

Abstract
The “element effect” in nucleophilic aromatic substitution reactions (SNAr) is characterized by the leaving group order, L = F > NO2 > Cl ≈ Br > I, in activated aryl substrates. A different leaving group order is observed in the substitution reactions of ring-substituted N-methylpyridinium compounds with piperidine in methanol: 2-CN ≥ 4-CN > 2-F ~ 2-Cl ~ 2-Br ~ 2-I. The reactions are second-order in [piperidine], the mechanism involving rate determining hydrogen-bond formation between piperidine and the substrate-piperidine addition intermediate followed by deprotonation of this intermediate. Computational results indicate that deprotonation of the H-bonded complex is probably barrier free, and is accompanied by simultaneous loss of the leaving group (E2) for L = Cl, Br, and I, but with subsequent, rapid loss of the leaving group (E1cB-like) for the poorer leaving groups, CN and F. The approximately 50-fold greater reactivity of the 2- and 4-cyano substrates is attributed to the influence of the electron withdrawing cyano group in the deprotonation step. The results provide another example of β-elimination reactions poised near the E2-E1cB mechanistic borderline.
INTRODUCTION
The nucleophilic aromatic substitution (SNAr) reactions of activated substrates follow a two-step, addition-elimination mechanism and have been well studied.1-8 They also continue to draw attention as a synthetic method.9 An order of halide leaving group (L) abilities, F > Cl ≈ Br > I, is often found in studies of rates of SNAr reactions of activated aryl halides. This is commonly referred to as the “element effect”, and is considered evidence for a mechanism in which the first step, addition of the nucleophile, is rate controlling.10-12 The factors that can account for the element effect and for SNAr reactivity in general are manifold, and have been thoroughly discussed in the literature.10-14 These factors include polar, polarizability, solvation, and negative hyperconjugative effects as well as polarity reversal of the C–L bond from reactant to transition state in the case of ArCl and ArBr compared to ArF.
In related studies, we have observed that 2-substituted pyridinium compounds are good substrates for SNAr reactions due to their electron-deficient nature.15,16 In this report, we examine experimentally the SNAr reactions of ring-substituted N-methylpyridinium substrates with piperidine.17 In addition, intermediates and transition states associated with the rate determining step were explored computationally.
EXPERIMENTAL RESULTS AND DISCUSSION
Third-order rate constants and activation parameters for the reaction of nucleophilic piperidine with 2-cyano-, 4-cyano-, 2-fluoro-, 2-chloro-, 2-bromo- and 2-iodo-N-methylpyridinium substrates in methanol are listed in Table 1. All the reactions are first-order in substrate and second-order in piperidine, and yield the corresponding piperidino-N-methylpyridinium ions as the only products.
Table 1.
Kinetic parameters for nucleophilic aromatic substitution reactions of 2-substituted N-methylpyridinium substrates with piperidine in methanol.a
| Substrates | Overall third- order rate constant (25° C, M−2s−1) |
Relative rate at 25 °C |
ΔG‡
(kcal/mol) |
ΔH‡
(kcal/mol) |
ΔS‡
(cal/mol K) |
|---|---|---|---|---|---|
| 2-cyano | 1.26 ×10 −3 | ~50 | 20.6 | 18.4 | −7.5 |
| 2-fluoro | 2.02 × 10−5 | ~1 | 22.5 | 19.8 | −9.1 |
| 2-chloro | 2.10 × 10−5 | ~1 | 22.4 | 16.3 | −20.6 |
| 2-bromo | 2.67 × 10−5 | ~1 | 22.3 | 20.9 | −4.8 |
| 2-iodo | 2.62 × 10−5 | ~1 | 22.3 | 15.7 | −22.2 |
| 4-cyano | 8.30 × 10−4 | ~30 | 20.9 | 20.0 | −2.9 |
Piperidine concentration is 0.5 M whereas initial substrate concentration is 10 mM. Kinetic parameters were calculated from temperature dependent studies using the Eyring equation. Estimated uncertainties in ΔH‡ and ΔS‡ are
Negative entropies of activation are found throughout the series, but are seen to be quite variable in magnitude, the TΔS‡ term contributing from 0.9 to 6.6 kcal/mol to the free energy of activation. The source(s) of the observed activation entropies are complex, and probably include differential solvation of reactants and transition states. For comparison we call attention to the fact that ΩS‡ values for a host of SN2 processes are known to be highly variable.18 The rate constant differences for the halide substrates are very small, and are associated with irregular changes in the enthalpies and entropies of activation. The cyano compounds are exceptional in that their reactivities are greater than those of the halides. Unlike the reactions of piperidine with 2,4-dintrophenyl halides there is no evidence of an “element effect”.
Reactivity
From Table 1 we see that reactivity is fairly high although less so than for the reactions of piperidine with 2,4-dinitrophenyl halides for which experimental activation enthalpies are much lower, but are partly compensated by more negative activation entropies.1 The reactivity of the cyanide substrates is noteworthy. There is precedent for this result in our previous study of the basic hydrolysis of ring-substituted N-methylpyridinium ions.15 Additionally Thompson and Huestis have recently observed loss solely of cyanide in SNAr reactions even in the presence of halides at reactive positions.19 We were surprised by these results, and considered the possibility that the cyano substrates react by a radical mechanism, e.g., the SRN1 pathway, since the cyano group is well known to stabilize free radicals.20,21 We examined the effect of two different radical scavengers, 2,2,6,6-tetramethylpiperidine oxide (TEMPO) and 2,6-di-t-butylphenol, on the rate of reaction of piperidine with all substrates, but found no inhibition at two or more scavenger concentrations. Hence, if radical intermediates are formed they are paired or otherwise inaccessible to the scavengers. However, the greater reactivity of the cyano substrates in these SNAr reactions is can perhaps be understood in a qualitative way on the basis of the greater total electron withdrawing effect of the cyano group relative to halogens as determined by NMR studies on ring-substituted N-methylpyridinium ions.22 This effect must be present in the rate-controlling step, shown below to be hydrogen-bond formation and proton transfer from the addition complex to a second piperidine molecule, activated by the electron-withdrawing cyano group.
Mechanism
The reaction mechanism could, in principle, be termolecular, but it is much more likely that more than one bimolecular step is involved. Given the second-order dependence of the rate on piperidine concentration we conclude that step 1, nucleophilic addition forming intermediate I-1 is not rate controlling for these SNAr reactions, but that a subsequent step, deprotonation of I-1 with or without departure of nucleofuge L−, both paths requiring a second piperidine molecule, determines the overall rate (see Figure 2). The results presented so far also explain the absence of the element effect, an effect identified with the addition step.10-14 An additional pertinent result is that there is no observation of an intermediate species at zero-time as monitored by 1HNMR or by UV spectrometry. Thus we treat the addition intermediate, I-1, as a reactive intermediate subject to the steady-state approximation, and formulate the rate law as equation (1). Taking k−1 >> k2[pip] then leads to equation (2)
Figure 2.

Proposed mechanism for reaction of piperidine with pyridinium substrates. A hydrogen-bonded complex between I-1 and the second piperidine is implied, but not shown.
| (1) |
| (2) |
Loss of the leaving group could occur from I-1 in a one-step E2 process, or in two steps: deprotonation of the piperidinium NH+ moiety giving intermediate I-2, followed by loss of the leaving group in step 3. Step 2, deprotonation to form intermediate I-2, could be rate controlling. Alternatively, the rate controlling step could be expulsion of the leaving group from I-2 (step 3).
Bernasconi has presented a thorough and persuasive analysis of the kinetic behavior of reactive tetrahedral intermediates.4 He concludes that in protic solvents the general-base catalyzed breakdown of such intermediates proceeds by rate-controlling deprotonation of the addition intermediate, I-1 in the reaction studied here. It might seem counter-intuitive that proton transfer between electronegative atoms, two nitrogens in this case, can be rate controlling. However, as Bernasconi points out, all that is necessary is that the subsequent step be much faster than the reverse of the deprotonation step. He also provides evidence that in general the secondary amine moiety in addition intermediate I-2 is less basic than the amine itself, hence that the reverse of step 2 will be endoergic. Finally, loss of L− is accompanied by re-aromatization of the heterocyclic ring, further accelerating the product-forming elimination in step 3. Thus we favor the pathway shown in Figure 2 with step 2, hydrogen-bond formation between I-2 and piperidine and/or deprotonation of the addition intermediate, rate controlling, whether accompanied by leaving group departure (E2) or not (E1cB-like) and in the latter case with step 3, loss of the leaving group, fast compared with step −2. Figure 2 is a summary of this proposal. The activating effect of the cyano group can now be understood as strengthening the acidity of the NH proton in the addition complex, I-1, thereby favoring formation of the hydrogen-bonded complex preceding the proton-transfer event, step 2. These details and the mechanism of the elimination step(s) will be further considered below.
In summary we propose that as the second, basic piperidine reactant approaches I-1 it undergoes hydrogen-bond formation with the δ-positive NH proton of the nucleophilic piperidine, that nitrogen becoming thereby more nucleophilic, thus bonding more strongly to electrophilic carbon-2. Proton transfer to the basic piperidine leads directly and irreversibly to a hydrogen-bonded complex between piperidinium cation and neutral intermediate I-2, thence to rapid loss of the leaving group and ultimate product formation. Thus, the mechanism is an example of catalysis by preassociation,23-25 and the greater reactivity of the cyano substrates is then associated with this set of events.
COMPUTATIONAL RESULTS AND DISCUSSION
The Elimination Step(s)
Computation provides useful information regarding the details of the elimination step(s). The calculated structures for I-2 differ in significant ways as the leaving group is changed. For the 2-cyano, 4-cyano, and 2-fluoro versions of I-2 the heterocyclic rings are puckered, the C-L bonds are slightly longer than normal length for sp3 carbon, especially the C-F bond, and, importantly, the N-C2 distances within the heterocycle are longer (1.460, 1.506, and 1.492 Å, respectively) than those in the original aromatic substrates (1.352, 1.340, and 1.329 Å, respectively). The latter lengths reflect the degree of aromaticity in the ring, and tell us that in these three cases I-2 is an ordinary molecule, not aromatic, albeit with some “hyperaromatic” character, particularly for L = F.26 In contrast I-2 for the 2-chloro and 2-bromo versions show flat heterocyclic rings, very long C-L bonds (2.949 and 3.045 Å, respectively) and short N-C2 bonds within the heterocycle: 1.365 and 1.367 Å, respectively; compare with 1.342 and 1.343 Å for the substrates. In fact for L− = chloride and bromide I-2 can be considered an ion-pair complex between product and L−, formed by departure of L− from I-2, and not a stable covalent neutral. This assessment is supported by the large negative npa charges on the chlorine and bromine in I-2: -0.95 in both cases. Structures constrained to have shorter C-Cl and C-Br bonds were of higher energy than those with the long C-L bonds.
On the basis of these differences we propose that the elimination stage for the poorer leaving groups, CN and F, uses an E1cB-like mechanism (probably the E1cB irreversible variant) while for the chloro, bromo and iodo leaving groups the elimination mechanism is E2, enforced by the instability of I-2 in those cases. We do not have experimental evidence regarding the exclusion of a reversible E1cB mechanism. A classic isotopic exchange experiment is not feasible for two reasons, either of which obviates the experiment. In the first place the N-H proton of piperidine exchanges essentially “immediately” with solvent methanol. Secondly, the pertinent intermediate, I-2, is too short-lived to be observed.27
Transition states and their precursor hydrogen-bonded complexes were also found for the deprotonation of I-1 by piperidine (see supporting information). The energies of the transition states and the H-bonded complexes are so similar as to suggest that the exothermic proton transfer event does not have a significant enthalpic barrier. Thus, we propose that the elimination stage of the overall reaction includes the critical free energy barrier (i.e., is in fact rate controlling), starting with hydrogen bond formation and continuing with a facile proton transfer and leaving group departure, the latter either concurrent with proton transfer (L = Cl, Br, I) or subsequent to it (L = F, CN). The loss of the leaving group from I-2 is expected to be rapid for F and CN, relative to reprotonation,4 supporting our suggestion that the elimination mechanism is probably the E1cBIRR variant in those two cases. Figure 3 shows calculated structures for two of the proton-transfer transition states and the corresponding neutral deprotonation products, I-2. We see that in I-2, L = Br, that the C-Br distance is very large compared with that in the ts. This result suggests that proton transfer and loss of leaving group, though very likely concerted for the better leaving groups, are not closely coupled.
Figure 3.

(a) proton-transfer transition state for deprotonation of the 2-bromo hydrogen-bonded complex; (b) the transition state for the 2-fluoro complex; (c) the neutral deprotonaed adduct (I-2) for the 2-bromo reaction; (d) the neutral I-2 for the 2-fluoro reaction.
These results add an example to the study of the mechanistic borderline in β-elimination reactions, a topic of continuing interest and activity.28,29 It is easy to imagine that a leaving group, intermediate in nucleofugacity between fluoride and chloride, would provide a mechanistically ambiguous case with, perhaps, a metastable neutral intermediate (I-2) and an almost barrier-free departure of the leaving group.
SUMMARY
The nucleophilic aromatic substitution reactions of 2-substituted N-methylpyridinium ions with piperidine in methanol do not proceed through rate controlling addition of the nucleophile, but by rate controlling deprotonation of the addition intermediate by a preassociation mechanism. Thus, the “element effect”10,11 commonly observed for activated SNAr reactions in which nucleophilic addition is rate controlling, is not observed. The reactivity order is 2-cyano ≥ 4-cyano > 2-fluoro ~ 2-chloro ~ 2-bromo ~2-iodo. The elimination of the leaving group, L−, is proposed to occur by a piperidine-catalyzed E1cBIRR-like mechanism for L = F and CN, but by a concerted E2 mechanism for the better leaving groups, L = Cl, Br and I. The data suggest that the cyano group’s ability to enhance the stability of the preassociation complex is a key driver in the higher substitution rates for these substrates.
EXPERIMENTAL SECTION
2-Cyano-N-methylpyridinium iodide as well as the 4-cyano-, 2-fluoro-, 2-chloro-, and 2-iodo iodides as well as 2-bromopyridinium bromide were prepared according to literature procedures.22, 30-32 The rate constants of the reactions were determined under pseudo-first order conditions (large excess of piperidine) by following the disappearance of substrate and appearance of products using NMR spectroscopy. The concentrations of the substrates and products were measured by the integration of signals of the aromatic protons.
COMPUTATIONAL METHODS
Structures were built and optimized at lower levels using the MacSpartan Plus software package,33 then optimized at HF/6-31+G* then MP2/6-31+G* using the Gaussian 03 quantum mechanical programs.34 Frequency and zero-point energy values were calculated at the HF/6-31+G* level, and the ZPVE values scaled as recommended by Scott and Radom.35 All structures reported here represent electronic energy minima, and all structures identified as transition states (tss) have one imaginary frequency, that corresponding to the reaction coordinate for the reaction event. The optimized MP2 geometries for substrates and transition states were used to obtain energies with the Polarizable Continuum Model (PCM), solvent = methanol (see Supporting Information).
Supplementary Material
Figure 1.
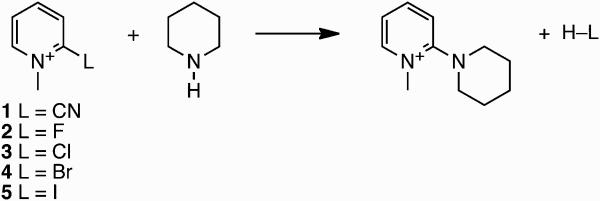
The overall result for reaction of piperidine with 2-substituted-N-methylpyridinium+ substrates.
ACKNOWLEDGEMENTS
This investigation was supported by the National Institutes of Health, Grant SC1 GM095419 and by a grant from the Center for Computing for Life Sciences at SFSU (WW). J.T.B. was supported by a Beckman Scholarship. We thank Dr. Robert Yen for obtaining the mass spectra. The Mass Spectrometry Facility was funded by National Science Foundation (CHE-1228656). The NMR facility was funded by the National Science Foundation (DUE-9451624 and DBI 0521342). SG acknowledges funding from the National Science Foundation (CHE-1300817).
Footnotes
SUPPORTING INFORMATION: Full reference 34; 1H and 13C NMR data as well as mass Spectrometry (MS) data for characterization of N-methyl-2-piperidinopyridinium products; calculated electronic energies and zero point vibrational energies for substrates are given in Table S1; calculated electronic energies, zero point vibrational energies and imaginary frequencies for the transition states and products for the addition of piperidine to substrates are in Table S2; calculated enthalpies of isomerization of 2-substituted to 4-substituted substrates and addition transition states are in Table S3; calculated enthalpies of solvation for substrates and transition states for the addition of piperidine to 2- and 4-substituted N-methylpyridinium ions, Table S4; calculated C-L and C-Nu distances in the transition states for addition of piperidine to 2- and 4-substituted N-methylpyridinium ions are in Table S5; calculated activation enthalpies for addition of piperidine to 2- and 4-substituted N-methylpyridinium ions are in Table S6; calculated npa charges for substrates and transition states for the addition of piperidine to substrates, Table S7; calculated energies and zero point vibrational energies for the hydrogen-bonded complexes formed by addition of piperidine to 2-substituted N-methylpyridinium ions and electronic energies for the subsequent proton transfer event, Table S8; enthalpies of association forming the H-bonded complexes between piperidine and the addition intermediate are in Table S9 along with activation enthalpies for proton transfer within this complex; enthalpies of association forming the H-bonded complexes from initial substrates and two piperidines are in Table S10, and Cartesian coordinates for computed structures in this study are in Table S11.
REFERENCES
- 1.Bunnett JF, Zahler RE. Chem. Rev. 1951;49:273–412. [Google Scholar]
- 2.Bunnett JF. Quart. Rev. (London) 1958;12:1–16. [Google Scholar]
- 3.Miller J. Aromatic Nucleophilic Substitution. Elsevier; Amsterdam: 1968. p. 941. [Google Scholar]; Nicholls B, Whiting MC. J. Chem. Soc. 1959:551. [Google Scholar]; Brown DA. J. Chem. Soc. 1963:4389. [Google Scholar]; Brown DA, Raju JR. J. Chem. Soc., A. 1966:40. [Google Scholar]
- 4.Bernasconi CF. Acc. Chem. Res. 1978;11:147–152. [Google Scholar]
- 5.Terrier F. Nuclear Aromatic Displacement: The Influence of the Nitro Group. VCH; New York: 1991. [Google Scholar]
- 6.Terrier F. Nucleophilic Aromatic Substitution. Wiley; Singapore: 2013. [Google Scholar]
- 7.Makosza M. Chem. Soc. Rev. 2010;39:2855–2868. doi: 10.1039/b822559c. [DOI] [PubMed] [Google Scholar]
- 8.Buncel E, Dust JM, Terrier F. Chem. Revs. 1995;95:2261–2280. [Google Scholar]
- 9.Walsh K, Sneddon HF, Moody CJ. ChemSusChem. 2013;6:1455–1460. doi: 10.1002/cssc.201300239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bunnett JF, Garbisch EW, Jr., Pruitt KM. J. Am. Chem. Soc. 1957;79:385–391. [Google Scholar]
- 11.Bunnett JF. J. Am. Chem. Soc. 1957;79:5969–5974. [Google Scholar]
- 12.Bunnett JF, Merritt WD. J. Am. Chem. Soc. 1957;79:5967. [Google Scholar]
- 13.Bartoli G, Todesco PE. Acc. Chem. Res. 1977;10:125. [Google Scholar]
- 14.Senger NA, Bo B, Cheng Q, Keeffe JR, Gronert S, Wu W. J. Org. Chem. 2012;77:9535–9540. doi: 10.1021/jo301134q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang S, Wong FM, Gassner GT, Wu W. Tetrahedron Lett. 2011;52:3960–3962. doi: 10.1016/j.tetlet.2011.05.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan RC, Vien JQT, Wu W. Bioorg. Med. Chem. Lett. 2012;22:1224–1225. doi: 10.1016/j.bmcl.2011.11.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roe A, Hawkins GF. The 4-halopyridinium substrates were not investigated due to the instability of 4-halopyridines. J. Am. Chem. Soc. 1947;69:2443–2444. [Google Scholar]
- 18.Bordwell FG, Brannen WT., Jr. Activation entropies in nucleophilic substitutions (SN2) have been shown to be sensitive to a variety of reaction conditions. J. Am. Chem. Soc. 1964;86:4645–4650. [Google Scholar]
- 19.Thompson AD, Huestis MP. J. Org. Chem. 2013;78:762–769. doi: 10.1021/jo302307y. [DOI] [PubMed] [Google Scholar]
- 20.Berkowitz J, Ellison GB, Gutman D. The bond dissociation energy of the C-H bond in acetonitrile is smaller than that in methane by ten kcal/mol. J. Phys. Chem. 1994;98:2744–2764. [Google Scholar]
- 21.Pirnot MT, Rankic DA, Martin DBC, MacMillan DWC. Science. 2013;339:1593–1596. doi: 10.1126/science.1232993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang S, Wong JCS, Leung AKC, Chan YM, Wong L, Fernendez MR, Miller AK, Wu W. Tetrahedron Lett. 2009;50:5018–5020. doi: 10.1016/j.tetlet.2009.06.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jencks WP, Salvesen K. J. Am. Chem. Soc. 1971;93:1419–1427. [Google Scholar]
- 24.Schowen RL, Kershner LD. J. Am. Chem. Soc. 1971;93:2014–2024. [Google Scholar]
- 25.Jencks WP. Acc. Chem. Res. 1976;9:425–432. [Google Scholar]
- 26.Kudavalli JS, Rao SN, Bean DE, Sharma ND, Boyd DR, Fowler PW, Gronert S, Kamerlin SCL, Keeffe JR, More O’Ferrall RA. J. Am. Chem. Soc. 2012;134:14056–14069. doi: 10.1021/ja304366j. [DOI] [PubMed] [Google Scholar]
- 27.Grunwald E, Eustace D. A reaction solution, less the substrate, shows no trace of the N-H proton at times less than ten minutes. In: Gold V, Caldin EF, editors. Proton Transfer Reactions. Chapman and Hall; London: 1975. ch. 4. [Google Scholar]
- 28.Kim Y, Mohrig JR, Truhlar DG. J. Am. Chem. Soc. 2010;132:110711–11082. doi: 10.1021/ja101104q. [DOI] [PubMed] [Google Scholar]; Mohrig JR. Acc. Chem. Res. 2013;46:1407–1416. doi: 10.1021/ar300258d. [DOI] [PubMed] [Google Scholar]
- 29.Duarte G, Gronert S, Kamerlin SCL. J. Org, Chem. 2013;78:1280–1288. doi: 10.1021/jo402702m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huang S, Miller AK, Wu W. Tetrahedron Lett. 2009;50:6584–6585. doi: 10.1016/j.tetlet.2009.09.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muscio O. J., Jr., Rutherford DR. J. Org. Chem. 1987;52:5194–5198. [Google Scholar]
- 32.Ammon HL, Erhardt WD. J. Org. Chem. 1980;45:1914–1918. [Google Scholar]
- 33.Wavefunction, Inc. 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612.
- 34.Frisch MJ, et al. GAUSSIAN 03, Revision B.04. Gaussian, Inc.; Pittsburgh PA: 2003. See Supporting Information for full reference. [Google Scholar]
- 35.Scott AP, Radom L. J. Phys. Chem. 1996;100:16502–16513. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.