

Abstract
Array-based comparative genomic hybridization (aCGH) chromosomal analysis facilitates rapid detection of cytogenetic abnormalities previously undetectable by conventional cytogenetics. In this study, we analyze 48 uniformly treated acute myeloid leukemia (AML) patients by 44K aCGH and correlated the findings with clinical outcome. aCGH identified previously undetected aberrations, as small as 5 kb, of currently unknown significance. The 36.7 Mb minimally deleted region on chromosome 5 lies between 5q14.3 to 5q33.3 contains 634 genes and 15 microRNAs whereas loss of chromosome 17 spans 3,194 kb involves 342 genes and 12 microRNAs. Loss of 155 kilobase (kb) region on 5q33.3 (p<0.05) is associated with achievement of complete remission. In contrast, loss of 17p11.2-q11.1 was associated with lower CR rate and poorer overall survival (Kaplan-Meier analysis, p<0.0096). aCGH detected loss of 17p in 12/48 patients as compared to 9/48 by conventional karyotyping. In conclusion, aCGH analysis adds to the prognostic stratification of AML patients.
Keywords: Array comparative genomic hybridization, AML, chromosome regions 5q and 17p, RNF145, TP53
Introduction
Assessment of chromosomal abnormalities is essential to the diagnostic workup of patients with acute myeloid leukemia (AML). Cytogenetic data in AML patients contribute to diagnosis and provide prognostic parameters useful for disease stratification and treatment evaluation [1,2]. Based on cytogenetic results, most often derived by using conventional karyotypic analysis or by fluorescence in situ hybridization, AML patients can be stratified into “favorable,” “intermediate,” and “poor” or “unfavorable” prognostic groups. Certain cytogenetic abnormalities also influence the choice of therapy as some targeted therapeutic regimens are available for AML patients [3,4]
Detection of chromosomal aberrations by conventional karyotypic analysis is typically performed at an average resolution of 6–10 megabases on a 300–500 G-band level. Technical factors, however, such as chromosomal condensation, imperfect banding, the requirement for dividing cells (metaphases), and the well-recognized difficulties in detecting small aberrations or aberrations at the tips of chromosomes can limit resolution to some extent [5]. Fluorescence in situ hybridization (FISH) is a targeted method that enhances analytical resolution to 300–800 kilobases (kb) and allows the analysis of interphase nuclei as well as metaphases. Using FISH, however, requires prior knowledge of chromosomal region(s) of interest to detect gene/locus-specific rearrangements and cannot provide a genome wide screen to search for unknown aberrations [1,6,7].
Array-based comparative genomic hybridization (aCGH) methods provide an attractive complement to the conventional cytogenetic analysis and FISH for the investigation of cancer genomes [4]. The higher resolution and throughput, robustness, simplicity, high reproducibility, shorter turnaround time and precise mapping of aberrations, while avoiding the need for cell culture and dividing cells, offers substantial advantages over conventional cytogenetic and FISH methods [6,8,9]. Although aCGH has some limitations, for example, aCGH cannot detect recurrent balanced reciprocal translocations or low-level secondary clonal abnormalities, overall aCGH analysis provides detailed genomic features of simple and complex chromosomal abnormalities and cryptic aberrations otherwise not detectable by conventional G-band and FISH assays [10]. Array platforms such as oligonucleotide and single nucleotide polymorphism (SNP) microarrays which utilize short sequences of newly synthesized fragments of DNA (oligonucleotides) of 40 or 60 base pair length (40-mer or 60-mer oligos) as targets for hybridization provide dense genome wide coverage at very high resolution. In cases assessed by conventional cytogenetic analysis of FISH, aCGH can show additional genomic imbalances emphasizing the advantages of a whole-genome approach [1,4,5,11–13]. Despite the great potential of aCGH to assess cases of AML, relatively few studies of AML patients assessed by aCGH have been published [14–16].
In this study, our goal was to assess value of aCGH as a clinical tool in detecting somatic chromosomal and segmental copy number alterations (CNAs) in uniformly treated patients with AML. We also correlated the aCGH results with clinical features and outcome. The higher resolution provided by aCGH highlights the genomic complexity of AML cases. In diploid AML, aCGH showed a high frequency of chromosomal deletions, duplications, and amplifications. Furthermore, in cases of AML in which conventional cytogenetic analysis showed a complex karyotype, aCGH identified these aberrations in more detail abnormalities involving loci that likely harbor candidate genes involve in leukemogenesis.
Materials and Methods
Study Group
The study group was composed of 48 patients with AML diagnosed and treated at The University of Texas MD Anderson Cancer Center (Houston, TX). All patients were uniformly treated with idarubicin 12mg/m2 IV daily for 3 days and cytarabine 1.5g/m2 by continuous infusion daily for 4 days; however, cytarabine was administered for 3 days in patients older than 60 years. The diagnosis of AML was based the criteria of the current World Health Organization classification. The study was approved by the Institutional Review Board (IRB).
DNA Purification and Labeling
Isolation and labeling of genomic DNA was performed as described previously [17,18]. Briefly, genomic DNA (gDNA) from bone marrow (BM) aspirate specimens was isolated using the Autopure extractor (Qiagen/Gentra, Valenica, CA). 500 ng of genomic DNA was digested with Alu and RsaI restriction enzymes for 2 hours at 37°C. Digested genomic DNA fragments from patients and reference DNA with neutral copy number (human female DNA, Promega Corporation, Madison, WI) were labeled with Cy5-dUTP and Cy3-dUTP, respectively, using the Agilent Genomic DNA labeling kit plus (Agilent Technologies, Polo Alto, CA). Labeled DNA was purified using Micron YM-30 columns (Millipore, Billerica, MA) and the volume was adjusted by 1 x Tris-EDTA buffer (pH 8.0) to 20 μl. Target yield and specific activity were quantified using Nanodrop ND-1000 (Thermo Fisher Scientific, Wilmington DE)
Genomic Array Design and Hybridization
A custom-designed, 4 x 44K, 60-mer oligonucleotide genomic array designed using array software (Agilent Technologies), with gene-centric full genome coverage augmented with high density probes, was used. For hybridization, labeled patient genomic and reference genomic DNA were mixed and co-precipitated with 5 μg of human Cot-1 DNA (Invitrogen, Carlsbad, CA) using 11 μl of 10X blocking reagent and 55 μl of 2X hybridization buffer (Agilent Technologies) in a total volume of 110 μl. After denaturing at 93°C for 3 minutes, the mixture was incubated at 37°C for 30 minutes. Hybridization was performed at 65°C for 40 hours in a rotating oven (Robbins Scientific, Mountain View, CA) at 10 rpm. After hybridization, slides were washed in oligo-aCGH wash Buffer 1 (Agilent Technologies) at room temperature, followed by washes for 1 minute at 37°C in oligo-aCGH wash Buffer 2 (Agilent Technologies), for 1 minute at room temperature in acetonitrile (Sigma-Aldrich, St Louis, MO), and a final 30 seconds wash in stabilization and drying solution (Agilent Technologies). Arrays were scanned using an Agilent 2565BA DNA microarray scanner.
aCGH Data Analysis
Data were normalized using the Feature Extraction Software version 9.5.3.1 (Agilent Technologies), and analyzed by Nexus Copy number 5 (Biodiscovery Inc, EL Segundo, CA). Previously developed analysis protocols were applied [17,18]. For the Nexus copy number analysis, the Rank Segmentation algorithm with a significance threshold of 1.0 × 10−5 was used. The settings for aberration calls in Nexus copy number were: 0.15 for gain, 0.04 for high gain, −0.1 for loss and −0.4 for high loss. Human genome assembly hg18 (NCBI Build 36.1) was applied for this analysis.
Quality Control Measures and Assay Performance Parameters
Only samples with a post-labeling yield of 5 to 7 μg DNA and a specific activity of 25 to 40 pmol/μg for Cy3 and 20 to 35 pmol/μg for Cy5 were used for array hybridization. Following hybridization the signal intensity, signal to noise ratio, background noise, the derivative log ratio spread, and the reproducibility were evaluated using the Feature Extraction software version 9.5.3.1 (Agilent Technologies), with cutoffs for sample rejection used according to the manufacturer’s recommendations.
Cytogenetics and FISH
Conventional cytogenetics and FISH analyses were performed on cultures of whole BM or blood samples as described earlier [17]. Briefly, 20 metaphase cells were used for conventional cytogenetic analysis using standard Giemsa-banding techniques. Locus-specific FISH probes to BCR-ABL, CEBP, CBFB, PML-RARA, MLL, 5, 7 and −5q. Four hundred interphase nuclei were examined and counted for each probe.
Copy Number Analysis by Real Time qPCR
To validate aCGH findings for chromosome loci 5q and 17p, qPCR assays were performed using TaqMan Copy Number Assays (Applied Biosystems, Carlsbad, CA) for EBF1 exon 1, EBF1 exon 14 and RNF 145 exon 11, TP53, NCOR1, RFFL, MRPS 23 and ACOX1 according to manufacturer’s instructions. The RPP40 gene, which is known to exist in two copies in a diploid genome, was used as the endogenous copy number reference in multiplex reactions. Healthy female genomic DNA from Promega was applied as a diploid control. The PCR reactions were performed in triplicate using 50 ng of genomic DNA, 1X TaqMan Universal PCR master mix, 1x TaqMan Copy Number Assay mix in a total volume of 20 μl per each reaction. PCR was performed on a 7900HT Sequence Detection System (Applied Biosystems) in a 96-well format, and amplification was achieved using a standard amplification protocol as follows: 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Post-PCR copy number analysis was performed by Applied Biosystems CopyCaller Software v1.0 (Applied Biosystems), which performs a comparative CT (ΔΔCT) relative quantitation analysis of the real-time data using RPP40 and Promega female DNA as controls. The software uses the statistical model ΔCT = K-log 1+E CN, where K is a constant, E is the PCR efficiency of the assay of interest, and CN is copy number with the range [1,¥].
Statistical Analysis
The Fisher exact and chi-square tests were applied for categorical variables. Patient survival was estimated by using the Kaplan–Meier method from the date of BM diagnosis until death from any cause or until last patient follow-up. Survival curves were statistically compared using the log-rank test. Differences between groups were considered statistically significant if p-values were less than 0.05 in a two-tailed test.
Results
Characterization of Study Group
The clinical and pathological characteristics of the study group patients are listed in Table I. There were 23 men and 25 women with a median age of 52 years (range, 25–73 years). The median leukocyte count was 4.8 × 109/L (range, 0.3 to 70.6 × 109/L). Thirty-one (65%) cases were classified as AML not otherwise specified, of which the most common type was AML with maturation. FLT3-ITD was the most common gene mutation identified, in 5 of 46 (11%) patients tested.
Table I.
Clinicopathological data for 48 AML patients taken in a study
| Number of patients | 48 |
| Gender(M/F) | 23/25 |
| Age of study, range (median) | 25–73 (52year) |
| Peripheral blood values | |
| WBC (K/μl) range (median) | 0.3–70.6 (4.8) |
| Bone marrow blasts, range (median) | 5–94% (29%) |
| WHO | |
| AML –t (9:11)(p22;q23) | 2 |
| AML-inv(16)(p13.1;q22) | 1 |
| AML, NOS | 31 |
| AML-t | 3 |
| AML-MDS | 9 |
| Other | 2 |
| FAB | |
| M0 | 2 |
| M1 | 7 |
| M2 | 13 |
| M4 | 4 |
| M5 | 5 |
| M6 | 3 |
| RAEB-T | 7 |
| Other/NA | 6 |
| Mix | 1 |
| Cytogenetic findings | |
| Recurrent genetic abnormalities | 3 |
| Diploid | 18 |
| 1–2 Abnormalities | 11 |
| Complex (>3 abnormalities) | 16 |
| Mutation Status (Positive/Tested) | |
| NRAS | 4/40 (10%) |
| KRAS | 2/40 (5%) |
| KIT | 1/24 (4 %) |
| FLT3-D835 | 3/46 (7%) |
| FLT3-ITD | 5/46 (11%) |
| NPM1 | 1/4 (25%) |
Conventional Cytogenetics Findings
Twenty-nine (60%) of 48 patients had various structural and numerical chromosomal abnormalities, 18 (38%) patients had a normal karyotype while in one patient, the results of conventional cytogenetics were not available due to unsuitability of metaphase cells for analysis (patient #16). Stratification of patients into cytogenetic groups showed 25 patients with poor-risk, 22 patients with intermediate-risk, and 1 patient with favorable-risk. Sixteen (33%) of 48 patients had complex karyotypes with greater than 3 abnormalities, 11 patients showed 1–2 abnormalities, 18 patients showed a normal karyotype (diploid) and 3 patients showed recurrent genetic abnormalities. Common aberrations detected by conventional cytogenetic analysis included: del(5q13-q33), del(5q31-q35), der(5), del(5), del(7), der(17p) and del (17) (Table II). Only one AML patients (patient #48) was favorable-risk with inv(16)(p13.1q22).
Table II.
Comparison of Karyotype and aCGH findings in uniformly treated AML Patients.
| S.No | Cytocomment | Karyotype | aCGH findings |
|---|---|---|---|
| 1 | DIP, -Y | 46XY, -9,+MAR[1]; 46XY[19] | No significant finding |
| 2 | DIP, -Y | 46,XX[20] | No significant finding |
| 3 | IM | 46XX[9] | No significant finding |
| 4 | MISC | 47XY+13[14]; 47IDEM,I(17)(Q10)[2]; 46XY[4] | −12p(13.31-13.1), +13q12.11-q21.32, −13q21.32-q34, −17p13.2-p11.2 |
| 5 | −5, −7 | 41–44XY,DEL(3)(P13P21), -5-7+8,DEL(12)(P12P13),DER(14;22)(Q10;Q10),DER(17;18)(Q10;Q10)[CP20] | del3p26.3-p13, −5q11.1-q11.2 |
| 6 | DIP, -Y | 46,XY[21] | No significant finding |
| 7 | DIP, -Y | 46XY[20] | +13q34 |
| 8 | DIP, -Y | 46,XX[20] | No significant finding |
| 9 | MISC | 47XY,+10,DEL(12)(P11.2)[18]; 46XY[2] | +10p14-p11.2, +10q21.1-q21.3, +10q23.1-q26.3, −12p13.31 – p12.3, −13q34 |
| 10 | DIP, -Y | 46XX[20] | −11q13.2-q13.3, −13q34 |
| 11 | −5, −7 | 46XX,DER(7)T(1;7)(Q23;Q22),T(15;16)(Q24;Q22)[20] | +1q31.1-q44, −7q31.31-q35 |
| 12 | +8 | 48XY,+8+ADD(8)(Q24.3)[11]; 46XY[8] | +8p21.3-p11.2, +8q11.1-q24.3, −9q34.13-q34.2, +11q22.3, −17p13.1, +17q23.2-q25.1 |
| 13 | +8 | 47XY,+8[1]; 46XY[18] | No significant finding |
| 14 | −5, −7 | 43–44XY,DEL(5)(Q13Q33), -6-7-18,ADD(19)(Q13.1)[CP16]; 46XY[4] | +5p13.33-p11, −5q11.2-q35.3, −6p25.3-p22.1, +6p22.1-p21.1, −7p22.3-p11.2, +7q11.23-q21.11, −7q21.11-q35, −12q24.11-q24.33, −18p11.32-q11.1-q11.2, −18q11.2-q23q23 |
| 15 | MISC | 46XX,DER(2)T(2;11)(Q31;Q23),T(6;11)(Q27;Q23)[20] | −2q22.1-q35, −2q36.1-q37.3, +6q22.33-q27, +11q23.3-q25 |
| 16 | DIP, -Y | No significant finding | |
| 17 | −5, −7 | 45XX, -7[20] | −7p22.3-p11.2, −7q11.21-q35 |
| 18 | 11Q | 46XX,T(9;11)(P22;Q23)[20] | No significant finding |
| 19 | −5, −7 | 43–46XY,INS(5;?)Q, -7-9,D(9)T9;11Q,11P+,14Q-,-17-22+2-7MAR[CP18]; 46XY[2] | −5p15.1-p13.3, +5p13.2-p13.1, −7p15.2-p15.1, −7p14.2-p14.1, − 9q33.2-q34.11, −14q11.2-q12, −20q11.23-q13.2 |
| 20 | DIP, -Y | 46XX[14] | No significant finding |
| 21 | +8 | 47XY,+8[13]; 48XY,+8,+8[1]; 46XY[6] | +8p12-p11.21, +8q11.23-q12.3, +8q23.33-24.3 |
| 22 | DIP, -Y | 46XY[20] | No significant finding |
| 23 | −5, −7 | 47XX, DEL(7)(Q22Q34)+8[1]; 46XX″-10[15]; 46XX[4] | +7p22.3-p11.2, −7q11.2-q35, +8p21.3-p11.21, +8q11.1-q12.3, +8q12.3-q24.3, −10p15.3-p12.31, −12q11.23 |
| 24 | DIP, -Y | 46,XY[20] | No significant finding |
| 25 | DIP, -Y | 46 XX [20] | No significant finding |
| 26 | −5, −7 | 46XX -7 +R[7] 46XX [12] | −7p22.3-p12.2, −7q35-q36.3 |
| 27 | −5, −7 | 46XX DER(5)DEL(5)(Q13;Q33) T(5;13)(Q33;Q12) DER(13)T(5;13) [20] | −5q14.3-q34, −6q23.2-q23.3 |
| 28 | −5, −7 | 45XX, -5+8-9,6P+DER7;17PQ,INV9PQ,12Q+,T13;18Q,15Q-16Q+20Q-,+M[15]; 46–47″+M[CP2]; 46XX[3] | −2p25.1-p24.1, −5q23.1-q33.3, −6p25.3-p21.33, +6p21.32-p12.1, −7q32.1-q33, +8p21.3-p11.21, +8q11.1-q24.3, −12q24.11-q24.31, −13q12.3-q13.2, −15q22.31-q23, −16q11.2-q24.3, −17p13.3-p13.1, −18q21.1 -20q12-q13.2 |
| 29 | −5, −7 | 44XY,DEL(5)(Q13Q33), -11,ADD(11)(Q25), -13-17-18,ADD(20)(Q13.3),+2MAR[18]; 46XY[2] | −5q14.3-q15, −5q21.3-q33.3, +11q22.1, +11q22.3-q25, +13q22.3-q33.1, −18p11.23-p11.21, −18q12.1-q22.1, −20q11.21-q11.23, −20q13.12, −21q22.11-q22.2 |
| 30 | −5, −7 | 44XY,DER(5)T(5;13)(Q15;Q12), -13, -18[2]; 43″-3-6-7-17,21P+,+3M[2]; 46XY[16] | +1p36.21-p36.12, +2q35, −3p 24.3-p22.2, −3p 21.1-p14.3, −3q11.2-q13.11, −3q23-q26.2, −5q15-q34, −5p25.3-p22.1, +6p22.1-p21.1, −6p12.3-p12.1, −7p14.2-p14.1, −7p12.3-p12.2, −7q11.25-q36.1, +11p11.2, +11q11-q13.3, −11q22.3, +11q23.3, +11q24.2, +12q13.13-q13.2, +12q24.13-q24.23, +13q34, +14q32.2, −17p13.1-p12, −18p11.32-p11.21, +19p13.2-q13.32 |
| 31 | +8 | 53 +X DER(1)t(1;1)(p36.3;q12) +2 +4 +5 +12 +16[4] 54 SL +I(8)(q10)[2] 54 SL +13 -21[1] 46XX[13] | +1q21.3-q23.3, +1q42.13-q42.2, +2p 25.3-p25.1,+2p14-p13.3, +2q35, +4q12-q13.1, +4q28.1-q28.3, +5q21.3-22.2, +5q31.2-q35.3, +8p22-21.3, +8q12.1-q12.3, +8q12.3-q24.3, +12q13.11-q13.2, +12q24.11-q24.31, +16q12.1,+16q13 |
| 32 | −5, −7 | 46XX,DER(5;17)(P10;Q10)+M[3]; 52XX″+X+4-7+8+9+14+22+2M[16] | +1p35.2-q13.3, +4p15.1-p11, +4q12-q34.3, +5p15.33-q15, −5q15-q35.3, +8p21.3-p11.21, +8q11.1-q24.3, +9q12-q34.3, +14q11.1-32.33 |
| 33 | +8 | 43–46,X, -Y,+3,INV(3)(P13Q29),+8,DER(12)T12;17)(P11.2),+15,DER(15;15)(Q10;Q10), -17[CP6] | +3p26.3-p11.1, +3q11.2-q29, +8q21.3-q24.3 |
| 34 | −5, −7 | 41–45,XX,ADD(2)(P11.2),DEL(5)(Q13Q33), -9, -11,ADD(12)(P13),ADD(15)(Q25), -17, -18, -20,+1-2MAR[CP8] | −5q23.2, −5q31.1-q33.3, +20q11.21 |
| 33 | DIP, -Y | 46XY [20] | No significant finding |
| 36 | DIP, -Y | 46XY[20] | No significant finding |
| 37 | −5, −7 | 46XX,DEL(5)(Q13Q33)[1]; 43XX″,DUP(1)(P32P34.3) -4-7-17,ADD(18)(Q23)[18] | −4q13.3-q34.3, −5q13.3-q33.3, −18p11.32-p11.21, −18q11.1-q23 |
| 38 | −5, −7 | 45XY, 2P+7Q-17P+20Q-DER5,T5;17Q,-3-9-12-17+R+2M[18]; 45XY-14[1] | +1p36.33-p36.31, −2p25.3-p23.3, -3p14.2-p11.1, −3q11.2-q13.31, −3q22.2-q29, −5q13.3- -q35.2, −7q21.3-q35, −9q22.31-q32, −12p13.31-p12.3, −12q21.31-q22, −17p13.2-p13.1, −20q11.23-q13.2 |
| 39 | DIP, -Y | 46X X[20] | No significant finding |
| 40 | DIP, -Y | 46XY[19] | No significant finding |
| 41 | DIP, -Y | 47XY,+MAR[1]; 46XY[19] | No significant finding |
| 42 | DIP, -Y | 46 XY [19] | No significant finding |
| 43 | DIP, -Y | 46 XX [20] | No significant finding |
| 44 | DIP, -Y | 46 XY [28] | No significant finding |
| 45 | −5, −7 | 45 -5(Q31Q35) -7 INV9(P11Q12)[1] 45 -4(Q25Q33) ” +2MAR[2] 42–48 ″ +1-4MAR[CP10] 51–53 ″ [CP3] 46 [4] | −5q31.2-q35.1, −7q11.23-q31.1, −7q35-q36.1, +11q22.3-q23.3 |
| 46 | +8 | 47XX,+8,T(9;11)(P22;Q23)[23] | + 8p21.3-p11.21, +8q12.1-q24.3 |
| 47 | −5, −7 | 44–45,-X, -3,DEL(5)(Q22Q35),DEL(6)(P21.3P23), -7,ADD(10)(P13),ADD(11)(P15),ADD(12)(P13),+2-4MAR[CP7] | −3p24.3-p21.3, −3p14.1-p12.1, −5q23.3-q33.1, +6p21.31-p12.3, −7p22.3-p11.2,-7q11.21-36.3, −10p14-p12.33 +11q22.3, −12p13.33-p11.23, −12q21.32-q24.32, −17p13.2-p13.1, −17q21.32-q21.33, +21q21.33-q22.3 |
| 48 | INV 16 | 46,XX,INV(16)(P13.1Q22)[20] | No significant finding |
Additional findings of Karyotype and aCGH are shown as bold values
Comparison of aCGH and Conventional Cytogenetics/FISH
There was excellent agreement between the results of conventional cytogenetics/FISH and aCGH in AML cases (Table II). The cumulative distribution of copy number abnormalities in 48 AML patients analyzed by aCGH is shown in Figure 1. All non-balanced chromosomal alterations detected by conventional karyotyping were confirmed by aCGH. In addition, aCGH provided more precise localization of chromosomal breakpoints. For example, aCGH confirmed and refined heterogeneous breakpoints in 5q that included: 5q11.1-q13.2, −5q11.2-q35.3, −5q13.2-q33.3, −5q23.1-q35.3, −5q13.2-q35.1, −5q31.1-q35.1, −5q14.3-q34, +5q34-q35.3, −5q14.3-q15. The increased resolution of aCGH also identified the origin of markers, the composition of additions and deletions to chromosomes, and unsuspected chromosomal rearrangements within single cytogenetically defined aberrations. aCGH analysis was able to detect aberrations in samples containing blast counts as low as 5% (patient #27). Some of the additional findings that were detected by only aCGH are listed in Table II (bold values show additional findings). Certain discrepancies, such as +8[1], t(11:19), +2p11.2, −9, −11, +12p13, +15q25, −17, −18[cp8] were observed. Deletions of 5q and chromosome 7 were confirmed by FISH with 100% concordance in three cases. In addition, aCGH identified previously undetected aberrations, as small as 5 kb, of currently unknown significance. As expected, balanced translocations were not detected by aCGH. Chromosomal aberrations present in a limited number of metaphases also were not detected by aCGH (Patient# 1, 13, 5, 33). Interestingly, in cases showing complete loss chromosome 17 by conventional cytogenetics, only the loss of 17p was confirmed by aCGH (discussed below).
Figure 1.

An overview of distribution of copy number abnormalities in 48 AML patients analyzed by array complete genomic hybridization. Losses are shown on left side with red color and gains on right of each chromosome ideogram. The thick line of the bar represents the samples with aberrations.
Detection of Retained Genetic Material from Chromosome 17p
Precise mapping of chromosomal breakpoints allowed identification of cases with loss of TP53 (Figure 2A). Complete loss of chromosome 17 [del(17)] was detected by conventional karyotyping in 9 of 48 (19%) cases. aCGH detected the presence of 17q genomic DNA in all of these cases suggesting that genomic material was retained, but could not be identified morphologically by conventional karyotyping. Presence of a marker chromosome was detected in 12 (25%) of 48 cases with del(17), which could harbor the 17q genomic DNA. To test this hypothesis, we performed real-time PCR-based DNA copy number assays for 5 genes distributed across chromosome 17, of which TP53 (17p13.1) and NCOR (17p11.2) are located on 17p whereas, RFFL(17q12), MRPS23(17q22) and ACOX1 (17q25.1) are located on 17q. Loss of 17p and retention of 17q DNA was confirmed in all 11 cases tested with predicted one copy loss in 9 of 11 cases and homozygous loss in 1 of 11 for TP53, one copy loss in 7 of 11 for NCOR, 4 of 11 for RFFL, 1 of 11 for MRPS23 and no loss for ACOX1 located at 17q25.1 (Figure 2B)
Figure 2.
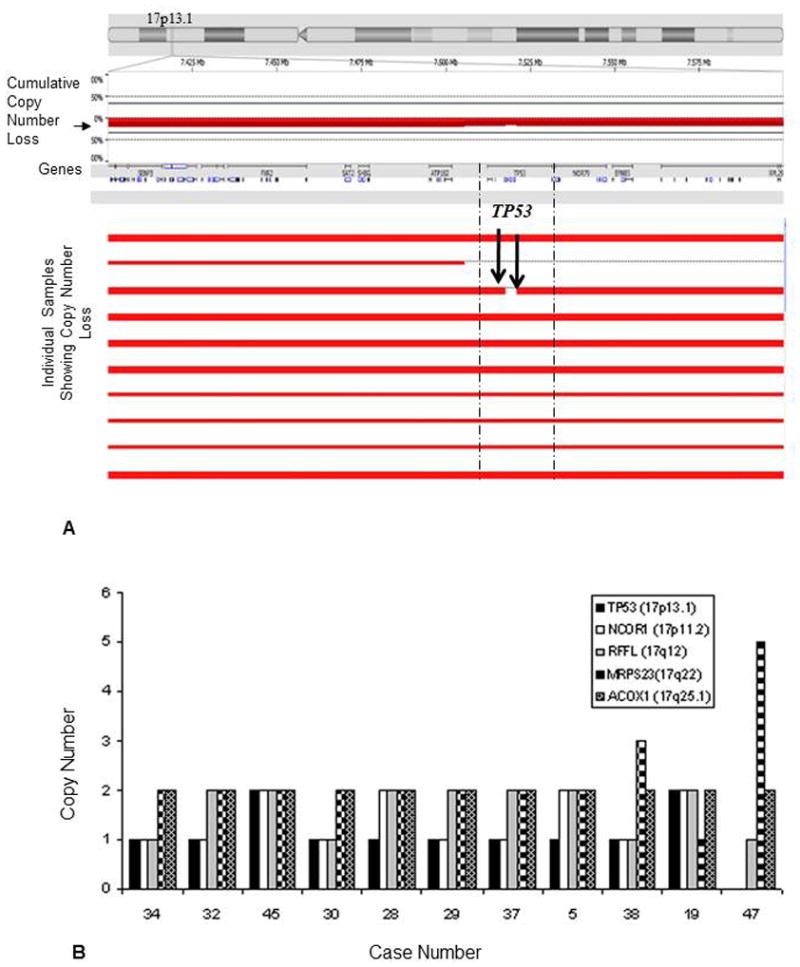
(A) Delineation of 17p boundaries revealed by aCGH in prominent AML cases showing loss of chromosome 17p. The top panel shows an ideogram of chromosome 17 with the p-arm to the left and q-arm to the right. The section of chromosome marked with vertical line is expanded to show the plot of array CGH data for the 17p13.1 sub-band. Precise mapping of this chromosomal breakpoint revealed AML cases with loss of TP53 gene which was shown to be as complete loss of chromosome 17 [del(17)] by conventional karyotyping. Loss was shown in red color. (B) Validation of 11 cases showing one copy loses in 8 of 11 cases and homozygous loss in 1 of 11 cases at 17p for candidate genes TP53 (17p13.1) for TP53, 7 of 11 with one copy loss and 1 of 11 with homozygous loss for NCOR (17p11.2) gene, 4 of 11 for RFFL (17q12), 1 of 11 for MRPS 23 17q22 and no loss of ACOX1 (17q25.1) by RT-PCR based copy number analysis.
Delineation of del(5q) Endpoints by aCGH
Given the ability of aCGH to map chromosomal breakpoints with precision, we evaluated deletions of chromosome 5. Del(5q) were noted in 9 (19%) of 48 samples (Table II; Figure 3). The 5q deletions ranged from position 5q11.2 to 5q34. The 36.7 Mb minimally deleted region lies between 5q14.3 to 5q33.3, and this region contains 634 genes and 15 microRNAs.
Figure 3.
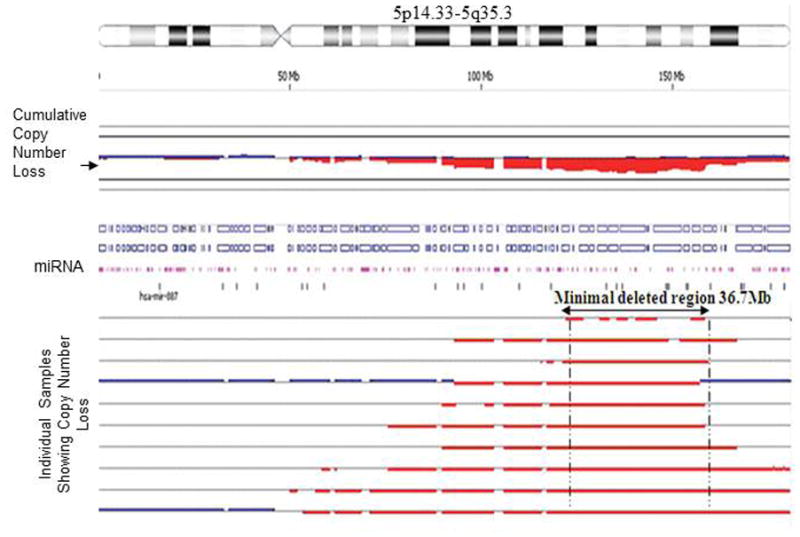
Delineation of del(5q) endpoints in individual AML cases run on 4 × 44 K oligonucleotide array CGH. Detection of 36.7 Mb minimally deleted region on chromosome 5 lies between 5q14.3 to 5q33.3 containing 634 genes and 15 microRNAs. The thick red line below the central line represents the samples showing 5q33.3 loss.
Cryptic Aberrations in Diploid AML Detected by aGCH
In 18 patients, aCGH detected aberrations in AML patients that were considered to be diploid by conventional cytogenetic analysis. In total, aCGH identified 97 chromosomal aberrations in diploid AML cases (average, 5.1 per case). These abnormalities included 41 single copy gains, 43 single copy losses, 2 high copy gains and 11 high copy losses. There were no recurrent cryptic aberrations observed in diploid AML patient samples using the predefined criteria of significance level of p<0.05 and less than 50% overlap with known copy number variants (Table II).
Response to Induction Therapy and Overall Survival According to Cytogenetic Risk
Follow up and therapy information were available for all patients. The median overall survival (OS) was 63.5 weeks and the median progression-free survival was 27 weeks. Complete remission was achieved in 34 (71%) patients and the CR rate varied among known cytogenetics risk groups, ranging from 72% (18 of 25) for poor-risk to 65.2% (15 of 23) for intermediate-risk patients. In the group of 34 patients who had a CR, 24 patients had sustained remission beyond 1 year with a median of 89 weeks (range, 58–132 weeks) whereas 10 patients developed relapse within a year with a median of 18.5 weeks (range, 1–44 weeks). Twenty-seven patients died, including 3 patients during CR. Overall survival varied significantly according to cytogenetic risk status. The estimated relative risk (RR) of death for the poor-risk group was 0.32 and for intermediate-risk group, 0.56.
Chromosomal Aberrations Detected by aCGH Correlated with Clinical Outcome
A total of 170 significant chromosomal aberrations (including microdeletions) were observed with submicroscopic aberrations being more frequent than whole chromosomal changes. An average of 5.7 copy number alterations was found per patient with 71 regions showing gains and 99 regions showing losses Table II. The most common whole chromosome gains were chromosome 8 (22.9%), chromosome 3 (2.2%) and chromosome 4 (2.2%) and the most common whole chromosome losses were chromosome 7 (11%) and chromosome 18 (2.2%). AML-associated submicroscopic deletions were detected at chromosomal loci 5q31.1 (27.1%), 3q11.2 (22.9%), 3q26.1 (18.8%), 4q28.3 (14.6%), 6p25.3 (20%), and 9q34.13-q34.3 (10.4%). Well documented AML-associated additions included chromosome 1q23.2 (10.4%), 6 p21.1 (12.5%), and 8q12.3 (22.9%). The distributions of significant recurrent chromosome aberrations observed in the study group are summarized in Table III.
Table III.
Distribution of significant* recurrent aCGH- detected aberrations in AML (n=48)
| Chromosome | Cytoband | Event | Frequency (%) |
|---|---|---|---|
| Chr1 | q23.2 | CN Gain | 10.4 |
| Chr3 | q26.1 | CN Loss | 18.8 |
| Chr3 | q11.2 | CN Loss | 22.9 |
| Chr4 | q28.3 | CN Loss | 14.6 |
| Chr5 | q31.1 | CN Loss | 27.1 |
| Chr6 | p21.1 | CN Gain | 12.5 |
| Chr6 | p25.3 | CN Loss | 20 |
| Chr8 | q12.3 | CN Gain | 22.9 |
| Chr9 | q34.13-q34.3 | CN Loss | 10.4 |
aCGH: array based comparative genomic hybridization, AML: acute myeloid leukemia, CN: copy number,
Aberrations observed in >10% patient samples at significance level of p<0.05, <50% overlap with known copy number variants
Chromosomal Aberrations Associated with Poorer Overall Survival
A 3.2 megabase loss at chromosome 17p11.2-q11.1 was associated with poorer overall survival (p<0.009). In addition, 5 deletions detected by aCGH at the 17p boundaries were identified 17p13.3-13.2 (25%), 17p13.2 (25%), 17p13.2-13.1(29%), 17p13.1(27%) and 17p13.1-p12(25%) at p value <0.02 that were missed by conventional karyotyping (Figure 4A). Correlation between aCGH findings associated with overall survival performed by Kaplan Meir test showed lower CR rate and poor overall survival rate at p value 0.0096 (Figure 4B).
Figure 4.
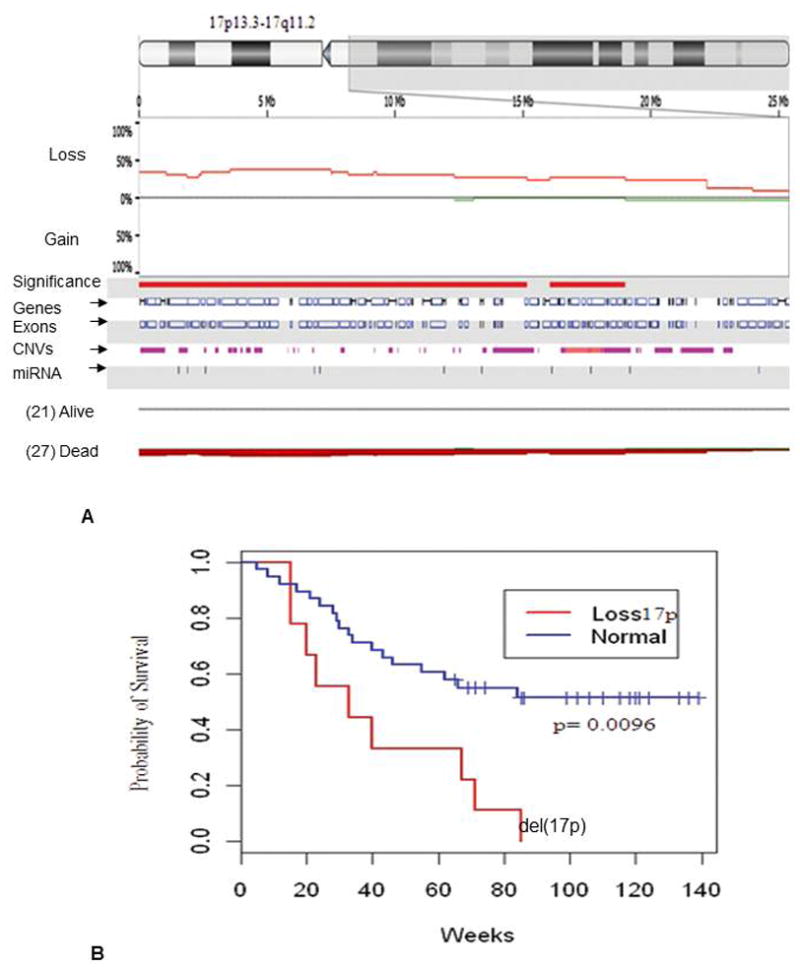
(A) aCGH based detection of loss of chromosomal region at 17p13.3-17q11.2 was associated with poor survival in AML patients. This 3.2Mb region loss spans 342 genes and 12 microRNAs. (B) Kaplan meir test showing correlation between aCGH detected aberrations at 17p and overall survival at p value 0.0096 in AML patients
Association of Copy Number Loss with Complete Remission
A 155 kb microdeletion at chromosome 5q33.3 was noted in 9 (27%) of 34 patients who achieved complete remission. The region was found to be intact in all 14 patients who were resistant to chemotherapy. The 5q33.3 region is known to harbor the candidate genes early B-cell factor 1 (EBF1) and ring finger protein 145 (RNF145) (Figure 5A). The association of loss of EBF1 and RNF145 with achievement of CR was found to be statistically significant (P<0.05). Nexus analysis for copy number changes of EBF1 and RNF145 showed 8.3% copy number loss, 4.2% copy number gain for each gene whereas 12.5% and 10.42% homozygous copy number loss was observed for EBF1 and RNF145, respectively. We confirmed this finding by performing copy number assay by real-time PCR. The predicted loss was confirmed by real-time PCR-based copy number assessment in 7 of 7 cases for RNF145 exon 11 and 4 (57%) of 7 tested cases for EBF1 exons 1 and 14 (Figure 5B).
Figure 5.
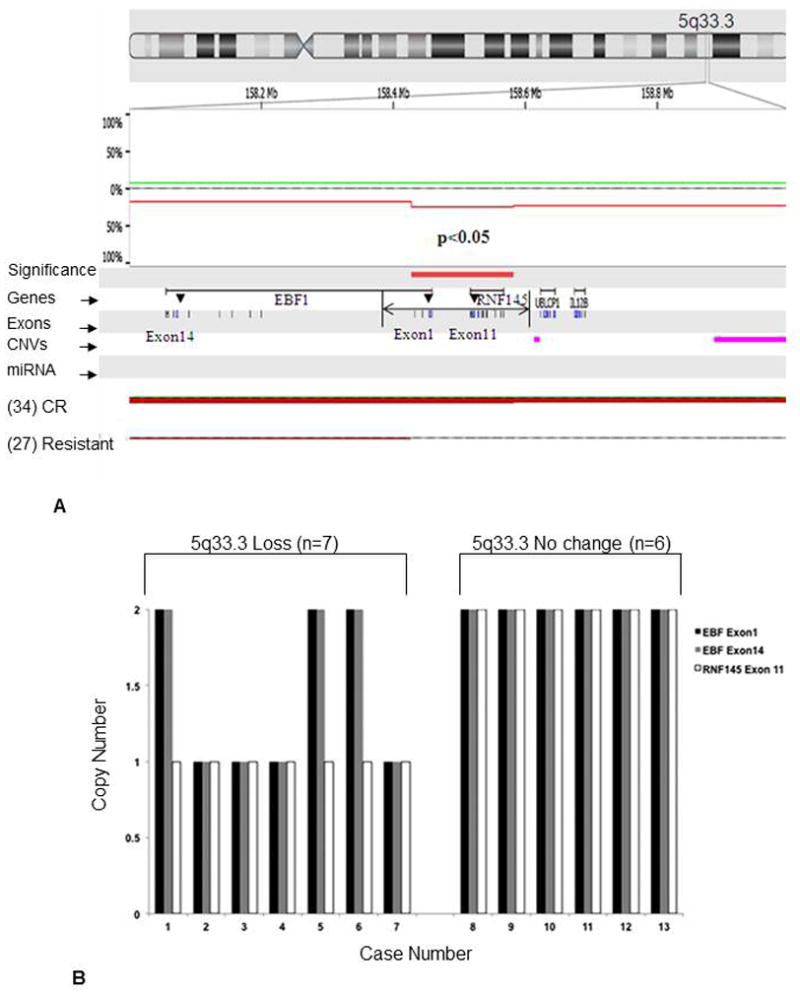
(A) Oligonucleotide based aCGH detection of candidate genes RNF145 and EBF1 residing in deleted 5q33.3 locus was found to be associated with AML patients achieving complete remission (27%). These aberrations were not detected in resistant patients. Loss of EBF1 and RNF145 was found to be statistically significant (P<0.05) CR: complete remission. Loss was shown in red. (B) Validation of 7 cases by RT-PCR based copy number analysis showing loses at 5q33.3 in 7 of 7 cases for RNF145 exon 11 and 4 of 7 tested cases for EBF1 exons 1 and 14 whereas 6 cases with no loss at 5q33.3 showed no change for candidate genes EBF1 (exon1 and exon14) and RNF145 (exon 11)
Discussion
During the last two decades, a number of clinicopathologic and genetic features have been shown to impact the prognosis of AML patients, leading risk-adapted therapeutic approaches. Conventional cytogenetics and FISH are excellent techniques to identify many chromosomal abnormalities and therefore are highly important in the classification of AML as well as in stratifying AML patients into “favorable-risk,” “intermediate-risk,” and “poor-risk” or “unfavorable risk” prognostic groups [1,3,4,19,20]. Chromosomal abnormalities such as t(15;17), t(8;21), inv(16), or 11q23 are included in the favorable-risk group, whereas abnormalities included in intermediate-risk group are heterogeneous and include a large subset of patients with diploid karyotypes. Therefore, more extensive molecular analyses may help to identify prognostic markers for stratification and detection of minimal residual disease in AML patients. aCGH overcomes the limitations of conventional cytogenetic analysis by allowing for a more in depth assessment of the genome in AML patients. These findings will likely better characterize the intermediate- and poor-risk categories of AML and will facilitate detection of additional abnormalities in favorable-risk AML patients that may further aid in risk stratification, as a subset of patients in the favorable-risk group respond well to therapy [5,13,14,16,21,22].
In this study, we investigated the utility of aCGH for detection of chromosomal aberrations in uniformly treated AML patients that were assessed by conventional cytogenetic analysis. Overall, conventional cytogenetic analysis and aCGH showed 94% concordance for the commonly observed aberrations in AML patients. Additionally, aCGH provided more precise details of the chromosomal alterations, including genomic size, gene content and finally mapped breakpoints which was found to be concordant with previously published reports [16]. Certain discrepancies were observed which can be explained by the limits of aCGH, those being an inability to detect a high-level contaminating normal population or balanced chromosome rearrangements [15,16,23]. Therefore an integrated cytogenomic approach using conventional cytogenetic analysis, FISH and aCGH may be ideal, improving the detection rate as well as the resolution of chromosomal abnormalities in AML patients [1]. The data in our study support this statement, as the aCGH platform detected of 39 CNA in 18 patients, many of which were either classified incompletely or missed by conventional cytogenetic analysis. Interstitial deletions of chromosome 5q observed in study are recurrent abnormalities mainly associated with acute leukemia and myelodysplastic syndromes.
The chromosome 5q region contains many genes that are involved in the regulation of hematopoiesis. The clustering of hematologic genes, including cytokines and their receptors, cell cycle regulators, transcription factors, and signaling mediators between 5q13 and 5q33 suggests that a tumor suppressor gene, which is important in hematological transformation, resides on 5q. In this study, we found variable deletion endpoints with some clustering of recurrent proximal and distal sites. Proximal breakpoints were identified more precisely at chromosome loci 5q13.3, 5q14.3~q21.2, 5q21.3-23.1, and 5q23.1-q34. We also showed that loss of 5q33.3 detected by aCGH was associated with achievement of CR in about one quarter of AML patients, but not in patients resistant to therapy. Loss at 5q has been associated with two candidate genes, EBF1 and RNF145. EBF1 is a transcription factor that is a member of EBF family and is important for lineage commitment and differentiation of B lymphocytes. During B-cell development, EBF1 is required for the expression of Pax5, an essential factor for the production of antibody-secreting cells. Therefore, inactivation of EBF1 could block these developmental processes and progenitor cells fail to express classical markers of B cells, including immunoglobulins, resulting in neoplastic phenotypes [24,25]. Accumulating evidence indicates that genomic deletions of the EBF1 gene contribute to pathogenesis, drug resistance, and relapse in AML. RNF145 gene expressed in T-lymphocytes and its expression is altered in acute myelomonocytic and acute promyelocytic leukemias [24,25]. RNF145 showed more consistent results in qPCR. However, this study included only a small number of cases and our findings need to be confirmed in a large patient cohort.
Another recurrent finding detected by aCGH in this study was loss of a 3.2 megabase region of 17p11.2-p13. Patients showing this loss had a median overall survival of 34 weeks. The aCGH analysis and subsequent confirmation by PCR-based copy number assay showed retention of 17q genomic DNA in cases that showed loss of entire chromosome by 17 by conventional karyotyping. This finding was consistent with a previous report by Jerez et al.[26]. Many of these cases showed the presence of marker chromosomes suggesting that the retained genetic material is contained in these marker chromosomes. This finding highlights the value of aCGH in evaluating the loss or gain of DNA copy number. The loss of 17p in AML is often accompanied by a TP53 mutation resulting in a loss of heterozygosity. Loss of p53 is predominantly attributed to an interstitial deletion that presumably is too small to be detected on metaphase chromosomes and is associated with significantly shortened survival. The loss of TP53 function could cause cell-cycle arrest in a very primitive stage of maturation. Inversely, 81.8% of the patients with loss of 17p sequences also have deletions of chromosome 5, which is consistent with data reported by others [27].
Although previous CNA studies in AML [14, 22] had identified similar chromosomal aberrations (5q31.1, 7q, 18p11, 21q22), no other study has evaluated the CNA in uniformly treated AML patients. In our cohort, the loss of 155 kilobase (kb) region at 5q33.3 was associated with complete remission, whereas loss of chromosome 17p11.2-q11.1 was associated with lower CR rate and poor overall survival. Walter et al. performed a genome wide copy number analysis in 86 adult AML samples (primarily M6 and M7 AML cases) using a high resolution SNP array which was confirmed by 135 K aCGH array. No CNV was detected in 50% patients. In contrast, our study includes just 3 cases of M6 accounting for the differences in the frequency and distribution of mutations [14]. Yasar et al. reported CNV in 41 adult leukemic cases constituting a mixture of ALL (majority of cases) and AML (minority of cases) compared to a population of AML in our study. Importantly, lack of cytogenetic information in many cases makes the comparison challenging. Costa et al. identified CNV in 8 AML cases with recurrent abnormalities using a low resolution aCGH chipTm 2600 from Perkin Elmer with a lower cut off compared to the present study [15]. Cases of AML with a normal karyotype in this study showed a remarkable heterogeneity of genetic mutations at the molecular level and an intermediate response to therapy. Identification of cryptic lesions in these patients, coupled with an increased understanding of the biological consequences of these lesions, could lead to more targeted approaches to develop more effective treatments [6,11,19,28]. Walter et al. previously reported 24 % of normal karyotype AML showing CNVs. We detected unbalanced chromosome aberrations in 18 patients with diploid AML as assessed by conventional cytogenetics yet no recurrent cryptic aberrations observed using the predefined criteria of significance level of p<0.05 and less than 50% overlap with known copy number variants. In summary, we have presented data to show that aCGH is valuable in the workup of AML patients. aCGH provided more accurate molecular characterization of recurrent chromosomal regions and evidence of new abnormalities. aCGH provided further insight into the size, genomic position, and gene content and provided higher resolution of the breakpoints producing these abnormalities. The results also show that genomic changes in AML patients are often more complex than was known previously using less robust methods of analysis. We suggest that precise descriptions of the genetic alterations in AML by using aCGH may lead to improved molecular diagnosis, better prognostic stratification of affected patients, and may facilitate the development of targeted therapies.
References
- 1.Bajaj R, Xu F, Xiang B, et al. Evidence-based genomic diagnosis characterized chromosomal and cryptic imbalances in 30 elderly patients with myelodysplastic syndrome and acute myeloid leukemia. Mol Cytogenet. 2011;4:3. doi: 10.1186/1755-8166-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen CC, Yang CF, Lee KD, et al. Complex karyotypes confer a poor survival in adult acute myeloid leukemia with unfavorable cytogenetic abnormalities. Cancer Genet Cytogenet. 2007;174:138–146. doi: 10.1016/j.cancergencyto.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 3.Nahi H, Lehmann S, Bengtzen S, et al. Chromosomal aberrations in 17p predict in vitro drug resistance and short overall survival in acute myeloid leukemia. Leuk Lymphoma. 2008;49:508–516. doi: 10.1080/10428190701861645. [DOI] [PubMed] [Google Scholar]
- 4.Veigaard C, Norgaard JM, Kjeldsen E. Genomic profiling in high hyperdiploid acute myeloid leukemia: a retrospective study of 19 cases. Cancer Genet. 2011;204:516–521. doi: 10.1016/j.cancergen.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 5.Casas S, Aventin A, Fuentes F, et al. Genetic diagnosis by comparative genomic hybridization in adult de novo acute myelocytic leukemia. Cancer Genet Cytogenet. 2004;153:16–25. doi: 10.1016/j.cancergencyto.2003.12.011. [DOI] [PubMed] [Google Scholar]
- 6.Rabin KR, Man TK, Yu A, et al. Clinical utility of array comparative genomic hybridization for detection of chromosomal abnormalities in pediatric acute lymphoblastic leukemia. Pediatr Blood Cancer. 2008;51:171–177. doi: 10.1002/pbc.21488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schoch C, Haferlach T, Bursch S, et al. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: a detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Genes Chromosomes Cancer. 2002;35:20–29. doi: 10.1002/gcc.10088. [DOI] [PubMed] [Google Scholar]
- 8.Shinawi M, Cheung SW. The array CGH and its clinical applications. Drug Discov Today. 2008;13:760–770. doi: 10.1016/j.drudis.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 9.Oostlander AE, Meijer GA, Ylstra B. Microarray-based comparative genomic hybridization and its applications in human genetics. Clin Genet. 2004;66:488–495. doi: 10.1111/j.1399-0004.2004.00322.x. [DOI] [PubMed] [Google Scholar]
- 10.Bejjani BA, Shaffer LG. Application of array-based comparative genomic hybridization to clinical diagnostics. J Mol Diagn. 2006;8:528–533. doi: 10.2353/jmoldx.2006.060029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paulsson K, Heidenblad M, Strombeck B, et al. High-resolution genome-wide array-based comparative genome hybridization reveals cryptic chromosome changes in AML and MDS cases with trisomy 8 as the sole cytogenetic aberration. Leukemia. 2006;20:840–846. doi: 10.1038/sj.leu.2404145. [DOI] [PubMed] [Google Scholar]
- 12.Suela J, Alvarez S, Cigudosa JC. DNA profiling by arrayCGH in acute myeloid leukemia and myelodysplastic syndromes. Cytogenet Genome Res. 2007;118:304–309. doi: 10.1159/000108314. [DOI] [PubMed] [Google Scholar]
- 13.Slovak ML, Smith DD, Bedell V, et al. Assessing karyotype precision by microarray-based comparative genomic hybridization in the myelodysplastic/myeloproliferative syndromes. Mol Cytogenet. 2010;3:23. doi: 10.1186/1755-8166-3-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walter MJ, Payton JE, Ries RE, et al. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci U S A. 2009;106:12950–12955. doi: 10.1073/pnas.0903091106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Costa AR, Belangero SI, Melaragno MI, de Chauffaille ML. Additional chromosomal abnormalities detected by array comparative genomic hybridization in AML. Med Oncol. 2012;29:2083–2087. doi: 10.1007/s12032-011-0108-5. [DOI] [PubMed] [Google Scholar]
- 16.Yasar D, Karadogan I, Alanoglu G, et al. Array comparative genomic hybridization analysis of adult acute leukemia patients. Cancer Genet Cytogenet. 2010;197:122–129. doi: 10.1016/j.cancergencyto.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 17.Sargent R, Jones D, Abruzzo LV, et al. Customized oligonucleotide array-based comparative genomic hybridization as a clinical assay for genomic profiling of chronic lymphocytic leukemia. J Mol Diagn. 2009;11:25–34. doi: 10.2353/jmoldx.2009.080037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma D, Chen Z, Patel KP, et al. Array comparative genomic hybridization analysis identifies recurrent gain of chromosome 2p25. 3 involving the ACP1 and MYCN genes in chronic lymphocytic leukemia. Clin Lymphoma Myeloma Leuk. 2011;11 (Suppl 1):S17–24. doi: 10.1016/j.clml.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheung SW, Shaw CA, Scott DA, et al. Microarray-based CGH detects chromosomal mosaicism not revealed by conventional cytogenetics. Am J Med Genet A. 2007;143A:1679–1686. doi: 10.1002/ajmg.a.31740. [DOI] [PubMed] [Google Scholar]
- 20.Slovak ML, Kopecky KJ, Cassileth PA, et al. Karyotypic analysis predicts outcome of preremission and postremission therapy in adult acute myeloid leukemia: a Southwest Oncology Group/Eastern Cooperative Oncology Group Study. Blood. 2000;96:4075–4083. [PubMed] [Google Scholar]
- 21.Rucker FG, Schlenk RF, Bullinger L, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114–2121. doi: 10.1182/blood-2011-08-375758. [DOI] [PubMed] [Google Scholar]
- 22.Evers C, Beier M, Poelitz A, et al. Molecular definition of chromosome arm 5q deletion end points and detection of hidden aberrations in patients with myelodysplastic syndromes and isolated del(5q) using oligonucleotide array CGH. Genes Chromosomes Cancer. 2007;46:1119–1128. doi: 10.1002/gcc.20498. [DOI] [PubMed] [Google Scholar]
- 23.Strefford JC, Worley H, Barber K, et al. Genome complexity in acute lymphoblastic leukemia is revealed by array-based comparative genomic hybridization. Oncogene. 2007;26:4306–4318. doi: 10.1038/sj.onc.1210190. [DOI] [PubMed] [Google Scholar]
- 24.Liao D. Emerging roles of the EBF family of transcription factors in tumor suppression. Mol Cancer Res. 2009;7:1893–1901. doi: 10.1158/1541-7786.MCR-09-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukin K, Fields S, Hartley J, Hagman J. Early B cell factor: Regulator of B lineage specification and commitment. Semin Immunol. 2008;20:221–227. doi: 10.1016/j.smim.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jerez A, Sugimoto Y, Makishima H, et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood. 2012;119:6109–6117. doi: 10.1182/blood-2011-12-397620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jerez A, Gondek LP, Jankowska AM, et al. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J Clin Oncol. 2012;30:1343–1349. doi: 10.1200/JCO.2011.36.1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuneo A, Bigoni R, Cavazzini F, et al. Incidence and significance of cryptic chromosome aberrations detected by fluorescence in situ hybridization in acute myeloid leukemia with normal karyotype. Leukemia. 2002;16:1745–1751. doi: 10.1038/sj.leu.2402605. [DOI] [PubMed] [Google Scholar]