

Abstract
Fixed-charge empirical force fields have been developed and widely used over the past three decades for all-atom molecular simulations. Most simulation programs providing these methods enable only one set of force field parameters to be used for the entire system. While this is generally suitable for single-phase systems, the molecular environment at the interface between two phases may be sufficiently different from the individual phases to require a different set of parameters to be used to accurately represent the system. Recently published simulations of peptide adsorption to material surfaces using the CHARMM force field have clearly demonstrated this issue by revealing that calculated values of adsorption free energy substantially differ from experimental results. While nonbonded parameters could be adjusted to correct this problem, this cannot be done without also altering the conformational behavior of the peptide in solution, for which CHARMM has been carefully tuned. We have developed a dual-force-field approach (Dual-FF) to address this problem and implemented it in the CHARMM simulation package. This Dual-FF method provides the capability to use two separate sets of nonbonded force field parameters within the same simulation: one set to represent intra-phase interactions and a separate set to represent inter-phase interactions. Using this approach, we show that interfacial parameters can be adjusted to correct errors in peptide adsorption free energy without altering peptide conformational behavior in solution. This program thus provides the capability to enable both intra-phase and inter-phase molecular behavior to be accurately and efficiently modeled in the same simulation.
1. INTRODUCTION
The simulation of large molecular systems typically involves the application of empirical force field-based molecular simulation methods. As the name indicates, the force field parameters that are used with these types of methods are generally established for specific types of molecular systems by empirical adjustment until desired physical and thermodynamic properties such as liquid density, solvation enthalpy, or molecular conformation, become acceptably close to targeted values.
The empirical nature of the development of parameters for empirical force fields raises the important issue of force field transferability, or the ability of a set of parameters to accurately represent the behavior of a system in an environment different from which it was parameterized for. This issue has been nicely demonstrated by van Gunsteren and coworkers,1 who found that it was not possible to obtain a unique set of partial charges for a designated group of molecules with polar functional groups that would satisfactorily reproduce the thermodynamic quantities of the pure liquids formed from these compounds while also correctly reproducing solvation enthalpy for the simulation of these compounds in aqueous solution. This problem was primarily attributed to the use of a nonpolarizable fixed-charge force field in which the partial charges of the functional groups in the system could not adjust themselves to different molecular environments. To address this problem, it was determined that two separate force field parameter sets were required—one to be used when these polar molecules were represented as homogeneous liquids and another to be used when they were dissolved in aqueous solution.1
A similar issue occurs for the simulation of mixed-phase systems, where the molecular environment at the interface between two separate phases may be substantially different from the bulk of either phase. Thus, for a two-phase system, a set of force field parameters that correctly represents the behavior of a given atom type in a given phase may not accurately represent the behavior of the same atom type and phase when an atom is located near the boundary of the other phase.
This problem is of particular concern for the simulation of peptide and protein adsorption to solid material surfaces,2 which is an important for many applications in biotechnology, and biomedical science and engineering.3 Simulations of peptide or protein adsorption behavior have typically been performed using a single protein force field, such as AMBER,4 CHARMM,5–7 or OPLS-AA,8 to represent the entire system (solid phase, liquid phase, and interactions at the solid-liquid interface). However, protein force fields have been primarily developed and validated to represent the conformational behavior of peptides and proteins in aqueous solution without consideration for their interactions with material surfaces, thus raising the issue of their transferability to this type of application. One compounding factor that has limited the ability to address this issue is the lack of quantitative experimental data on peptide and protein adsorption, which has hindered attempts to assess and validate existing protein force fields for this kind of applications.2
In order to address this problem, Wei and Latour recently conducted a study with the objective of generating a large benchmark experimental data set that could be used to directly assess the ability of an empirical force field to accurately represent peptide adsorption behavior in physiological saline solution (approximately 140 mM Na+/Cl−).9 In their study, the free energy of adsorption (ΔGads) was determined using surface plasmon resonance spectroscopy for a set of host-guest peptides (TGTG-X-GTGT; T=threonine, G=glycine, X=variable guest residue) with twelve different guest residues adsorbed to alkanethiol self-assembled monolayer (SAM) surfaces functionalized with nine different organic functional groups, thus providing 108 different peptide-surface combinations. The free energies of adsorption determined from these experiments varied between 0.0 and −5.0 kcal/mol. Biased-energy replica-exchange molecular dynamics (REMD)10–12 simulations were then conducted by Vellore et al.13 using the CHARMM force field for a set of 38 of these same peptide-SAM systems in CHARMM’s TIP3P water with 140 mM Na+/Cl−. Adsorption free energies were calculated and compared with the experimental data set. While the CHARMM force field was able to reproduce experimental values within 1.0 kcal/mol for 24 of these 38 systems, 14 of the calculated values differed by more than 1.0 kcal/mol, with some values being in error by as much as 4 kcal/mol. Table 1 presents the results from two of these systems, TGTG-V-GTGT (V = Valine) on SAM surfaces functionalized with methyl (CH3) and hydroxyl (OH) groups. Using the CHARMM force field, the calculated free energy of adsorption on the CH3-SAM surface predicted adsorption affinity to be too weak by 2.84 kcal/mol while the peptide adsorption affinity on the OH-SAM surface was predicted to be about 0.77 kcal/mol too strong.
Table 1.
Free energies of adsorption (Gibbs free energies for experimental data, ΔGads, Helmoltz free energies for values from the simulations, ΔAads) for a TGTG-V-GTGT peptide over CH3- and OH- SAM surfaces: Comparison between experimental values and values obtained by simulation using the CHARMM force field.9,13 (Mean ± 95% confidence interval). Due to the near incompressibility of water, differences between ΔGads and ΔAads can be considered to be negligible for adsorption processes in aqueous solution.
| SAM | ΔGads (kcal/mol) ΔAads
|
|
|---|---|---|
| Experiment | Simulation | |
| OH-SAM | 0.0 ± 0.4 | −0.77 ± 0.32 |
| CH3-SAM | −4.4 ± 0.5 | −1.56 ± 0.56 |
The errors in adsorption free energy shown in Table 1 represent imbalances in the relative attraction between the peptide and water for the functional groups of the SAM surface. The behavior of the ions was not considered to significantly influence peptide adsorption behavior for these SAMs because of the lack of charged groups on the surface. As noted by van Gunsteren and coworkers,1 the apparent imbalances in force field parameterization for these systems may be a result of the inability of the atoms in the system to be polarized in response to differences in the molecular environment at the interface compared to bulk solution conditions. However, these differences could also be due to relatively small errors in the Lennard-Jones (L–J) terms of the CHARMM protein force field that become amplified in the simulation of peptide adsorption behavior, which is dominated by the balance of the nonbonded parameters responsible for peptide and TIP3P water interactions with the surface.
While the identified errors in peptide adsorption behavior could be corrected by adjusting the CHARMM nonbonded parameters for the peptide and TIP3P water, these changes would alter the conformational behavior of the peptide in solution, which the CHARMM force field has been carefully tuned to represent. Thus, modification of the CHARMM force field to more closely match peptide adsorption free energies is not a viable solution to this problem, since it is desirable to accurately represent both the conformational behavior of a peptide in solution as well as its interactions with a surface. Alternatively, the MSCALE facility in CHARMM, which has recently been introduced by Woodcock et al.,14 could be used to address this problem, which utilizes a separate set of nonbonded force field parameters to represent interfacial behavior while still using regular CHARMM parameters to represent molecular behavior in bulk solution. However, the use of MSCALE for this type of system would be computationally inefficient due to the high communication costs required to perform the simulations. Finally, while the use of a polarizable force field may help reduce these errors, the incorporation of polarizability requires a longer simulation times compared to fixed charge force field simulations. This is because either smaller timesteps are required for polarizable simulations to conserve energy or additional MD iterations are required per timestep in order to equilibrate the charge distribution or dipole moment of the functional groups in the system. Furthermore, if the errors in the free energies of peptide adsorption are due to imbalances in L–J parameters, the use of a polarizable force field would not correct the problem because current polarizable force fields only address electrostatic effects.
As an alternative means to address this issue, we have developed an approach that uses a completely independent set of nonbonded force field parameters to represent the interactions between atoms of one phase with atoms of another phase (i.e., inter-phase behavior) while retaining the use of single-phase force fields (such as CHARMM) to represent the molecular interactions of the atoms within each individual phase of the system (i.e., intra-phase interactions). This approach has been implemented using the CHARMM program. We refer to this as the dual force field (Dual-FF) approach, and the modified program as Dual-FF CHARMM.
In this paper, we first present a description of the strategy and code modifications that were implemented to develop Dual-FF CHARMM. We then illustrate the development of an interfacial force field, designed for interactions between aqueous peptide solutions and the distinctly different surfaces represented by CH3- and OH-SAMs. This modified interfacial force field parameter set is then used to obtain adsorption free energy values that are in much closer agreement with the experimental data without altering the conformation behavior of the peptide in solution. These newly developed capabilities provide a novel path toward accurate simulation of protein adsorption behavior.
2. MATERIALS AND METHODS
2.1. Program Modifications to Implement Dual-FF CHARMM
In order to describe a solid-liquid multiphase system with a tunable interfacial force-field within the CHARMM molecular simulation program, we defined solid phase (phase I) and liquid phase (phase II) subsystems represented with CHARMM force-field parameters while representing interactions between phase I and II atoms by a different set of nonbonded parameters (i.e., interfacial force field parameters). For this initial implementation, we require there be no covalently bonded interactions between the atoms of the two phases. Under this assumption, the interfacial force field requires only an additional set of parameter values for (i) the atomic partial charges used to account for Coulombic interactions between phase I and II atoms and (ii) the L–J parameters used to define the van der Waals attractions and Pauli-repulsive interactions between phase I and II atoms.
The additional set of partial charges needed to describe the Coulombic interactions between phase I and phase II atoms are included in the input residue topology file, alongside the standard partial charges used by the CHARMM force field. Thus, each atom has two partial charge entries in the input topology files (e.g., a CHARMM.rtf file). These interfacial partial charges are also stored in memory at run time.
Likewise, two additional parameters ε and Rmin/2 must be specified for each atom type in the input parameter files (e.g., a CHARMM. prm file), representing the L–J interfacial interactions between phase I and phase II atoms.
These modifications thus simply alter the interactions between atoms across the interface, which are regulated by the thermostat in a normal manner, thus providing a means to adjust interfacial behavior independently of either of the individual phases of a molecular system.
These changes have been implemented in version c34b2 of the CHARMM program, and details of the code modifications that were made to implement the Dual-FF capability in CHARMM are provided in the Supplementary Material provided for this paper. In the current implementation in CHARMM, the Dual-FF approach can be used with only electrostatic cutoffs, including REMD simulations with the MMTSB package, but not when using Ewald summation. Incorporating the Ewald functionality of the CHARMM code would improve the usefulness of the approach, and an effort to do so is currently underway.
2.2. Relationships Between Nonbonded Parameters and Interfacial Behavior
In order to gain insight into the effects of nonbonded force field parameters on interfacial behavior, two sets of studies were conducted. Umbrella sampling simulations were first conducted using Dual-FF CHARMM to generate potential of mean force (PMF) profiles as a function of the surface separation distance (SSD) between the center of mass of a TGTG-V-GTGT peptide and the average position of heavy atoms in the surface groups of CH3- and OH-functionalized SAM surfaces. Drastic changes to the nonbonded components of the interfacial force field were made in order to separate the contributions of the electrostatic and L-J nonbonded interactions. A second set of simulations was then conducted using the original CHARMM program but with TIP3P water replaced with polarizable TIP4P-FQ water in order to assess the degree in which conditions over the CH3- and OH-SAM surfaces influence the dipole moment of water (i.e., via changes in the O and H partial charges) compared to bulk solution conditions. The results from these studies were then used to provide direction regarding how the nonbonded parameters of the peptide and water could be modified to substantially increase peptide binding affinity to the CH3-SAM surface while decreasing binding affinity to the OH-SAM surface, making them more consistent with the experimental results.
2.2.1. Influence of Electrostatic and L–J Parameters on the PMF vs. SSD Profiles
2.2.1.i. Model System
Similar to our prior studies, for these simulations the SAM surfaces consisted of an assembly of alkanethiol chains with a structure of HS-(CH2)11-R, with R representing the surface functional group. A methyl-terminated SAM (R=CH3) and a hydroxyl-terminated SAM (R=OH) were constructed to represent hydrophobic and hydrophilic SAM surfaces, respectively. To mimic an alkanethiol SAM surface on Au (111), 90 chains in a 9×10 array were aligned in a geometry with 5 Å spacing and were tilted initially to the orientation specified by Vericat and coworkers.15 Detailed descriptions of the construction of the SAM surfaces are provided by Vellore et al.13 In order to maintain the structure of the SAM in the absence of an underlying gold surface, all the thiol groups in the SAM chains were kept fixed during the simulations, while the rest of each chain was free to move dynamically. Similar to the experimental studies performed by Wei and Latour,9,16 a peptide model with zwitterionic end groups was used in the simulations, which had an amino acid sequence of TGTG-V-GTGT, where T, G, and V represent threonine, glycine, and valine residues, respectively. The explicitly represented saline solution was constructed from a water box containing 2,241 TIP3P water molecules17,18′ with the dimensions of the box chosen so as to match the dimensions of the SAM surface in the XY plane. Six sodium (Na+) and six chloride (Cl−) ions were added to the system to approximate a 140 mM physiological saline solution by randomly replacing water molecules with each ion type. CHARMM22/CMAP force field parameters were used to represent solution behavior and CGENFF parameters19 were used for modeling the SAM surfaces.
The peptide was placed in the water box and any water molecules found to be within 2.2 Å of the peptide were deleted. The height of the water box was then adjusted to provide 1 atm pressure based on a virial-based pressure optimization algorithm,20 which is useful when the presence of fixed-atom constraints causes the reported pressure to be erroneous. To avoid the problem of the peptide interacting with the nonpolar bottom layer of the SAM surface when using three-dimensional (3-D) periodic boundary conditions, a 14 Å layer of bulk saline solution was placed at the top of the unit cell and fixed in place during the simulations.13 The complete 3-D periodic unit cell was orthogonal in shape with the dimensions of 44.3 × 45.0 × 64.7 Å3 for the CH3-SAM surface (see Figure 1) and 44.3 × 45.0 × 62.8 Å3 for the OH-SAM surface. Once constructed, each of our model systems was prepared for molecular dynamics (MD) simulations with 500 steps of steepest-descent and 200 steps of conjugate-gradient energy minimization. MD simulations were conducted under the canonical ensemble, using the Nosé-Hoover thermostat for temperature control.21,22 The RATTLE algorithm23 was used to constrain bonds involving hydrogen atoms and the velocity-Verlet (VV2) algorithm24 was used to integrate equations of motion with a 2 fs timestep. Interactions through periodic boundaries were handled using the CHARMM crystal facility with van der Waals (vdW) interactions represented using the 12–6 Lennard-Jones (L–J) potential with a group-based force-switched cutoff that was invoked at 8 Å and terminated at 12 Å with a non-bonded pair-list generation cutoff at 14 Å. Because the Dual-FF CHARMM program has not yet been parameterized to represent electrostatic interactions using Ewald summation, electrostatic interactions were represented using the same cutoff parameters as were used for L–J interactions. This resulted in slightly different values of ΔGads from previous results using PME, but values were still within 0.5 kcal/mol of previous results. The systems were then each heated up to 298 K and thermally equilibrated for 100 ps. All systems were constructed using the academic version c34b2 of CHARMM5–7 and simulated using the Dual-FF program.
Figure 1.

A molecular model of the TGTG-V-GTGT peptide over a hydrophobic CH3-SAM surface in TIP3P water with 140 mM NaCl (visualized using Visual MD software (VMD25)). The peptide, the SAM surface, Na+ (yellow) and Cl− (cyan) ions in solution, and the fixed layer of water at the top of the unit cell are shown as space-filled atoms. The mobile bulk water molecules are represented by space-filled atoms that have been made translucent for clarity. The total molecular assembly consists of 12,850 atoms.
2.2.1. ii. Assessment of Electrostatic and L–J Contributions for Peptide Adsorption
As previously noted, use of the CHARMM force field substantially underestimates peptide adsorption affinity for a hydrophobic CH3-SAM surface and slightly over-estimates binding affinity for a hydrophilic OH-SAM surface.13 In order to more clearly understand the factors governing the interfacial region, we sought to assess the specific contributions of the different types of nonbonded interactions that influence the relative strength of interaction between the peptide and water with these surfaces. This can be achieved by isolating the contributions of electrostatic and vdW interactions to the adsorption profile using the Dual-FF CHARMM program. To accomplish this, the interface region was modeled using three different schemes, (i) normal CHARMM parameters, (ii) L–J interactions only, and (iii) electrostatics interaction only. For scheme (ii), the partial charges for the SAM atoms were set to zero for the interfacial interactions, and for scheme (iii), the L–J well depths (εi) were reduced to 10−4 kcal/mol instead of zero in order to effectively eliminate attractive vdW interactions while maintaining overlap repulsion at short distances, thus avoiding the complete collapse of atoms due to electrostatic attraction. Using these schemes, umbrella sampling simulations were performed to study the influence of L–J vs. electrostatic interaction on the adsorption process by comparing the PMF vs. SSD profiles. Details of the umbrella sampling procedures that we used for these simulations are documented in our prior publications.11,13
2.2.2. Influence of the Interface on the Dipole Moment of Water
To understand how the properties of interfacial water above the SAM surfaces differ from those of bulk water, and to use those results as guidance for the development of force field parameters to represent interfacial behavior, we employed a polarizable water model, TIP4P-FQ26 over the CH3- and OH-terminated SAMs without the presence of the peptide or ions in the water. Simulations were also performed with standard TIP3P water17,18 for comparison. TIP4P-FQ has the same rigid geometry as the TIP4P model,26 but includes explicit polarization through a fluctuating-charge method. The partial charges on this model respond to changes in the local electrostatic field, inducing molecular electronic polarization.
All simulations were performed in the canonical (NVT) ensemble at 298 K with a 20 Å (z-height) vacuum layer above the water (thus ensuring that the pressure is P°, the saturation vapor pressure). Each structure contained 90 SAM chains and 2,059 TIP3P waters or 1,984 TIP4P-FQ waters. All heavy atoms of the SAMs except the top-most surface CH3 or OH groups were restrained with a force constant of 1,000 kcal mol−1 Å−2. Temperature control was achieved with the Nosé-Hoover thermostat.21,22 The SHAKE algorithm was used to constrain bonds between hydrogen and heavy atoms.27 The leapfrog Verlet algorithm was used to integrate equations of motion. Time steps of 0.5 fs and 2 fs were used in the polarizable and fixed-charge simulations, respectively. Orthorhombic simulation boxes (43.3 × 45.0 × 68.2 Å3) with periodic boundary conditions were used with particle mesh Ewald summation28 for the electrostatic interactions. An atom-based, energy-switched cutoff between 9 and 13 Å was employed to truncate the van der Waals interactions.29 The simulations were run for 5 ns with the last 1.5 ns used for analysis to evaluate the influence of the SAM surface upon the water structure and its dipole moment (i.e., shift in partial charge distribution) at the interface.
2.3. Interfacial Force Field Parameterization
Based on the individual contributions from L–J and electrostatic interactions to peptide adsorption behavior and the dipole shift in interfacial water that were determined by the above-described set of studies, adjustments in the nonbonded parameters of the peptide and water were made to tune the interfacial force field. This approach enabled the relative attraction of the atoms of the peptides and water for a surface to be directly adjusted. In addition to this approach, SAM parameter adjustment can also be used to tune the interfacial force field to alter specific interactions arising from the surface or to further address any remaining imbalances.
The interfacial force field was initially modeled using CHARMM22 force field parameters, with the objective of minimizing the changes in these parameters for the peptide and TIP3P water while optimizing agreement of peptide adsorption free energy values obtained from the simulations to the experimental data set.9 Initial screening of force field parameter changes was done using umbrella sampling to evaluate if the parameter adjustments resulted in the desired shifts in the PMF vs. SSD profiles. More accurate biased-energy REMD simulations10,11,13 were then performed to more carefully assess the influence of parameter adjustment on adsorption free energy (ΔGads). The interfacial force field was manually adjusted in this manner until the calculated ΔGads values were well within 1.0 kcal/mol of the experimental values for both the CH3- and OH-SAM surfaces.
3. RESULTS AND DISCUSSION
3.1. Relationships Between Nonbonded Parameters and Interfacial Behavior
3.1.1. Influence of Electrostatic and L–J Parameters on the PMF vs. SSD Profiles
In order to provide direction regarding how the nonbonded parameters influences peptide adsorption behavior, umbrella sampling simulations were conducted using the standard CHARMM parameters under the Dual-FF approach to separate the contributions of Lennard-Jones (L–J) and the electrostatic parameters on peptide adsorption behavior by setting the phase I and phase II L–J and electrostatics interactions alternately to zero. The results of these simulations are presented in Figure 2.
Figure 2.
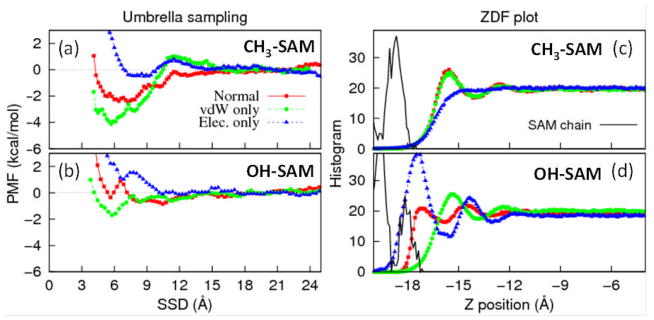
PMF vs. SSD profiles generated using umbrella sampling of the TGTG-V-GTGT peptide over the (a) CH3-SAM and (b) OH-SAM surfaces using Dual-FF CHARMM. Corresponding plots of vertical density profiles (ZDF) for both water and SAM are shown for the (c) CH3-SAM and (d) OH-SAM surfaces. The interactions in the interface region were modeled by three different schemes: normal CHARMM parameters (red squares), L–J parameters alone (green circles), and electrostatic parameters alone (blue triangles). The density profiles for the SAM surfaces are shown by the black curves in (c) and (d).
As can be seen from these PMF plots (Figures 2a and 2b) and their corresponding density profiles (ZDFs) (Figures 2c and 2d), the vdW and electrostatic interfacial parameters have profoundly different effects on adsorption, thus providing direction regarding how these parameters could be adjusted in order to improve peptide adsorption behavior. Figure 2a indicates that above the CH3-SAM surface, vdW contributions to the peptide-SAM interactions favor strong adsorption of the peptide, i.e. the peptide is able to strongly out-compete water for adsorption sites under influence of the vdW interactions alone. Electrostatic interactions with this hydrophobic surface, on the other hand, are fairly evenly balanced between peptide-surface and water-surface, with little preference for peptide adsorption. Also, from the corresponding density profiles plot (ZDF, Figure 2c), it can be seen that the vdW interactions are primarily responsible for the development of the water structure over this SAM surface. Based on these results, it is evident that the driving force for adsorption on this hydrophobic surface is the vdW interactions. To strengthen peptide adsorption to the hydrophobic CH3-SAM surface, the L–J interactions between the peptide and surface could be increased, while those between water and surface could be decreased. Over the OH-SAM surface, the behavior is entirely different. The peptide PMF profiles demonstrate little preference for peptide adsorption using CHARMM force field parameters while the purely electrostatic interfacial interactions favor displacement of the peptide by the water. This behavior is also evident from the density profiles (Figure 2d), which indicate that the water is more tightly bound to the surface by electrostatic interactions. In contrast to this behavior, the peptide does exhibit some preference, compared to water, for attraction to the surface under vdW interactions alone, with the water structure (Figure 2d) under this condition appearing very similar to the CH3-SAM surface (Figure 2c) under these same conditions. Thus the weak adsorption in this case is driven primarily by electrostatic interactions, and changes to the vdW interactions should have relatively little effect on peptide adsorption.
3.1.2. Influence of the Interface on the Dipole Moment of Water
The two sets of water-SAM simulations, i.e. with polarizable and nonpolarizable water, were analyzed to determine the influence of the interface on water dipole moment distributions and density profiles over the surface.
The influence of the SAM surfaces on water structure as characterized by the density profiles as shown in Figures 3a and 3b for the CH3-SAM and the OH-SAM systems, respectively. The interface occurs at a height of approximately −18 Å in both systems. The discrete layers of the carbon backbone of the restrained SAM surfaces are very prominent at coordinates below the interface, with slight variations in the structural profile of the unrestrained top surface functional groups. The water model influences the functional group geometry considerably more at the OH-SAM surface. Both models also show that the water is structured by the SAM surface within a few molecular layers of the interface, with no structure induced by the surface beyond about 10 Å from the interface. The TIP3P and TIP4P-FQ water structures are surprisingly similar over the nonpolar CH3-SAM surface, while exhibiting substantial differences in structure above the OH-SAM. Comparing the effect of the surface, it appears that the OH-SAM organizes a very narrow and well-structured first molecular layer, but with rather weak structure beyond this first layer. The CH3-SAM, on the other hand induces order in two or three distinct molecular layers of water. The water penetrates less deeply into the surface of the nonpolar CH3-SAM surface as well, with a larger separation between the water and functional group layers. Similar types of water structuring over hydrophobic and hydrophilic surfaces have been previously reported from both experimental studies30–32 and molecular simulations,33 with water structure peaks forming more closely to hydrophilic surfaces due to their ability to directly hydrogen bond with surface functional groups compared to a hydrophobic surface.
Figure 3.
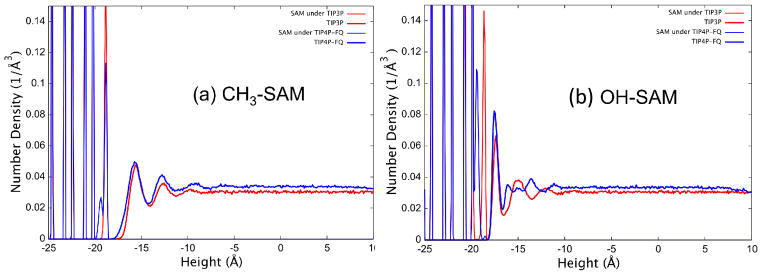
Density profile for heavy atoms (i.e., carbons in SAM chains, oxygen in water) in the direction normal to the surface plane for (a) the hydrated CH3-SAM system and (b) the hydrated OH-SAM system.
The SAM surfaces have an effect on the charge distribution of the polarizable water model, as well as their structure. The dipole moment distributions for TIP4P-FQ water molecules in the first layer above each SAM surface are compared to that in pure TIP4P-FQ water in Figure 4. The dipole moment of water is decreased over both surfaces, although the depolarization is greater over the nonpolar CH3-SAM (14% decrease in induced dipole) than over the OH-SAM surface (10% decrease in induced dipole). These results suggest that water is depolarized when structured over organized SAM surfaces (even hydrophilic ones), and that the partial charges in a given water model that are suitable for the bulk can be expected to be too large to accurately represent conditions at the liquid-solid interface for these surfaces.
Figure 4.

Dipole moment distributions of plain TIP4P-FQ water in bulk solution (pure TIP4P-FQ, solid line) and adjacent to the OH (long dashed line) and CH3 SAM (dotted line) surfaces. The (constant) dipole moment of TIP3P water is indicated by the vertical dot-dash line.
3.2. Interfacial Force Field Parameterization
Based upon the errors in peptide adsorption behavior indicated in Table 1 and the results from the interfacial studies shown in Figures 2–4, modifications were made to the CHARMM nonbonded parameters for TIP3P water and the peptide atom types to tune the interfacial force field in order to provide closer agreement with experimental peptide adsorption free energy values. Use of the regular CHARMM force field parameters results in a substantial under-estimation of peptide adsorption affinity to the hydrophobic CH3-SAM surface, along with a slight over-prediction of peptide adsorption affinity to the OH-SAM surface as indicated by the values presented in Table 1. The PMF adsorption profiles (Figure 2) indicate that the overly weak adsorption of the peptide to the CH3-SAM surface could be strengthened by increasing the strength of the peptide-surface L–J interactions relative to the water-surface L–J interactions and by decreasing the electrostatic attraction of water for the surface groups relative to the peptide. The depolarization of water over the SAMs (Figure 4) lends additional support to the conclusion that the electrostatic interactions with water should be weakened for these systems. These changes, of course, would also tend to increase peptide adsorption affinity to the OH-SAM surface, which could be countered by decreasing the partial charges on the peptide based on the results presented in Figure 2b. Several iterations of parameter modifications consistent with these goals were evaluated for their effect on adsorption free energy, with the additional constraint of making minimal alterations to the original CHARMM parameters.
Based on the assessment of the needed corrections as addressed in the preceding paragraph, the final set of parameters for our interfacial force field is presented in Table 2. The value of εi (the L–J well depth parameter) was increased by 25% for the carbon atoms present in the CH2 and CH3 groups that appear in the amino acid libraries of each of the peptides while concomitantly decreasing εi of the oxygen and hydrogen atoms of water by 20%. The atomic radii were not altered due to the fact that adjustments of the well-depth parameters were deemed sufficient to correct the adsorption behavior. This change in the L–J interactions was accompanied by a 20% reduction in the magnitude of the partial charges of both the carbon and hydrogen atoms in the terminal CH3 groups for each peptide and the oxygen and hydrogen atoms of the water. These modifications were assessed by conducting 5 ns biased-energy REMD simulations10,11,13 of the peptide over these two contrasting surfaces (CH3- and OH-SAMs) to assess the resulting changes in the calculated values of adsorption free energy compared to the experimental values. As shown in Table 3 and Figure 5, the use of these interfacial force field parameters with the Dual-FF approach implemented in CHARMM resulted in the ability to closely match experimental results. Peptide adsorption affinity to the CH3-SAM surface was substantially increased, while maintaining low adsorption on the OH-SAM surface. The errors in ΔGads were reduced to less than 0.3 kcal/mol in both cases. It should be noted that the about 0.5 kcal/mole differences from the CHARMM FF values shown here compared to those shown in Table 1 are a result of the use of a cut-off instead of PME for the calculation of electrostatic interactions for these simulations as required with our current version of the Dual-FF program.
Table 2.
Optimized interfacial parameters for Dual FF program.
| Residue | Atom Type | Partial charge (e) | Epsilon (kcal/mol) | ||
|---|---|---|---|---|---|
| CHARMM FF | Interfacial FF | CHARMM FF | Interfacial FF | ||
| TIP3P | OT | −0.834 | −0.6672 | −0.1521 | −0.12168 |
| HT | 0.417 | 0.3336 | −0.0460 | −0.0368 | |
|
| |||||
| Amino acid | CT3 (CH3) | −0.270 | −0.216 | −0.0800 | −0.1000 |
| HA (CH3) | 0.090 | 0.072 | -- | -- | |
|
| |||||
| CT2 (CH2) | -- | -- | −0.0550 | −0.06875 | |
Table 3.
Free energy of adsorption (Gibbs free energies for experimental data, ΔGads, Helmoltz free energies for values from the simulations (original values using CHARMM and new values using Dual-FF), ΔAads) for a TGTG-V-GTGT peptide over the CH3- and OH-SAM surfaces using Dual-FF CHARMM from biased-REMD simulations (mean ± 95% confidence interval). Due to the near incompressibility of water, differences between ΔGads and ΔAads can be considered to be negligible for adsorption processes in aqueous solution.
| SAM | ΔGad (kcal/mol) | ΔAads (kcal/mol) | ΔAads (kcal/mol) |
|---|---|---|---|
|
| |||
| Experiment | Original Simulations | Dual-FF Simulation | |
| OH-SAM | 0.0 ± 0.4 | −0.77 ± 0.32 | −0.20 ± 0.21 |
| CH3-SAM | −4.4 ± 0.5 | −1.56 ± 0.56 | −4.13 ± 0.93 |
Figure 5.
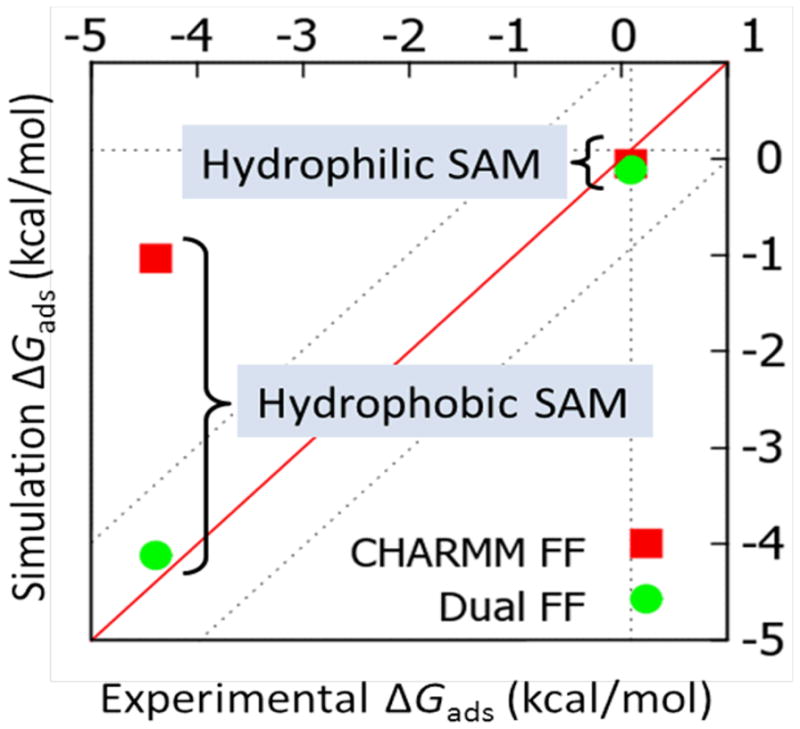
Comparison of calculated mean ΔGads values to experimental values, with calculated values predicted using both the regular CHARMM22 force field (red squares) and the Dual-FF CHARMM program with the tuned interfacial force field (green circles).
In order to confirm that the use of the Dual-FF CHARMM program enables the behavior at the liquid-solid interface to be adjusted without altering the conformational behavior of the solution phase of the system, the conformational behavior of the peptide was examined with the peptide restrained to remain sufficiently far from the CH3-SAM interface to provide bulk solution conditions (i.e., about 25 Å above the SAM surface).13 Ramachandran plots representing the dihedral angle sampling of the peptide backbone are presented in Figure 6 for three different parameter sets: (i) CHARMM for all atoms, (ii) Dual-FF CHARMM with the tuned interfacial force field, and (iii) CHARMM but with nonbonded parameters of the peptide and water set to the values shown in Table 2. As presented in Figure 6, the conformational behavior of the peptide in solution is shown to be essentially identical for sets (i) and (ii), while set (iii) provides substantially perturbed conformational behavior. These results verify that the implementation of a separate interfacial force field using Dual-FF CHARMM modifies peptide adsorption behavior to the SAM surfaces while allowing the conformational behavior of the peptide in solution away from the influence of the interface to be controlled by the CHARMM force field as intended. The dihedral angles of the amino acid residues were also analyzed when present at the interface of each SAM surface (SSD < 8 Å, Figure 7), which shows that the DUAL-FF simulations populated different regions of the Ramachandran plot when compared to the CHARMM force field, especially for valine as the host residue. Unfortunately, experimental data is not available for comparison with the predicted conformational behavior of these peptides when adsorbed to SAM surfaces, thus the accuracy of these conformational results cannot be assessed at this time. We speculate, however, that the conformational behavior of the peptides at the interface using the Dual-FF interfacial parameters should be closer to reality given that the free energies of adsorption are more closely aligned with experimental values.
Figure 6.
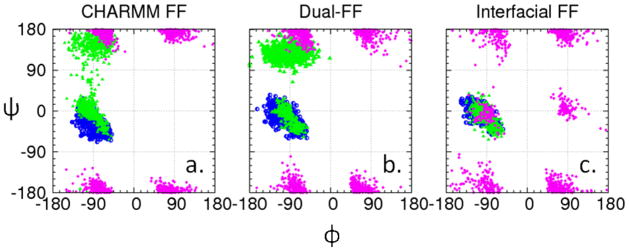
Ramachandran plots (glycine=purple, threonine=green, valine=blue) for the peptide in solution above the SAM surface using (a) CHARMM parameters, (b) Dual-FF CHARMM parameters, and (c) CHARMM parameters but with the nonbonded parameters of the peptide and water set to the values shown in Table 2 (c).
Figure 7.

Comparison of Ramachandran angles for the host amino acid residues (glycine, threonine, and valine) when present in the interfacial region (SSD < 8 Å) above the (a) CH3-SAM and (b) OH-SAM surfaces using CHARMM parameters (top row) and DUAL force field (bottom row).
4. CONCLUSIONS
A new approach has been developed that enables an independent set of nonbonded force field parameters to be used to represent inter-phase behavior between two separate phases in a multiphase system while enabling separate sets of force field parameters to represent intra-phase molecular behavior. This approach, which has been implemented in CHARMM, has been demonstrated for the case of peptide adsorption behavior to functionalized material surfaces. The interfacial force field parameters have been successfully tuned to correct previously identified errors in peptide adsorption energies.
By providing the means to separately adjust interphase interactions, the Dual-FF method provides a versatile approach to accurately and efficiently simulate many different types of multiphase systems. Further studies are underway to apply this approach to the simulation of other peptide-surface combinations in order to evaluate the transferability of the developed interfacial force field parameters to a broader range of adsorption systems. Additional studies are then planned to apply the Dual-FF approach for the simulation of protein adsorption behavior. Results from the protein adsorption simulations will be validated against matched experimental studies that have been designed to characterize adsorbed protein orientation and conformational behavior. The Dual-FF method should be generally applicable to any interfacial system that requires separate tuning of interfacial behavior while representing solution-phase and solid-phase behavior with other established force fields.
Supplementary Material
Acknowledgments
This work was funded under NIH Grant No. R01 EB00616 and also received support from the Defense Threat Reduction Agency-Joint Science and Technology Office for Chemical and Biological Defense (Grant no. HDTRA1-10-1-0028). Partial support was also provided by “RESBIO-The National Resource for Polymeric Biomaterials” funded under NIH Grant No. P41 EB001046. The authors thank Dr. J. Barr von Oehsen and Ms. Corey Ferrier for support of the Palmetto Cluster computational resources at Clemson University. We also acknowledge Dr. Alex MacKerell for his generosity in providing us with the CGenFF parameters, which were used for modeling our SAM surfaces.
References
- 1.Oostenbrink C, Villa A, Mark AE, van Gusteren WF. J Comput Chem. 2004;25:1656–1676. doi: 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- 2.Latour RA. Biointerphases. 2008;3(3):FC2–FC12. doi: 10.1116/1.2965132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Latour RA. In: In The Encyclopedia of Biomaterials and Bioengineering. 2. Wnek GE, Bowlin GL, editors. Informa Healthcare; New York, NY: 2008. pp. 270–284. [Google Scholar]
- 4.Weiner SJ, Kollman PA, Case DA, Singh UC, Ghio C, Alagona G, Profeta JS, Weiner P. J Am Chem Soc. 1984;106:765–784. [Google Scholar]
- 5.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4(2):187–217. [Google Scholar]
- 6.Brooks BR, Brooks CL, Mackerell AD, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. J Comput Chem. 2009;30(10):1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.MacKerell AD, Bashford D, Bellott M, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. J Phys Chem B. 1998;102(18):3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 8.Kaminski GA, Friesner RA, Tirado-Rives J, Jorgensen WL. J Phys Chem B. 2001;105:6474–6487. [Google Scholar]
- 9.Wei Y, Latour R. Langmuir. 2009;25(10):5637–5646. doi: 10.1021/la8042186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang F, Stuart SJ, Latour RA. Biointerphases. 2008;3:9–18. doi: 10.1116/1.2840054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Brien CP, Stuart SJ, Bruce DA, Latour RA. Langmuir. 2008;24(24):14115–14124. doi: 10.1021/la802588n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vellore NA, Yancey JA, Collier G, Latour RA, Stuart SJ. Langmuir. 2010;26(10):7396–7404. doi: 10.1021/la904415d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodcock HL, Miller BT, Hodoscek M, Okur A, Larkin JD, Ponder JW, Brooks BR. J Chem Theory Comput. 2011;7(4):1208–1219. doi: 10.1021/ct100738h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141–151. [Google Scholar]
- 15.Vericat C, Vela ME, Benitez GA, Gago JAM, Torrelles X, Salvarezza RC. J Phys- Condens Mat. 2006;18(48):R867–R900. [Google Scholar]
- 16.Wei Y, Latour RA. Langmuir. 2008;24(13):6721–6729. doi: 10.1021/la8005772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jorgensen WL. J Chem Phys. 1982;77(8):4156–4163. [Google Scholar]
- 18.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. J Chem Phys. 1983;79(2):926–935. [Google Scholar]
- 19.Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, Shim J, Darian E, Guvench O, Lopes P, Vorobyov I, Mackerell AJ. J Comput Chem. 2010;31(4):671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yancey JA, Vellore NA, Collier G, Stuart SJ, Latour RA. Biointerphases. 2010;5(3):85–95. doi: 10.1116/1.3493470. [DOI] [PubMed] [Google Scholar]
- 21.Nosé S, Klein ML. Mol Phys. 1983;50(5):1055–1076. [Google Scholar]
- 22.Nosé S. Mol Phys. 1984;52(2):255–268. [Google Scholar]
- 23.Andersen HC. J Comput Phys. 1983;52(1):24–34. [Google Scholar]
- 24.Martyna GJ, Tuckerman ME, Tobias DJ, Klein ML. Mol Phys. 1996;87(5):1117–1157. [Google Scholar]
- 25.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 26.Rick SW, Stuart SJ, Berne BJ. J Chem Phys. 1994;101:6141–6156. [Google Scholar]
- 27.Ryckaert JP, Ciccotti G, Berendsen HJC. J Comput Phys. 1977;23:327–341. [Google Scholar]
- 28.Darden T, York D, Pedersen L. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 29.Steinbach P, Brooks BR. J Comput Chem. 1994;15:667–683. [Google Scholar]
- 30.Yalamanchili MR, Atia AA, Miller JD. Langmuir. 1996;12:4176–4184. [Google Scholar]
- 31.Subramanian S, Sampath S. J Colloid Interf Sci. 2007;313:64–71. doi: 10.1016/j.jcis.2007.04.019. [DOI] [PubMed] [Google Scholar]
- 32.Acuña SM, Toledo PG. Langmuir. 2008;24:4881–4887. doi: 10.1021/la703866g. [DOI] [PubMed] [Google Scholar]
- 33.Yang AC, Weng CI. J Chem Phys. 2008;129:1547101–9. doi: 10.1063/1.2996179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.