZ235 alone or in combination did not induce significant apoptosis in primary uninfected mouse N-TVA/Ink4a/Arflox/lox astrocytes (M.A., Figure 4).
Figure 4. Apoptosis induced by inhibition of MEK and PI3K/mTOR signaling in mouse astrocytes.

Cells were infected with viruses containing the genes indicated. After confirmation of gene expression, cells were treated in triplicate with either DMSO as a control, 0.5 μmol/L PD0325901 (901), 0.5 μmol/L NVP-BEZ235 (BEZ), or both (BEZ/901) in combination for 48 hours and apoptosis was quantitated using the Guava Nexin® assay using a Guava EasyCyte flow cytometer. Data were normalized to the controls and are represented as mean ± SEM.
MEK and PI3K/mTOR inhibition induces variable apoptosis in human glioma cell lines
To evaluate the role of RAS/MAPK signaling in human glioma maintenance, we treated 12 glioma cell lines and normal human astrocytes (H.A.) with PD0325901. Single dose treatment of 1 μmol/L resulted in complete ERK inactivation for the experimental duration as measured by Western blot for phosphorylated ERK (Supplementary Figure 4) and resulted in varying apoptotic responses after 48 hours (Figure 5a). While no increase in apoptosis was detected in normal human astrocytes (H.A.), treatment with PD0325901 was most effective at inducing apoptosis in LN229 cells and least effective at inducing apoptosis in U-118 cells (Figure 5a–b). Phosphorylated ERK levels correlated with apoptotic response to PD0325901 in LN229, which was the most sensitive and had the highest levels of P-ERK, and U-118, which was the most resistant and had the lowest P-ERK levels (Figure 5b); however, this effect was not consistent in the majority of the cell lines. Interestingly, cell lines possessing intact PTEN were more responsive to single agent MEK inhibition as a group compared to cell lines lacking PTEN. To investigate if targeting the RAS/MAPK and PI3K/mTOR signaling pathways simultaneously would increase cell death, we treated the panel of 12 glioma cell lines with 1 μmol/L PD0325901, 1 μmol/L NVP-BEZ235, or 1 μmol/L NVP-BEZ235 in combination with 1 μmol/L PD0325901. We observed significant AKT and ERK inhibition after 48 hours as verified by Western blot (Supplementary Figure 4). Importantly, enhanced cell death was observed in nearly all of the glioma cell lines when these inhibitors were used in combination regardless of PTEN status and no significant effect was observed on the level of apoptosis in primary human astrocytes (Figure 5a). To assess whether the increase in apoptosis following the combined therapy was synergistic, additive or antagonistic, we treated two cell lines containing wt PTEN (LN18 and SF268) and two cell lines containing known PTEN mutations (SF539 and U251) with 1 μmol/L PD0325901 and 1 μmol/L NVP-BEZ235 alone or in combination for 72 hours (Supplementary Figure 5). The treatments were performed in at least quadruplicate to ensure that the statistical findings would be accurate and reliable. The test for the interaction term was statistically significant in cell lines LN18 (P < 0.001), SF268 (P = 0.003), and U251 (P < 0.001) indicating departure from additivity; however, additivity between NVP-BEZ235 and PD0325901 was not statistically significantly rejected in cell line SF539 (P = 0.215). The combination effect was calculated for cell lines LN18, U251, and SF268 and results showed that the apoptotic effect of the combination of PD0325901 and NVP-BEZ235 was synergistic (Supplementary Figure 5). The degree of synergy was stronger in LN18 and U251 as compared with SF268.
Figure 5. Cytotoxic effect of the MEK inhibitor PD0325901 and PI3K/mTOR inhibitor NVP-BEZ235 in a panel of glioma cell lines.

a. Apoptotic sensitivity of normal human astrocytes (H.A.) and glioma cell lines to treatment with 1 μmol/L PD0325901 (901), 1 μmol/L NVP-BEZ235 (BEZ), or both (BEZ/901) in combination for 48 hours. Cells were treated in triplicate and DMSO treated samples served as controls. Apoptosis was quantitated using the Guava Nexin® assay using a Guava EasyCyte flow cytometer. Data were normalized to the controls and are represented as mean ± SEM. Cell lines are grouped according to PTEN mutation status. b. Phosphorylated ERK levels correlate with apoptotic response to PD0325901. LN-229 and U-118 cells were treated with 1 μmol/L PD0325901 for 48 hours followed by assessment of apoptosis using the Guava Nexin® assay using a Guava EasyCyte flow cytometer. Cells staining positive for both Annexin V and 7-AAD represent apoptotic cells and are in the upper right quadrant. The numbers in the upper right quadrant reflect the percentage of cells that have undergone apoptosis. Western blot analysis of treated LN-229 and U-118 cells was used to assess phosphorylated and total ERK levels. Blots were reprobed with α-tubulin to confirm equal loading.
A negative feedback loop exists between MEK and PI3K signaling
Following treatment with PD0325901, we observed increased AKT phosphorylation in several of the cell lines analyzed. Increased ERK phosphorylation was also observed in several of the cell lines evaluated upon administration of NVP-BEZ235 (Supplementary Figure 4). We confirmed these findings using an additional MEK inhibitor, U0126, demonstrating that this phenomenon is not the result of off-target effects of one drug. In 10% serum, this effect could not be completely abolished through inhibition of both pathways (Supplementary Figure 6); however, we found that in 2% serum activation of AKT by MEK inhibition and activation of ERK by PI3K/mTOR inhibition was abolished by treatment with both NVP-BEZ235 and PD0325901 (Supplementary Figure 4).
MEK and PI3K/mTOR inhibition is more effective than temozolomide at inducing cell death in human glioma cells
Chemotherapeutic use of temozolomide requires a prolonged treatment to allow for a build up of DNA adducts and a number of different temozolomide regimens and dosing schedules have been described (Newlands et al., 1997; Stupp et al., 2009). To assess whether combined MEK and PI3K/mTOR inhibition is equally or more effective in inducing cell death than temozolomide, the 12 glioma cell line panel and normal human astrocytes were treated with 200 μmol/L temozolomide for 7 days (Beier et al., 2008). To provide a direct comparison, we also treated the same panel of glioma cell lines with 1 μmol/L PD0325901 and 1 μmol/L NVP-BEZ235 in combination for 7 days. At 200 μmol/L, temozolomide induced only modest amounts of cell death. SF295 cells were the most sensitive with ~30% cell death. The combined inhibition of MEK and PI3K/mTOR proved much more effective at inducing cell death than temozolomide. LN229 and LN18 were the most sensitive to the combination therapy and demonstrated the highest levels of cell death at 94% and 83%, respectively. U373 was the most resistant at ~40% cell death. Importantly, less than 20% cell death was observed following treatment with the combined therapy in the normal human astrocytes (Figure 6).
Figure 6. MEK and PI3K/mTOR inhibition more effectively induces cell death compared with temozolomide.
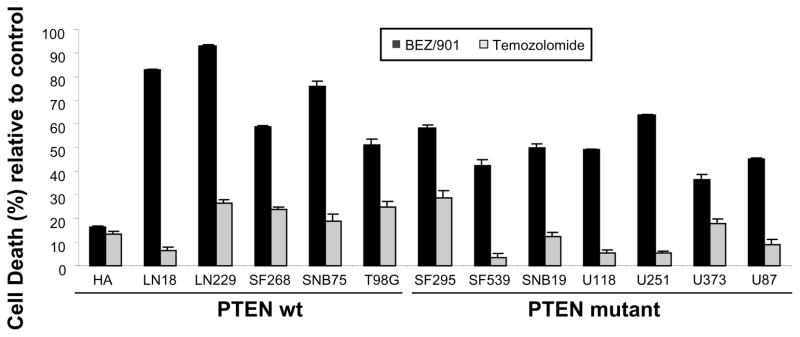
Cell death in human cell lines in response to treatment with 1 μmol/L PD0325901 (every 72 hours) in combination with 1 μmol/L NVP-BEZ235 (dosed daily) for seven days compared with 200 μmol/L temozolomide (dosed daily) for seven days. Cell death was quantitated by ViaCount® assay using a Guava EasyCyte flow cytometer. Each experiment was carried out in triplicate. Data were normalized to the controls and are represented as mean ± SEM.
Discussion
High grade gliomas have been found to possess multiple genetic abnormalities. Incorporation of genomic analyses into major signaling pathways implicated in this disease revealed alterations in three major pathways encompassing RTK signaling to RAS/PI3K/AKT and the tumor suppressor pathways linked to TP53 and RB. RTK/RAS/PI3K signaling was altered in 88%, the TP53 pathway was altered in 87%, and the RB pathway was altered in 78% of the samples analyzed. Interestingly, 74% of tumor samples had alterations in all three pathways further highlighting their importance in high-grade gliomas (TCGA, 2008); however, the key elements that are required for continued tumor growth and maintenance remain to be defined such that more effective therapies can be designed. We previously demonstrated that expression of BRAFV600E in cooperation with activation of AKT or loss of the Ink4a/Arf tumor suppressor genes can generate high-grade gliomas in mice (Robinson et al., 2010). In this study, we have evaluated signaling downstream of BRAF and observed that activated MEK, a RAF effector, is capable of inducing high-grade gliomas in the same context. These data reveal that activation of the classical RAS/MAPK pathway, which is mediated through RAF and MEK, is critical for the development of high grade gliomas.
Four molecular subtypes of gliomas have recently been classified based on gene expression signatures. These include Classical, Mesenchymal, Proneural, and Neural (Brennan et al., 2009; Verhaak et al., 2010). MEK-induced tumors most closely reflect the Classical and Proneural subtypes, which display the highest activity of the PI3K and MAPK pathways; however, it should be noted that elevated levels of both of these signaling pathways were detected in nearly all tumors analyzed (Brennan et al., 2009). Behaviorally, the MEK-induced tumors were more invasive than the tumors arising from BRAFV600E expression in the context of Ink4a/Arf loss that we have previously described; the gliomas were most similar to those induced by KRASG12D or BRAFV600E expression in the context of AKT activation (Robinson et al., 2010). In contrast to the BRAF-driven tumors where clear differences were observed in the context of Ink4a/Arf loss or AKT activation, no distinguishing features defined the MEK-driven tumors in either context. These differences may instead result from either the infection of nestin-expressing cells committed to different lineages or variation in the level of expression of the delivered genes. The latency and incidence of the MEK-driven tumors was most similar to that observed with BRAF and AKT, BRAF and Cre, and KRAS and AKT. In contrast, both latency and incidence were significantly higher in tumors induced with KRAS and Cre (Robinson et al., 2010).
Based on these findings, it is logical to assume that inhibition of the MAPK pathway alone or in combination with PI3K inhibition may be an effective strategy for the treatment of high-grade gliomas. A current challenge facing development of targeted therapies is selection of the appropriate patient population for clinical trial evaluation. Combination therapy that targets common downstream mediators of growth factor signaling such as MEK, PI3K, and mTOR should be effective in the Classical, Mesenchymal, and Proneural glioma subtypes that harbor mutations in upstream signaling components. Many inhibitors targeting the RAS/MAPK signaling pathway are currently in clinical trials, including inhibitors of RAS, RAF, and MEK (Roberts and Der, 2007). Two novel, orally bioavailable MEK inhibitors (PD0325901 and ARRY-142886) demonstrated increased potency against MEK (IC50: 1–10 nM) and had better biopharmaceutical properties including improved bioavailability, lower metabolic clearance, and longer-lasting target suppression compared with first and second generation MEK inhibitors (Wang et al., 2007). PD0325901 has shown promising preclinical activity in vitro and in vivo against a broad spectrum of tumors and objective patient responses have been reported in melanoma; however, neurotoxicity was observed in a subset of patients (LoRusso et al., 2010). While this effect might be considered detrimental for some tumor types, it is necessary and potentially beneficial for the treatment of brain metastases and gliomas. Importantly, NVP-BEZ235 has been shown to reduce PI3K signaling in an in vivo glioma model when administered orally (Liu et al., 2009).
PI3K pathway activation is known to mediate resistance to MEK inhibitors in RAS mutant cancers (Wee et al., 2009) and accordingly we observed greater resistance to MEK inhibition in tumor cells with high levels of PI3K/AKT signaling. Interestingly, we also observed negative regulatory feedback between MEK and AKT that amplify the signal and promote resistance to the inhibitors. This cross-talk may explain why only moderate levels of cell death were observed with single agent treatment. We found that combined inhibition of MEK and PI3K/mTOR in low serum eliminated the cross-talk and synergistically induced apoptosis in the majority of glioma cell lines tested. Importantly, the combination therapy did not significantly increase the level of apoptosis in normal human astrocyte controls but was more effective than temozolomide at inducing cell death in a large panel of human glioma cell lines. In addition to maximizing response, this combination may reduce or prevent the occurrence of both primary and acquired resistance, which remains a significant obstacle to the successful attainment of a targeted agents’ therapeutic potential. Conversely, it is possible that this multi-targeted approach may produce unacceptable side effects that limit the therapeutic benefit of the combination. This will need to be determined empirically through additional in vivo studies. Our data clearly demonstrate the potential power of targeted molecular therapy for the treatment of glioma and highlights the necessity of targeting multiple growth pathways to induce apoptosis. Combined treatment with PD0325901 and NVP-BEZ235 has the potential to improve overall patient survival and progression free survival compared to the current standard of care. Altogether these findings have important implications for the design of therapeutic strategies aimed at this disease.
Materials and Methods
Transgenic mice
Nestin-TVA/Ink4a/Arflox/lox mice have been described (Robinson et al., 2010). All experiments were performed in compliance with the “Care and Use of Animals” (Council et al., 1996) and were approved by the IACUC prior to experimentation.
Vector constructs
The retroviral vectors used in this study were replication-competent avian leukosis virus (ALV) long terminal repeat (LTR), splice acceptor, and Bryan polymerase-containing vectors of envelope subgroup A [designated RCASBP(A)] (Federspiel and Hughes, 1997). RCASBP(A)CRE, RCASBP(A)BRAFV600E, RCASBP(A)CRAFΔ22W, RCASBP(A)KRASG12D and RCASBP(A)AKTΔ11-60 have been previously described (Holland et al., 2000; Holmen and Williams, 2005; Lyustikman et al., 2008; Robinson et al., 2010). The N-terminus of KRAS has a FLAG epitope tag and AKT contains an HA epitope tag at the C-terminus (Holland et al., 2000). A constitutively active human gain of function (GF) MEK1 mutant (MEK-GF) lacking residues 32–51 (ΔN3) and with S218E and S222D substitutions has been described (Mansour et al., 1996). Details regarding RCASBP(A)MEK-GF cloning are available upon request.
Cell culture
DF-1 cells were maintained as described (Schaefer-Klein et al., 1998). Primary mouse astrocytes have been described (Robinson et al., 2010). Human glioma cell lines were maintained in RPMI with 5% FBS and 1 × penicillin/streptomycin (Invitrogen, Carlsbad, CA). Normal human astrocytes were maintained in astrocyte media (AM) containing astrocyte growth supplement (AGS) (ScienCell, Carlsbad, CA) with 2% FBS and 1 × penicillin/streptomycin (Invitrogen, Carlsbad, CA).
Virus propagation and infection
Viral propagation and infection was performed as previously described (Robinson et al., 2010).
Detection of RCASBP(A)MEK-GF integration
Detection of RCASBP(A)MEK-GF integration into the genome of the tumor cells was performed by PCR. Brain tissue from injected and control mice was paraffin embedded and 5-μm sections were adhered to glass slides. The location of the tumors on the slides was confirmed by histopathology on corresponding sections. The slides were de-waxed in xylene and rehydrated through graded alcohols. Tissue from the tumors was collected using a sterile needle and DNA was extracted using the Qiagen DNAeasy mini kit per the manufacturer’s specifications (Qiagen, Valencia, CA). Details of the PCR are available upon request.
Western blotting
Western blotting was performed using standard procedures. Details regarding the specific antibodies used are provided in Supplementary Information.
Immunohistochemistry (IHC)
Dissected brains were fixed in 10% formalin, embedded in paraffin, cut into 5 μm sections and adhered to glass slides. The sections were stained with hematoxylin and eosin (H&E) or left unstained for IHC. All immunostaining was carried out using the Ventana Benchmark or Discovery automated staining apparatus (Ventana Medical Systems, Tuscon, AZ) following the manufacturer’s guidelines. Details regarding the specific antibodies used are provided in Supplementary Information.
Drug treatments and cell viability
All cell viability assays were initiated 24 hours post cell line seeding and were carried out for the times indicated in media containing 2% serum. Single treatment dosing of human and mouse astrocytes and glioma cell lines with PD0325901 (Selleck Chemicals, Houston, TX), U0126 (Cell Signaling, Boston, MA), NVP-BEZ235 (Novartis, Groton, CT) was based on titration studies (0 – 5 μmol/L) examining phospho-ERK and phospho-AKT levels. Human astrocytes and glioma cell lines were also treated with 200 μmol/L of temozolomide (Santa Cruz, Santa Cruz, CA). Vehicle control samples were treated with an equal volume of DMSO. Cell death was quantitated using the Guava ViaCount® assay (Guava Technologies, Hayward, CA) and apoptosis was quantitated using the Guava Nexin® Annexin V assay (Guava Technologies, Hayward, CA). The Nexin assay uses two dyes: 7-AAD, a cell impermeant dye, as an indicator of membrane structural integrity and Annexin V-PE to detect phosphatidylserine (PS) on the external membrane of apoptotic cells. Samples were prepared per the manufacturer’s specifications at the indicated time points and assayed promptly.
Statistical analysis
Censored survival data was analyzed using a log-rank test of the Kaplan-Meier estimate of survival for the n indicated. In vitro experiments were performed in duplicate unless otherwise stated and data are presented as the mean with standard error of the mean (S.E.M.). To assess if combination therapy with PD0325901 and NVP-BEZ235 produced a synergistic response, direct measurements of apoptosis were obtained in replicates of 4 to 6 for each group. After log transformation, results were analyzed using analysis of variance techniques for 2 by 2 factorial designs. The statistical significance of the interaction term was tested at the two-sided alpha = 0.05 level to evaluate the additivity of the apoptotic effects of PD0325901 and NVP-BEZ235. Statistically significant interaction terms indicate a departure from additivity and that the effect of the combination of PD0325901 and NVP-BEZ235 was either synergistic or antagonistic. For cell lines with statistically significant interaction terms the following calculation was made to assess the synergistic effect of the combination: where A901, ABEZ, and A901+BEZ are the mean apoptosis values for PD0325901, NVP-BEZ235, and the combination, respectively. S is the combination effect, and values less than one indicate synergy while values greater than one indicate antagonism (S = 1 indicates additivity).
Supplementary Material
a. Western blot analysis of N-TVA/Ink4a/Arflox/lox primary astrocytes infected with viruses containing the genes indicated. (*) The blots were probed with either a pan-RAS antibody to detect KRASG12D, a RAF-1 antibody to detect CRAFΔ22W, a V5 antibody to detect BRAFV600E, or a phosphorylated MEK (Ser221) antibody to detect phosphorylated MEK (P-MEK). Cells infected with a virus containing only GFP served as negative controls. Blots were reprobed with α-tubulin to confirm equal loading. b. Anchorage-independent growth of N-TVA;Ink4a/Arflox/lox astrocytes in soft agar following infection with viruses containing the genes indicated. Colony formation was assessed after 7 days by counting ten fields at 10 x magnification. Each condition was performed in triplicate and the data represent mean ± SEM. Western blot analysis of phosphorylated Akt, total Akt, Akt-HA, phosphorylated Erk, total Erk and Ras expression in N-TVA/Ink4a/Arflox/lox primary astrocytes infected with viruses containing the genes indicated below the bar graph. Blots were reprobed with α-tubulin to confirm equal loading.
a. Immunohistochemical confirmation of AKT-HA expression in gliomas arising in N-TVA;Ink4a/Arflox/lox mice infected with RCASBP(A) MEK-GF and AKT. IHC was carried out with primary antibodies against the HA epitope tag on AKT in gliomas induced by expression of MEK-GF+ AKT. b. MEK-GF PCR analysis of tumor samples from mice infected with RCASBP(A)MEK-GF and RCASBP(A)AKT (T1-3) and RCASBP(A)MEK-GF and RCASBP(A)CRE (T4-6). DNA extracted from the brains of uninfected mice served as the negative control (N) and 4ng of RCASBP(A)MEK-GF plasmid DNA served as the positive control (+). c. Nestin expression in tumors induced by expression of MEK-GF + AKT and MEK-GF + CRE. The scale bars represent 200 μm.
Mouse astrocytes infected with RCASBP(A)MEK-GF and RCASBP(A)CRE were treated with 0, 0.04, 0.20, 1.0 and 5.0 μmol/L of PD0325901 or NVP-BEZ235 for 48 hours. Human glioma cell lines (SF268, SF295, SF239 and SNB19) were treated with 0, 0.001, 0.01, 0.1 and 1.0 μmol/L of PD0325901 or NVP-BEZ235. Western blot analysis of treated cells was used to assess phosphorylated and total ERK and AKT levels. α-Tubulin was used to confirm equal loading. Single dosing of 1 μmol/L for PD0325901 and daily dosing of 1 μmol/L NVP-BEZ235 was required for inhibition of MAPK and AKT signaling, respectively. 30 μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated and total ERK and AKT.
Cells were treated with 1 μmol/L of PD0325901 (901) and/or 1 μmol/L NVP-BEZ235 (BEZ). Human glioma cell lines were maintained in RPMI supplemented with 2% FBS and were treated for 48 hours. DMSO is an equal volume vehicle control. Thirty μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated and total ERK and AKT.
Apoptotic sensitivity of four glioma cell lines after treatment with 1 μmol/L PD0325901 (901) and/or 1 μmol/L NVP-BEZ235 (BEZ) for 72 hours. Apoptosis was quantitated using the Guava Nexin® assay using a Guava EasyCyte flow cytometer. Each experiment was repeated four to six times and the data are represented as the mean ± SEM. Interaction p-values < 0.05 indicate significant departure from additivity. Combined treatment led to a synergistic apoptotic response in 3 out of 4 the cell lines tested. * P < 0.001; ** P = 0.003.
Human glioma cell lines were maintained in RPMI supplemented with 10% FBS and were treated for 48 hours. DMSO is an equal volume vehicle control. Final concentration of each compound was 1 μmol/L (BEZ235) and 10 μmol/L (U0126). Thirty μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated ERK and AKT.
Acknowledgments
We thank Han-Mo Koo, Eric Holland, and Ronald DePinho for reagents and advice. We thank James Symanowski for assistance with statistical analysis of the data. We also thank Novartis Pharmaceuticals for providing the NVP-BEZ235. This work was supported by the Nevada Cancer Institute, the National Brain Tumor Foundation, and RSG-06-198-01-TBE from the American Cancer Society.
Footnotes
Conflict of interest
The authors declare no conflict of interest
Supplementary Information is available at Oncogene’s website.
Literature Cited
- Balmanno K, Cook SJ. Tumour cell survival signalling by the ERK1/2 pathway. Cell Death Differ. 2009;16:368–77. doi: 10.1038/cdd.2008.148. [DOI] [PubMed] [Google Scholar]
- Beier D, Rohrl S, Pillai DR, Schwarz S, Kunz-Schughart LA, Leukel P, et al. Temozolomide preferentially depletes cancer stem cells in glioblastoma. Cancer Res. 2008;68:5706–15. doi: 10.1158/0008-5472.CAN-07-6878. [DOI] [PubMed] [Google Scholar]
- Brennan C, Momota H, Hambardzumyan D, Ozawa T, Tandon A, Pedraza A, et al. Glioblastoma subclasses can be defined by activity among signal transduction pathways and associated genomic alterations. PLoS One. 2009;4:e7752. doi: 10.1371/journal.pone.0007752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin L, Pomerantz J, DePinho RA. The INK4a/ARF tumor suppressor: one gene--two products--two pathways. Trends Biochem Sci. 1998;23:291–6. doi: 10.1016/s0968-0004(98)01236-5. [DOI] [PubMed] [Google Scholar]
- Council NR, Research IoLA, Sciences CoL. Guide for the Care and Use of Laboratory Animals. 7. National Academy Press; Washington, D.C: 1996. p. 140. [Google Scholar]
- Federspiel M, Hughes S. Retroviral gene delivery. In: Emerson C, Sweeney H, editors. Methods in Cell Biology: Methods in Muscle Biology. Academic Press; San Diego: 1997. pp. 179–214. [PubMed] [Google Scholar]
- Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN. Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet. 2000;25:55–7. doi: 10.1038/75596. [DOI] [PubMed] [Google Scholar]
- Holmen SL, Williams BO. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005;65:8250–5. doi: 10.1158/0008-5472.CAN-05-1173. [DOI] [PubMed] [Google Scholar]
- Hu X, Pandolfi PP, Li Y, Koutcher JA, Rosenblum M, Holland EC. mTOR promotes survival and astrocytic characteristics induced by Pten/AKT signaling in glioblastoma. Neoplasia. 2005;7:356–68. doi: 10.1593/neo.04595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura K, Schmidt EE, Goike HM, Collins VP. Human glioblastomas with no alterations of the CDKN2A (p16INK4A, MTS1) and CDK4 genes have frequent mutations of the retinoblastoma gene. Oncogene. 1996;13:1065–72. [PubMed] [Google Scholar]
- Jiang BH, Aoki M, Zheng JZ, Li J, Vogt PK. Myogenic signaling of phosphatidylinositol 3-kinase requires the serine-threonine kinase Akt/protein kinase B. Proc Natl Acad Sci U S A. 1999;96:2077–81. doi: 10.1073/pnas.96.5.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res. 2008;68:8673–7. doi: 10.1158/0008-5472.CAN-08-2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ashmun RA, et al. Tumor suppression at the mouse INK4a locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–59. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- Liu TJ, Koul D, LaFortune T, Tiao N, Shen RJ, Maira SM, et al. NVP-BEZ235, a novel dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor, elicits multifaceted antitumor activities in human gliomas. Mol Cancer Ther. 2009;8:2204–10. doi: 10.1158/1535-7163.MCT-09-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoRusso PM, Krishnamurthi SS, Rinehart JJ, Nabell LM, Malburg L, Chapman PB, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral MAPK/ERK kinase inhibitor PD-0325901 in patients with advanced cancers. Clin Cancer Res. 2010;16:1924–37. doi: 10.1158/1078-0432.CCR-09-1883. [DOI] [PubMed] [Google Scholar]
- Lyustikman Y, Momota H, Pao W, Holland EC. Constitutive activation of Raf-1 induces glioma formation in mice. Neoplasia. 2008;10:501–10. doi: 10.1593/neo.08206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour SJ, Candia JM, Matsuura JE, Manning MC, Ahn NG. Interdependent domains controlling the enzymatic activity of mitogen-activated protein kinase kinase 1. Biochemistry. 1996;35:15529–36. doi: 10.1021/bi961854s. [DOI] [PubMed] [Google Scholar]
- Newlands ES, Stevens MF, Wedge SR, Wheelhouse RT, Brock C. Temozolomide: a review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat Rev. 1997;23:35–61. doi: 10.1016/s0305-7372(97)90019-0. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell. 1995;83:993–1000. doi: 10.1016/0092-8674(95)90214-7. [DOI] [PubMed] [Google Scholar]
- Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- Robinson JP, Vanbrocklin MW, Guilbeault AR, Signorelli DL, Brandner S, Holmen SL. Activated BRAF induces gliomas in mice when combined with Ink4a/Arf loss or Akt activation. Oncogene. 2010;29:335–44. doi: 10.1038/onc.2009.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF. The INK4 family of cell cycle inhibitors in cancer. Oncogene. 1999;18:5311–7. doi: 10.1038/sj.onc.1202998. [DOI] [PubMed] [Google Scholar]
- Schaefer-Klein J, Givol I, Barsov EV, Whitcomb JM, VanBrocklin M, Foster DN, et al. The EV-O-derived cell line DF-1 supports the efficient replication of avian leukosis-sarcoma viruses and vectors. Virology. 1998;248:305–11. doi: 10.1006/viro.1998.9291. [DOI] [PubMed] [Google Scholar]
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–7. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–66. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- TCGA . Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–8. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhrbom L, Dai C, Celestino JC, Rosenblum MK, Fuller GN, Holland EC. Ink4a-Arf loss cooperates with KRas activation in astrocytes and neural progenitors to generate glioblastomas of various morphologies depending on activated Akt. Cancer Res. 2002;62:5551–8. [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–67. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- Wang D, Boerner SA, Winkler JD, LoRusso PM. Clinical experience of MEK inhibitors in cancer therapy. Biochim Biophys Acta. 2007;1773:1248–55. doi: 10.1016/j.bbamcr.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Wee S, Jagani Z, Xiang KX, Loo A, Dorsch M, Yao YM, et al. PI3K pathway activation mediates resistance to MEK inhibitors in KRAS mutant cancers. Cancer Res. 2009;69:4286–93. doi: 10.1158/0008-5472.CAN-08-4765. [DOI] [PubMed] [Google Scholar]
- Wellbrock C, Karasarides M, Marais R. The RAF proteins take centre stage. Nat Rev Mol Cell Biol. 2004;5:875–85. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell. 1998;92:725–34. doi: 10.1016/s0092-8674(00)81401-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
a. Western blot analysis of N-TVA/Ink4a/Arflox/lox primary astrocytes infected with viruses containing the genes indicated. (*) The blots were probed with either a pan-RAS antibody to detect KRASG12D, a RAF-1 antibody to detect CRAFΔ22W, a V5 antibody to detect BRAFV600E, or a phosphorylated MEK (Ser221) antibody to detect phosphorylated MEK (P-MEK). Cells infected with a virus containing only GFP served as negative controls. Blots were reprobed with α-tubulin to confirm equal loading. b. Anchorage-independent growth of N-TVA;Ink4a/Arflox/lox astrocytes in soft agar following infection with viruses containing the genes indicated. Colony formation was assessed after 7 days by counting ten fields at 10 x magnification. Each condition was performed in triplicate and the data represent mean ± SEM. Western blot analysis of phosphorylated Akt, total Akt, Akt-HA, phosphorylated Erk, total Erk and Ras expression in N-TVA/Ink4a/Arflox/lox primary astrocytes infected with viruses containing the genes indicated below the bar graph. Blots were reprobed with α-tubulin to confirm equal loading.
a. Immunohistochemical confirmation of AKT-HA expression in gliomas arising in N-TVA;Ink4a/Arflox/lox mice infected with RCASBP(A) MEK-GF and AKT. IHC was carried out with primary antibodies against the HA epitope tag on AKT in gliomas induced by expression of MEK-GF+ AKT. b. MEK-GF PCR analysis of tumor samples from mice infected with RCASBP(A)MEK-GF and RCASBP(A)AKT (T1-3) and RCASBP(A)MEK-GF and RCASBP(A)CRE (T4-6). DNA extracted from the brains of uninfected mice served as the negative control (N) and 4ng of RCASBP(A)MEK-GF plasmid DNA served as the positive control (+). c. Nestin expression in tumors induced by expression of MEK-GF + AKT and MEK-GF + CRE. The scale bars represent 200 μm.
Mouse astrocytes infected with RCASBP(A)MEK-GF and RCASBP(A)CRE were treated with 0, 0.04, 0.20, 1.0 and 5.0 μmol/L of PD0325901 or NVP-BEZ235 for 48 hours. Human glioma cell lines (SF268, SF295, SF239 and SNB19) were treated with 0, 0.001, 0.01, 0.1 and 1.0 μmol/L of PD0325901 or NVP-BEZ235. Western blot analysis of treated cells was used to assess phosphorylated and total ERK and AKT levels. α-Tubulin was used to confirm equal loading. Single dosing of 1 μmol/L for PD0325901 and daily dosing of 1 μmol/L NVP-BEZ235 was required for inhibition of MAPK and AKT signaling, respectively. 30 μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated and total ERK and AKT.
Cells were treated with 1 μmol/L of PD0325901 (901) and/or 1 μmol/L NVP-BEZ235 (BEZ). Human glioma cell lines were maintained in RPMI supplemented with 2% FBS and were treated for 48 hours. DMSO is an equal volume vehicle control. Thirty μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated and total ERK and AKT.
Apoptotic sensitivity of four glioma cell lines after treatment with 1 μmol/L PD0325901 (901) and/or 1 μmol/L NVP-BEZ235 (BEZ) for 72 hours. Apoptosis was quantitated using the Guava Nexin® assay using a Guava EasyCyte flow cytometer. Each experiment was repeated four to six times and the data are represented as the mean ± SEM. Interaction p-values < 0.05 indicate significant departure from additivity. Combined treatment led to a synergistic apoptotic response in 3 out of 4 the cell lines tested. * P < 0.001; ** P = 0.003.
Human glioma cell lines were maintained in RPMI supplemented with 10% FBS and were treated for 48 hours. DMSO is an equal volume vehicle control. Final concentration of each compound was 1 μmol/L (BEZ235) and 10 μmol/L (U0126). Thirty μg of each cell lysate was separated by SDS-PAGE and assessed for phosphorylated ERK and AKT.