

Abstract
We propose a novel statistical approach to improve the reliability of 1H NMR spectral analysis in complex metabolic studies. The Statistical HOmogeneous Cluster SpectroscopY (SHOCSY) algorithm aims to reduce the variation within biological classes by selecting subsets of homogeneous 1H NMR spectra that contain specific spectroscopic metabolic signatures related to each biological class in a study. In SHOCSY, we used a clustering method to categorize the whole data set into a number of clusters of samples with each cluster showing a similar spectral feature and hence biochemical composition, and we then used an enrichment test to identify the associations between the clusters and the biological classes in the data set. We evaluated the performance of the SHOCSY algorithm using a simulated 1H NMR data set to emulate renal tubule toxicity and further exemplified this method with a 1H NMR spectroscopic study of hydrazine-induced liver toxicity study in rats. The SHOCSY algorithm improved the predictive ability of the orthogonal partial least-squares discriminatory analysis (OPLS-DA) model through the use of “truly” representative samples in each biological class (i.e., homogeneous subsets). This method ensures that the analyses are no longer confounded by idiosyncratic responders and thus improves the reliability of biomarker extraction. SHOCSY is a useful tool for removing irrelevant variation that interfere with the interpretation and predictive ability of models and has widespread applicability to other spectroscopic data, as well as other “omics” type of data.
Nuclear magnetic resonance (NMR)1 and/or mass spectroscopy (MS)2,3 based metabolic profiling studies are usually analyzed by multivariate statistical methods that have been developed to identify specific metabolic signatures contributing to different biological classes within a data set such as disease versus healthy. Typically, unsupervised approaches such as principal component analysis (PCA)4 are used for identifying outliers and detecting analytical variation/drift within data sets. The PCA scores plot indicates similarities/dissimilarities between samples, and the loadings plot identifies the metabolites that contribute most to the clustering pattern. Subsequently, supervised algorithms such as orthogonal partial least square discrimination analysis (OPLS-DA)5 are then applied to optimize the classification and extract potential biomarkers for each class. To assess the OPLS-DA model and to prevent overfitting, 7-fold cross validation and permutation testing are often used. The 7-fold cross-validation Q2 statistic is calculated by leaving every seventh sample out and predicting them back in the model; thus, Q2 measures the similarity between the predicted data and the real data. Permutation tests randomly assign samples to classes and recalculate the model: the random reassignments of samples to classes are repeated for a large number of times in order to ascertain the likelihood of the actual results being obtained by chance. As a rule of thumb, the closer the Q2 value is to 1, the better the predictive ability of the OPLS-DA model, and the model actual Q2 value should be significantly higher than the Q2 obtained by permutation test.
Although there are numerous examples of successful applications of OPLS-DA1,2,6 and related techniques for metabonomic data sets,7 the complexity of biological data, particularly for human studies with multiple sources of environmental and genetic variation, can compromise the analysis. Similarly, for animal studies, the diversity of response to stimuli may vary even when studies are carried out in a highly homogeneous environment and in animals of the same genetic strain. Recent publications have demonstrated considerable variation in responses to drug treatment in both animal8,9 and human10,11 studies. Some individuals have been shown to be more susceptible to drug toxicity8 and some respond better or more quickly to drug treatment than others.12 This phenomenon prompted the evolution of pharmacometabonomics:8 the prediction of response to an intervention based on their predose metabolic profiles.10,13 In these scenarios, OPLS-DA modeling may generate suboptimal results, as the samples in each class are usually assumed to be homogeneous.
One method of addressing inhomogeneity is to use autoclustering methods such as K-means,14 self-organizing mapping (SOP),15 and nearest-neighbor clustering,16 where these approaches group the samples based on their similarity. Although these methods have been employed in “omics” studies,17−22 two issues are yet to be rectified: first, clusters of homogeneous samples may not be relevant to the biological question of interest; and second, the identity of each cluster, which may constitute the homogeneous core of a biological class, is not specifically determined by the clustering algorithm. Moreover, the clustering methods applied previously in metabonomics studies were mainly used to aid the extraction of metabolic information and to identify molecules of interest with regard to defining a particular condition. For example, Robinette et al.23 developed CLuster Analysis Statistical SpectroscopY (CLASSY), which aims to cluster the peaks from the same molecule by the correlation of the spectroscopic variables, whereas Blaise et al.24 used the ratio of covariance and correlation of the variables to achieve it. Statistical TOtal Correlation SpectroscopY (STOCSY)25 has been used to recover structural metabolic information, and its extension, SubseT Optimization by Reference Matching (STORM),26 utilizes an iterative selection of homogeneous subsets of spectra to improve structural elucidation by reducing variation across inhomogeneous spectral data sets.
Here we adopt a similar principle to STORM in combination with OPLS-DA and an enrichment test to address the issues associated with the autoclustering methods stated above to reduce the variation of the data set and enhance robust biomarkers selection. In our proposed algorithm, Statistical HOmogeneous Cluster SpectroscopY (SHOCSY), OPLS-DA is first applied to identify the potential common spectral features related to different biological classes of interest. We then employ the K-means clustering approach to cluster the spectra based on the potential discriminatory biomarkers. This ensures the K-means clusters have similar metabolic features to the biological classes. An enrichment test27−29 that evaluates which biological class is over-represented in each K-means cluster is then employed. The enrichment test thus associates clusters to specific biological classes and identifies the samples that constitute homogeneous cores for the specific biological class. The whole procedure is performed iteratively, and in each iteration, a new set of common spectral features are obtained. This enables identification of the “true” metabolic characteristics representative of each biological class within the data set to be uncovered. The algorithm converges when the cross-validation Q230 of the OPLS-DA model based on the homogeneous representatives of biological classes is maximal. An OPLS-DA model based on these core homogeneous subsets, without spectra contributing idiosyncratic responses, will have an improved predictive ability and potentially a more robust selection of biomarkers.
Initially, we evaluate the SHOCSY algorithm using a simulated 1H NMR spectral data set and then exemplify the method using a rodent hepatoxicology study, in which treated animals were known to show variable degrees of response.31
MATERIALS AND DATA ANALYSIS
Simulated NMR Spectra
We used simulated NMR spectra designed to emulate Paraquat toxicity, reflecting damage to the renal proximal tubules. To evaluate our algorithm, we generated a total of 12 data sets with different sizes (N = 30, 100, and 500 spectra in each biological class) and different proportions of idiosyncratic responders. Compared to the control class, the Paraquat toxicity spectra were designed with increased signals intensity for lactate (δ 1.32, doublet (d), δ 4.10, quartet (q)), and l-alanine (δ 1.46, d, δ 3.76, q) and reduced signal intensities for creatinine (δ 3.03, singlet (s), δ 4.05, s) and citrate (δ 2.53, d, δ 2.65, d). To introduce variable responses within the Paraquat toxicity class, 5%, 10%, 33.3%, and 50% spectra within the Paraquat toxicity class were designed to mimic resistance to the poisoning intervention. This was achieved by setting the intensities of the above four biomarkers to similar intensities to those in the control class. In addition, we also simulated metabolic variation across the whole data set that was irrelevant to the response to Paraquat toxicity. Variation was introduced in the intensities of signals relating to hippurate (δ 3.96, d, δ 7.54, triplet (t), δ 7.62, t, δ 7.82, double doublet (dd)), glycine (δ 3.54, s), trimethylamine-N-oxide (δ 3.25, s), l-histidine (δ 3.16, dd, δ 3.23, dd, δ 3.98, dd, δ 7.09, d, δ 7.90, d), and phenylacetylglutamine (δ 1.92, multiplet (m), δ 2.11, m, δ 2.26, t, δ 3.66, q, δ 4.19, m, δ 7.37, t, δ 7.43, t, δ 7.90, d). The variation was evenly distributed in both control and Paraquat toxicity classes. An illustration of the means and variances in signal intensities for these metabolites are shown in Supporting Information Table S-1 for N = 30, and these were proportionally expanded for all other data sets. All spectral data sets were generated using MetAssimulo software.32 Within the software package, the HMDB database33 and a local NMR standard spectra database were used to extract information for 1H NMR metabolite signals. The concentrations of the remaining metabolites were simulated using the same Gaussian distributions for the whole data set. Each simulated spectrum covered a chemical shift region of δ 0–10 with 27 679 spectral variables. The peak shift Δδ was set by using the default parameters of the software package and signal-to-noise ratio (S/N) was set to 100. The simulated spectral data were mean centered and scaled to unit variance prior to normalization by the probabilistic quotient normalization (PQN) method using the median spectrum from the whole data set as a reference.34
Hydrazine Toxicology Study
This study formed part of the Consortium for Metabonomic Toxicology (COMET) project. Details of this study can be found in Lindon et al.35 Briefly, three groups of male Sprague–Dawley rats, N = 50 in each group, were administered a single dose of either saline (control) or hydrazine hydrochloride in saline at 90 mg/kg or 30 mg/kg (high and low dose interventions, respectively). Urine samples collected over the following nine time periods were analyzed by 1H NMR spectroscopy: t1 = −8 to 0 h, t2 = 0 to 8 h, t3 = 8 to 24 h, t4 = 24 to 48 h, t5 = 48 to 72 h, t6 = 72 to 96 h, t7 = 96 to 120 h, t8 = 120 to 144 h, and t9 = 144 to 168 h. Half of the rats were killed at 48 h postdose for histology, leaving 25 rats in each group after t5.
NMR Spectroscopy
Each 200 μL of urine sample was added to 200 μL of phosphate buffer containing 10% deuterium oxide. 1H NMR spectra of urine were acquired on a Bruker DRX600 spectrometer operating at 600.13 MHz 1H observation frequency using the BEST (Bruker Efficient Sample Transfer) 5 mm flow-injection probe for sample delivery and analysis. One-dimensional (1D) NMR spectra of urine samples were acquired using a solvent presaturation pulse sequence to suppress the residual water resonance. Sixty-four free induction decays (FIDs) were collected into 64 k data points, at 300 K, using a spectral width of 12019 Hz, with an acquisition time of 2.04 s, giving a total pulse recycle delay of 3.04 s. These data were zero-filled by a factor of 2, and the FIDs were multiplied by an exponential weighting function equivalent to a line broadening of 0.3 Hz prior to Fourier transformation (FT).
Pretreatment of NMR Spectra
The raw spectral data were loaded using the BATMAN R package.36 The spectra were referenced to an internal standard (sodium 3-trimethylsilyl-[2,2,3,3-2H4]-propionate, TSP). The spectral regions containing water and urea resonances were excluded, leaving chemical shift ranges from δ 0.24–4.48 and δ 5.96–9.96. The spectra were visually checked, and those with poor water suppression and hence distorted baselines were excluded. The resulting numbers of spectra included in the multivariate analysis are given in Table S-2 in Supporting Information. Each spectrum consisted of 27 679 variables with a bin width 0.0003 ppm. As with the simulated data, these spectral data were mean-centered and scaled to unit variance before normalization using the median PQN method.
Data Analysis
Using the simulated data set, we aimed to evaluate whether the correct discriminatory variables for “toxicity” could be identified. Here, we considered a spectral variable to be discriminatory if the p-value of the correlations between the spectral variable and the OPLS-DA scores vector37 was smaller than μp and the OPLS-DA loading weight6 was larger than μw. As there are initially no established values for μp and μw, we used 1.85 × 10–6 for μp (corresponding to 0.05 after Šidák correction38), and we used the simulated data sets to establish an appropriate μw value. We subsequently applied the same μw in the hydrazine toxicity study. Using these criteria, we evaluated if we could correctly identify the homogeneous and idiosyncratic responders within the simulated data sets and compared the performance of SHOCSY to standard OPLS-DA method for their sensitivity, specificity, and overall accuracy. PCA scores plots were employed to provide an overview of the whole data set as well as to map the homogeneous and idiosyncratic subsets individually. The predictive ability of the OPLS-DA models built on the homogeneous subsets were compared to the model based on the whole data set, using 7-fold and double cross-validation Q2.39 Permutation tests were calculated by randomly assigning classes to the samples, and remodelling was repeated 50 times. To establish the model significance, the Q2 statistics for the actual model was compared to the empirical null distribution obtained from the permuted Q2.
Statistical HOmogeneous Cluster SpectroscopY (SHOCSY)
The proposed algorithm consists of four steps, in which Step 1–3 are performed iteratively: (i), identifying discriminatory spectral variables by OPLS-DA; (ii), clustering of spectra based on the discriminatory variables by K-means; (iii), identifying homogeneous subsets using an enrichment test; and (iv) extracting discriminating metabolic features based on the homogeneous subsets.
Step 1: Standard OPLS-DA analysis is performed to identify potential discriminatory spectral variables. A spectral variable is classified as discriminatory when the p-value of the correlations between the spectral variable and the OPLS-DA scores <1.85 × 10–6 and at the same time, its OPLS-DA loading weight >0.3, where this cutoff point was found to be appropriate for all the simulated data sets. In SHOCSY, the whole data set is only used in Step 1 of the first iteration. Subsequent iterations use subsets of the data set showing similar metabolic features.
Step 2: K-means method is applied to categorize the data set into a few clusters. We assume each biological class could potentially consist of two clusters—one cluster showing the dominant metabolic features as identified in step 1 and another lacking these metabolic features. We therefore cluster the data set to model the dominant metabolic features by the “K”-means method. In K-means clustering, the NMR spectral data are grouped to minimize within-group diversity but maximize between-groups diversity by Euclidean distance. Thus, for a biological class that is highly homogeneous, all the spectra can be clustered in one cluster showing the dominant metabolic features. However, for a biological class that is heterogeneous, two clusters can be formed (i.e., a cluster of spectra showing the dominant metabolic features and another cluster of spectra lacking these metabolic features). Consequently, for a data set consisting of two biological classes, a total of up to four clusters may be generated.
Step 3: A hypergeometric enrichment test is used to identify the dominant cluster for each biological class and thus associates clusters to specific biological classes within the data set. This enables the identification of spectral data with dominant metabolite features. Here, the cluster is considered dominant for a biological class when p ≤ 0.05 and thus considered as the homogeneous subset for that specific biological class. The other clusters with p > 0.05 are therefore considered idiosyncratic responders. However, when both clusters of a biological class show p > 0.05, the algorithm ceases to work, and this indicates that no dominant metabolic feature can be identified for this biological class. The identified homogeneous subsets for each biological class from the first iteration are fed back to Step 1 of the algorithm. In the subsequent iteration, a new set of discriminatory spectral variables are identified using these homogeneous subsets from each biological class. Subsequently, new K-means clusters are formed to generate updated homogeneous subsets. When the “truly” homogeneous subsets have been identified, the iteration will stop as the predictive ability of the OPLS-DA model is maximized (Figure S-1).
Step 4: The homogeneous subsets with maximal Q2 are used to identify the potential discriminatory variables. The OPLS-DA model based on these homogeneous spectra will enable a more robust selection of biomarkers.
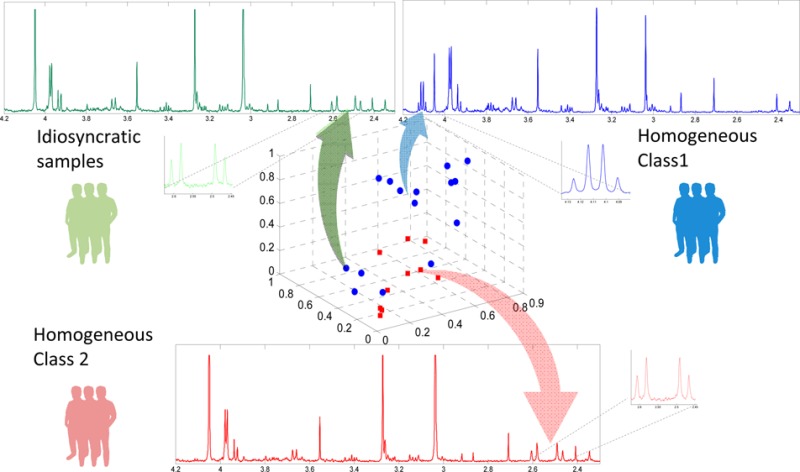
A schematic diagram describing the SHOCSY algorithm, for a data set containing two biological classes, is shown in Figure 1. All calculations and the SHOCSY algorithm were written in MATLAB (R2012a, Mathworks, Natick, U.S.A.) environment. The matlab code identifying the homogeneous and idiosyncratic responders is available on request.
Figure 1.

Schematic diagram of the SHOCSY algorithm for a data set consisting two biological classes. The closed circles and rectangles denote homogeneous samples; and the open circles and rectangles denote idiosyncratic samples.
Results and Discussion
Evaluation of the Performance of the SHOCSY Algorithm on a Simulated NMR Spectral Data Set
The SHOCSY algorithm was evaluated by varying the μw while fixing μp = 1.85 × 10–6 (0.05 after Šidák correction). Using the simulated data sets, we found that a μw value ranging from 0.2 to 0.6 was able to correctly identify all four metabolite signals corresponding to “Paraquat toxicity”. When the μw value was increased to >0.7, these models only identified three metabolites as biomarkers. This demonstrates that a high μw value increases the risk of excluding genuine biomarkers. We therefore adopted a conservative cutoff value, μw = 0.3, for the remaining analyses to ensure reliable biomarker signature selection. However, the standard OPLS-DA only identified creatinine and citrate even a low μw of 0.2 was used when the proportion of idiosyncratic spectra is >33.3%.
The PCA scores plot based on the whole data set (N = 30 in each biological class with 50% idiosyncratic spectra) shows that some spectra in the simulated Paraquat group cocluster with the control class (Figure 2a). We then evaluated the ability of the SHOCSY algorithm to correctly identify the homogeneous and idiosyncratic subsets within the data set. SHOCSY was found to be highly reliable (Figure 2b) and more robust than standard OPLS-DA approach. SHOCSY showed an overall accuracy of ≥0.94, sensitivity ≥0.94, and specificity ≥0.80 irrespective of data set size and the extent of idiosyncratic responders. This was not the case for the standard OPLS-DA approach, where the overall accuracy fell to 0.75 and with poor specificity (Supporting Information Table S-3). The median 1H NMR spectra of the homogeneous control, idiosyncratic responders, and homogeneous responders are shown in Figure 2c. It can be clearly seen that the idiosyncratic responders had similar spectral features to those of the control. In addition, the homogeneous responders manifested an increase in lactate and l-alanine but a reduction in the intensity of creatinine and citrate signals. When the proportion of idiosyncratic responders is low (e.g., 5%), both 7-fold and double cross-validation methods show comparable results for the standard OPLSDA approach (Supporting Information Table S-4). As the proportion of idiosyncratic responders increases, the Q2 decreases progressively. However, when the homogeneous subgroups were used, both the 7-fold and double cross-validation generated comparable Q2 values and remained high (>0.78, permutation test of p < 10–5 for all data sets). These high Q2 values are due to the exclusion of idiosyncratic responders from the data set, which removes irrelevant responses from the model and thus improves the predictivity of the model. Moreover, with 50% idiosyncratic spectra, the l-alanine and lactate signals in the standard OPLSDA were obscured by the interference samples in the data sets and thus did not significantly differentiate the toxicity class from control. The loading coefficient plots (Figure 2d,e) show that the loading weights for these four metabolite signals were higher in the model based on homogeneous subsets (r2 > 0.7) than those of the whole data set (r2 > 0.5, N = 30).
Figure 2.
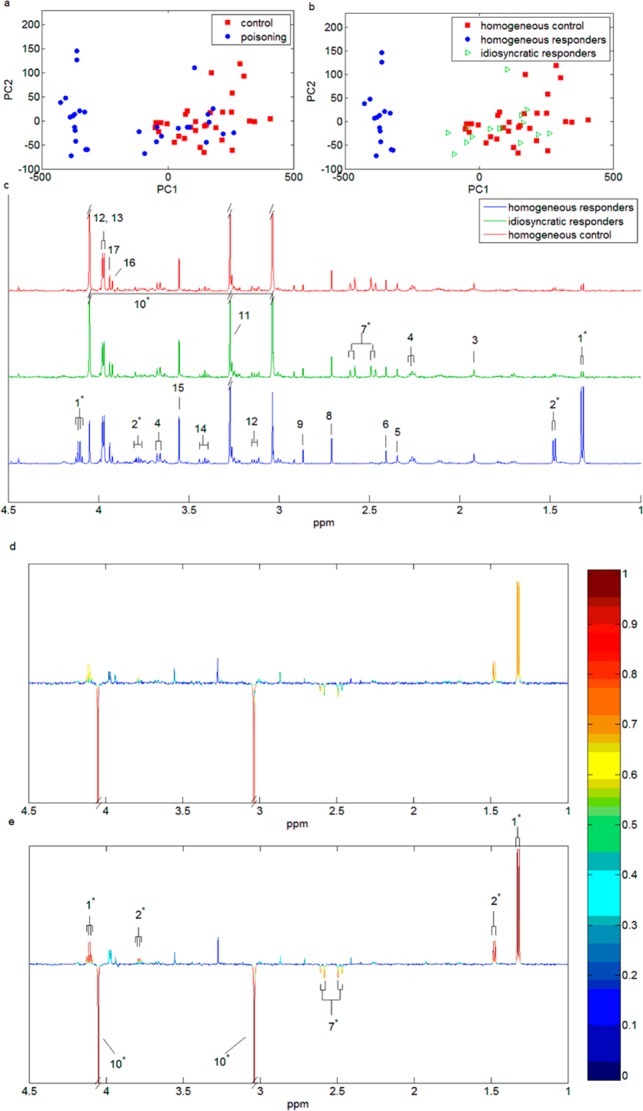
PCA scores plots for principal components 1 and 2 using the simulated data set of 30 spectra in each biological class with 50% idiosyncratic responders for (a) the whole data set and (b) the homogeneous and idiosyncratic subsets identified by the SHOCSY algorithm. (c) The median 1H NMR spectral profiles of homogeneous control (N = 30, red), idiosyncratic responders (N = 15, green), and homogeneous responders (N = 15, blue) for aliphatic regions of the spectrum. The OPLS-DA loading plots of the whole data set (d) and the homogeneous data set (e). The metabolite signals pointing upward correspond to those metabolites up-regulated in Paraquat toxicity, and conversely, metabolite signals pointing downward correspond to those metabolites down-regulated in Paraquat toxicity. The color bar defines the weights of the corresponding discriminating biomarkers between the control and Paraquat toxicity with “hotter” colors indicating a higher correlation with class discrimination. Key: 1, lactate; 2, l-alanine; 3, acetic acid; 4, phenylactylglutamine; 5, p-cresl sulfate; 6, succinic acid; 7, citrate; 8, dimethylamine; 9, trimethylamine; 10, creatinine; 11, trimethylamine-N-oxide; 12, l-histidine, 13, hippurate; 14, taurine; 15, glycine; 16, creatine; 17, glycolic acid. * indicates the metabolic biomarker signals that correspond to Paraquat toxicity.
Application of SHOCSY to a Rat Hydrazine Toxicity Study
The SHOCSY algorithm was used to identify homogeneous subsets from a hydrazine study in rats. The responses were highly homogeneous for all time points except t7 (96–120 h) and t8 (120–144 h) for high dose and t5 (48–72 h) for low dose. SHOCSY was able to identify four and nine spectra as idiosyncratic responders from the high dose hydrazine for t7 and t8, respectively. Furthermore, nine spectra from the hydrazine low dose data at t5 were identified as “idiosyncratic” responders. No idiosyncratic animals were identified in the control groups. We therefore focused our subsequent analyses using the time points where variation in responses to hydrazine toxicity was more evident (t7 and t8 for high dose; and t5 for low dose). As we have shown that 7-fold and double cross-validation methods obtained comparable results using the simulated data sets, we therefore only applied the 7-fold cross-validation for the analysis of the hydrazine toxicity study.
The Q2 value of the OPLS-DA model obtained from the whole data set of the high dose hydrazine class was 0.88 for t7 and 0.77 for t8 (permutation test p < 105). The model statistics improved to 0.94 for t7 and 0.96 for t8 (permutation test p < 10–5) when homogeneous subsets were used rather than the whole class. For the low dose, the Q2 for t5 data slightly improved from 0.75 based on whole data set to 0.77 based on the homogeneous subsets (permutation test p < 10–5).
The OPLS-DA model coefficient plots of the whole data set and the homogeneous subsets at t8 after administration of a high dose of hydrazine showed both endogenous metabolites as well as hydrazine-related metabolites as potential discriminatory biomarkers (Figure 3). On the basis of the defined criteria (i.e., p < 1.85 × 10–6 and loading weight >0.3), the following metabolites were identified from the whole data set: N-α-acetyl-citrulline (δ 4.13, m, δ 3.11, t, δ 2.03, s, δ 1.8, m, δ 1.66, m, δ 1.52, m), 2-aminoadipic acid (δ 3.77, t, δ 2.26, t, δ 1.89, m, δ 1.64, m), citruline (δ 3.76, t, δ 3.15, quintet, δ 1.88, m, δ 1.52, m), 2-oxoglutarate (δ 3.01, t, δ 2.45, t), creatinine (δ 4.05, s, δ 3.04, s), creatine (δ 3.94, s, δ 3.04, s), glycine (δ 3.54, s), and hippurate (δ 7.73, d, δ 7.64, t, δ 7.55, t, δ 3.97, d). Two additional discriminatory biomarkers were also uncovered when the OPLS-DA model based on homogeneous subsets was used (Figure 3c,d). These were diacetyl hydrazine (δ 2.07, s) and beta-alanine (δ 3.19, s, δ 2.56, t). This was made possible as the exclusion of idiosyncratic responders reduced the variability of the data set and thus potential discriminatory biomarkers were not obscured by overlapping signals in confounded spectra. The use of the subset optimization algorithm therefore provided a clearer description of the potential discriminatory biomarkers. As the focus of our current work is to demonstrate how SHOCSY works, the biological interpretation of these metabolite signatures is beyond the scope of this paper but relates to improved capture both of hydrazine metabolism (diacetyl hydrazine) and the endogenous response to liver toxicity (beta-alanine). Readers are referred to previous publications.40,41
Figure 3.

OPLS-DA loading coefficient plots comparing control and hydrazine 90 mg/kg at 120–144 h for the following: (a) the aliphatic spectral region and (b) the aromatic spectral region; for the whole data set (N = 23 for control and N = 18 for hydrazine class); (c) the aliphatic spectral region and (d) the aromatic spectral region; for the homogeneous subsets (N = 23 for control and N = 9 for hydrazine class). Key: 1, N-α-acetyl-citrulline; 2,2-aminoadipic acid; 3, citruline; 4, diacetyl-hydrazine; 5, succinate; 6, 2-oxoglutarate; 7, citrate; 8, creatinine; 9, creatine; 10, beta-alanine; 11, taurine; 12, glycine; 13, hippurate. * indicates the metabolic signals were identified as “biomarkers” of response to hydrazine based on the defined criteria.
The median 1H NMR spectra showing the characteristics of each homogeneous subset as well as the idiosyncratic responders is shown in Supporting Information Figure S-2. From the loading coefficient plots (Figure 3) and the median spectra plots (Figure S-2), it can be seen that the homogeneous responders showed metabolite signatures indicative of hydrazine toxicity. The metabolic profiles of the idiosyncratic responders were more similar to those in the control, as evident by the high level of metabolites such as succinate and 2-oxoglutarate (Figure S-2). The PCA scores plots of the median metabolic time trajectory for these rats showed that these idiosyncratic responders recovered from the hydrazine insult quicker than the others (Figure 4). A similar trend was found for the high dose t7 and low dose t5 data.
Figure 4.

PCA scores plots showing the metabolic trajectory in animals dosed with hydrazine at 90 mg/kg (a and b) and 30 mg/kg (c and d). The metabolic time trajectory was calculated by averaging the PC1 and PC2 scores, respectively, for each time point data. t1: −8–0 h; t2: 0–8 h; t3: 8–24 h; t4: 24–48 h; t5: 48–72 h; t6: 72–96 h; t7: 96–120 h; t8: 120–144 h; t9: 144–168 h.
Benefits of the SHOCSY Algorithm
The SHOCSY algorithm is efficient: an output for the simulated and hydrazine data set was obtained in less than 60 s when we used a computer with 16GB RAM with a Xeon 2.4 GHz preprocessor. The algorithm converged within three iterations for all data sets. This time taken for SHOCSY is comparable to a standard OPLS-DA approach. However, the double cross-validation approach took significantly longer (>15 h, Table S-4).
We also investigated the proportion of idiosyncratic responders in a biological class that the algorithm can tolerate. Using the simulated data sets, we varied the proportion of idiosyncratic responders from 5% to 50%. We found the SHOCSY algorithm could tolerate up to 50% of the spectra showing idiosyncratic responses, without compromising the validity of the model (Table S-3 and S-4). However, this was not the case for standard OPLS-DA approach. Our results showed that our algorithm will work despite extreme condition, and therefore, it will be suitable for use in most biological data sets, particularly those with high variation of nonresponders.
Having developed the SHOCSY algorithm, we applied it successfully to a rodent NMR-based metabonomics study. However, we envisage that the algorithm will be particularly appropriate for spectral data generated from human studies, where varying responses to intervention, such as drugs treatments42−44 and susceptibility to side effects of drugs,13,45 have been observed. As the key goal of SHOCSY is for data variation reduction to improve the reliability of multivariate modeling and to enhance robust biomarkers selection, the mathematical criteria can also be applied to other “omics”-based data sets as well as other data types of spectroscopic data (e.g., GC-MS or LC-MS). Furthermore, the proposed algorithm could also be applied in combination with various data analyses methods other than OPLS-DA.
Conclusion
The reliability of modeling and the extraction of biomarkers are both critical aspects of metabolic profiling studies. Established methods such as OPLS-DA can fail to correctly identify relevant biomarkers when there is variation in responses to biological studies. We have shown how SHOCSY can improve the model classification ability for NMR spectra, while tolerating high variation of responses or idiosyncratic behavior in up to 50% of the data set. Unlike currently used methods, SHOCSY iteratively “learns” the metabolic features best representing the biological classes in the data set and identifies irrelevant samples lacking these features. This enhances the robustness of the biomarker selection process. Moreover, SHOCSY has wide applicability and is not limited to analysis of NMR spectroscopic data.
Acknowledgments
R.L.L. acknowledges support from the MRC New Investigator Grant Award (G1002151) and X.Z. is a postdoc working on the grant. The authors thank the many colleagues who performed the experiments and collected the NMR data for the COMET study. We also thank Dr. M. Rantalainen for his OPLS-DA matlab code and Dr. J. Hao for the help on the use of BATMAN R package.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Holmes E.; Loo R. L.; Stamler J.; Bictash M.; Yap I. K.; Chan Q.; Ebbels T.; De Iorio M.; Brown I. J.; Veselkov K. A.; Daviglus M. L.; Kesteloot H.; Ueshima H.; Zhao L.; Nicholson J. K.; Elliott P. Nature 2008, 453, 396–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan E. C. Y.; Koh P. K.; Mal M.; Cheah P. Y.; Eu K. W.; Backshall A.; Cavill R.; Nicholson J. K.; Keun H. C. J. Proteome Res. 2009, 8, 352–361. [DOI] [PubMed] [Google Scholar]
- Zhou B.; Xiao J. F.; Tuli L.; Ressom H. W. Mol. BioSyst. 2012, 8, 470–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenring J. R. J. Classif. 1993, 10, 131–132. [Google Scholar]
- Bylesjo M.; Rantalainen M.; Cloarec O.; Nicholson J. K.; Holmes E.; Trygg J. J. Chemom. 2006, 20, 341–351. [Google Scholar]
- Cloarec O.; Dumas M. E.; Trygg J.; Craig A.; Barton R. H.; Lindon J. C.; Nicholson J. K.; Holmes E. Anal. Chem. 2005, 77, 517–526. [DOI] [PubMed] [Google Scholar]
- Nevedomskaya E.; Pacchiarotta T.; Artemov A.; Meissner A.; van Nieuwkoop C.; van Dissel J. T.; Mayboroda O. A.; Deelder A. M. Metabolomics 2012, 8, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton T. A.; Lindon J. C.; Cloarec O.; Antti H.; Charuel C.; Hanton G.; Provost J. P.; Le Net J. L.; Baker D.; Walley R. J.; Everett J. R.; Nicholson J. K. Nature 2006, 440, 1073–1077. [DOI] [PubMed] [Google Scholar]
- Coen M.; Goldfain-Blanc F.; Rolland-Valognes G.; Walther B.; Robertson D. G.; Holmes E.; Lindon J. C.; Nicholson J. K. J. Proteome Res. 2012, 11, 2427–2440. [DOI] [PubMed] [Google Scholar]
- Winnike J. H.; Li Z.; Wright F. A.; Macdonald J. M.; O’Connell T. M.; Watkins P. B. Clin. Pharmacol. Ther. 2010, 88, 45–51. [DOI] [PubMed] [Google Scholar]
- Wikoff W. R.; Frye R. F.; Zhu H.; Gong Y.; Boyle S.; Churchill E.; Cooper-Dehoff R. M.; Beitelshees A. L.; Chapman A. B.; Fiehn O.; Johnson J. A.; Kaddurah-Daouk R. PLoS One 2013, 83e57639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudkowska I.; Paradis A. M.; Thifault E.; Julien P.; Barbier O.; Couture P.; Lemieux S.; Vohl M. C. Genes Nutr. 2013, 8, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backshall A.; Sharma R.; Clarke S. J.; Keun H. C. Clin. Cancer Res. 2011, 17, 3019–3028. [DOI] [PubMed] [Google Scholar]
- Dasgupta S.; Freund Y. IEEE Trans. Inf. Theory 2009, 55, 3229–3242. [Google Scholar]
- Kohonen T. Biol. Cybern. 1982, 43, 59–69. [Google Scholar]
- Bubeck S.; von Luxburg U. J. Mach. Learn Res. 2009, 10, 657–698. [Google Scholar]
- Gasch A. P.; Eisen M. B. Genome Biol. 2002, 3, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuperlovic-Culf M.; Belacel N.; Culf A. S.; Chute I. C.; Ouellette R. J.; Burton I. W.; Karakach T. K.; Walter J. A. Magn. Reson. Chem. 2009, 47, S96–S104. [DOI] [PubMed] [Google Scholar]
- O’Sullivan A.; Gibney M. J.; Connor A. O.; Mion B.; Kaluskar S.; Cashman K. D.; Flynn A.; Shanahan F.; Brennan L. Mol. Nutr. Food Res. 2011, 55, 679–690. [DOI] [PubMed] [Google Scholar]
- Sato S.; Arita M.; Soga T.; Nishioka T.; Tomita M. BMC Syst. Biol. 2008, 2, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumpula L. S.; Makela S. M.; Makinen V. P.; Karjalainen A.; Liinamaa J. M.; Kaski K.; Savolainen M. J.; Hannuksela M. L.; Ala-Korpela M. J. Lipid Res. 2010, 51, 431–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckonert O.; Bollard M. E.; Ebbels T. M. D.; Keun H. C.; Antti H.; Holmes E.; Lindon J. C.; Nicholson J. K. Anal. Chim. Acta 2003, 490, 3–15. [Google Scholar]
- Robinette S. L.; Veselkov K. A.; Bohus E.; Coen M.; Keun H. C.; Ebbels T. M. D.; Beckonert O.; Holmes E. C.; Lindon J. C.; Nicholson J. K. Anal. Chem. 2009, 81, 6581–6589. [DOI] [PubMed] [Google Scholar]
- Blaise B. J.; Shintu L.; Elena B.; Emsley L.; Dumas M. E.; Toulhoat P. Anal. Chem. 2009, 81, 6242–6251. [DOI] [PubMed] [Google Scholar]
- Cloarec O.; Dumas M. E.; Craig A.; Barton R. H.; Trygg J.; Hudson J.; Blancher C.; Gauguier D.; Lindon J. C.; Holmes E.; Nicholson J. Anal. Chem. 2005, 77, 1282–1289. [DOI] [PubMed] [Google Scholar]
- Posma J. M.; Garcia-Perez I.; De Iorio M.; Lindon J. C.; Elliott P.; Holmes E.; Ebbels T. M.; Nicholson J. K. Anal. Chem. 2012, 84, 10694–10701. [DOI] [PubMed] [Google Scholar]
- Gusev Y. Methods 2008, 44, 61–72. [DOI] [PubMed] [Google Scholar]
- Bessarabova M.; Ishkin A.; JeBailey L.; Nikolskaya T.; Nikolsky Y. BMC Bioinf. 2012, 13, S13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X.; Dawson N.; Macintyre L.; Morris B. J.; Pratt J. A.; Watson D. G.; Higham D. J. BMC Syst. Biol. 2011, 5, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trygg J.; Wold S. J. Chemom. 2003, 17, 53–64. [Google Scholar]
- Cunningham K.; Claus S. P.; Lindon J. C.; Holmes E.; Everett J. R.; Nicholson J. K.; Coen M. J. Proteome Res. 2012, 11, 4630–4642. [DOI] [PubMed] [Google Scholar]
- Muncey H. J.; Jones R.; De Iorio M.; Ebbels T. M. BMC Bioinf. 2010, 11, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Jewison T.; Guo A. C.; Wilson M.; Knox C.; Liu Y.; Djoumbou Y.; Mandal R.; Aziat F.; Dong E.; Bouatra S.; Sinelnikov I.; Arndt D.; Xia J.; Liu P.; Yallou F.; Bjorndahl T.; Perez-Pineiro R.; Eisner R.; Allen F.; Neveu V.; Greiner R.; Scalbert A. Nucleic Acids Res. 2013, 41, D801–D807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterle F.; Ross A.; Schlotterbeck G.; Senn H. Anal. Chem. 2006, 78, 4281–4290. [DOI] [PubMed] [Google Scholar]
- Lindon J. C.; Nicholson J. K.; Holmes E.; Antti H.; Bollard M. E.; Keun H.; Beckonert O.; Ebbels T. M.; Reily M. D.; Robertson D.; Stevens G. J.; Luke P.; Breau A. P.; Cantor G. H.; Bible R. H.; Niederhauser U.; Senn H.; Schlotterbeck G.; Sidelmann U. G.; Laursen S. M.; Tymiak A.; Car B. D.; Lehman-McKeeman L.; Colet J. M.; Loukaci A.; Thomas C. Toxicol. Appl. Pharmacol. 2003, 187, 137–146. [DOI] [PubMed] [Google Scholar]
- Hao J.; Astle W.; De Iorio M.; Ebbels T. M. Bioinformatics 2012, 28, 2088–2090. [DOI] [PubMed] [Google Scholar]
- Wiklund S.; Johansson E.; Sjostrom L.; Mellerowicz E. J.; Edlund U.; Shockcor J. P.; Gottfries J.; Moritz T.; Trygg J. Anal. Chem. 2008, 80, 115–122. [DOI] [PubMed] [Google Scholar]
- Chadeau-Hyam M.; Ebbels T. M.; Brown I. J.; Chan Q.; Stamler J.; Huang C. C.; Daviglus M. L.; Ueshima H.; Zhao L.; Holmes E.; Nicholson J. K.; Elliott P.; De Iorio M. J. Proteome Res. 2010, 9, 4620–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma S.; Simon R. BMC Bioinf. 2006, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard M. E.; Keun H. C.; Beckonert O.; Ebbels T. M.; Antti H.; Nicholls A. W.; Shockcor J. P.; Cantor G. H.; Stevens G.; Lindon J. C.; Holmes E.; Nicholson J. K. Toxicol. Appl. Pharmacol. 2005, 204, 135–151. [DOI] [PubMed] [Google Scholar]
- Bollard M. E.; Contel N. R.; Ebbels T. M.; Smith L.; Beckonert O.; Cantor G. H.; Lehman-McKeeman L.; Holmes E. C.; Lindon J. C.; Nicholson J. K.; Keun H. C. J. Proteome Res. 2010, 9, 59–69. [DOI] [PubMed] [Google Scholar]
- Trupp M.; Zhu H. J.; Wikoff W. R.; Baillie R. A.; Zeng Z. B.; Karp P. D.; Fiehn O.; Krauss R. M.; Kaddurah-Daouk R. PLoS One 2012, 7, e38386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y.; Hebbring S.; Zhu H.; Jenkins G. D.; Biernacka J.; Snyder K.; Drews M.; Fiehn O.; Zeng Z.; Schaid D.; Mrazek D. A.; Kaddurah-Daouk R.; Weinshilboum R. M. Clin. Pharmacol. Ther. 2011, 89, 97–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaddurah-Daouk R.; Boyle S. H.; Matson W.; Sharma S.; Matson S.; Zhu H.; Bogdanov M. B.; Churchill E.; Krishnan R. R.; Rush A. J.; Pickering E.; Delnomdedieu M. Transl. Psychiatry 2011, 1, 10.1038/tp.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keun H. C.; Sidhu J.; Pchejetski D.; Lewis J. S.; Marconell H.; Patterson M.; Bloom S. R.; Amber V.; Coombes R. C.; Stebbing J. Clin. Cancer Res. 2009, 15, 6716–6723. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.