

Background: Ser/Thr protein kinases (STPKs) form multilayered signaling networks that mediate cellular responses in eukaryotes and prokaryotes.
Results: A preliminary interaction network for the STPKs in Mycobacterium tuberculosis is described.
Conclusion: STPKs that cross-phosphorylate are often co-localized, suggesting multiple activation mechanisms.
Significance: The initial map of this prokaryotic STPK network provides a framework for defining the logic of M. tuberculosis signaling pathways.
Keywords: Bacterial Protein Kinase, Mycobacterium tuberculosis, Prokaryotic Signal Transduction, Serine/Threonine Protein Kinase, Signaling
Abstract
Many Gram-positive bacteria coordinate cellular processes by signaling through Ser/Thr protein kinases (STPKs), but the architecture of these phosphosignaling cascades is unknown. To investigate the network structure of a prokaryotic STPK system, we comprehensively explored the pattern of signal transduction in the Mycobacterium tuberculosis Ser/Thr kinome. Autophosphorylation is the dominant mode of STPK activation, but the 11 M. tuberculosis STPKs also show a specific pattern of efficient cross-phosphorylation in vitro. The biochemical specificity intrinsic to each kinase domain was used to map the provisional signaling network, revealing a three-layer architecture that includes master regulators, signal transducers, and terminal substrates. Fluorescence microscopy revealed that the STPKs are specifically localized in the cell. Master STPKs are concentrated at the same subcellular sites as their substrates, providing additional support for the biochemically defined network. Together, these studies imply a branched functional architecture of the M. tuberculosis Ser/Thr kinome that could enable horizontal signal spreading. This systems-level approach provides a biochemical and spatial framework for understanding Ser/Thr phospho-signaling in M. tuberculosis, which differs fundamentally from previously defined linear histidine kinase cascades.
Introduction
Although reversible protein phosphorylation is ubiquitous in all kingdoms of life, different organisms use chemically distinct modifications to coordinate cellular signals. Higher eukaryotes primarily use relatively stable Ser/Thr or Tyr modifications, whereas prokaryotes rely more heavily on histidine kinases (HK)2 (1). In addition to the chemical differences between these pathways, current data indicate that the network architecture of the cascades may also be fundamentally distinct. Prokaryotic HKs generally have a single preferred substrate, often a transcription factor (i.e. “response regulator”), that is activated by phosphorylation (2). In contrast, eukaryotic Ser/Thr kinases (STPK) often have many substrates, including additional kinases, that act to spread signals (3). These observations imply that HK and STPK signaling networks could have different architectures, which would rationalize the simultaneous presence of both systems in many bacteria and fungi. However, the complete structure of a prokaryotic STPK network has not yet been defined, leaving this hypothesis untested.
In addition to 11 paired HK-response regulator systems, the Mycobacterium tuberculosis genome encodes an equal number of STPKs that function as important nodes of the environment sensing and response network and enable bacterial survival within the human host (Fig. 1A) (4–7). The retention of these genes in the M. tuberculosis genome suggests an important role in pathogenesis or transmission. Indeed, proteomic and transcriptomic studies of multiple M. tuberculosis strains indicate that at least eight members of this signaling family are expressed during infection (7–9). PknA and PknB, the two M. tuberculosis STPKs essential for survival in vitro, are implicated in controlling cell growth and division by regulating cell wall synthesis, transcription, translation, septum formation, and other processes (5, 10–18). In contrast, PknH controls expression of genes involved in arabinogalactan biosynthesis and response to nitric oxide stress, PknG regulates carbon metabolism, and PknD has been implicated in regulating the stressosome in response to changes in osmolarity (17, 19–22, 29). Although the cellular roles of these individual STPKs are beginning to be defined, the networks that are responsible for integrating cellular signals remain obscure.
FIGURE 1.
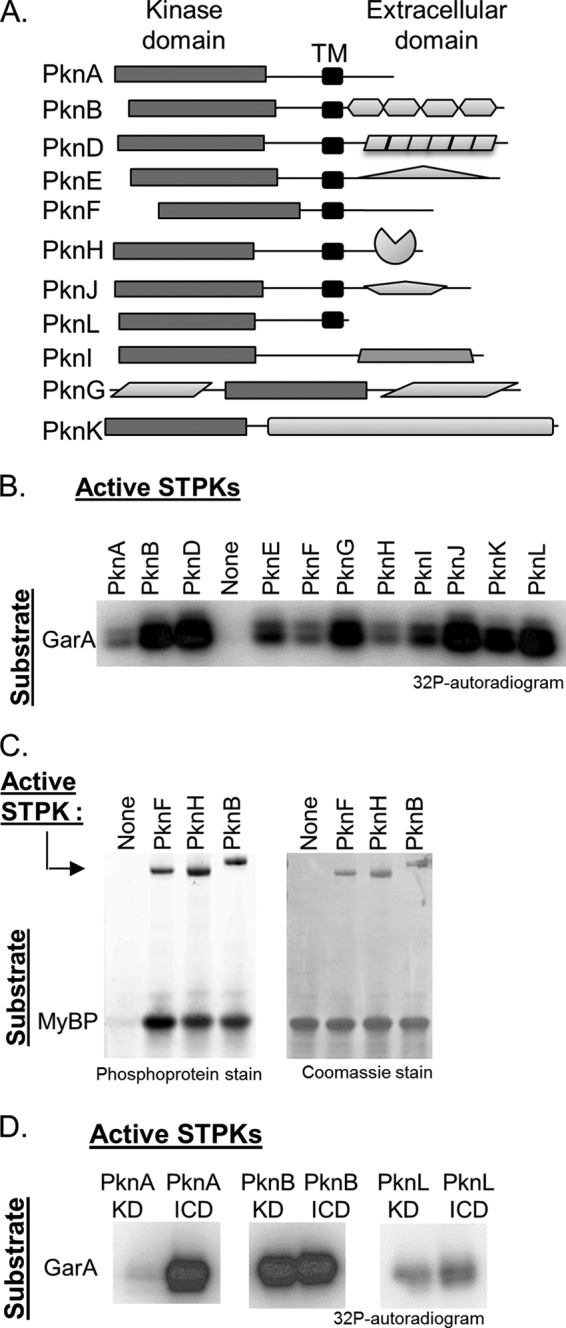
11 M. tuberculosis STPKs display trans-phosphorylation activity on multiple substrates. A, domain architecture of the 11 M. tuberculosis STPKs. Soluble, active KDs were obtained for PknA, PknB, PknD, PknE, PknF, PknH, PknJ, PknK, and PknL. PknG and PknI are only active as full-length soluble enzymes. Shapes indicate predicted or experimentally verified folded domains (42). TM, transmembrane. B, autoradiogram showing the reactions of the 11 STPK constructs incubated with the substrate protein GarA and [γ-32P]ATP (top). The STPK constructs phosphorylate GarA. C, PknF and PknH efficiently phosphorylate the model substrate MyBP in comparison with PknB. Phosphoproteins were visualized using Pro-Q Diamond Phosphoprotein Gel Stain. Active STPK autophosphorylation is visible (upper bands) as is trans-phosphorylation on the MyBP substrate; equal protein loading was confirmed by Coomassie stain. D, trans-phosphorylation activity of KD and full-length ICD constructs was compared using GarA as a substrate. The autoradiogram confirms that the PknA ICD is more active than the PknA KD as reported previously (38), but that activity does not differ for the two PknB and PknL constructs.
Like many eukaryotic protein kinases, the M. tuberculosis STPKs are generally stimulated by phosphorylation of the activation loop, a conserved peptide bordered by the DFG and APE tripeptides (23–25). Studies of PknD (26) and PknB (27) show that this process can be initiated by intermolecular autophosphorylation reactions. Interactions across two interfaces mediate PknB phosphorylation. An allosteric “back-to-back” N-lobe interface stabilizes the active conformation, and transient “front-to-front” contacts through the G-helix in the kinase C-lobe are required for efficient intermolecular autophosphorylation (26–28). These two activation mechanisms result in an autophosphorylated active kinase. In contrast, PknG contains phosphorylation sites in the C-terminal domain that may be necessary for activity (30, 31). These biochemical and structural insights suggest a general model for activation in which ligand binding to the extracellular domain promotes KD dimerization and intermolecular autophosphorylation (26, 32).
In addition to autophosphorylation, inter-kinase cross-phosphorylation of PknA and PknB has been reported in Mycobacterium smegmatis (15). Kinase cross-phosphorylation, such as that observed in the signaling cascade that controls development in Myxococcus xanthus (33), potentially adds a level of regulatory control and integration to the system. However, it is not clear to what extent kinase cross-talk acts as an alternative activation pathway and whether prokaryotic STPK networks share the multiply branched structure that is characteristic of eukaryotic kinase cascades.
The linear structures of bacterial HK pathways have been experimentally defined by determining the intrinsic biochemical substrate specificity of the kinases. The resulting pattern of efficient in vitro phosphorylation of response regulators parallels the physiological activation pathways in vivo (2). This rationale also has been used to identify potential signaling partners of eukaryotic kinases by defining intrinsic biochemical preferences, and this approach has been especially useful in pathways that lack scaffolding proteins that restrict kinase specificity (34). These precedents suggest that the efficiency of kinase reactions in vitro can provide candidates for the physiological interactions that comprise signaling networks in vivo. The regulation of the bacterial STPKs by activation loop phosphorylation (23, 24, 35) and the discovery of functional kinase cross-phosphorylations in M. tuberculosis and M. xanthus (15, 33) suggested that bacterial STPKs may form a hierarchical network that could be defined based on the kinetic efficiency with which each kinase modified the others (36).
Focusing on the kinase domains (KDs), we used this strategy to initially discover all of the efficient in vitro auto- and cross-STPK phosphorylations. The 11 STPKs expressed by M. tuberculosis provide a tractable family with sufficient complexity to enable this comprehensive approach. The interaction pattern was remarkably restricted, dominated by autophosphorylation and a small number of intermolecular phosphorylations. These reactions targeted the conserved Thr residues in the activation loop where phosphorylation stimulates kinase activity. This global network represents a functional map of potential STPK activation. Using STPKs fused to fluorescent tags in M. smegmatis, we found that each kinase was specifically concentrated in polar, septal, cytoplasmic, or diffuse membrane-associated locations. Remarkably, the interaction map is consistent with the cellular distribution of functionally interacting kinases. This systems level analysis implies a branching network that differs from the linear signaling pathways mediated by HKs.
EXPERIMENTAL PROCEDURES
STPK Cloning and Mutagenesis
KD boundaries were determined using the PHYRE homology prediction server (32). M. tuberculosis H37Rv genomic DNA was used as the PCR template for cloning. KDs with a tobacco etch virus protease cleavage site were cloned into the pHMGWA or the pET28b expression vectors (EMD Millipore) (43). Site-directed mutants were constructed using the two-primer method (Invitrogen). Construct details are listed in Table 1.
TABLE 1.
Protein construct details for the STPKs utilized in this study
HMBP indicates His6-maltose-binding protein.
| Protein | Length (amino acids) | Mutation | Tag |
|---|---|---|---|
| PknA | 1–279 | HMBP | |
| PknA | 1–279 | T172A/T174A | HMBP |
| PknA | 1–279 | D141N | His |
| PknA | 1–279 | D141N/T172A/T174 | His |
| PknA | 1–336 | HMBP | |
| PknB | 1–291 | HMBP | |
| PknB | 1–291 | T171A/T173A | HMBP |
| PknB | 1–279 | D138N | His |
| PknB | 1–279 | D138N/T171A/T173A | His |
| PknB | 1–330 | HMBP | |
| PknD | 1–292 | HMBP | |
| PknD | 1–292 | T169A/T171A | HMBP |
| PknD | 1–292 | D138N | HMBP |
| PknD | 1–292 | D138N/T169A/T171A | HMBP |
| PknE | 1–279 | HMBP | |
| PknE | 1–279 | T170A/T175A | HMBP |
| PknE | 1–279 | D139N | HMBP |
| PknE | 1–279 | D139N/T170A/T175A | HMBP |
| PknF | 1–280 | HMBP | |
| PknF | 1–280 | T173A/T175A | HMBP |
| PknF | 1–280 | D137N | HMBP |
| PknF | 1–280 | D137N/T173A/T175A | HMBP |
| PknG | 144–403 | HMBP | |
| PknG | 144–403 | D137N | HMBP |
| PknG | 1–750 | D143N | His |
| PknG | 1–750 | HMBP | |
| PknH | 1–280 | HMBP | |
| PknH | 1–280 | T170A/T174A | HMBP |
| PknH | 1–280 | D139N | HMBP |
| PknH | 1–280 | D139N/T170A/T174A | HMBP |
| PknI | 1–265 | HMBP | |
| PknI | 1–585 | HMBP | |
| PknI | 1–585 | D137N | HMBP |
| PknJ | 1–286 | HMBP | |
| PknJ | 1–286 | D125N | HMBP |
| PknJ | 1–286 | T171S/S172S/T173A | HMBP |
| PknJ | 1–286 | D125N/T171A/S172A/T173A | HMBP |
| PknK | 1–290 | HMBP | |
| PknK | 1–290 | T179A/T181A | HMBP |
| PknK | 1–290 | D149N | HMBP |
| PknK | 1–290 | D149A/T179A/T181A | HMBP |
| PknL | 1–302 | HMBP | |
| PknL | 1–302 | T173A/T175A | HMBP |
| PknL | 1–302 | D142N | HMBP |
| PknL | 1–302 | D142N/T173A/T175A | HMBP |
| PknL | 1–366 | HMBP |
STPK Expression and Purification
Proteins were expressed in Escherichia coli BL21 CodonPlus (Agilent) cells using autoinduction (44). Cultures were grown at 37 °C for 8 h and shifted to 18–22 °C for 18–24 h. Following harvest by centrifugation and storage at −80 °C, each pellet was resuspended in 300 mm NaCl, 25 mm HEPES, pH 8.0, 25 mm imidazole, 0.5 mm tris(2-carboxyethyl)phosphine, and 10% (v/v) glycerol with 250 μm 4-(2-aminoethyl)benzenesulfonyl fluoride and lysed using sonication. The cleared lysate was run on a 10-ml nickel-HiTrap column (GE Healthcare) and eluted with 300 mm NaCl, 25 mm HEPES, pH 8.0, 300 mm imidazole, 0.5 mm tris(2-carboxyethyl) phosphine, and 10% (v/v) glycerol. Where desired, tobacco etch virus protease cleavage followed by a second nickel immobilized metal-affinity chromatography step was performed. Proteins were further purified using size-exclusion chromatography with a HiLoad 26/60 Superdex S75 (GE Healthcare) column in size-exclusion chromatography buffer (150 mm NaCl, 25 mm HEPES, pH 8.0, 0.5 mm tris(2-carboxyethyl)phosphine and 20% (v/v) glycerol).
Kinase Assays
Reactions were run in size-exclusion chromatography buffer with 25 μm substrate protein and 1 μm active kinase. The reaction was initiated by the simultaneous addition of 1 μCi of [γ-32P]ATP (6000 Ci/mmol and 10 mCi/ml; MP Biomedicals), ATP (Sigma), and MnCl2 to final concentrations of 50 nCi/μl, 50 μm, and 50 μm, respectively. After 30 min at room temperature, reactions were quenched with SDS-PAGE loading dye, separated by SDS-PAGE, and radioactivity was detected with a Typhoon 860 phosphorimager from GE Healthcare.
For the assays of the full-length PknI and PknG constructs, the His6-MBP tags were cleaved prior to running the reactions on SDS-PAGE. The reactions were quenched by the simultaneous addition of EDTA to 20 mm and 6 μg of tobacco etch virus protease. After 2 h, the reactions were separated by SDS-PAGE and imaged as described above. All autoradiographs were quantified using ImageJ, and intensity values were normalized for each kinase to the activity detected for autophosphorylation.
For Western blots and Pro-Q Diamond phosphoprotein gel stain (Invitrogen), 25 μm substrate protein and 1 μm active kinase were incubated with 1 mm ATP (Sigma) and 1 mm MnCl2. Reactions were separated by SDS-PAGE. Diamond staining was completed according to the manual and imaged on a Typhoon 860 imager from GE Healthcare. Samples for Western blotting were transferred to a nitrocellulose membrane. The membranes were blocked for 4 h in 3% (w/v) BSA in PBS with 0.2% Tween (PBST and then incubated with (1:1000) phosphothreonine antibody (9381, Cell Signaling Technology) overnight in 3% (w/v) BSA in PBST. After washing with PBST, Cy3-labeled anti-rabbit secondary antibody (Invitrogen) at 1:5,000 dilution in PBST was added and incubated for 30 min. Blots were imaged on a Typhoon 860 imager from GE Healthcare.
Liquid Chromatography-Mass Spectrometry
Prior to LC-MS analysis, 25 μm substrate protein and 1 μm active kinase were incubated with 1 mm ATP (Sigma) and 1 mm MnCl2 for 12–20 h at 4 °C. Intact protein constructs were analyzed using an Agilent 1200 series liquid chromatograph (LC) connected in-line with an LTQ Orbitrap XL hybrid mass spectrometer equipped with an Ion Max electrospray ionization source (Thermo Fisher Scientific). The LC was equipped with a C8 column (Poroshell 300SB-C8, 5 μm, analytical 75 mm × 0.5 mm, Agilent). Solvent A was 0.1% formic acid, 99.9% water (v/v), and solvent B was 0.1% formic acid, 99.9% acetonitrile (v/v). For each sample, ∼100 pmol of analyte was injected onto the column. Following sample injection, analyte trapping was performed for 5 min with 99.5% (v/v) A at a flow rate of 90 μl/min. The elution program consisted of a linear gradient of 35–95% (v/v) B over 34 min, isocratic conditions at 95% (v/v) B for 5 min, a linear gradient to 0.5% (v/v) B over 1 min, and then isocratic conditions at 0.5% (v/v) B for 14 min at a flow rate of 90 μl/min.
External mass calibration was performed prior to analysis using the standard LTQ calibration mixture. Mass spectra were recorded in the positive ion mode over the range m/z = 500–2000. Raw mass spectra were processed using Xcalibur software (version 2.0.7 SP1, Thermo), and measured charge-state distributions of proteins were deconvoluted using ProMass software (version 2.5 SR-1, Novatia).
Construction of Fluorescent Strains and Confirmation of Expression
Vectors expressing fluorescent protein-tagged STPKs were constructed using multisite Gateway cloning (Invitrogen). Each reporter fusion was under the control of the P16 promoter and contained an N-terminal FLAG-mVenus tag in the episomal expression vector, pDE43-MEK, or in the integrating vector, pDE43-MEK (kindly provided by D. Schnappinger). Plasmids were transformed into M. smegmatis MC2155. To confirm expression, each strain was lysed by beadbeating in PBS with 2% (w/v) SDS. The soluble fraction of the lysate was quantified for total protein content using a BCA assay (Pierce) and separated by SDS-PAGE. Western blots were performed with anti-FLAG M2 primary antibody (Sigma) and Cy-5 anti-mouse secondary antibody (Invitrogen), and then imaged on a Typhoon 860 imaging system (GE Healthcare).
Fluorescence Microscopy
M. smegmatis strains were grown in 7H9/ADS/Tween medium to an A600 of 0.8 and imaged on a DeltaVision microscope (Applied Precision) mounted on agar pads. Bacteria were stained with FM4-64 (Invitrogen) at a final concentration of 1 μg/ml for 90 min at room temperature before imaging. Images were deconvolved, and statistical analysis was completed using softWoRx (Applied Precision) and Microsoft Excel.
RESULTS
STPK Intermolecular Phosphorylation Follows Specific Patterns
To explore the intrinsic specificity of the M. tuberculosis STPKs, we expressed and purified a complete set of kinase domains. The catalytic activity of several M. tuberculosis STPK KDs has been demonstrated previously (35, 37). We focused on the minimal KD constructs, because these segments contain conserved activation loop Thr residues that must be phosphorylated to elicit activity. Although the STPKs also contain juxta-membrane phosphorylation sites implicated in regulation or binding accessory proteins (38, 39), phosphorylation of the activation loop Thr is the primary mechanism of activation. Moreover, the in vitro specificity of a number of KDs has been shown to recapitulate functional substrate phosphorylations in vivo (40). Although other factors such as co-expression, co-localization, and phosphatase activity can influence functional phosphorylations in vivo, the biochemical specificity of the KDs provides a provisional network architecture.
KD constructs (Table 1) were designed based on sequence alignments, secondary structure prediction, and available crystal structures (41–43). His-MBP tags were maintained on the active kinases throughout all experiments to enable size separation of STPKs from untagged or minimally tagged (His6) substrates. In contrast to the other STPKs, the PknG and PknI KDs are not active. Consequently, the full-length PknG and PknI proteins were included in the comprehensive set of active M. tuberculosis STPKs.
Each of the STPKs was found to phosphorylate the M. tuberculosis substrate protein GarA (Fig. 1B). GarA contains a forkhead-associated domain known to bind phosphopeptides and has previously been reported as a PknB and PknG substrate (19). As PknF and PknH displayed reduced activity on GarA, we confirmed that these two STPKs were active on myelin basic protein (MyBP), a model substrate previously reported to be phosphorylated by these kinases (44, 45). PknF and PknH display equal trans-phosphorylation activity on MyBP in comparison with PknB when imaged by Pro-Q Diamond phosphoprotein gel stain (Invitrogen) (Fig. 1C).
The PknA kinase domain also displayed reduced activity on GarA. As the full-length intracellular domain (ICD) of PknA has been previously reported to be necessary for full activity (38), we tested the activity of the PknA KD and PknA ICD constructs on GarA (Fig. 1D). The two PknA constructs display significantly different activity; therefore, both versions of this kinase were used in later experiments. We also tested the relative activity of KD and ICD constructs for PknB and PknL as these two STPKs are closely related to PknA by sequence homology (46). No significant difference in activity was noted for PknB and PknL (Fig. 1D); thus the PknB and PknL KD constructs were used for all assays. Substrates for intermolecular cross-phosphorylation assays were created by generating inactive variants of all 11 kinase constructs. To eliminate activity in each kinase, an Asp-to-Asn substitution was introduced in the catalytic HRD motif (17).
Using this complete set of active and inactive kinase constructs, we assayed all pairwise combinations for autophosphorylation (i.e. modification of homologous inactive STPK) and cross-phosphorylation (i.e. modification of heterologous inactive STPK). Each substrate was trans-phosphorylated by a distinctive subset of the active kinase constructs (Fig. 2A). Seven of the STPKs catalyzed intermolecular autophosphorylation. The PknA and PknL KDs did not efficiently autophosphorylate. Likewise, the PknA ICD does not efficiently phosphorylate the PknA KD (Fig. 2B). Instead, PknA and PknL were only efficiently phosphorylated by heterologous kinases. Notably, the PknA ICD construct does not display increased inter-kinase phosphorylation compared with PknA KD, indicating that the kinase-kinase interactions are specific and not promoted by increased enzymatic activity. In addition, strong and specific inter-STPK preferences were apparent. Quantification of the autoradiographs reveals that each active kinase phosphorylated 0–3 heterologous KDs (Fig. 2D). To confirm the specificity of the observed inter-STPK phosphorylation pattern, we assayed specific interactions at a series of time points (Fig. 3). Despite reaction times of up to 24 h in the presence of excess ATP and MnCl2, promiscuous phosphorylation was not observed. The small number of efficient inter-STPK phosphorylations suggested the presence of a signaling network defined at least in part by the substrate specificity inherent in the kinase domains.
FIGURE 2.

M. tuberculosis STPKs display specific intermolecular phosphorylation patterns. A, autoradiograms showing the reactions of each active STPK construct (top) with the 11 inactivated substrate kinases (left). Each inactive Asp-to-Asn mutant kinase construct was incubated with each of the 11 M. tuberculosis STPKs in [γ-32P]ATP transfer assays. His-MBP tags on the active kinases and tagless inactive kinases enable separation of the proteins in each reaction by SDS-PAGE. Assays were imaged by autoradiography in parallel. Products on the diagonal reflect efficient autophosphorylation. Off-diagonal bands in each column indicate cross-phosphorylation. B, PknA ICD was tested against the 11 Asp-to-Asn mutant kinases. No additional inter-kinase interactions were observed. C, PknG and PknI KDs were not phosphorylated by any of the other kinases. The full-length PknG and PknI constructs are cross-phosphorylated by multiple kinases, presumably on sites outside of the catalytic domain. D, autoradiographs were quantified in ImageJ and normalized to the autophosphorylation signal observed for each kinase. PknA and PknL do not efficiently autophosphorylate; therefore, these two kinases are not included in this graph. The PknH-PknE interaction was the lowest detected by LC-MS and was used as the lower boundary. Low intensity interactions at or below this cutoff are depicted by gray columns.
FIGURE 3.

M. tuberculosis STPK interactions are substrate-specific. Inter-kinase phosphorylation occurs in a substrate-specific pattern and is not due to differences in enzyme activity. PknB, PknD, and PknH, three STPKs with differing activity, were incubated with the PknB and PknD Asp-to-Asn mutant kinases. Time points taken at 30, 60, and 120 min and 24 h after reaction initiation display the same relative inter-kinase phosphorylation patterns (lower band) when Western-blotted with a phosphothreonine antibody. Active STPK autophosphorylation was similar at each time point (upper bands). Coomassie-stained gels demonstrate equal substrate protein concentrations in all reactions.
The PknG and PknI KDs were not substrates for trans-phosphorylation (Fig. 2A). We tested catalytically inactive, full-length mutants of these two kinases as substrates and discovered widespread trans-phosphorylation by a number of the STPKs (Fig. 2C). Given that PknG and PknI lack threonines in the activation loop and are therefore activated through an alternative mechanism other than STPKs, it is possible that the phosphorylation on other regulatory domains alters activity.
Inter-STPK Phosphorylation Targets Regulatory Activation Loop Thr Residues
To investigate the functional significance of cross-phosphorylation, we first determined whether the activation loop Thr residues are involved in activating each STPK. Activation loop Thr residues essential for kinase activity in PknB and PknH are conserved in all of the M. tuberculosis STPKs except PknG and PknI (Fig. 4A) (23, 25, 35, 47). In all nine active KDs, alanine substitutions of the conserved pair of threonines abolished kinase activity on GarA (Fig. 4B). These results suggest that activation loop Thr phosphorylation is necessary for function of the nine canonical STPKs and that PknG and PknI are activated by alternative mechanisms. As a result, the conserved threonines were candidates for the sites of modification observed in the auto- and cross-phosphorylation reactions of the active kinases.
FIGURE 4.

Activation loop threonines are the primary sites of intermolecular phosphorylation. A, sequence alignment (47) of the M. tuberculosis STPK activation loop region highlights the conserved Thr residues (boxed). B, mutation of the conserved activation loop threonines in nine of the STPKs markedly reduces or abolishes phospho-transfer activity. Reactions and autoradiography were performed in parallel and under identical conditions to active control kinase in Fig. 1B. C, autoradiograms showing representative autophosphorylation (left) and cross-phosphorylation (right) reactions of substrate (D-N) and activation loop double Thr mutants (D-N/T-A). The activation loop substitutions reduced or abolished phosphorylation.
To survey the importance of these sites, we measured the phosphorylation of representative inactive substrate kinases containing the Asp-to-Asn HRD mutation in which the activation loop threonines were also mutated to alanine. The activation loop mutations significantly reduced or abolished intermolecular phosphorylation (Fig. 4C). These results suggest that most intermolecular phosphorylation of KDs occurs specifically on the activation loop residues that are essential for STPK activity.
LC-MS measurements of several cross-phosphorylated STPKs revealed that these reactions produced residue-specific modifications with distinct stoichiometries. Under the defined conditions used in our assay, for example, the inactive PknD substrate kinase was phosphorylated on up to two sites by active PknB and PknD (Fig. 5B). In contrast, the active PknD KD purified from E. coli was a heterologous mixture modified from 3 to 11 phosphoryl groups, likely due to robust autophosphorylation during expression of the protein (Fig. 5A). Like PknD, PknK exhibited similarly limited patterns of in vitro cross-phosphorylation. PknB and PknJ phosphorylated only a single residue on the inactive PknK substrate KD, whereas PknK autophosphorylation involved the addition of two to three phosphates (Fig. 5C). Consistent with biochemical results, the PknA KD did not autophosphorylate but was phosphorylated on one site by PknB (Fig. 5D). PknE efficiently autophosphorylated, with the addition of a single phosphoryl group (Fig. 5D). These examples illustrate that under defined conditions, STPK cross-phosphorylation involves the addition of a limited number of phosphates on the substrate STPK. Because the Thr mutations in the substrate kinase abolished phosphorylation, these interactions appear to specifically target the activation loop.
FIGURE 5.

Inter-STPK phosphorylation is highly site-specific. Mass spectra of STPK constructs following auto- and cross-phosphorylation reactions. The unphosphorylated substrate (D-N) mass distribution is shown in black in each panel. A mass increase of 80 Da is equivalent to one phosphoryl group. The reactions are labeled with the substrate (D-N)/active STPK constructs. A, compared with the unmodified substrate PknD KD, the wild-type, active PknD KD purified from E. coli is a heterogeneous mixture of autophosphorylation states, ranging from 3 to 11 phosphates. B, PknD KD autophosphorylates PknD D-N at one or two sites (blue). PknB cross-phosphorylates PknD D-N at 0–2 sites (orange). C, PknK KD autophosphorylates 2–3 sites (left, blue). The PknJ (left, orange) and PknB (right, green) KDs cross-phosphorylate PknK D-N at one site. D, PknA KD does not autophosphorylate (left), but is cross-phosphorylated on one site by PknB (middle, orange). E, PknE KD autophosphorylates in trans at a single site.
Provisional Kinase Network Is Consistent with the Cellular Distribution of STPKs
To explore whether the observed specificity of in vitro STPK KD interactions was recapitulated in vivo, we investigated whether the STPKs shared the same subcellular localization with their preferred substrates. N-terminal mVenus STPK fusions were expressed in M. smegmatis under the control of a relatively weak promoter. Reporter fusions for PknA and PknB were constructed using catalytically inactive Asp-to-Asn mutants to reduce the toxic effects of constitutive expression (15). Expression of four fusion proteins (PknF, PknG, PknI, and PknK) could not be detected by Western blotting or fluorescence microscopy. The other seven STPK fusions, however, were expressed at equivalent levels (Fig. 6A).
FIGURE 6.
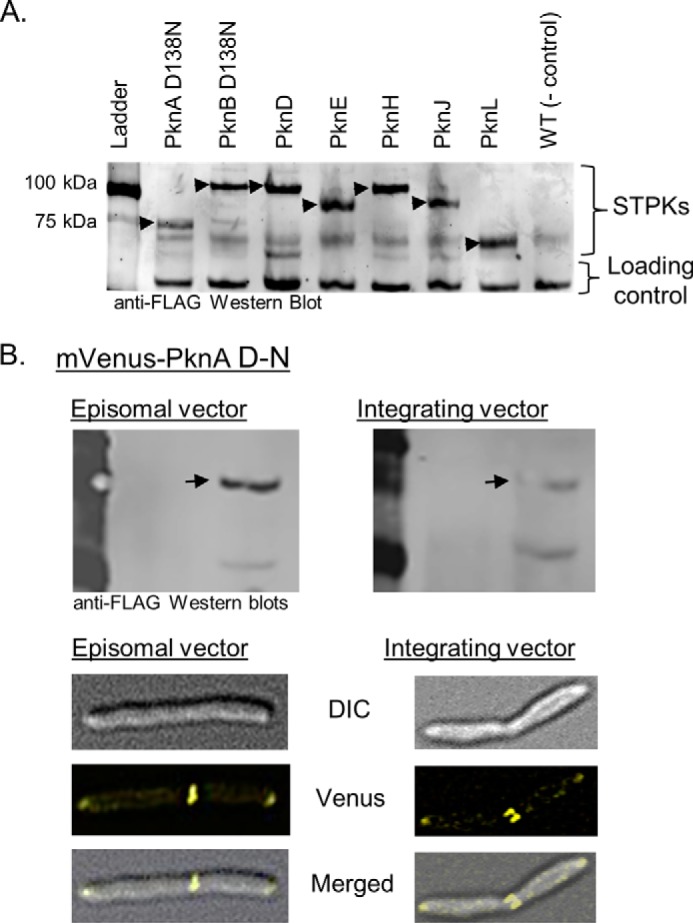
Expression of FLAG-mVenus-STPK reporter fusions in M. smegmatis. A, anti-FLAG Western blot of M. smegmatis lysates containing each indicated full-length STPK fused to FLAG-mVenus expressed in the pDE43-MEK episomal vector. The common band at the bottom of the blot serves as a loading control. Arrows indicate mVenus-kinase fusions. B, Western blots (top) and microscopy (bottom) that compare protein expression levels for the FLAG-Venus-PknA D-N reporter construct in an episomal (left) and integrating vector (right). Western blots were exposed in parallel. Differences in expression level do not change the apparent localization at the poles and septum. DIC, differential interference contrast.
To control for the possible effects of gene copy number, expression was compared using either a low copy number episome or a single copy integrating plasmid. Fusions that failed to express in one vector also failed to express in the other. PknA provides a representative example of the effects of switching vectors. The level of the PknA fusion was reduced using the integrating vector (Fig. 6B). The differences in expression, however, did not change the localization pattern of any of the STPK fusions (e.g. Fig. 6B). The results indicate that in the ranges tested, expression levels did not influence localization. Based on these controls, we conclude that intrinsic localization signals determine the observed cellular distributions of the mVenus-STPK fusions.
The seven expressed STPK fusions were differentially localized in three distinct patterns. PknA and PknB were found exclusively at the poles and the septa, along with the kinase most similar in primary sequence, PknL (Fig. 7A) (46). PknD, PknE, and PknH were more evenly distributed along the cell membrane (Fig. 7B). Finally, PknJ was only discretely localized at the midplane of a small percentage of cells (Fig. 7C). Subtle differences were apparent within these three groups. For example, although all of the kinases were localized to the midplane of some dividing bacteria, the percentage of cells with fluorescent STPK located at this site varied from 6.0% (PknJ) to 65.2% (PknA) (Fig. 7). These results suggested that STPK localization was temporally regulated during cell division.
FIGURE 7.

Subcellular localization of M. tuberculosis STPKs in M. smegmatis. Differential interference contrast (DIC), fluorescence (mVenus and FM4-64) microscopy and merged images for the M. smegmatis strains carrying mVenus fusions of the indicated full-length STPKs. Tables present statistical analyses of kinase localization. The STPKs localize in three distinct patterns in vivo. A, PknA, PknB, and PknL localize to the poles and midplane (green). The individual kinases persist at the midplane/septum for varying lengths of time throughout the cell cycle, as represented by the percentage of cells with a fluorescent signal at the septa compared with the cells without visible septa. FM4-64 staining for septa confirms that, with the exception of mVenus-PknA D-N, expression of the STPKs does not alter the cell cycle. B, PknD, PknE, and PknH localize to the peripheral membrane and the midplane (blue). C, PknJ is only found at the midplane of a small percentage of cells (orange).
To control for the possible effects of STPK expression on the cell cycle, the reporter strains were stained with FM4-64 membrane dye to enable visualization of the septum. Only the PknA Asp-to-Asn inactive construct caused an increase in cells with discernible septa (Fig. 7A). When expressed as a ratio of the FM4-64-positive septa, STPK localization to this site differed from 15% for PknJ to 121% for PknA. Thus, PknA appears to mark the midplane before membrane deposition at the nascent septum, whereas the other STPKs occupy this location for a shorter and variable portion of the cell cycle. Importantly, these data indicate that the STPK signaling pathways are not distributed randomly in the cell. Remarkably, the STPKs that cross-phosphorylate were concentrated at similar subcellular sites, indicating that these preferred biochemical reactions have the opportunity to occur in vivo.
DISCUSSION
Bacterial STPKs were first identified over 2 decades ago (48), but the overall architecture of a prokaryotic Ser/Thr signaling network has not been defined (18). Here, we used a complete set of M. tuberculosis STPK constructs to comprehensively map the preferred substrates and cellular localizations that comprise the relatively complex M. tuberculosis signaling network. At the biochemical level, we found that the pattern of preferred STPK phosphorylation is very limited. Most STPK KDs showed in vitro auto- and cross-phosphorylation reactions of similar efficiencies, as expected for functionally cognate donor-acceptor pairs. The activation loop threonines, which are required for activation, were the specific targets of auto- and cross-phosphorylation reactions for the majority of the intermolecular interactions mapped. The biochemical specificity for these sites matches the expectation for functional interactions. The provisional map of STPK cross-talk recapitulates reported interactions in vivo between PknA and PknB in M. smegmatis (15) and the autophosphorylation of PknD in M. tuberculosis (17).
At the cellular level, the STPKs fused to mVenus were distributed in M. smegmatis in three patterns as follows: at the pole and midplane (PknA, PknB, and PknL), in the cell body membrane and at the midplane (PknD, PknE, and PknH), and solely at the midplane (PknJ). The cellular location of several kinases is also consistent with their functions. PknA and PknB are essential for cell growth and are found exclusively at the pole and septum, sites of new cell wall synthesis for elongation and division (13). It is at these sites where the peptidoglycan-binding extracellular domain of PknB is likely to receive its activating signal (32). Conversely, PknD, PknE, and PknH are broadly distributed along the cell membrane and are not required for cell growth, suggesting that these kinases may respond to condition-specific environmental cues. The convergence of the STPKs at the poles and septum suggest that Ser/Thr phospho-signaling plays a particularly important role at these sites. Rather than being spread uniformly throughout the cell, the STPK network is spatially segregated indicating that kinase localization could play a role in substrate selection.
This concept is consistent with the biochemical specificity of the kinases. PknB, for example, co-localizes with its substrates, PknA and PknL. This pattern provides a potential mechanism to activate PknA and PknL, because these kinases do not efficiently autophosphorylate. Likewise, PknH co-localizes with its substrates. The consistency between the experimentally defined biochemical network and the subcellular distribution of the STPKs indicates that the cognate kinase-substrate pairs are present at the same site. Therefore, these efficient phosphorylation events have the opportunity to occur in vivo.
The provisional STPK signaling network shows three striking features. First, autophosphorylation is the dominant reaction. Seven of the STPKs (excluding PknA, PknG, PknI, and PknL) efficiently phosphorylate otherwise identical inactive KD constructs. Autophosphorylation is a mechanism of efficient signal amplification. A single activated STPK can rapidly phosphorylate identical molecules (26, 28).
Second, each STPK phosphorylates a different subset of the provisional kinase network (Fig. 2D). These data suggest that different signals produce distinct phosphorylation states of the kinome, leading to the activation or potentiation of different response pathways in vivo. The coordinated phosphorylation of multiple STPKs allows an integrated response to external signals. This multilevel response may complicate substrate identification as downstream proteins can be phosphorylated in vivo not in response to direct activation of the immediate upstream kinase, but instead through an alternative STPK-STPK activation reaction driven by the integration of multiple signals.
Third, the M. tuberculosis STPKs fall into three distinct functional classes based on the pattern of in vitro phosphorylation as follows: master regulator kinases, signal transduction kinases, and substrate kinases (Fig. 8). Two kinases, PknB and PknH, exclusively undergo autophosphorylation. This requirement for an autoactivating signal suggests that PknB and PknH occupy the role of master regulators. As expected for kinases that sit atop the signaling hierarchy, both STPKs contain folded extracellular sensor domains (ECDs). Importantly, other kinases with folded ECDs, PknD, PknE, and PknJ, are targets of cross-phosphorylation, indicating that domain architecture alone is insufficient to assign network position. By activating additional STPKs, the signals that control the master regulators can coordinate more complex cellular processes than signals that activate downstream kinases.
FIGURE 8.
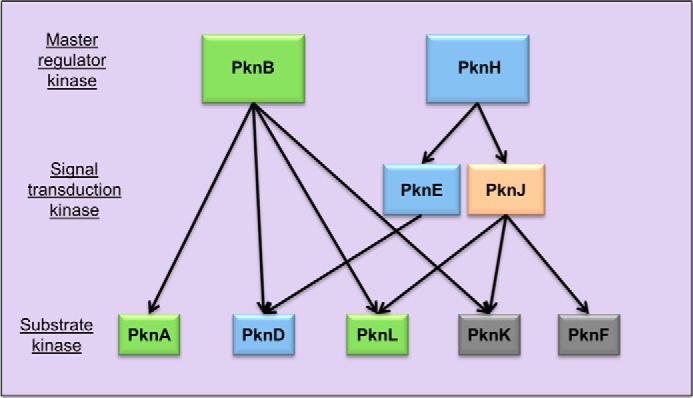
Provisional M. tuberculosis STPK interaction network. Arrows indicate intermolecular phosphorylation in vitro. STPKs with polar and midplane localization are depicted in green. STPKs with membrane and midplane localization are colored blue. PknJ, localizing only transiently to the septa, is colored orange. PknF and PknK, depicted in gray, were characterized biochemically, but expression of the mVenus fusions in M. smegmatis was not detected. The master STPKs, PknB and PknH, only undergo autophosphorylation and contain folded extracellular domains that receive environmental signals. In contrast, PknA and PknL not only fail to autophosphorylate, they also lack folded extracellular receptor domains. However, PknB efficiently phosphorylates the PknL and PknA KDs, and these three kinases co-localize in vivo. PknH localizes along the cell membrane and also initiates a cascade of phosphorylation of other STPKs, PknE and PknD, that also reside along the cell membrane. PknD contains a folded extracellular domain, but phosphorylation by other kinases suggests that it may be activated by other kinases as well as external signals that trigger autophosphorylation.
The second tier of the provisional STPK hierarchy consists of signal transduction kinases that undergo auto- and cross-phosphorylation and also phosphorylate downstream kinases. PknH cross-phosphorylates PknE and PknJ, which in turn phosphorylate PknD, PknF, PknK, and PknL. This pattern of cross-phosphorylation may allow signals to propagate through specific STPKs that regulate distinct sets of downstream cellular proteins. These interactions could function as part of regulatory circuits that spread extracellular signals to intracellular substrates of multiple kinases.
The base of the hierarchy is occupied by the substrate STPKs PknA, PknD, PknF, and PknL. These kinases do not transfer phosphates to any other STPK catalytic domains. Notably, PknA and PknF contain extracellular domains that are predicted to be unfolded, and PknL lacks an ECD altogether. Thus, these kinases apparently lack the machinery to respond directly to an extracellular signal, and the master kinases spread physiological adaptations through cross-phosphorylation of these STPKs (Fig. 8). In contrast, PknD, which contains a β-propeller ECD, not only senses osmolarity but also is positioned in the network to regulate the stressosome in response to upstream STPKs (17, 22, 49).
The pattern of intermolecular in vitro STPK phosphorylation and cellular localization suggests new hypotheses for the spread of signals through the M. tuberculosis kinome. It must be stressed that other factors, such as molecular scaffolding and co-expression, may influence the cross-phosphorylation patterns in vivo. Moreover, the constructs used for in vitro biochemistry lacked possible regulatory sites in the juxtamembrane segments (24, 38), potentially masking additional functional relationships. In addition, the network architecture may be modified by the temporally restricted activity of the STPKs mediated, in part by localization or regulation of the antagonistic phosphatase (50). Nonetheless, the exquisite biochemical specificity of each KD and the distinct cellular compartmentalization that coincides with the phosphorylation of signaling partners imposes a fundamental structure on the M. tuberculosis STPK network onto which these additional complexities will be layered. The distinct architecture of HK and STPK cascades that occur in this single organism indicate that these systems integrate signals and outputs through different mechanisms. Linear HK pathways rely on downstream transcriptional changes to achieve a coordinated response, whereas the hierarchical structure of the STPK network may allow the integration of multiple signals into distinct phosphorylation patterns that post-translationally coordinate the activities of disparate cellular enzymes and transcriptional regulators.
This work was supported, in whole or in part, by National Institutes of Health Grants AI095208 (to C. S.) and GM070962 (to T. A.).
This article was selected as a Paper of the Week.
- HK
- histidine kinase
- STPK
- Ser/Thr protein kinase
- ICD
- intracellular domain
- KD
- kinase domain
- MyBP
- myelin basic protein
- ECD
- extracellular sensor domain
- MBP
- maltose-binding protein.
REFERENCES
- 1. Wehenkel A., Bellinzoni M., Graña M., Duran R., Villarino A., Fernandez P., Andre-Leroux G., England P., Takiff H., Cerveñansky C., Cole S. T., Alzari P. M. (2008) Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Biochim. Biophys. Acta 1784, 193–202 [DOI] [PubMed] [Google Scholar]
- 2. Skerker J. M., Prasol M. S., Perchuk B. S., Biondi E. G., Laub M. T. (2005) Two-component signal transduction pathways regulating growth and cell cycle progression in a bacterium: a system-level analysis. PLoS Biol. 3, e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chang L., Karin M. (2001) Mammalian MAP kinase signalling cascades. Nature 410, 37–40 [DOI] [PubMed] [Google Scholar]
- 4. Cole S. T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S. V., Eiglmeier K., Gas S., Barry C. E., 3rd, Tekaia F., Badcock K., Basham D., Brown D., Chillingworth T., Connor R., Davies R., Devlin K., Feltwell T., Gentles S., Hamlin N., Holroyd S., Hornsby T., Jagels K., Krogh A., McLean J., Moule S., Murphy L., Oliver K., Osborne J., Quail M. A., Rajandream M. A., Rogers J., Rutter S., Seeger K., Skelton J., Squares R., Squares S., Sulston J. E., Taylor K., Whitehead S., Barrell B. G. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393, 537–544 [DOI] [PubMed] [Google Scholar]
- 5. Fernandez P., Saint-Joanis B., Barilone N., Jackson M., Gicquel B., Cole S. T., Alzari P. M. (2006) The Ser/Thr protein kinase PknB is essential for sustaining mycobacterial growth. J. Bacteriol. 188, 7778–7784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sassetti C. M., Rubin E. J. (2003) Genetic requirements for mycobacterial survival during infection. Proc. Natl. Acad. Sci. U.S.A. 100, 12989–12994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rachman H., Strong M., Ulrichs T., Grode L., Schuchhardt J., Mollenkopf H., Kosmiadi G. A., Eisenberg D., Kaufmann S. H. (2006) Unique transcriptome signature of Mycobacterium tuberculosis in pulmonary tuberculosis. Infect. Immun. 74, 1233–1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bell C., Smith G. T., Sweredoski M. J., Hess S. (2012) Characterization of the Mycobacterium tuberculosis proteome by liquid chromatography mass spectrometry-based proteomics techniques: a comprehensive resource for tuberculosis research. J. Proteome Res. 11, 119–130 [DOI] [PubMed] [Google Scholar]
- 9. Kruh N. A., Troudt J., Izzo A., Prenni J., Dobos K. M. (2010) Portrait of a pathogen: the Mycobacterium tuberculosis proteome in vivo. PLoS One 5, e13938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Molle V., Kremer L. (2010) Division and cell envelope regulation by Ser/Thr phosphorylation: Mycobacterium shows the way. Mol. Microbiol. 75, 1064–1077 [DOI] [PubMed] [Google Scholar]
- 11. Barthe P., Mukamolova G. V., Roumestand C., Cohen-Gonsaud M. (2010) The structure of PknB extracellular PASTA domain from Mycobacterium tuberculosis suggests a ligand-dependent kinase activation. Structure 18, 606–615 [DOI] [PubMed] [Google Scholar]
- 12. Dasgupta A., Datta P., Kundu M., Basu J. (2006) The serine/threonine kinase PknB of Mycobacterium tuberculosis phosphorylates PBPA, a penicillin-binding protein required for cell division. Microbiology 152, 493–504 [DOI] [PubMed] [Google Scholar]
- 13. Gee C. L., Papavinasasundaram K. G., Blair S. R., Baer C. E., Falick A. M., King D. S., Griffin J. E., Venghatakrishnan H., Zukauskas A., Wei J. R., Dhiman R. K., Crick D. C., Rubin E. J., Sassetti C. M., Alber T. (2012) A phosphorylated pseudokinase complex controls cell wall synthesis in mycobacteria. Sci. Signal. 5, ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jones G., Dyson P. (2006) Evolution of transmembrane protein kinases implicated in coordinating remodeling of Gram-positive peptidoglycan: inside versus outside. J. Bacteriol. 188, 7470–7476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kang C. M., Abbott D. W., Park S. T., Dascher C. C., Cantley L. C., Husson R. N. (2005) The Mycobacterium tuberculosis serine/threonine kinases PknA and PknB: substrate identification and regulation of cell shape. Genes Dev. 19, 1692–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kang C. M., Nyayapathy S., Lee J. Y., Suh J. W., Husson R. N. (2008) Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology 154, 725–735 [DOI] [PubMed] [Google Scholar]
- 17. Greenstein A. E., MacGurn J. A., Baer C. E., Falick A. M., Cox J. S., Alber T. (2007) M. tuberculosis Ser/Thr protein kinase D phosphorylates an anti-anti-σ factor homolog. PLoS Pathog. 3, e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pereira S. F., Goss L., Dworkin J. (2011) Eukaryote-like serine/threonine kinases and phosphatases in bacteria. Microbiol. Mol. Biol. Rev. 75, 192–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. England P., Wehenkel A., Martins S., Hoos S., André-Leroux G., Villarino A., Alzari P. M. (2009) The FHA-containing protein GarA acts as a phosphorylation-dependent molecular switch in mycobacterial signaling. FEBS Lett. 583, 301–307 [DOI] [PubMed] [Google Scholar]
- 20. O'Hare H. M., Durán R., Cerveñansky C., Bellinzoni M., Wehenkel A. M., Pritsch O., Obal G., Baumgartner J., Vialaret J., Johnsson K., Alzari P. M. (2008) Regulation of glutamate metabolism by protein kinases in mycobacteria. Mol. Microbiol. 70, 1408–1423 [DOI] [PubMed] [Google Scholar]
- 21. Schultz C., Niebisch A., Schwaiger A., Viets U., Metzger S., Bramkamp M., Bott M. (2009) Genetic and biochemical analysis of the serine/threonine protein kinases PknA, PknB, PknG and PknL of Corynebacterium glutamicum: evidence for non-essentiality and for phosphorylation of OdhI and FtsZ by multiple kinases. Mol. Microbiol. 74, 724–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hatzios S. K., Baer C. E., Rustad T. R., Siegrist M. S., Pang J. M., Ortega C., Alber T., Grundner C., Sherman D. R., Bertozzi C. R. (2013) An osmosensory signaling pathway in Mycobacterium tuberculosis mediated by a eukaryotic-like Ser/Thr protein kinase. Proc. Natl. Acad. Sci. U.S.A. 110, E5069–E5077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boitel B., Ortiz-Lombardía M., Durán R., Pompeo F., Cole S. T., Cerveñansky C., Alzari P. M. (2003) PknB kinase activity is regulated by phosphorylation in two Thr residues and dephosphorylation by PstP, the cognate phospho-Ser/Thr phosphatase, in Mycobacterium tuberculosis. Mol. Microbiol. 49, 1493–1508 [DOI] [PubMed] [Google Scholar]
- 24. Molle V., Zanella-Cleon I., Robin J. P., Mallejac S., Cozzone A. J., Becchi M. (2006) Characterization of the phosphorylation sites of Mycobacterium tuberculosis serine/threonine protein kinases, PknA, PknD, PknE, and PknH by mass spectrometry. Proteomics 6, 3754–3766 [DOI] [PubMed] [Google Scholar]
- 25. Molle V., Girard-Blanc C., Kremer L., Doublet P., Cozzone A. J., Prost J. F. (2003) Protein PknE, a novel transmembrane eukaryotic-like serine/threonine kinase from Mycobacterium tuberculosis. Biochem. Biophys. Res. Commun. 308, 820–825 [DOI] [PubMed] [Google Scholar]
- 26. Greenstein A. E., Echols N., Lombana T. N., King D. S., Alber T. (2007) Allosteric activation by dimerization of the PknD receptor Ser/Thr protein kinase from Mycobacterium tuberculosis. J. Biol. Chem. 282, 11427–11435 [DOI] [PubMed] [Google Scholar]
- 27. Lombana T. N., Echols N., Good M. C., Thomsen N. D., Ng H. L., Greenstein A. E., Falick A. M., King D. S., Alber T. (2010) Allosteric activation mechanism of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknB. Structure 18, 1667–1677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mieczkowski C., Iavarone A. T., Alber T. (2008) Auto-activation mechanism of the Mycobacterium tuberculosis PknB receptor Ser/Thr kinase. EMBO J. 27, 3186–3197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Papavinasasundaram K. G., Chan B., Chung J. H., Colston M. J., Davis E. O., Av-Gay Y. (2005) Deletion of the Mycobacterium tuberculosis pknH gene confers a higher bacillary load during the chronic phase of infection in BALB/c mice. J. Bacteriol. 187, 5751–5760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cowley S., Ko M., Pick N., Chow R., Downing K. J., Gordhan B. G., Betts J. C., Mizrahi V., Smith D. A., Stokes R. W., Av-Gay Y. (2004) The Mycobacterium tuberculosis protein serine/threonine kinase PknG is linked to cellular glutamate/glutamine levels and is important for growth in vivo. Mol. Microbiol. 52, 1691–1702 [DOI] [PubMed] [Google Scholar]
- 31. Scherr N., Honnappa S., Kunz G., Mueller P., Jayachandran R., Winkler F., Pieters J., Steinmetz M. O. (2007) Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 104, 12151–12156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mir M., Asong J., Li X., Cardot J., Boons G. J., Husson R. N. (2011) The extracytoplasmic domain of the Mycobacterium tuberculosis Ser/Thr kinase PknB binds specific muropeptides and is required for PknB localization. PLoS Pathog. 7, e1002182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nariya H., Inouye S. (2005) Identification of a protein Ser/Thr kinase cascade that regulates essential transcriptional activators in Myxococcus xanthus development. Mol. Microbiol. 58, 367–379 [DOI] [PubMed] [Google Scholar]
- 34. Canman C. E., Lim D. S., Cimprich K. A., Taya Y., Tamai K., Sakaguchi K., Appella E., Kastan M. B., Siliciano J. D. (1998) Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 281, 1677–1679 [DOI] [PubMed] [Google Scholar]
- 35. Durán R., Villarino A., Bellinzoni M., Wehenkel A., Fernandez P., Boitel B., Cole S. T., Alzari P. M., Cerveñansky C. (2005) Conserved autophosphorylation pattern in activation loops and juxtamembrane regions of Mycobacterium tuberculosis Ser/Thr protein kinases. Biochem. Biophys. Res. Commun. 333, 858–867 [DOI] [PubMed] [Google Scholar]
- 36. Chao J., Wong D., Zheng X., Poirier V., Bach H., Hmama Z., Av-Gay Y. (2010) Protein kinase and phosphatase signaling in Mycobacterium tuberculosis physiology and pathogenesis. Biochim. Biophys. Acta 1804, 620–627 [DOI] [PubMed] [Google Scholar]
- 37. Prisic S., Dankwa S., Schwartz D., Chou M. F., Locasale J. W., Kang C. M., Bemis G., Church G. M., Steen H., Husson R. N. (2010) Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thakur M., Chaba R., Mondal A. K., Chakraborti P. K. (2008) Interdomain interaction reconstitutes the functionality of PknA, a eukaryotic type Ser/Thr kinase from Mycobacterium tuberculosis. J. Biol. Chem. 283, 8023–8033 [DOI] [PubMed] [Google Scholar]
- 39. Roumestand C., Leiba J., Galophe N., Margeat E., Padilla A., Bessin Y., Barthe P., Molle V., Cohen-Gonsaud M. (2011) Structural insight into the Mycobacterium tuberculosis Rv0020c protein and its interaction with the PknB kinase. Structure 19, 1525–1534 [DOI] [PubMed] [Google Scholar]
- 40. Av-Gay Y., Everett M. (2000) The eukaryotic-like Ser/Thr protein kinases of Mycobacterium tuberculosis. Trends Microbiol. 8, 238–244 [DOI] [PubMed] [Google Scholar]
- 41. Rost B., Yachdav G., Liu J. (2004) The PredictProtein Server. Nucleic Acids Res. 32, W321–W326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kelley L. A., Sternberg M. J. (2009) Protein structure prediction on the web: a case study using the Phyre server. Nat. Protoc. 4, 363–371 [DOI] [PubMed] [Google Scholar]
- 43. Young T. A., Delagoutte B., Endrizzi J. A., Falick A. M., Alber T. (2003) Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases. Nat. Struct. Biol. 10, 168–174 [DOI] [PubMed] [Google Scholar]
- 44. Sharma K., Chandra H., Gupta P. K., Pathak M., Narayan A., Meena L. S., D'Souza R. C., Chopra P., Ramachandran S., Singh Y. (2004) PknH, a transmembrane Hank's type serine/threonine kinase from Mycobacterium tuberculosis, is differentially expressed under stress conditions. FEMS Microbiol. Lett. 233, 107–113 [DOI] [PubMed] [Google Scholar]
- 45. Koul A., Choidas A., Tyagi A. K., Drlica K., Singh Y., Ullrich A. (2001) Serine/threonine protein kinases PknF and PknG of Mycobacterium tuberculosis: characterization and localization. Microbiology 147, 2307–2314 [DOI] [PubMed] [Google Scholar]
- 46. Narayan A., Sachdeva P., Sharma K., Saini A. K., Tyagi A. K., Singh Y. (2007) Serine threonine protein kinases of mycobacterial genus: phylogeny to function. Physiol. Genomics 29, 66–75 [DOI] [PubMed] [Google Scholar]
- 47. Chenna R., Sugawara H., Koike T., Lopez R., Gibson T. J., Higgins D. G., Thompson J. D. (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 31, 3497–3500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muñoz-Dorado J., Inouye S., Inouye M. (1991) A gene encoding a protein serine/threonine kinase is required for normal development of M. xanthus, a Gram-negative bacterium. Cell 67, 995–1006 [DOI] [PubMed] [Google Scholar]
- 49. Good M. C., Greenstein A. E., Young T. A., Ng H. L., Alber T. (2004) Sensor domain of the Mycobacterium tuberculosis receptor Ser/Thr protein kinase, PknD, forms a highly symmetric β propeller. J. Mol. Biol. 339, 459–469 [DOI] [PubMed] [Google Scholar]
- 50. Sajid A., Arora G., Gupta M., Upadhyay S., Nandicoori V. K., Singh Y. (2011) Phosphorylation of Mycobacterium tuberculosis Ser/Thr phosphatase by PknA and PknB. PLoS One 6, e17871. [DOI] [PMC free article] [PubMed] [Google Scholar]