

Abstract
The gene expression of human embryonic stem cells (hESC) is a critical aspect for understanding the normal and pathological development of human cells and tissues. Current bulk gene expression assays rely on RNA extracted from cell and tissue samples with various degree of cellular heterogeneity. These ‘cell population averaging’ data are difficult to interpret, especially for the purpose of understanding the regulatory relationship of genes in the earliest phases of development and differentiation of individual cells. Here, we report a microfluidic approach that can extract total mRNA from individual single-cells and synthesize cDNA on the same device with high mRNA-to-cDNA efficiency. This feature makes large-scale single-cell gene expression profiling possible. Using this microfluidic device, we measured the absolute numbers of mRNA molecules of three genes (B2M, Nodal and Fzd4) in a single hECS. Our results indicate that gene expression data measured from cDNA of a cell population is not a good representation of the expression levels in individual single cells. Within the G0/G1 phase pluripotent hESC population, some individual cells did not express all of the 3 interrogated genes in detectable levels. Consequently, the relative expression levels, which are broadly used in gene expression studies, are very different between measurements from population cDNA and single-cell cDNA. The results underscore the importance of discrete single-cell analysis, and the advantages of a microfluidic approach in stem cell gene expression studies.
Introduction
The fundamental challenge for stem cell gene expression studies is that stem cells are extremely rare in the context of numerous differentiated and mature cells, and there is no definitive marker for the isolation of a homogenous pure stem cell population. There is especially a lack of markers to distinguish closely related stem cells and progenitor cells. Many stem cell gene expression profiling studies have by default used the heterogeneous populations of stem cells and progenitor cells. Data obtained from these population-averaging expression profiles reflect the sum of all the sub-populations, the population-averaging expression. Without knowing the relative percentages of stem cells and progenitor cells in the interrogated population, these gene expressions profiles, which are the sum of all expression profiles from various cell types, are very difficult to interpret and not sufficiently informative. Even if the percentages are known, variation in the expression profiles among individual stem cells in different phases of the cell cycle would not be detectable by these current methods. Single-cell transcriptome analysis can overcome this hurdle and provide precise information of stem cell gene regulation as revealed in individual separately analyzed cells.
Single-cell gene expression profiling from early embryos has suggested the transient expression of critical regulatory genes, again underscoring the importance of systematical single-cell expression profiling.1,2 Currently, multiple color fluorescent-activated cell sorting (FACS) has a limited utility for isolation of pure stem cell populations due to a lack of discriminating cell markers. Single-cell gene expression profiling studies with laser capture microdissection (LCM),3–12 patch-clamp analysis,2,13,14 as well as in situ mRNA amplification13,15 have been reported, but analysis of a large number of cells has proven to be very difficult with these methods. Single-cell whole genome microarray gene expression screening4,5,16 and single-cell cDNA library construction1,17,18 also have been conducted on a limited number of cells. While these studies demonstrate the potential value of single-cell gene expression profiling, they also showed the limitation of these methods in processing a large number of samples. Material loss and low biochemical reaction efficiency (mRNA capture and RT reaction) are other major challenges for single-cell analysis. A single mammalian cell contains 20–40 pg of total RNA,19,20 but only 0.5–1.0 pg of mRNA (105 to 106 mRNA molecules).21 Therefore, detecting single-cell mRNA is difficult with current methods.
The microfluidic device described here significantly increases the mRNA-to–cDNA processing efficiency ∼5 fold to 54% compared to bulk reactions (∼12%).22,23 With this device, we measured the absolute copy number of three genes in individual single hESCs and compared these data to those obtained from the cDNA of FACS sorted hESCs in the G0/G1 phase. Our results indicate that the hESC colony is a heterogeneous cell population and many single cells do not express all three of the interrogated genes. This result suggests that pluripotent hESC colonies are not homogeneous cell populations, rather they are highly heterogeneous cell populations regulated by different gene networks. Besides stochastic factors of individual cells, heterogeneity of cells with respect to cell cycle and other factors may be a major contributor to observed variations in mammalian single-cell gene expression.
Materials and methods
Mold fabrication
All photomasks were designed with AutoCAD software, and printed at a resolution of 20 000 dots per inch on transparency films (CAD/Art services). We used both the control mold and flow mold to define device features. The control mold with 24 μm high features was fabricated with a single step using SU8-2025 (Microchem, USA). The flow mold was fabricated with three lithographic steps. First we defined the 10 μm high column construction flow channels with SU8-2010 (Microchem, USA). Then the 12 μm high output, bead and buffer delivery channels were fabricated using SPR220-7 (Shipley, USA). In addition, a hard bake process at 200 °C facilitating channel rounding was necessary for valve closure. The last step was to construct the 40 μm high cell loading channels with AZ-50 (Clariant). In all optical lithography processes, mold exposures were under UV light on a MJB mask aligner (7 mW cm−2).
Device fabrication
The microfluidic devices were fabricated by multilayer soft lithography with the silicone elastomer polydimethylsiloxane (PDMS, RTV615, General Electric, USA). Each device employs push-up valve geometry and consists of a three layer elastomeric structure.
The molds were first exposed to chlorotrimethylsilane (TMCS, Aldrich) vapor for 2 min to promote elastomer release after the baking steps. For the flow layer of the device, a mixture of PDMS (5 parts RTV615A : 1 part RTV615B) was poured onto the flow mold. After degassing, the flow molds were baked for 45 min at 80 °C. For the control layer of the device, a mixture of PDMS (20 parts RTV615A : 1 part RTV615B) was spun on the control mold at 1800 rpm and baked for 30 min at 80 °C. The flow layer was separated from the flow mold and flow channel access holes were then punched. Next, the flow and control layers were aligned and baked for 45 min at 80 °C. The two layer structure was peeled from the control mold with control channel access holes punched, and was mounted to another thin PDMS layer made by spinning 20 : 1 PDMS mixture on a blank wafer. After baking for 3 hours at 80 °C, the three layer structure was bonded to a clean microscope slide and baked overnight at 80 °C.
Microfluidic station
Fig. 1 shows the setting of the microfluidic system and the hESC colonies for microfluidic analysis. The microfluidic valves within the device are controlled by individual pressure regulators (Fluidigm, USA) and are interfaced via 23 gauge stainless steel tubing (New England Small Tube) and tygon tubing (VWR). An NI-DAQ card through a Labview interface (National Instruments) was used to control the pressure regulators.
Fig. 1.

The setting of the microfluidic device for single hESC mRNA extraction. A. The system includes a microscope, a computer to control air pressure with pressure regulators, and a heating stage to heat the microfluidic chip to desire temperatures. B. A typical microfluidic chip. C. Merged image of immunofluorescent stained (Oct-3/4) and light microscope images from a pluripotent hESC colony. The hESC colony was labeled with mouse α human Oct-3/4 IgG and PE-conjugated rabbit anti-mouse IgG antibodies. Only cells in the center of the hESC colony expressed Oct-3/4. The intensity of the labeling indicates the Oct-3/4 positive cells expressed Oct-3/4 at different levels. The spontaneously differentiated cells around the colony do not express Oct-3/4.
hESC culture
The hESC line (H9) was obtained from the WiCell Research Institute (Madison, WI) and maintained as instructed.24 Undifferentiated hESCs were cultured on an irradiated (5500 rads) layer of mouse embryonic fibroblast (MEF) feeder cells in 6-well plates or in matrigel-coated plates with MEF conditioned medium. To avoid contamination of mouse cells from the MEF, hESC colonies were dissected and transferred to matrigel-coated plates before using for micro-fluidic processing.
MEF were prepared from the embryos of 13–14 day pregnant CF-1 mice (Charles River Labs) and stocks were cryopreserved until required for culture of hESCs. hESCs were consistently observed as large clumps that appeared on the matrigel-coated surface, consistent with the published observations of others. Periodic karyotyping confirmed their human diploid chromosomal character. After injection into severe combined immunodeficient (SCID) mice, these cells produced teratomas which include tissues of ectodermal, mesodermal and endodermal origin. Immunostaining of the hESC colonies for alkaline phosphatase and with antibodies specific for SSEA-4, TRA-1-60, TRA-1-81 or Oct-3/4 confirmed the pluripotency of these cells.
Synthesis of cDNA from hESCs
The centers (100 to 200 cells) of hESC colonies from feeder-free cultures (matrigel) were mechanically picked up with a 25 gauge needle. The cells were disassociated into single-cell suspension with trypsin. After being labeled with a DNA-selective dye for living cells, Vybrant DyeCycle Green (Invitrogen, USA), FACS was performed to isolate cells in the G0/G1 phase based on DNA content. These isolated cells were pooled to extract total RNA with Trizol LS (Invitrogen, USA), or loaded into the microfluidic device for processing. Typically, 150 000 FACS sorted cells were pooled for Trizol RNA extraction, and 2000 to 5000 cells were used for micro-fluidic experiments. To compare the efficiency of bench-top bulk assays to microfluidic assay, the same biochemical reagents were used for the bulk assay and the microfluidic device.
Single-cell lysis, mRNA capture and RT were performed in the same microfluidic device to convert mRNA into cDNA. Dynabeads with oligo (dT)25 (Invitrogen, USA) were used to capture mRNA. Sensiscript RT kit (Qiagen, USA) was used for converting captured mRNA to cDNA. The oligo (dT)25 sequence in Dynabeads serves as both mRNA capture sequence and primer for cDNA synthesis. After oligo (dT)25 beads with attached cDNA were flushed to the collection wells, the wells were cut off from the chip. Centrifugation was used to transfer the beads from individual wells into PCR tubes. These beads with attached cDNA from individual cells were subjected to realtime qPCR for measurement of the number of molecules of interrogated mRNA in IQ5 (Bio-Rad, USA).
Microfluidic device and single-cell process
The basic components of a microfluidic processor are shown in Fig. 2. The processor captures mRNA from 20 single-cells separately and simultaneously, then converts it into individual cDNA. The lysis buffer was loaded into the flow channels until it reached the waste outlets, so as to leave no air bubbles in the channel. Oligo (dT)25 beads (Invitrogen, USA) were then loaded and columns were built serially by addressing flow lines individually with the multiplexer control channels, while keeping the sieve valve actuated. Once columns were built, excess beads still present in the flow channels were flushed with the lysis buffer to the constructed columns. A single-cell suspension was then loaded. By adjusting cell concentration and flow rate to obtain optimal distance between two floating cells, single hESC was captured in individual cell lysis module (approximately 80% of modules captured only one cell). Microscopic examination was performed to ensure that only cDNA from modules that captured one cell will be used for PCR analysis. Cells were then lysed chemically by mixing cells with the lysis buffer in the ∼10 nL ring (Fig. 2 insert 1). Mixing occurred by executing a peristaltic pump sequence25,26 with control channels. Cell lysates were then pushed via pneumatic pressure over the affinity columns to captured mRNA with oligo (dT)25 beads. After washing the columns with a first strand synthesis buffer, the reverse transcription (RT) master mix was introduced. Once the RT master mix filled the flow channels, first strand synthesis was then carried out by heating the processor to 40 °C on a thermal microscope stage. The oligo (dT)25 beads served as both primers (oligo (dT)25 sequences and a solid phase support. The RT reaction mixture (99 μL) was flown over the columns for 45 min until the RT reaction was completed at a flow rate of ∼20 mm s−1. Upon completion of the RT reaction, the waste valves were closed, and collection valves were opened. The beads were sent to collection wells by opening the sieve valves and flowing columns of the processor in a serial manner with a PCR buffer. The fluid multiplexer was used to push beads in each of the 20 reaction channels individually. Beads were collected by cutting the collection wells off the device and centrifuging the beads into PCR tubes.
Fig. 2.
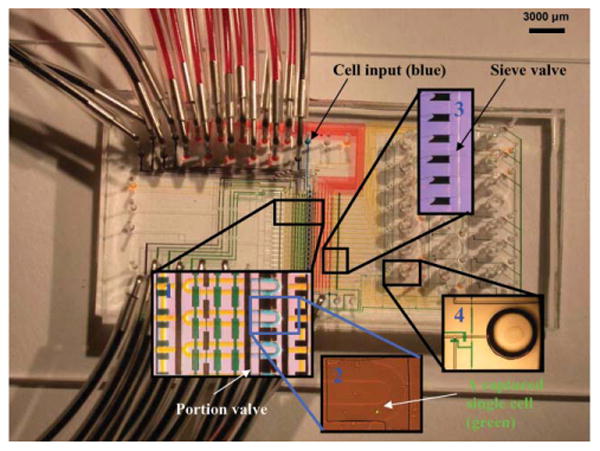
Single-cell mRNA extraction microfluidic device filled with food dye for illustration. All flow channels are filled with yellow food dye, multiplexer control channels are filled with red dye, collection and waste channels are in blue. The inserts show enlargements of four important areas of the chip. After loading cell suspension from the cell input inlet, single-cells are captured in cell lysis module (Insert 1) within the flow channels (blue). The pump valves are green. The separation valve is black. The lysis buffer is yellow. A captured single hESC is labeled with a fluorescent dye (green) and shown in Insert 2. Cell lysis is performed by opening the portion valve and pumping to mix lysis buffer (yellow) with the captured cell. The resulting cell lysate is pushed through oligo-dT bead columns for mRNA capture. Oligo-dT beads are stacked into columns by closing the sieve valve while loading bead suspension. Insert 3 shows six stacked oligo-dT bead columns next to the sieve valve. After washing beads with buffers, RT reaction master mix is flown through the bead columns to synthesize cDNA from the captured mRNA at 40 °C. After RT reaction, beads with attached cDNA are pushed to collection wells (Insert 4) by opening the sieve valve. The beads are recovered by cutting the wells off the chips and centrifuging a flipped-well in a microcentrifuge tube.
Standard curves
Human fetal brain cDNA (Invitrogen, USA) and cDNA generated from poly-A RNA control kit (Affymetrix, USA) were used as templates to obtain quantitative PCR amplicons. Quantification of the PCR amplicons was performed by gel densitometry with DNA ladders (Norgen, Canada). These amplicons contain the majority of the cDNA sequence of the respective genes and were used for generating standard curves for qPCR. The lys standard curve was generated with a known amount of a 846 bp fragment of the lys cDNA. The primers for the amplification were: cagtcaacccttaccgcatt (forward) and acatggacaggaggcatttc (reverse).
Three sets of primers were used to amplify 905 bp, 982 bp and 890 bp fragments of B2M, Nodal and Fzd4 respectively from human brain cDNA (Invitrogen, USA). The primer sets were: B2M: ggcattcctgaagctgaca (forward) and ccagattaaccacaaccatgc (reverse); Nodal: cttcctgagccaacaagagg (forward) and cagactccactgagcccttc (reverse); Fzd4: gggacgtctaaaatcccaca (forward) and ggcagtggagatgaaacaca (reverse)
Quantitative PCR
Four sets of multiplex Taqman primers and probes were designed with Beacon Designer (Premier Biosoft International, USA). B2M: aattgctatgtgtctgggtttcatcc (forward), gcttacatgtctcgatcccacttaac (reverse) and acaaagtcacatggttcacacggcaggca (probe-FAM); Nodal: catacatccagagtctgctgaaacg (forward), atcagaggcacccacattcttcc (reverse) and cccaccgagtcccttccacttgttgtgcc (probe-Cy5);Fzd4: cgaccccatccgcatctcc (forward), acattggcacataaacagaacaaagg (reverse) and ccagaacctcggctacaacgtgaccaaga (probe-Hex); Lys: ggccggttttgtgttagcag (forward), gcggttcatcatcttccgtataac (reverse) and ccgaaacctcctccaagattcagcacct (probe-FAM). Multiplex quantitative PCR was performed with IQ-5 (bio-rad, USA) and Quantitect Taqman PCR kit (Qiagen, USA) for B2M, Nodal and Fzd4. The qPCR of lys was performed independently from the multiplex qPCR.
Results and discussion
Device efficiency
Single-cell measurement of mRNA is difficult. One difficulty results from the loss of material during the steps of single-cell capture, lysis, mRNA isolation and cDNA synthesis. Unlike the DNA molecule, mRNA is very susceptible to degradation by widely existing RNase. Therefore, it is essential to carry out the cDNA first strand synthesis on the same device immediately after mRNA capture. Another difficulty is the low mRNA-to-cDNA efficiency, mRNA capture (∼40–50%) and RT reaction (∼20%)22,23 in bulk assays.
In order to measure the absolute number of molecules of mRNA with standard curves, the mRNA-to-cDNA efficiency of the processor must be obtained. An artificial Poly-A RNA standard (Affymetrix, USA) was used to compare the input mRNA copy number to the cDNA copy number which was measured by qPCR at the end of the processes. This RNA standard contains known amounts of artificial RNAs with poly-A tails from 4 B. subtilis genes, lys, phe, thr and dap. The lys gene was used as RNA standard for calculation of mRNA-to-cDNA efficiency. A standard curve was generated with a known amount of lys DNA at 10 fold dilutions. The mRNA capture and RT reaction were carried out at both conventional micro-litre level and at nano-litre scale with the microfluidic processor.
In the conventional reaction, a 50 μL RT reaction was carried out after capturing 4 μL of artificial mRNA (7.3 nM) with 30 μL oligo (dT)25 beads as instructed by the manufacturer. One micro litre of the RT product (corresponding to 2.16 × 108 molecules of lys artificial mRNA) was used for realtime quantitative PCR (qPCR) detection. Based on the standard curve, the final detected number of molecules was 2.5 ± 0.5 × 107 molecules (Table 1). The processing efficiency of conventional micro-litre scale reaction is 12%.
Table 1.
Microfluidic device increasing mRNA capture and reverse transcription (RT) efficiency. Known amounts of artificial Lys mRNAs in a mixture with 3 other poly-A artificial RNAs were processed with traditional microliter scale reaction or with microfluidic device in nanoliter scale. A standard curve was generated with known amount of lys DNA for the detection of the number of molecules of cDNA (y = −3.5547x + 39.538 R2 = 0.99). The molecule number of lys cDNA was calculated from the standard curve with real-time PCR threshold cycles. Processing efficiencies were calculated by dividing the measured number of cDNA molecules with the number of input mRNA molecules
| Input Lys RNA (molecule number) | Threshold Cycle | Detected cDNA (molecule number) | Efficiency (%) | |
|---|---|---|---|---|
| Microlitre Reaction (Bench-top) | 2.16 × 108 | 13.22 ± 0.37 (n = 20) | 2.6 ± 0.6 × 107 | 12 |
| Nanolitre Reaction (Microfluidic) | 1.84 × 106 | 18.15 ± 0.46 (n = 20) | 1.0 ± 0.3 × 106 | 54 |
To compare the efficiency of conventional and microfluidic approaches, the same artificial RNA sample was introduced into the cell capturing chamber, processed into cDNA and detected with realtime qPCR. Each of the 20 cell-capture chambers in the microfluidic device has a volume of 3.4 nL resulting in 1.84 × 106 molecules of input lys artificial mRNA. The absolute lys cDNA molecule number detected by qPCR with the standard curve is 1.0 + 0.3 × 106. The total processing efficiency was 54% which is approximately 5 times the efficiency of the conventional approach. A series of 10 fold dilutions of the artificial RNA were used and they verified that the processing efficiencies of both approaches do not vary significantly in different concentrations of the input artificial RNA. The standard deviations of both bench-top and microfluidic reactions were also calculated and are similar (Table 1). The small standard deviation of the 20 reactions in the same device indicates consistent processing efficiency among the 20 reaction chambers.
Gene expression of single hESCs
Conventional gene expression studies were carried out with RNA extracted from cell populations. To investigate whether the gene expression of cell population is a reasonable sum of the expressions of individual cells, the expression levels of three genes were measured by two approaches: with pooled cDNA from the hESC population and with cDNA from individual hESCs.
Based on the mRNA-to-cDNA efficiency calculated above, the numbers of three mRNAs in a single hESC were measured. Standard curves were constructed with known amounts of full length B2M, Fzd 4 and Nodal cDNA. The three standard curves overlap each other, indicating similar qPCR efficiency for each set of the primers (Fig. 3A). These standard curves have a range of 2 to 2 × 106 molecules that allow the direct comparison of the expression levels of these three genes from their amplification curves (Fig. 3B). The minimal detectable level is 4 molecules (2/54%) for our microfluidic processor, and 17 molecules (2/12%) for bulk assays with these standard curves.
Fig. 3.
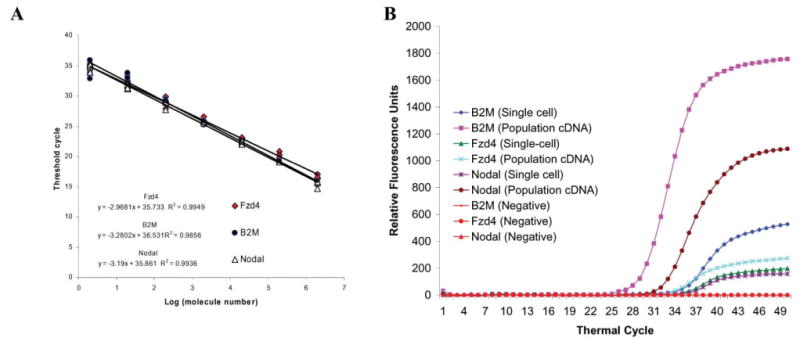
Measuring the absolute molecule number of the three genes in a single hESC with multiplex quantitative PCR. A. Standard curves are generated with known amounts of plasmid DNA containing the full sequence of the genes. The curves cover from 2 to 2 × 106 copies of the respective genes. With our primer design, all the curves overlap each other, and indicate similar PCR efficiency. B. The multiplex quantitative PCR amplification curves obtained from cDNA of hESC colonies are plotted with curves obtained from cDNA of a representative hESC. Because the standard curves of the three genes are very similar, these amplification curves show that the expression ratio of B2M and Nodal is similar in population cDNA and this single-cell cDNA. However, the expression of Fzd4 and Nodal is very similar in this particular single hESC, but very different in the hESC population. Unlike this single hESC, some single hESC do not express all three genes. This result suggests the heterogeneity of hESC and underscores the importance of single-cell analysis.
The pluripotent hESC colonies are often considered and treated as a homogenous population. However, our data indicate that this is not the case. As Fig. 3B shows, the absolute expression levels (mRNA molecule number) measured from cDNA equivalents to 1000 cells are not even close to 1000 fold of the levels measured from a typical single-cell. With cDNA equivalent to 1000 G1/G0 phase hESC, the absolute molecule numbers of B2M, Nodal and Fzd4 are 6,034 ± 660, 402 ± 55 and 69 ± 19 respectively. However, the distributions of the molecule numbers of these 3 genes are diverse in single hESCs (Fig. 4). Among the 54 interrogated single hESCs, 14.8%, 37% and 37% of hESC did not express detectable levels (less than 4 copies) of B2M, Fzd4 and Nodal respectively. The expression of B2M ranged from 4 to 76 copies, with the majority between 30 and 50 copies. The expression of Fzd4 and Nodal ranged from 6 to 548 copies, and 22 to 504 copies respectively. All cells expressed at least one of the 3 genes.
Fig. 4.
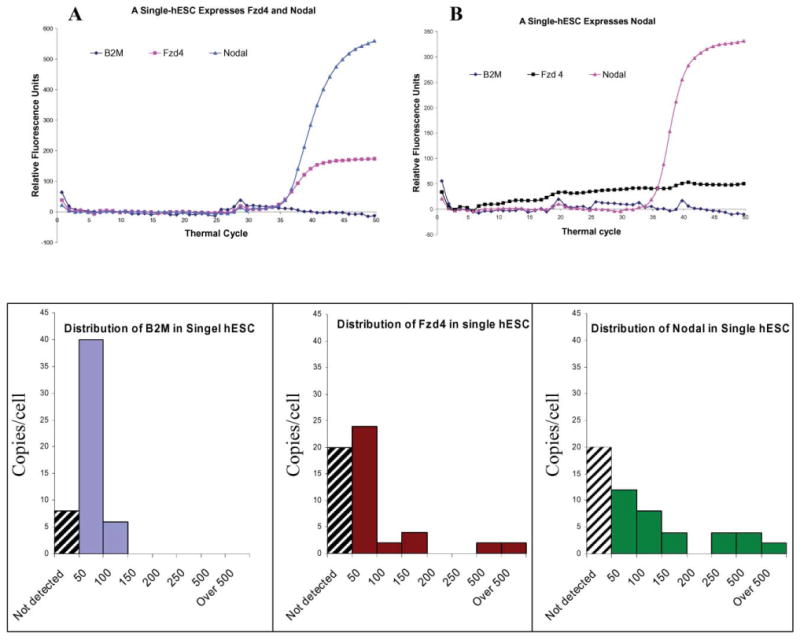
Expression of B2M, Nodal and Fzd4 in single hESCs. A. A single-hESC expresses both Nodal and Fzd4, but does not express B2M in a detectable level. B. A single hESC expresses only Nodal. The mRNAs of B2M and Fzd4 are undetectable. C, D and E shows the distribution of B2M, Fzd4 and Nodal in single hESCs. Among the 54 interrogated single hESCs, 14.8%, 37% and 37% of cells (indicated by black strait pattern) do not express detectable levels (less than 4 copies) of B2M, Fzd4 and Nodal respectively. The distribution pattern of expressions is narrower for B2M compared to the other 2 genes. The discontinuous distribution of Nodal and Fzd4 suggest a high heterogeneity of the 54 cells.
Many gene expression studies, such as microarray or quantitative PCR, are based on the relative expression level of genes with normalization to a house keeping gene which is assumed to have a similar expression in all samples. Our data show that even the relative expression level of the three genes are very different between measurements derived from cDNA of a cell population and separately from individual cells. In the measurement from population cDNA, the expression level of Fzd4 is approximately 8 fold lower than the expression of Nodal after being normalized to B2M. However, in a representative single hESC, the expression levels of Nodal and Fzd4 are very similar after normalization with B2M (Fig. 4).
Conclusions
Microfluidic devices that are designed to manipulate nanolitre amounts of reagents provide a desirable platform for single-cell gene expression processing. As our data indicate, capturing mRNA and converting it to cDNA in a microfluidic device not only consumes significantly less reagents, but most importantly increases the efficiency of biochemical reactions. The microfluidic device described has a mRNA-to-cDNA efficiency 5 times better than the corresponding bench top reactions. Previous studies showed a 40–55% mRNA capture efficiency of oligo (dT)25,22 and ∼20% RT efficiency23 in conventional bench top reactions. The 12% overall efficiency of our bulk assay is consistent with these previous studies (55% × 20%). With our microfluidic device, the mRNA-to-cDNA processing efficiency is 54% (5 fold better than bulk assay). This finding indicates a very efficient mRNA-to-cDNA processing that may be due to the nanolitre scale reaction. This mRNA-to-cDNA efficiency is similar to the previously reported efficiency from other microfluidic devices.27
Due to the nanolitre scale nature of the microfluidic device, the input number of mRNA in conventional reaction and microfluidic reaction is ∼100 fold different when same RNA standard is used. To rule out the possibility of RNA concentration in processing efficiency, serial 10-fold dilutions of the RNA standard were used for the same experiments. The mRNA-to-cDNA efficiencies are not significantly different for both bench-top and microfluidic reactions. Therefore, the high mRNA-to-cDNA efficiency is a desirable characteristic of microfluidic devices. With this 54% processing efficiency, the microfluidic device described potentially can detect 2 mRNA copies of interrogated genes.
In our device, there are 20 individual single-cell processing reactors for simultaneously processing. The standard deviation of threshold cycles of the 20 reactors is small (0.37) and similar to those from conventional reactions (∼0.5). These data indicate that the processing efficiencies among the 20 reactors are very similar. Similar results are also obtained in multiple lots of devices. This aspect is critical for comparing results from individual cells intra- and inter-devices.
With this device, we profiled the expression of three genes in hESC. Our data indicated that even for FACS-sorted G1/G0 phase hESC, the expression data from population cDNA can not be used to estimate the gene expression level in individual cells. Therefore, the population averaging effect distorts the expression levels of the three genes in bulk assays conducted with population cDNA from hESC colonies. Because the population averaging effects are not the same for all three genes, the relative expression levels are also distorted. The qPCR with cDNA from 1000 hESC indicates that Nodal is expressed approximately 8 times more than Fzd4 after normalization to B2M. However, in individual cells the expressions of these genes are very diverse. The difference observed in pooled cDNA may be due to the different number of cells expressing the respective genes.
It has been reported that gene expression is highly variable in the single-cell levels.28,29 A speculation is that the high variability is due to experimental variation. In the present study, we showed that experimental variation may not be the main factor. The stochastic gene expression behavior of single-cells (biological noise) has been reported in various studies of prokaryote cells.30–34 Biochemical processes such as transcription, translation, RNA and protein degradation have been thought of as the primary contributors. However, heterogeneity of the mammalian cell populations may be a factor related to the observed expression variations in single-cell analysis of mammalian cells. When gene expression studies are conducted at the single-cell level, we must recognize that no two cells are identical. In a particular mammalian cell population, two cells always are different in cell cycle, differentiation stages, and environmental stimulation. The variation of gene expression at the single-cell levels is expected, and the reason may be due to stochastic expression fluctuation, or due to heterogeneity of the cell populations. In mammalian cells which are regulated by more complex gene networks than yeast and bacteria, cell heterogeneity may be the major contributor of variation of gene expression at single-cell levels.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH 1RO1 HG002644-01A1) and the Nancy Davis Foundation for Multiple Sclerosis. The authors thank Dr Richard Vestewig for his suggestions and proof reading of the manuscript. The authors also thank the laboratory of Dr Melvin Simon and the laboratory of Dr Vijay Kalra for their assistance in real-time PCR.
References
- 1.Adjaye J, Daniels R, Bolton V, Monk M. Genomics. 1997;46:337–344. doi: 10.1006/geno.1997.5117. [DOI] [PubMed] [Google Scholar]
- 2.Rambhatla L, Patel B, Dhanasekaran N, Latham KE. Mol Reprod Dev. 1995;41:314–324. doi: 10.1002/mrd.1080410306. [DOI] [PubMed] [Google Scholar]
- 3.Chiang MK, Melton DA. Dev Cell. 2003;4:383–393. doi: 10.1016/s1534-5807(03)00035-2. [DOI] [PubMed] [Google Scholar]
- 4.Kamme F, Erlander MG. Curr Opin Drug Discov Dev. 2003;6:231–236. [PubMed] [Google Scholar]
- 5.Luo L, Salunga RC, Guo H, Bittner A, Joy KC, Galindo JE, Xiao H, Rogers KE, Wan JS, Jackson MR, Erlander MG. Nat Med. 1999;5:117–122. doi: 10.1038/4806. [DOI] [PubMed] [Google Scholar]
- 6.Tietjen I, Rihel JM, Cao Y, Koentges G, Zakhary L, Dulac C. Neuron. 2003;38:161–175. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- 7.Rappolee DA, Wang A, Mark D, Werb Z. J Cell Biochem. 1989;39:1–11. doi: 10.1002/jcb.240390102. [DOI] [PubMed] [Google Scholar]
- 8.Trumper LH, Brady G, Bagg A, Gray D, Loke SL, Griesser H, Wagman R, Braziel R, Gascoyne RD, Vicini S. Blood. 1993;81:3097–3115. [PubMed] [Google Scholar]
- 9.Brady G, Billia F, Knox J, Hoang T, Kirsch IR, Voura EB, Hawley RG, Cumming R, Buchwald M, Siminovitch K. Curr Biol. 1995;5:909–922. doi: 10.1016/S0960-9822(95)00181-3. [DOI] [PubMed] [Google Scholar]
- 10.Dixon AK, Richardson PJ, Lee K, Carter NP, Freeman TC. Nucl Acids Res. 1998;26:4426–4431. doi: 10.1093/nar/26.19.4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Emmert-Buck MR, Bonner RF, Smith PD, Chuaqui RF, Zhuang Z, Goldstein SR, Weiss RA, Liotta LA. Science. 1996;274:998–1001. doi: 10.1126/science.274.5289.998. [DOI] [PubMed] [Google Scholar]
- 12.Suarez-Quian CA, Goldstein SR, Pohida T, Smith PD, Peterson JI, Wellner E, Ghany M, Bonner RF. Biotechniques. 1999;26:328–335. doi: 10.2144/99262rr03. [DOI] [PubMed] [Google Scholar]
- 13.Eberwine J, Yeh H, Miyashiro K, Cao Y, Nair S, Finnell R, Zettel M, Coleman P. Proc Natl Acad Sci U S A. 1992;89:3010–3014. doi: 10.1073/pnas.89.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt-Ott KM, Tuschick S, Kirchhoff F, Verkhratsky A, Liefeldt L, Kettenmann H, Paul M. J Cardiovasc Pharmacol. 1998;31:S364–366. doi: 10.1097/00005344-199800001-00102. [DOI] [PubMed] [Google Scholar]
- 15.Crino PB, Trojanowski JQ, Dichter MA, Eberwine J. Proc Natl Acad Sci U S A. 1996;93:14152–14157. doi: 10.1073/pnas.93.24.14152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamme F, Salunga R, Yu J, Tran DT, Zhu J, Luo L, Bittner A, Guo HQ, Miller N, Wan J, Erlander M. J Neurosci. 2003;23:3607–3615. doi: 10.1523/JNEUROSCI.23-09-03607.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ying SY, Lui HM, Lin SL, Chuong CM. Biotechniques. 1999;27:410–412. doi: 10.2144/99273bm02. [DOI] [PubMed] [Google Scholar]
- 18.Trumper LH, Brady G, Vicini S, Cossman J, Mak TW. Ann Oncol. 1992;4:25–26. doi: 10.1093/annonc/3.suppl_4.s25. [DOI] [PubMed] [Google Scholar]
- 19.Roozemond RC. Histochem J. 1976;8:625–638. doi: 10.1007/BF01003963. [DOI] [PubMed] [Google Scholar]
- 20.Uemura E. Brain Res Bull. 1980;5:117–119. doi: 10.1016/0361-9230(80)90182-3. [DOI] [PubMed] [Google Scholar]
- 21.Brady G. Yeast. 2000;17:211–217. doi: 10.1002/1097-0061(20000930)17:3<211::AID-YEA26>3.0.CO;2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hamaguchi Y, Aso Y, Shimada H, Mitsuhashi M. Clin Chem. 1998;44:2256–2263. [PubMed] [Google Scholar]
- 23.Zhang J, Byrne CD. Biochem J. 1999;337:231–241. [PMC free article] [PubMed] [Google Scholar]
- 24.Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, Jones JM. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- 25.Chou HP, Unger MA, Quake SR. Biomed Microdev. 2001;3:323–330. [Google Scholar]
- 26.Marcus JS, Anderson WF, Quake SR. Analyt Chem. 2006;78:956–958. doi: 10.1021/ac0513865. [DOI] [PubMed] [Google Scholar]
- 27.Warren L, Bryder D, Weissman IL, Quake SR. Proc Natl Acad Sci U S A. 2006;103:17807–17812. doi: 10.1073/pnas.0608512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- 30.Newman JR, Ghaemmaghami S, Ihmels J, Breslow DK, Noble M, DeRisi JL, Weissman JS. Nature. 2006;441:840–846. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 31.Swain PS, Longtin A. Chaos. 2006:16. doi: 10.1063/1.2213613. [DOI] [PubMed] [Google Scholar]
- 32.Suel GM, Kulkarni RP, Dworkin J, Garcia-Ojalvo J, Elowitz MB. Science. 2007;315:1716–1719. doi: 10.1126/science.1137455. [DOI] [PubMed] [Google Scholar]
- 33.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 34.Ryley J, Pereira-Smith OM. Yeast. 1065;23:1065–1073. doi: 10.1002/yea.1412. [DOI] [PubMed] [Google Scholar]