

Abstract
A large variety of functional self-assembled supramolecular nanostructures have been reported over recent decades.1 The experimental approach to these systems initially focused on the design of molecules for specific interactions that lead to discrete geometric structures.1–4 Recently, kinetics and mechanistic pathways of self-assembly have been investigated,6,7 but there remains a major gap in our understanding of internal conformational dynamics and their links to function. This challenge has been addressed through computational chemistry with the introduction of molecular dynamics (MD) simulations, which yield information on molecular fluctuations over time.5–7 Experimentally, it has been difficult to obtain analogous data with sub-nanometer spatial resolution. Thus, there is a need for experimental dynamics measurements, to confirm and guide computational efforts and to gain insight into the internal motion in supramolecular assemblies. Using site-directed spin labeling and electron paramagnetic resonance (EPR) spectroscopy, we measured conformational dynamics through the 6.7 nm cross-section of a self-assembled nanofiber in water and provide unique insight for the design of supramolecular functional materials.
Supramolecular assemblies may exhibit a wide range of dynamics within different domains of their internal structure. This complexity is ubiquitous in nature, where hydrophilic amino acid residues positioned in a region of a protein with no secondary structure may be highly dynamic, exhibiting greater fluctuations in space as compared to slow-moving residues locked into a hydrogen bonded, β-sheet domain of the same protein.8 This example illustrates the strong correlation between the dynamics at specific protein sites and the localized dominant intermolecular interactions.9–11 Similar to the range of conformational dynamics within a protein, supramolecular nanostructures are also likely to demonstrate a range of conformational dynamics concordant with the strength of local intermolecular interactions. Such variation in conformational dynamics could be an important element to the design of their functions.12 For example, the dynamics at the surface of a supramolecular nanofiber may have significant consequences to cell signaling functions, while nanofiber stiffness is expected to influence the mechanobiology of cell differentiation13 and maturation.14 Experimental dynamics measurements with sub-nanometer resolution have not yet been demonstrated.
We examined the dynamics through the cross-section of a self-assembled nanofiber whose intermolecular interactions are designed to differ on the nanometer length scale.15 The range of internal motion is expected to be diverse due to the modularity of the self-assembling subunits in a nanofiber constructed of peptide amphiphiles (PAs).1,2,7 As illustrated in Fig. 1a, the PA molecule is defined by its three structural components: (1) a hydrophobic alkyl tail, (2) a structural peptide sequence often designed to exhibit a high propensity for β-sheet formation, and (3) a terminal sequence that contains charged residues and sometimes a bioactive moiety. In water, hydrophobic collapse of the aliphatic tails induces self-assembly into high-aspect-ratio nanofibers in which the aliphatic core is partitioned from the peptide domain. When the sequence closest to the core is composed of residues with a high propensity for β-sheet formation, such as valines and alanines, β-sheets are expected to persist down the long axis of the fiber.16,17 Conversely, these β-sheets can be disrupted by replacing the amide hydrogen atoms involved in hydrogen bonding with methyl groups.18 We investigated cross-sectional dynamics by covalently bonding to specific sites of the PA molecules spin label probes containing small and unobtrusive radical electron moieties fused directly to the peptide backbone or alkyl tail of a PA molecule (Fig. 1),19,20 and used quantitative EPR spectroscopy to analyze local molecular motion at each site.
Figure 1.

Peptide amphiphiles self-assemble into high-aspect-ratio nanofibers. a) Chemical structures of the PA molecules investigated where the aliphatic region is shown in black, structural sequence shown in red, and charged residues in blue. PA 1 contains six residues VVAAEE, where the valines are expected to contribute to intermolecular hydrogen bonding. PA 2 contains an N-methylated valine, VN(me)VAAEE, to reduce intermolecular hydrogen bonding propensity. PAs 1a–1e are labeled with radical electron spin labels (radicals shown in green); b) Molecular graphics rendering of a coassembled supramolecular nanofiber with PA molecules containing spin labels at the innermost residue corresponding to PA 1/1c; c) Cryogenic transmission electron micrograph of PA 1 in water reveals high-aspect-ratio nanofibers with diameters of approx. 7 nm; the area outlined with a square is shown magnified in the inset.
For this work, PA 1 was chosen because the alkyl core, structural sequence, and headgroup domains embody three different intermolecular interactions: van der Waals dominate in the core, β-sheet hydrogen bonding dominates the structural domain, and the surface is governed by electrostatic repulsion. This internal complexity allows the observation of a range of dynamics through the PA 1 cross-section. To provide contrast to the internal dynamics of PA 1 nanofibers, chemical modification of a residue in the structural sequence was carried out to decrease intermolecular hydrogen bonding propensity. Specifically, incorporation of an N-methylated residue in the PA structural sequence (PA 2) has been shown to hinder internal β-sheet formation, and thus a nanofiber composed of both PA 1 and 2 is a weak β-sheet analogue of PA 1.18 We therefore pursued measurements of site-specific dynamics through the cross-section of PA 1 nanofibers and nanofibers formed by co-assembly of PA 1 and 2 (PA 1/2). Each of these systems form nanofibers with static structures that cannot be distinguished using conventional techniques, such as electron microscopy or small angle x-ray scattering (SAXS), that characterize only geometry, dimensions, and aspect-ratio (Fig. 1c and Supplementary Section S2).
EPR spectra of nitroxide spin probes contain information of local molecular motion. This information arises from the interaction between the radical electron of the nitroxide probe and an applied magnetic field. When EPR spectra are carried out at X band (9.8 GHz), correlation times on the order of 10−6 to 10−9 s can be resolved (corresponding to rotational diffusion rates in the range of MHz to GHz). Site-specific dynamics within PA nanofibers were investigated by incorporation of nitroxide spin labels (PAs 1a–1e) into PA 1 and PA 1/2 nanofibers. The spin label moieties on PAs 1a–1e were positioned to survey the full dynamic range of the nanofiber cross-section. Cryo-TEM and SAXS confirm that the incorporation of spin labels does not perturb the canonical PA nanofiber structure (see Supplementary Section S2). EPR spectroscopy of the hybrid nanofibers (each of PA 1 and PA 1/2 combined with 0.4 % of PAs 1a–1e) was carried out to obtain dynamics information from each site containing the spin label (Fig. 2). In this methodology, narrow EPR line shape indicates fast motion as expected when the region of the PA molecule containing the nitroxide probe experiences weak intermolecular interactions, and broadened spectra correspond to slow motion as in the case of a spin probe buried in a region of the PA nanofiber that exhibits high propensity for hydrogen bonding. This effect can be seen by eye in Fig. 2, where the narrowest spectra are observed when PA 1a, the spin label at the surface, and 1e, the spin label in the core domain, are incorporated into the nanofibers. In addition, the breadth of spectra at each site (1a–1e) of the PA 1 nanofibers (Fig. 2a) is greater than that of each corresponding site of PA 1/2 composite nanofibers (Fig. 2b).
Figure 2.
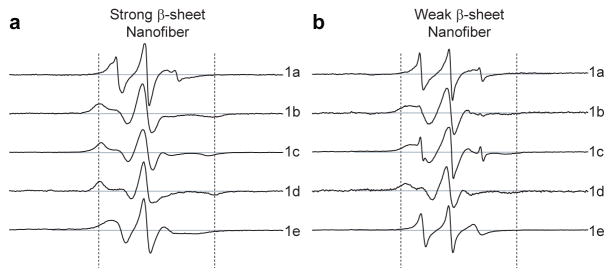
Electron paramagnetic resonance spectra at specific sites through the cross-section of peptide amphiphile nanofibers. a) EPR spectra of strong β-sheet nanofiber composed of 99.6% PA 1 and 0.4% of each spin labeled PA (1a–1e). Spectra of nanofibers produced by co-assembly of PA 1 with PA 1a (top) through PA 1e (bottom) correspond to the cross-section from the nanofiber surface to the core, respectively; b) weak β-sheet nanofiber composed of 49.8% PA 1, 49.8% PA 2, and 0.4% of each PA 1a–1e. Sweep widths shown correspond to 150 Gauss. Dashed lines indicate a separation of 65 Gauss.
EPR simulations of the spectra shown in Fig. 2 were performed using a non-linear least squares analysis based on the microscopic order macroscopic disorder (MOMD) model pioneered by the Freed laboratory.21 In this model, PA nanofibers are randomly oriented in the sample and nanofiber diffusion is taken to be negligible on the timescale of the molecular motion captured by the spin probes. Thus, the spin probes reflect molecular motion with respect to the stationary nanofiber frame of reference. Details of the fitting procedure can be found in Supplementary Section S3. From the EPR simulations, rotational diffusion rates (kr) were extracted from the fitting parameters and were plotted against radial position within the nanofiber (Fig. 3a).
Figure 3.

Internal dynamics through the cross-section of peptide amphiphile nanofibers are obtained with sub-nanometer resolution and shown to depend on the presence of β-sheet promoting amino acids. a) Rotational diffusion rates (kr) extracted from EPR spectral simulations (Fig. S4) with each spin label (PAs 1a–1e) integrated into the nanofiber, plotted against theoretical radial position (calculated from a fully extended PA molecule) and radial distance. The purple line corresponds to PA 1 nanofibers designed to possess strong β-sheet character, and the green line to equimolar coassemblies of PA 1 and 2 designed to possess weak β-sheet character. The three domains of the nanofiber cross-section are indicated by I. aliphatic core, II. structural domain shell, and III. charged corona. b) ThT assay showing the decrease in β-sheet character with increasing incorporation of PA 2 into PA 1 nanofibers. When the composition of the nanofiber contains < 10% PA 2, ThT fluorescence intensity remains high because β-sheet hydrogen bonding is present to a substantial extent. At 20–50% incorporation of PA 2, the nanofibers’ β-sheet character has been significantly reduced. c) Circular dichroism spectrum of PA 1 (purple) indicates strong β-sheet character, and of PA 1/2 coassembly (green) showing that after incorporation of PA 2, β-sheet character is no longer dominant.
The rotational diffusion rates (kr), shown in Fig. 3a, span a range of nearly two orders of magnitude. This broad range confirms the anticipated complexity in internal dynamics of the PA nanofiber and indicates that the nanofiber simultaneously expresses liquid-like and solid-like dynamics through the small (6.7 nm) cross-section. In addition, these measurements reveal substantial differences in dynamics in the structural domain of the nanofiber depending on the presence of low β-sheet propensity PA molecules (PA 2). At the surface of the PA 1 nanofiber (radial position 1) the dynamics are commensurate with a viscous fluid, with a rotational diffusion rate of 105 MHz. As a point of reference, freely dissolved spin labeled small molecules exhibit rotational diffusion rates on the order of hundreds of MHz. When PA 2 is incorporated into the nanofiber, the surface dynamics are unchanged, indicating that the internal N-methyl modification does not significantly affect the PA surface.
The innermost residue is known to contribute as a critical component to the β-sheet structure in the canonical PA nanofiber, and so this site is expected to exhibit slow dynamics.18 Fig. 3a shows that indeed, the inner structural sequence of the PA 1 nanofiber (at radial position 0.54) exhibits very slow motion of kr=6.5 MHz. Interestingly, the rotational diffusion rate at the region of the alkyl core adjacent to the innermost residue (at radial position 0.35) also shows very slow dynamics (kr=5.4 MHz). This site is separated by a distance of only about 5 Å from the structural sequence, and thus the hydrogen bonding of the innermost residues may impose order, and consequently reduced dynamics, at a moderate depth into the nanofiber core. In contrast to the slow dynamics at the innermost residue of PA 1, the PA 1/2 nanofiber exhibits much faster dynamics at the same radial position. This is the result of the reduced hydrogen bonding propensity arising from the incorporation of an N-methylated valine in the structural sequence, an effect that unambiguously shows the relationship between strength of intermolecular interaction and internal site-specific dynamics. At radial position 0 of the PA 1 nanofiber, viscous fluid-like, dynamics are observed. While we acknowledge that the spin label at radial position 0 might be capable of surveying the entire core, and even possibly folding back into the peptide domain, it is still not surprising that the dynamics of this spin label are much faster than at radial position 0.35 since only a weak correlation to the structural sequence exists at a site that is separated by 16 as opposed to five carbon atoms. Similarly, at radial position 0 of the PA 1/2 nanofiber, viscous fluid-like dynamics are detected, but with faster motion than at the same site of the PA 1 nanofiber. This observation suggests that less order is present when internal hydrogen bonding is precluded by the incorporation the N-methyl substituted residues in PA 2.
The presence of solid-like motion in PA 1 nanofibers resembles the dynamics of protein sites in regions of persistent intermolecular hydrogen bonding, for example within a β-sheet. Therefore, β-sheet content within the PA 1 and PA 1/2 nanofibers was probed by thioflavin T (ThT) assay and circular dichroism spectroscopy. The ThT assay is a well established technique for characterization of amyloid fibrils, where high fluorescence intensity of ThT indicates the presence of β-sheets.22 Fig. 3b shows the fluorescence spectra of ThT with PA nanofibers composed of increasing ratios of PA 2 to PA 1. Fig. 3b shows that the fluorescence intensity of PA 1 nanofibers is high, as expected when β-sheet structure is dominant. When PA 2 is incorporated at 1–10%, the ThT fluorescence intensity remains high, suggesting that β-sheet structure is still favored. A sharp decrease in ThT fluorescence intensity occurs when the content of PA 2 reaches the range 20–50%. At these compositions, the nanofibers are expected to have significantly less β-sheet content. Another transition in ThT fluorescence intensity occurs between 50–100% PA 2. This transition likely corresponds to a geometric change in which monodisperse cylindrical nanofibers are no longer the main component (as supported by SAXS, see Supplementary Section S3). CD was used to further analyze the β-sheet content of PA 1 and PA 1/2 nanofibers. The CD spectrum of PA 1, shown in Fig. 3c, is characteristic of β-sheet structure with a minimum at 215 nm, whereas the spectrum of the PA 1/2 nanofibers is consistent with a combination of random coil or α-helical configurations. The observation of β-sheet structure by the ThT assay and CD spectra in conjunction with the solid-like internal dynamics revealed by EPR spectroscopy suggests immobilized β-sheet structure is present only in the case of PA 1 nanofibers. This indicates ordered and persistent hydrogen bonding locks the residues in the structural sequence into a supramolecular configuration that exhibits nearly negligible fluctuations about an average structure.
The molecular dynamics within a self-assembled nanofiber were experimentally quantified, revealing for the first time the complex nature of the molecular motion through a nanofiber cross-section at sub-nanometer length scales. We found that a significant contrast in dynamics exists within a supramolecular nanofiber which can be mediated by the hydrogen bonding propensity of the constituent molecules. Conformational dynamics of a covalently bound spin label probe ranging from rotational diffusion rates of 5 MHz to 100 MHz were observed within a single nanofiber, comparable to the motion of solids and viscous fluids, respectively. The slow dynamics observed at the innermost residue and adjacent sites can be defined as a solid-like shell surrounding the viscous core. This shell within the PA 1 nanofiber should affect nanofiber stiffness and cohesion, and should be important to the mechanobiology of artificial extracellular matrices created with PA fibers. In addition, the ability to characterize surface dynamics is important because the nanofibers may be functionalized with epitopes to introduce bioactivity. Cellular response to the epitopes displayed by the nanofibers may be controlled by tuning their dynamics through molecular design. The observed sensitivity of dynamics to the internal intermolecular interactions and the contrast in state of matter within the cross-section of a single nanofiber suggest that the measurements described here are a useful tool for the design of new biomaterials and other supramolecular materials with integrated functions.
Methods
Synthesis of PAs
PAs were synthesized using standard Fmoc-based solid-phase peptide synthesis. PAs 1a, 1b, and 1c were synthesized using Fmoc-2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid (Fmoc-TOAC), an unnatural(nitroxide-containing) amino acid.19 PAs 1d and 1e were made by off-resin coupling of the free amine of the H2N-VVAAEE peptide in solution to a spin labeled alkyl tail consisting of NHS-derivatives of DOXYL-containing alkyl carboxylates. Full synthetic details can be found in the Supplementary Section S1.
Sample Preparation
PA samples were prepared by dissolving PA 1, PA 2, and/or PA 1a–1e in 5% NH4OH(aq). The solutions were mixed together to obtain equimolar ratios of PA 1/2 and/or final spin label concentrations of 0.4%. The mixtures were bath sonicated for 30 min, then lyophilized to white powders. Each powder mixture was dissolved at 2 wt% in pure water and pH-adjusted to 7.4 by addition of NaOH. 1M CaCl2 was added for final [Ca2+] of 100 mM and final PA concentrations of 1 wt%.
Cryogenic Transmission Electron Microscopy (Cryo-TEM)
CryoTEM was performed on a JEOL 1230 microscope with an accelerating voltage of 100 kV. A Vitrobot Mark IV equipped with controlled humidity and temperature was used for plunge freezing samples. A small volume (5–10 μL) of PA nanofiber suspension at 0.1% (w/v) in water was deposited on a copper TEM grid with holey carbon support film (Electron Microscopy Sciences) and held in place with tweezers mounted to the Vitrobot. The specimen was blotted in an environment with 90–100% humidity and plunged into a liquid ethane reservoir that was cooled by liquid nitrogen. The vitrified samples were transferred in a nitrogen environment into liquid nitrogen and transferred to a Gatan 626 cryo-holder using a cryo-transfer stage. Samples were imaged using a Gatan 831 bottom-mounted camera.
Electron Paramagnetic Resonance (EPR)
EPR spectroscopy was performed on a Bruker EMX with a 4119HS cavity at X band (9.8 GHz) with center field at 3400 G and a 200 G sweep width. Modulation amplitudes were kept below 0.2 times the peak-to-peak line widths. Each sample (3 μL) was injected into a 1.5 cm PTFE capillary (O.D. 1.58 mm, I.D. 0.76 mm), and capped on both ends with Critoseal. The capillaries were deposited into a quartz EPR tube that was inserted into a rectangular EPR resonator cavity; all spectra were collected at ambient temperature.
EPR Spectral Simulations
Spectral simulations were performed using the Acert software package (NLSL), with a microscopic order macroscopic disorder (MOMD) model._ENREF_1921 Frozen spectra (150 K) were fit to find A and g tensor components, which were input and fixed for the room temperature fitting procedure. From the output simulation parameters, the rotational diffusion coefficient, rbar, is extracted, from which the rotational diffusion rate, kr, is computed: kr = 10rbar. Details of the fitting procedure and model can be found in Supplementary Section S3.
ThT Assay
To characterize β-sheet formation, a thioflavin T (ThT) assay was performed. PA solutions were dissolved in buffer (70 mM NaCl, 2 mM KCl, 4 mM Na2HPO4, pH 7.4) at 10 mM and aged overnight. A freshly dissolved stock solution of ThT (Sigma-Aldrich) was prepared and added to PA solutions to give 1 mM PA with 100 μM ThT. Fluorescence spectra were monitored with an excitation wavelength of 440 nm and emission wavelengths 450 nm – 550 nm using a Molecular Devices SpectraMax M5 spectrometer.
Circular Dichroism
CD measurements were performed on a Jasco J-815 spectrometer using a quartz cuvette with a path length of 1 mm. A solution of 1% (w/v) PA was dissolved in 100 mM NaCl, pH 7.4 and aged overnight. Prior to CD experiments, a CaCl2 solution was added to the PA for a final concentration of 100 mM CaCl2 and 0.05% (w/v) PA. The data were averaged over five spectra for each sample and measured at room temperature.
Supplementary Material
Figure 4.

Schematic molecular graphics representation of peptide amphiphile nanofibers. Nanofiber composed of a) PA 1 and b) PA 1/2. The vertical bar indicates the gradient of solid-like to liquid-like dynamics (blue and red, respectively) through the nanofibers’ cross-sections. PA 1 has high β-sheet character resulting in solid-like dynamics in the structural sequence. By removing the β-sheet (by coassembly of PA 1 with PA 2), faster dynamics are observed as the PA structural sequences is no longer locked into hydrogen bonding networks.
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences and Engineering Division, of the U.S. Department of Energy under Award # DE-FG02-00ER45810. JBM acknowledges a postdoctoral fellowship through the National Institutes of Health National Research Service Award # 1F32AR06195501. JHO acknowledges an IBNAM-Baxter early career award. Electron paramagnetic resonance experiments were carried out at The Medical College of Wisconsin, National Biomedical EPR Center, supported by National Institutes of Health Grant P41 EB001980, and also at Northwestern University under support of the National Heart, Lung and Blood Institute of the National Institutes of Health (NIH-HL-13531). X-ray diffraction experiments were carried out at beamline 5ID-D of the Advanced Photon Source at Argonne National Laboratory. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This work also made use of Northwestern University’s Biological Imaging Facility (BIF) for electron microscopy and the Integrated Molecular Structure Education and Research Center (IMSERC) for mass spectrometry and FTIR spectroscopy. The authors acknowledge Prof. Songi Han for helpful discussions and Mark Seniw for illustrations.
Footnotes
Author Contribution Statement
JHO designed and performed experiments, and analyzed data. CJN synthesized chemicals, performed experiments, and analyzed data. JBM synthesized chemicals. LCP assisted in data analysis and writing. PED assisted in performing experiments. LCP, BMH, SIS, and CJN provided intellectual input. JHO and SIS wrote the manuscript.
References
- 1.Palmer LC, Stupp SI. Molecular self-assembly into one-dimensional nanostructures. Acc Chem Res. 2008;41:1674–1684. doi: 10.1021/ar8000926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hartgerink JD, Beniash E, Stupp SI. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 3.Whitesides GM, Grzybowski B. Self-assembly at all scales. Science. 2002;295:2418–2421. doi: 10.1126/science.1070821. [DOI] [PubMed] [Google Scholar]
- 4.Hill JP, et al. Self-assembled hexa-peri-hexabenzocoronene graphitic nanotube. Science. 2004;304:1481–1483. doi: 10.1126/science.1097789. [DOI] [PubMed] [Google Scholar]
- 5.Venable RM, Zhang Y, Hardy BJ, Pastor RW. Molecular Dynamics Simulations of a Lipid Bilayer and of Hexadecane: An Investigation of Membrane Fluidity. Science. 1993;262:223–226. doi: 10.1126/science.8211140. [DOI] [PubMed] [Google Scholar]
- 6.Klein ML, Shinoda W. Large-scale molecular dynamics simulations of self-assembling systems. Science. 2008;321:798–800. doi: 10.1126/science.1157834. [DOI] [PubMed] [Google Scholar]
- 7.Lee OS, Stupp SI, Schatz GC. Atomistic Molecular Dynamics Simulations of Peptide Amphiphile Self-Assembly into Cylindrical Nanofibers. J Am Chem Soc. 2011;133:3677–3683. doi: 10.1021/ja110966y. [DOI] [PubMed] [Google Scholar]
- 8.Shaw DE, et al. Atomic-Level Characterization of the Structural Dynamics of Proteins. Science. 2010;330:341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 9.Buchete NV, Tycko R, Hummer G. Molecular Dynamics Simulations of Alzheimer’s β-Amyloid Protofilaments. J Mol Biol. 2005;353:804–821. doi: 10.1016/j.jmb.2005.08.066. [DOI] [PubMed] [Google Scholar]
- 10.McHaourab HS, Lietzow MA, Hideg K, Hubbell WL. Motion of Spin-Labeled Side Chains in T4 Lysozyme. Correlation with Protein Structure and Dynamics. Biochemistry. 1996;35:7692–7704. doi: 10.1021/bi960482k. [DOI] [PubMed] [Google Scholar]
- 11.Török M, et al. Structural and dynamic features of Alzheimer’s Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J Biol Chem. 2002;277:40810–40815. doi: 10.1074/jbc.M205659200. [DOI] [PubMed] [Google Scholar]
- 12.Newcomb CJ, et al. Cell death versus cell survival instructed by supramolecular cohesion of nanostructures. Nature communications. 2014;5 doi: 10.1038/ncomms4321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gilbert PM, et al. Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science Signaling. 2010;329:1078. doi: 10.1126/science.1191035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sur S, et al. A hybrid nanofiber matrix to control the survival and maturation of brain neurons. Biomaterials. 2012;33:545–555. doi: 10.1016/j.biomaterials.2011.09.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberger JE, Berns EJ, Bitton R, Newcomb CJ, Stupp SI. Electrostatic Control of Bioactivity. Angew Chem Int Ed. 2011;50:6292–6295. doi: 10.1002/anie.201100202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velichko YS, Stupp SI, de la Cruz MO. Molecular Simulation Study of Peptide Amphiphile Self-Assembly. J Phys Chem B. 2008;112:2326–2334. doi: 10.1021/jp074420n. [DOI] [PubMed] [Google Scholar]
- 17.Paramonov SE, Jun HW, Hartgerink JD. Self-Assembly of Peptide–Amphiphile Nanofibers: The Roles of Hydrogen Bonding and Amphiphilic Packing. J Am Chem Soc. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 18.Paramonov SE, Jun HW, Hartgerink JD. Self-assembly of peptide-amphiphile nanofibers: the roles of hydrogen bonding and amphiphilic packing. J Am Chem Soc. 2006;128:7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 19.Toniolo C, Crisma M, Formaggio F. TOAC, a nitroxide spin-labeled, achiral Cα-tetrasubstituted α-amino acid, is an excellent tool in material science and biochemistry. J Peptide Sci. 1998;47:153–158. [Google Scholar]
- 20.Karim CB, Kirby TL, Zhang Z, Nesmelov Y, Thomas DD. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc Natl Acad Sci USA. 2004;101:14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Budil DE, Lee S, Saxena S, Freed JH. Nonlinear-least-squares analysis of slow-motion EPR spectra in one and two dimensions using a modified Levenberg-Marquardt algorithm. J Magn Reson, Ser A. 1996;120:155–189. [Google Scholar]
- 22.Levine H. Thioflavine T interaction with synthetic Alzheimer’s disease β-amyloid peptides: Detection of amyloid aggregation in solution. Protein Sci. 1993;2:404–410. doi: 10.1002/pro.5560020312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.