

Background: Toxin-antitoxin complexes autoregulate transcription depending upon growth conditions.
Results: DinJ-YafQ structure was determined, and minimal requirements for transcriptional autorepression were identified.
Conclusion: The dinJyafQ operon is regulated in a novel manner by either DinJ-YafQ- or LexA-mediated repression.
Significance: Our results reveal new mechanistic insights into the action of DinJ-YafQ as a transcriptional repressor.
Keywords: Bacterial Toxin, Energy Metabolism, Stress Response, Transcription Repressor, X-ray Crystallography, Ribbon-Helix-Helix Factor, Toxin-Antitoxin Complexes
Abstract
Bacteria encounter environmental stresses that regulate a gene expression program required for adaptation and survival. Here, we report the 1.8-Å crystal structure of the Escherichia coli toxin-antitoxin complex YafQ-(DinJ)2-YafQ, a key component of the stress response. The antitoxin DinJ dimer adopts a ribbon-helix-helix motif required for transcriptional autorepression, and toxin YafQ contains a microbial RNase fold whose proposed active site is concealed by DinJ binding. Contrary to previous reports, our studies indicate that equivalent levels of transcriptional repression occur by direct interaction of either YafQ-(DinJ)2-YafQ or a DinJ dimer at a single inverted repeat of its recognition sequence that overlaps with the −10 promoter region. Surprisingly, multiple YafQ-(DinJ)2-YafQ complexes binding to the operator region do not appear to amplify the extent of repression. Our results suggest an alternative model for transcriptional autorepression that may be novel to DinJ-YafQ.
Introduction
The ability of bacteria to quickly regulate metabolic processes in response to stress gives them an inherent advantage for survival. Such adaptive responses include the formation of biofilms and persister cells (1–3). Persisters are cells with noninherited epigenetic changes that exhibit antibiotic tolerance due to their metabolically dormant or slow growth state (3). Genes up-regulated in the persister state include those encoding toxin proteins from toxin-antitoxin (TA)3 operons (1). At least 19 type II TA systems have been identified in Escherichia coli and a putative 88 in Mycobacterium tuberculosis (4, 5). This expansion of TA systems in pathogens has been suggested to lead to increases in persistence, latency, and pathogenesis (3, 6).
Type II TA complexes are small protein-protein pairs that function as transcriptional repressors of their own and, in some cases, other genes under normal growth conditions (7–13). Antitoxin binding to the toxin also inhibits the activity of the toxin (8, 14–17). Upon activation of the stress response, cellular proteases such as Lon, ClpXP, or ClpAP selectively degrade antitoxin proteins (18–21). This allows the toxin to target important cellular processes such as DNA replication or protein translation, enabling cells to enter a metabolically dormant state until the stress is removed (21–23). Because TA systems are bacteriostatic and beneficial to bacteria, once the stress has passed and normal growth has resumed, toxin-antitoxin complexes return to their canonical function of transcriptional repression (24). The dual functions that TA pairs play as both transcriptional repressors and growth suppressors allow for an efficient switch regulated by the metabolic requirements under different growth environments (22).
The expression level of toxin and antitoxin proteins appears to fluctuate as a function of the growth state of the cell (25). During normal growth when the TA complex functions as a transcriptional autorepressor, little to no free toxin is present. Cellular stress leads to an increase in toxin protein accumulation, most likely from antitoxin degradation. Specific toxin proteins such as RelE, Doc, and CcdA have been proposed to function as either transcriptional co-repressors, as part of the TA complex, or as de-repressors, which are dependent upon changes in the toxin-antitoxin ratios (25–29). Transcriptional de-repression is thought to occur by a mechanism in which excess toxin interacts with and destabilizes the TA-DNA repressor complex (25, 26, 29). This conditional role of the toxin has now been observed for a number of TA systems such as CcdAB, RelBE, and PhD-Doc, but it is unclear whether this model accurately describes de-repression at all TA loci.
The E. coli DinJ-YafQ complex represents a novel TA system because of its potentially unique mechanism of transcriptional regulation. The dinJ promoter contains two palindromic operator regions, each containing two imperfect inverted repeats that overlap with the −35 and −10 promoter regions (30). However, in vitro binding studies show that the DinJ-YafQ complex binds to the first operator but not the second operator, although the SOS-activated, transcriptional repressor LexA binds to the second operator that also contains an overlapping LexA consensus sequence (30, 31). The possible dual regulation by the DNA damage response and stringent response represents a potentially unique repression mechanism that may constitute overlapping responses to stress. The molecular understanding of this repression and how it relates to other TA systems are currently unknown.
Despite extensive research aimed at understanding the molecular basis of transcriptional regulation by TA systems, a number of fundamental questions remain. Here, we describe the 1.8-Å crystal structure of the E. coli DinJ-YafQ complex. The toxin YafQ is a member of the RelE/YoeB superfamily of endonucleases (22, 32) and degrades mRNA at adenosine-rich codons, most likely while bound to the ribosome (31). The activity of YafQ is neutralized upon direct binding and active site occlusion by its cognate antitoxin, DinJ, a transcription factor of the ribbon-helix-helix (RHH) family. Additionally, the binding of a single DinJ-YafQ complex is sufficient for transcriptional repression, although attenuation of repression is not promoted by the binding of an additional TA complex.
EXPERIMENTAL PROCEDURES
Bacterial Strains and Plasmids
Strains and plasmids used in this study are listed in supplemental Table S1.
DinJ-YafQ Purification and Crystallization
E. coli BL21 Gold (DE3) pLysS cells transformed with pET-DinJ-(His6)YafQ were grown in Lysogeny Broth (LB) medium at 37 °C and induced with 0.5 mm isopropyl 1-thio-β-d-galactopyranoside at mid-log phase. After 3 h of induction, cells were harvested by centrifugation at 4,500 × g for 30 min at 4 °C. Cell pellets were resuspended in lysis buffer (20 mm Tris-HCl, pH 7.5, 250 mm KCl, 5 mm β-mercaptoethanol, 0.1% (v/v) Triton X-100, 0.1 mm benzamidine, and 0.1 mm phenylmethanesulfonylfluoride (PMSF) and lysed by sonication. The lysate was clarified by centrifugation at 40,000 × g for 30 min at 4 °C and filtered through a 0.45-μm membrane filter.
The cleared lysate was immediately loaded onto a 5-ml HisTrap FF CrudeTM nickel-Sepharose column (GE Healthcare) equilibrated with column buffer (40 mm Tris-HCl, pH 7.5, 10% glycerol, 250 mm KCl, 5 mm MgCl2, 5 mm β-mercaptoethanol, 5 mm imidazole), and the DinJ-YafQ complex was eluted using a linear gradient of 5–500 mm imidazole. The complex was further purified on a Superdex 200 16/60 column (GE Healthcare) equilibrated with column buffer without glycerol and imidazole. Fractions containing the complex were pooled, concentrated to 25 mg/ml, flash-frozen in liquid nitrogen, and stored at −80 °C. The final purified DinJ-YafQ was >95% homogeneous as determined by SDS-PAGE.
To generate the selenomethionine derivative of DinJ-YafQ, standard techniques for nonauxotrophic E. coli were used (33, 34). Selenomethionine-derived DinJ-YafQ was purified following the same protocol used for native DinJ-YafQ. Crystals of selenomethionine-derived DinJ-YafQ were obtained using sitting drop vapor diffusion at 4 °C with the mother liquor containing 1.9–2.3 m (NH4)2SO4. Crystals were cryoprotected by gradually exchanging the mother liquor with a cryoprotectant solution containing 20% glycerol in addition to the mother liquor and flash-frozen in liquid nitrogen.
DinJ-YafQ Structure Determination
A single anomalous dispersion dataset was collected at the Northeastern Collaborative Access Team 24-IDC facility at the Advanced Photon Source at the Argonne National Laboratory (Argonne, IL). The dataset was collected under cryogenic conditions (100 K) using 0.979 Å radiation.
The selenomethionine dataset was indexed, integrated, and scaled with HKL2000 to a maximum of 1.76 Å (35). Phase determination was performed by single anomalous dispersion, and heavy atom sites were identified by SHELX using HKL2MAP (36, 37). A total of 24 heavy atom sites were identified (22 from SHELX and 2 more from PHENIX AutoSol). Initial phasing and model building were carried out using AutoSol and AutoBuild of the PHENIX software suite (38). Multiple iterative rounds of manual model building and refinement, including energy minimization and B-factor refinement with NCS, were performed in Coot (39) and PHENIX, respectively. The first two residues of both DinJ and YafQ showed poor electron density; therefore, these residues were omitted from the final model. Water and ion molecules were added manually in Coot. The final model quality was determined using Molprobity of the PHENIX validation tools (40). Complete data collection and refinement statistics are shown in Table 1. All figures were generated using PyMOL (41).
TABLE 1.
Data collection and refinement statistics
Values in parentheses are for the highest resolution shell.
| Data collection statistics | |
| Space group | C2 |
| Unit cell dimension | |
| a, b, and c | 176.54, 120.92, 120.83 Å |
| α, β, and γ | 90.0, 130.8, 90.0° |
| Source | 24IDC |
| Wavelength | 0.979 Å |
| Resolution range | 43.91–1.76 Å (1.82–1.76 Å) |
| Total reflections observed | 501,571 |
| Unique reflections | 176,184 (17,512) |
| Completeness | 99.36% (99.11%) |
| Redundancy | 1.4 (1.3) |
| I/σ(I) | 5.01 (1.88) |
| Rmerge | 0.092 (0.614) |
| Refinement statistics | |
| Resolution range | 43.91 to 1.80 Å (1.86 to 1.80 Å) |
| Rworka | 18.29% (31.57%) |
| Rfree | 21.50% (35.98%) |
| Total no. of non-hydrogen atoms | 12,659 |
| Protein | 11,256 |
| Ions | 245 |
| Water | 1142 |
| Total no. of protein residues | 1390 |
| r.m.s.d. | |
| Bond length | 0.015 Å |
| Bond angle | 1.42° |
| Wilson B factors | 20.38 Å2 |
| Average B factors | 26.00 Å2 |
| Protein atoms | 25.70 |
| Solvent atoms | 27.70 |
a R factor calculated for all reflections (working + test set) is 18.32%.
β-Galactosidase Activity Assays
The dinJ promoter region spanning −259 to +50 was PCR-amplified from E. coli DH5α genomic DNA and cloned before the lacZ gene in the pQF50 plasmid (pQF50-PdinJ-lacZ; supplemental Table S1) (42). N-terminal DinJ truncation mutations at residues 12 and 44 were designed based upon our crystal structure, and DinJ R10A and R35A were designed based upon homology modeling. The inverted repeats of the PdinJ operator 1 and 2 were mutated to scramble the DinJ-YafQ recognition sequences in the pQF50-PdinJ-lacZ plasmid using site-directed mutagenesis (supplemental Table S1). For the first operator, the inverted repeat 1 (IR1) sequence of 5′-TGTTGCTCA-3′ was changed to 5′-AAGGTCTCA-3′ (mutated residues in boldface type). The second half of the operator, inverted repeat 1′ (IR1′) was mutated from 5′-TGAGCTACA-3′ to 5′-ACGGTTACA-3′. For the second operator, inverted repeat 2 (IR2) was changed from 5′-GCTGAATA-3′ to 5′-TACTGGTA-3′, and inverted repeat 2′ (IR2′) was mutated from 5′-TATACAGC-3′ to 5′-CGCGAGGC-3′. In all cases, E. coli BW25113 cells were co-transformed with a pBAD33 plasmid containing wild-type DinJ-YafQ, DinJ-YafQ mutants, DinJ alone, or LexA along with wild-type or mutated pQF50-PdinJ-lacZ. Cells were grown overnight at 37 °C in M9 minimal medium supplemented with 0.2% glucose. Fifty microliters of each overnight culture was used to inoculate 5 ml of fresh M9 medium supplemented with 0.2% glycerol. Protein expression was induced with 0.2% arabinose at an A600 of 0.2, and cells were grown for 4 h before harvesting by centrifugation at 3,000 × g for 20 min at 4 °C. Quantitative β-galactosidase activity assays were performed in triplicate using the Miller method (43). Uninduced DinJ-YafQ samples were used to monitor dinJ promoter activity, and background β-galactosidase activity was determined using BW25113 carrying the original promoter-less pQF50 plasmid.
Size Exclusion Chromatography
A Superdex S75 10/300 GL column (GE Healthcare) equilibrated with column buffer without glycerol and imidazole was used to determine the size and oligomeric state of wild-type DinJ-YafQ and DinJ-YafQ mutants (100 μm) in solution. Molecular weights were estimated by comparison of the elution volume with protein molecular weight standards (Bio-Rad).
Modeling of the YafQ-(DinJ)2-YafQ Tetramer to the dinJ Operator
To identify which DinJ residues interact with its DNA operator, the (RHH)2 motif of the DinJ-DinJ′ dimer was structurally aligned to the (RHH)2 motifs of FitA-DNA (PDB code 2BSQ) (44) and PutA-DNA complexes (PDB code 2RBF) (45) using the pairwise structural alignment program DaliLite (46).
To predict how two YafQ-(DinJ)2-YafQ complexes recognize two adjacent inverted repeats (IR1 and IR1′) of the dinJ operator, we used the structure of DNA-Arc repressor complex (PDB code 1PAR) (47) as a template because this structure contains two Arc dimers bound to its operator. First, we used DaliLite to align YafQ-(DinJ)2-YafQ to each of the two Arc dimers. Because the arc operator contains 11 bp between the two inverted repeats as compared with 10 in the dinJ promoter, we translated one DinJ-YafQ tetramer 1 bp closer to the other tetramer to more accurately reflect the correct spacing using least squares fitting in Coot (39).
Electrophoretic Mobility Shift Assay (EMSA)
The wild-type and mutated PdinJ regions were PCR-amplified from the pQF50 plasmid used in the β-galactosidase assays and agarose gel-purified for use in binding assays. DNA (200 pg/μl final concentration) was incubated at 37 °C for 20 min with increasing concentrations of purified DinJ-YafQ complex or purified DinJ alone (0–1.8 μm) in a buffer containing 50 mm Tris-HCl, pH 8.0, 50 mm KCl, 5 mm MgCl2, 1 mm EDTA, 10 μg/ml bovine serum albumin (BSA), 1 mm dithiothreitol (DTT), and 2.5% (v/v) glycerol. The wild-type and mutated PdinJ promoter regions were incubated with 3.6 μm purified DinJ-YafQ complex in the presence of 0, 1.6, or 3.6 μm purified YafQ following the same protocol. The samples were resolved by electrophoresis on a 6% polyacrylamide native gel run in 0.5× TBE buffer. Gels were run at 180 V for 40 min at 4 °C and then stained with 1× SYBRTM Green I in 0.5× TBE buffer. The DNA bands on the gel were visualized by fluorescence scanning using a Typhoon Trio (GE Healthcare) at 488 nm excitation and 520 nm emission settings.
RESULTS
Structure Determination
Crystals of selenomethionine-DinJ-YafQ were obtained, and the structure was solved at a resolution of 1.8 Å by selenium single-wavelength anomalous dispersion (Table 1). Crystals formed in the space group C2 with eight structurally identical DinJ-YafQ dimers in the asymmetric unit. A DinJ-YafQ tetramer (hereafter denoted as YafQ-(DinJ)2-YafQ to describe its modular domain structure) consists of two DinJ-YafQ heterodimers associated by the N-terminal dimerization of two DinJ antitoxins (Fig. 1, A and B). The final model contains 83 or 84 (4–86 or 3–86) of the 86 residues of DinJ and 90 or 91 (3–92 or 2–92) residues of the 92 residues of YafQ.
FIGURE 1.

Overall structure of the DinJ-YafQ complex. A, ribbon representation of the YafQ-(DinJ)2-YafQ tetramer with two YafQs depicted in light blue and navy, and two DinJs are in green and magenta. The boxed area is where DinJ L3 interacts with the proposed YafQ active site. Secondary structure elements and end termini are labeled. Colors are preserved throughout all figures. B, perpendicular view of A showing the (RHH)2 motif formed by the two N termini of DinJ. C, close-up view of the proposed catalytic site of YafQ that forms interactions with L3 of DinJ. Residues comprising this interaction are shown as sticks with a sulfate ion shown in magenta. D, hydrogen bond network between the C terminus of DinJ (α3, β2, and α4) and α1 and α3 of YafQ. Interacting residues are shown as sticks. E, anti-parallel β-strand interactions between DinJ β2 and YafQ β1. F, hydrophobic interface between α4 of DinJ and α1 and α2 of YafQ. DinJ residues involved are shown as sticks in magenta, and the YafQ hydrophobic surface is shown in white.
YafQ Adopts a Tertiary Structure Topologically Resembling Other Ribosome-dependent RNases of the RelE/YoeB Family
YafQ is a small globular protein (10.8 kDa) consisting of a four-stranded antiparallel β-sheet (β2↑β3↓β4↑β1↑) flanked by two α-helices (α1 and α2) on one side and a short α-helix (α3) nearly 180° on the opposite surface (Fig. 1A). Despite exhibiting low sequence identity with other ribosome-dependent RNases (3–13%), YafQ adopts a microbial RNase fold, specifically the RelE/YoeB toxin family tertiary fold that is characterized by a compact four- or five-stranded antiparallel β-sheet with at least one adjacent α-helix (48, 49). Three-dimensional homology searches using the Dali server revealed that YafQ is most similar to E. coli RelE (PDB code 4FXI, Z-score = 9.5, r.m.s.d. = 2.2 Å, 4.4% sequence identity) (50), YoeB (PDB code 2A6S, Z-score = 11.8, r.m.s.d. = 2.1 Å, 13.1% sequence identity) (51), and MqsR (PDB code 3HI2, Z-score = 5.1, r.m.s.d. = 2.7 Å, 3.3% sequence identity) (52–54).
Type II toxins that target RNA for degradation typically contain a distinctive concave surface where the RNA substrate is thought to be recognized. Our structure of YafQ indicates it adopts a similar tertiary structure encompassing a topologically comparable concave surface created by three β-strands (β2–4), a loop (between α2 and β2), and an unstructured C terminus. At this concave region of YafQ, residues proposed to be important for function cluster (Fig. 1C) (30, 31). Interestingly, a sulfate ion, likely from the crystallization solution, was visible in our electron density maps near highly conserved and proposed catalytic YafQ residues His-50, His-63, and His-87 (Fig. 1C). The negative sulfate ion may be bound at a similar location as the negative RNA phosphate backbone that YafQ recognizes and cleaves. There are variations in the identity of catalytic residues among toxin family members, and this may be the source for some toxins exhibiting codon dependence (50–52, 55, 56).
DinJ-DinJ′ Dimer Forms an (RHH)2 DNA-binding Motif
DinJ is a small protein (9.4 kDa) that has a distinct N-terminal DNA binding domain consisting of a single β-strand (β1) followed by two α-helices (α1 and α2) that together comprise the RHH motif (Fig. 1B). The toxin neutralization domain is located at the C terminus of DinJ and is composed of a β-strand (β2) flanked by two α-helices (α 3 and α4). These two modular domains are connected by a linker region (L3, residues 46–57) that partially covers the proposed YafQ active site (Fig. 1C). The lack of a hydrophobic core in the C-terminal toxin neutralization domain implies it probably adopts a more dynamic structure in the absence of YafQ.
Dimerization of DinJ with a second DinJ′ (where ′ denotes partner) forms a canonical DNA-binding motif that is hypothesized to regulate transcription as in the case of other antitoxins (Fig. 1B) (57). The two DinJ monomers are related by 2-fold symmetry and are held together via an extensive hydrogen bonding network between β1 and β1′ antiparallel strands and at the N terminus of DinJ (Figs. 1B and 2A). Overall, the most highly conserved DinJ residues among homologs are located within the entire RHH motif, and presumably this is for specific DNA operator recognition (Fig. 2B).
FIGURE 2.
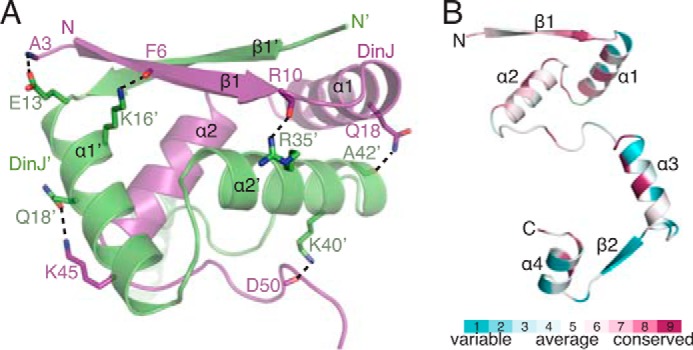
N-terminal DNA binding domain of DinJ is highly conserved. A, DinJ-DinJ′ (RHH)2 interaction is stabilized by a number of hydrogen bonds, depicted as sticks. B, one monomer of DinJ colored by amino acid conservation among DinJ homologs using the ConSurf server (68).
Three-dimensional structural searches using the Dali server reveal the RHH motif of DinJ is most similar to E. coli transcriptional repressor PutA (PDB code 2RBF, Z-score = 5.3, r.m.s.d. = 2.1 Å, 7% sequence identity) (45), E. coli Arc repressor (PDB code 1MYL, Z-score = 5.4, r.m.s.d. = 2.1 Å, 7% sequence identity) (58), Neisseria gonorrhoeae FitA antitoxin (PDB code 2BSQ, Z-score = 3.8, r.m.s.d. = 2.3 Å, 9.3% sequence identity) (44), and E. coli RelB antitoxin (PDB code 4FXE, Z-score = 5.9, r.m.s.d. = 1.1 Å, 30.2% sequence identity) (50, 53, 54).
C Terminus of DinJ Wraps around YafQ
The C terminus (α3, β2, and α4) of DinJ extends around YafQ and interacts at three distinct regions through a combination of van der Waals, hydrophobic, hydrogen bond, and salt bridge interactions (Fig. 1A). At the first interface, the linker L3 of DinJ, which connects the DinJ N-terminal DNA binding domain and the C-terminal toxin neutralization domain, packs directly against the proposed YafQ active site (Fig. 1C). YafQ His-50, a highly conserved residue among YafQ homologs and a proposed catalytic residue (30), contacts both DinJ Asp-50 and Phe-49 through salt bridge and π-stacking interactions, respectively (Fig. 1C). This suggests that toxin inactivation may occur by blocking access to the YafQ active site residue His-50 (Fig. 1C). Similar inhibitory interactions that bury the toxin active site when bound to its cognate antitoxin include YefM-YoeB and RelBE complexes (16, 50, 51). For example, the proposed general base in RelE, Lys-52, forms an interaction with Asp-55 of RelB (59). Additional interactions are seen between DinJ and YafQ at this same interface; DinJ α3 packs against the YafQ β-sheet comprising β1–2, YafQ α1, and YafQ α3, to form a number of hydrogen bond and salt bridge interactions (Fig. 1D). A second distinct YafQ-DinJ interface is the formation of an antiparallel two-stranded β-sheet between YafQ β1 and DinJ β2 (Fig. 1E). Finally, a hydrophobic interface is formed from a cluster of residues at the C-terminal α4 of DinJ (Leu-80, Phe-81, Leu-84, Gly-85, and Ile-86) that pack tightly against a YafQ hydrophobic patch formed by α1 and α3 (Ile-6, Tyr-12, Val-16, Leu-29, Leu-32, Ile-37, Leu-79, and Phe-81) (Fig. 1F). This interaction buries close to 860 Å2 of both the toxin and antitoxin surface. Both the β-sheet and the hydrophobic patch are also found in E. coli RelBE (50) (PDB code 4FXE) and M. tuberculosis RelJK (60) (PDB code 3OEI) complexes, implying a structural commonality in antitoxins that recognize RelE-like toxins.
N-terminal RHH Domain of DinJ Is Required for Transcriptional Autorepression
Our structure reveals that DinJ adopts an RHH DNA-binding motif consistent with structurally homologous antitoxins such as RelB (50). To test whether the intact N terminus of DinJ is required for transcriptional repression, we designed two N-terminal DinJ truncations based upon our structure and tested for their ability to repress lacZ transcription from the dinJ promoter (PdinJ) using β-galactosidase activity assays (Fig. 3, A and B) (43). The level of β-galactosidase activity was monitored, and the ability of the YafQ-(DinJ)2-YafQ complex to repress lacZ transcription was reported as the percentage of repression compared with the uninduced control. Our results show that PdinJ is constitutively active and is repressed by DinJ-YafQ (81% repression) (Fig. 3B). Both the DinJ(Δ1–12)-YafQ complex (lacking DinJ β1) and DinJ(Δ1–44)-YafQ complex (lacking full DinJ dimerization domain) repress lacZ expression to a much lesser extent than wild-type YafQ-(DinJ)2-YafQ (19 and 34% repression, respectively) (Fig. 3B). This indicates that the entire N-terminal DinJ domain is necessary for robust transcriptional repression. As a control, the DinJ truncations do not affect the expression level of DinJ mutants as seen by Western blot analysis (data not shown).
FIGURE 3.

Dimerization of two DinJ N termini is required for transcriptional repression. A, schematic representation of DinJ N-terminal truncations mapped onto the tertiary structure of YafQ-(DinJ)2-YafQ. B, transcriptional repression of the lacZ reporter gene by DinJ-YafQ. DinJ(Δ1–12)-YafQ, DinJ(Δ1–44)-YafQ, DinJ(R10A)-YafQ, and DinJ(R35A)-YafQ alleviate transcriptional repression. Error bars represent mean ± S.E. of the experiments performed in triplicate. C, size exclusion chromatography of wild-type DinJ-YafQ, DinJ(Δ1–12)-YafQ, and DinJ(Δ1–44)-YafQ. D, SDS-PAGE of DinJ(Δ1–12)-YafQ and DinJ(Δ1–44)-YafQ peak fractions from C indicates that the toxin and antitoxin co-elute as a complex.
To determine the oligomeric state of our DinJ truncation mutations, we subcloned each construct into pET overexpression vectors, purified each complex, and performed size exclusion chromatography (Fig. 3C). YafQ alone elutes at a volume representative of a monomer at ∼10 kDa, whereas wild-type YafQ-(DinJ)2-YafQ elutes as a tetramer at ∼56 kDa (Fig. 3C). DinJ(Δ1–44)-YafQ fails to form a tetramer and exists mainly as a dimer in solution, whereas DinJ(Δ1–12)-YafQ maintains its tetrameric state. SDS-PAGE analysis reveals that both DinJ and YafQ proteins are present in each complex (Fig. 3D).
Identification of Specific DinJ Residues Required for Transcriptional Control
To identify DinJ residues that interact with DNA, we structurally aligned the dimeric DinJ (RHH)2 motif to other (RHH)2 motif-containing repressors where a structure bound to DNA exists. The DinJ RHH motif is most structurally similar to PutA (PDB code 2RBF) (45); however, we also used the model of FitAB because it represented the most structurally homologous antitoxin protein where a structure bound to DNA also is known (PDB code 2BSQ) (44). From these structural alignments, we predict that DinJ residues Arg-10 and Arg-35 interact with DNA as these residues align well to key DNA-binding residues of both FitA and PutA (Fig. 4A). Highly conserved DinJ β1 residue Arg-10 structurally aligns to FitA Arg-7 and PutA Lys-9, both of which were found to be crucial for DNA recognition. Likewise, the position of highly conserved DinJ Arg-35 is structurally equivalent to FitA Arg-33 and PutA His-30/Lys-34, which form electrostatic interactions with the DNA phosphate backbone directly or via a water molecule (e.g. PutA Lys-34).
FIGURE 4.

DNA recognition by the N terminus of DinJ at the dinJ promoter. A, modeling of DinJ binding to DNA based on the structure of FitA-DNA complex (blue, PDB code 2BSQ) and PutA-DNA complex (purple, PDB code 2RBF). PutA and FitA residues required for DNA binding as well as structurally equivalent DinJ residues are displayed as sticks. B, EMSAs show that the wild-type DinJ-YafQ complex binds to its promoter, whereas DinJ(R10A)-YafQ and DinJ(R35A)-YafQ fail to bind DNA. Black-filled arrows and open arrows indicate unbound and TA-bound DNA, respectively. C, model of YafQ-(DinJ)2-YafQ bound to two inverted DNA repeats (DNA shown in gray). A minor steric clash occurs between DinJ α1 (magenta) and DinJ′ α1 (green) (outlined in red; zoomed in, right panel).
We next constructed alanine mutations of the predicted DNA-binding DinJ residues Arg-10 and Arg-35, and we tested their effects on binding to the endogenous dinJ operator in vitro by EMSA and their effects on transcriptional repression in vivo using the previously described β-galactosidase activity assay. Our EMSA results indicate that both DinJ R10A and R35A result in a complete loss of binding to the operator region compared with wild-type DinJ-YafQ (Fig. 4B). We confirmed these results in vivo as both fail to repress lacZ transcription (3.5 and 11.3% transcriptional repression, respectively versus 81% repression by wild-type YafQ-(DinJ)2-YafQ) (Fig. 3B). Taken together, both approaches support our prediction that DinJ Arg-10 and Arg-35 directly interact with the dinJ operator.
Modeling of YafQ-(DinJ)2-YafQ Bound to DNA Reveals Two YafQ-(DinJ)2-YafQ Tetramers Minimally Sterically Clash
Because transcriptional repression in TA systems has been directly correlated to the number of TA complexes that bind to DNA (25, 26), we were cognizant of the role that base pair spacing between inverted repeats may influence the spatial coexistence of multiple YafQ-(DinJ)2-YafQ complexes bound. Therefore, because the Arc-DNA complex contained two Arc dimers bound to repressor DNA sites, we used this structure as a guide (Fig. 4C). Modeling YafQ-(DinJ)2-YafQ onto two binding sites on the arc promoter revealed minimal overlap between DinJ-DinJ′ dimers (Fig. 4C). This is in contrast to when two structurally homologous RelBE tetramers are modeled onto the same Arc-DNA complex (50). In our study, we adjusted our model to allow for a 10-bp spacing between inverted repeats, which is consistent with the endogenous spacing of the dinJ operon (Fig. 5A). This result suggests that it may be physically possible for two YafQ-(DinJ)2-YafQ complexes to interact with each inverted repeat simultaneously with perhaps slight conformational rearrangements of either the DNA or each of the YafQ-(DinJ)2-YafQ tetramers to alleviate the small clash between DinJ-DinJ′ dimers. These modeling results indicate the YafQ-(DinJ)2-YafQ TA system may be regulated via a distinct mechanism and prompted us to identify the minimal DNA repressor sites sufficient to confer transcriptional repression by YafQ-(DinJ)2-YafQ.
FIGURE 5.

Transcriptional repression and direct binding of YafQ-(DinJ)2-YafQ at the dinJ promoter. A, wild-type promoter region of dinJ with each IR of operator 1 and 2, the −35 and −10 promoter regions, the ribosome-binding site (RBS), and the LexA consensus box indicated. Nucleotides depicted in red were mutated to test for transcriptional repression by YafQ-(DinJ)2-YafQ. B, β-galactosidase activity assays testing the repression at the dinJ operator by wild-type YafQ-(DinJ)2-YafQ. Error bars represent mean ± S.E. of experiments performed in triplicate. C, EMSAs with increasing concentrations of wild-type YafQ-(DinJ)2-YafQ binding to either wild-type or IR1′-2–2′ mutated dinJ promoter region.
Transcriptional Repression at the DinJ Promoter Is Mediated by Single-site Binding of the YafQ-(DinJ)2-YafQ Complex
To test the relative contributions of YafQ-(DinJ)2-YafQ binding to each inverted repeat, we mutated either the first inverted repeat (IR1) or the second inverted repeat (IR1′) of operator one (Fig. 5A). Each mutation imparted similar repression levels as the wild-type dinJ operator (83% repression for both mutants; 81% repression by wild type) (Fig. 5B). When both IR1 and IR1′ were mutated in combination, repression is significantly alleviated implying this is the main site for YafQ-(DinJ)2-YafQ repression. Although it was previously shown that YafQ-(DinJ)2-YafQ does not bind the second operator in vitro (30), we tested for the ability of YafQ-(DinJ)2-YafQ to repress this operator in vivo. Mutation of either both IR2 and IR2′ or the complete IR2/IR2′ deletion produced comparable repression levels as the wild-type dinJ operator (Fig. 5B and data not shown, respectively). This suggests the second operator contributes minimally to transcriptional regulation by the YafQ-(DinJ)2-YafQ complex.
Now that we established that YafQ-(DinJ)2-YafQ-mediated repression primarily occurs at operator one alone, we next asked whether IR1 or IR1′ of operator one is sufficient to illicit transcriptional repression. Using the IR2-IR2′ mutation background which did not alter YafQ-(DinJ)2-YafQ-mediated transcriptional repression, mutation of either IR1 or IR1′ produced the same level of transcriptional repression as the wild-type dinJ operator (Fig. 5B). These data strongly suggest that only single-site binding by one YafQ-(DinJ)2-YafQ complex is sufficient to repress transcription at PdinJ. Control experiments where expression levels of YafQ-(DinJ)2-YafQ were varied showed comparable repression trends at all arabinose concentrations tested (0.002–0.2%; data not shown). Likewise, when all four IRs of the two operators were mutated (i.e. IR1, IR1′, IR2, and IR2′), repression was alleviated (Fig. 5B). Furthermore, in vitro EMSAs show that YafQ-(DinJ)2-YafQ still binds to the promoter containing either the mutated IR1 or IR1′, albeit with a reduction in apparent binding affinities (Fig. 5C and data not shown). These results are consistent with the conclusion that binding of YafQ-(DinJ)2-YafQ to only one inverted repeat is sufficient to achieve transcriptional repression.
DinJ Alone Confers Full Transcriptional Repression at the DinJ Promoter
Antitoxin proteins alone have been reported to act as autorepressors of transcription; the levels of transcriptional repression is further enhanced when in complex with toxin proteins (22). Using β-galactosidase activity assays, we next tested whether DinJ alone is able to repress transcription at the PdinJ promoter as effectively as the YafQ-(DinJ)2-YafQ complex. DinJ alone repressed transcription to almost the full extent as YafQ-(DinJ)2-YafQ (85% repression by DinJ versus 81% repression by YafQ-(DinJ)2-YafQ; Fig. 6A). As seen with YafQ-(DinJ)2-YafQ, repression by DinJ is also mainly mediated by its binding to the first operator because mutating both IRs of the second operator, IR2 and IR2′, did not change the level of repression significantly (72% repression). Mutation of either IR1 or IR1′ with the mutated IR2-IR2′ background also showed a comparable level of repression by DinJ alone (73% for IR1-IR2-IR2′ mutation and 69% for IR1′-IR2-IR2′ mutation). Finally, mutation of both IR1 and IR1′ eliminates transcriptional repression and DNA binding (data not shown). As might be expected, mutation of all four IRs (IR1, IR1′, IR2, and IR2′) abolished DinJ-mediated transcriptional repression as well as binding to PdinJ (Fig. 6A and data not shown). In summary, DinJ alone represses transcription to a comparable level as YafQ-(DinJ)2-YafQ, and DinJ recognition of either IR1 or IR1′ is sufficient to mediate the same transcriptional repression.
FIGURE 6.

Transcriptional repression and direct binding of DinJ and LexA at the dinJ promoter. A, β-galactosidase assays using the same dinJ operator mutations as in Fig. 5B, but instead testing repression by DinJ alone. Mutation of all four IRs (IR1–1′ to 2–2′) resulted in a complete loss of transcriptional repression by DinJ and thus is depicted as 0% repression. B, EMSAs of increasing concentrations of wild-type DinJ binding to either wild-type or IR1–1′ to 2–2′ mutated dinJ promoter region, respectively. C, β-galactosidase assays testing repression by LexA. D, complexes of wild-type DinJ-YafQ-dinJ promoter were incubated with an increasing molar excess of YafQ as indicated. Black-filled arrows and open arrows indicate unbound and TA-bound DNA, respectively. Error bars represent mean ± S.E. of reactions performed in triplicate for assays in A and C.
LexA Represses Transcription at the Second Operator of the DinJ Promoter
The LexA repressor regulates the transcription of genes involved in the SOS response mediated by DNA damage (61). LexA binds directly to the second operator of the dinJ promoter region in vitro (31); however, it is unclear whether LexA-mediated repression occurs in vivo. When LexA is delivered in trans, LexA represses transcription from the PdinJ promoter (67% repression; Fig. 6C). Furthermore, deletion of the entire second operator (IR2 and IR2′) diminished the repression to almost uninduced levels (Fig. 6C). These data confirm that the second operator site of the dinJ promoter is essential for LexA-mediated transcriptional repression of the dinJ-yafQ operon.
Addition of the YafQ Toxin to the YafQ-(DinJ)2-YafQ-DNA Repressor Complex Does Not Alter Its Stability
One proposed additional role of toxin proteins is that upon increasing toxin levels during stress, the toxin functions as a transcriptional de-repressor via destabilization of the TA-DNA repressor complex (25–28). To address whether transcription of the dinJ-yafQ operon is regulated in the same manner, we performed competition EMSAs where we first formed a complex of YafQ-(DinJ)2-YafQ bound to PdinJ and then incubated with increasing concentrations of YafQ (Fig. 6D). In this assay, the following PdinJ variants were used: wild type, IR2-IR2′ mutant (two binding sites available), IR1′-IR2-IR2′ (one binding site available), or IR1-IR2-IR2′(one binding site available; data not shown). In all cases, YafQ was unable to compete off the YafQ-(DinJ)2-YafQ complex even when YafQ was in a 1 m excess of the antitoxin, in contrast to what has previously been shown for RelBE and CcdAB (25, 29). These results indicate YafQ-(DinJ)2-YafQ uses a distinct mechanism to regulate its own transcription.
DISCUSSION
Bacterial type II toxin-antitoxin systems form tight protein-protein complexes that function to repress their own transcription and inhibit the bacteriostatic activity of the toxin under steady state growth conditions. The transcriptional regulation of TA systems is essential for proper biological functions, such as persister cell formation (62). Collectively, our structural, biochemical, and functional data provide new mechanistic insights into the novel transcriptional regulation of the E. coli DinJ-YafQ TA system.
There are two major biological functions for the formation of a complex between toxins and antitoxins. The first is to sequester the toxin and inactivate its function. The second is for the complex to serve as transcriptional repressors of their own operons during steady state growth conditions. TA complexes bind directly to operator regions that overlap with the −35 and −10 promoter sites, repressing transcription by restricting RNA polymerase access. This regulation appears also to be subtly regulated by the relative ratios of toxins and antitoxins whose levels fluctuate rapidly as stress is encountered (25, 29, 63). In vitro studies of some TA systems show that excess toxin, resulting from antitoxin degradation, destabilizes the interaction between the TA pair-DNA operator complex (25, 26, 64). This regulation has been suggested to be controlled by conditional cooperativity although the exact details differ. In one interpretation, one TA complex can cooperatively enhance binding at the second inverted repeat; a slightly different interpretation is that the toxin enhances avidity for the inverted repeat DNA, in the context of the complex (25, 50). Modeling tetrameric TA complexes such as RelBE to its operator sequence shows there is a steric clash when two RelBE tetramers are bound to two adjacent inverted repeats. This suggests that both inverted repeats cannot be occupied simultaneously unless the oligomeric state of the TA complex is altered (50).
YafQ-(DinJ)2-YafQ appears to be transcriptionally regulated in a manner distinct from other TA systems studied to date. Only a single YafQ-(DinJ)2-YafQ complex bound to one inverted repeat mediates significant levels of repression (Fig. 5B). Furthermore, the extent of repression does not intensify upon two YafQ-(DinJ)2-YafQ complexes bound at both inverted repeats of the first operator (Fig. 5B). Additionally, although some antitoxins have been shown to bind more weakly to their operators in vitro than TA complexes, it is unclear whether this correlation is seen in terms of transcriptional repression. Our in vivo results indicate both DinJ alone and in complex with YafQ result in similar levels of transcriptional repression (Fig. 6A). Finally, our results indicate that the stability of either one or two YafQ-(DinJ)2-YafQ complexes bound to the dinJ promoter is unaffected by increasing levels of toxin (Fig. 6D). Possible explanations for why different transcriptional regulatory mechanisms may exist for some TA systems include diverse antitoxin DNA binding domains, recognition of different DNA operator sequences, or different base pair spacing between inverted repeats.
Toxin-antitoxins gene pairs are part of the stress response and, in some cases, the stringent response, both of which facilitate intricate regulatory gene expression pathways for bacterial survival. De-repression at TA loci during stress may be mediated by the alarmone (p)ppGpp, which is generated by RelA in response to deactylated tRNAs binding to the ribosome during nutrient deprivation (65). The increase of (p)ppGpp during the stringent response regulates several hundred genes, although it is unclear whether the effect is always direct (66). In fact, (p)ppGpp up-regulation was seen to increase levels of mRNA transcripts involved in both the stringent and SOS responses such as RelBE, DinJ, RecA, and LexA. Therefore, it is plausible that de-repression at TA gene pairs may be influenced by (p)ppGpp indirectly via RNA polymerase and possibly in conjunction with cofactor DskA, which can destabilize promoter complexes (67). The presence of the LexA consensus site in the dinJ promoter suggests a possible link between the SOS and the stringent response. Although LexA has been reported to interact with the dinJ promoter (31), our results extend this observation by demonstrating that LexA represses the dinJ operon in vivo (Fig. 6C). Whether LexA regulation at the dinJ operon is modulated by the physiological state of the bacteria requires further study.
The exciting possible connection between the SOS and the stringent response pathways may represent a more global regulatory network that is highly tuned to distinct stresses required for bacterial survival. Therefore, understanding the regulation of TA pairs and their possible role in persister cell formation is critical given their potential biomedical importance as antimicrobial targets (3).
Acknowledgments
We thank K. Rajashankar and S. Banerjee at the Northeastern Collaborative Access Team beamline for assistance during data collection and G. L. Conn and M. A. Schureck for helpful discussions throughout the project and critical reading of the manuscript. This work is based on research conducted at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines, National Institutes of Health Grant RR-15301 from NCRR, and at the SER-CAT beamline. Use of the Advanced Photon Source, an Office of Science User Facility operated for the United States Department of Energy Office of Science by Argonne National Laboratory, was supported by the United States Department of Energy under Contract DE-AC02-06CH11357.
This work was supported by National Science Foundation CAREER Award MCB 0953714 (to C. M. D.).

This article contains Table S1 and additional references.
The atomic coordinates and structure factors (code 4Q2U) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- TA
- toxin-antitoxin
- IR
- inverted repeat
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- RHH
- ribbon-helix-helix
- (p)ppGpp
- tetra or pentaguanosine phosphate.
REFERENCES
- 1. Keren I., Shah D., Spoering A., Kaldalu N., Lewis K. (2004) Specialized persister cells and the mechanism of multidrug tolerance in Escherichia coli. J. Bacteriol. 186, 8172–8180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lazazzera B. A. (2005) Lessons from DNA microarray analysis: the gene expression profile of biofilms. Curr. Opin. Microbiol. 8, 222–227 [DOI] [PubMed] [Google Scholar]
- 3. Lewis K. (2010) Persister cells. Annu. Rev. Microbiol. 64, 357–372 [DOI] [PubMed] [Google Scholar]
- 4. Ramage H. R., Connolly L. E., Cox J. S. (2009) Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 5, e1000767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yamaguchi Y., Inouye M. (2011) Regulation of growth and death in Escherichia coli by toxin-antitoxin systems. Nat. Rev. Microbiol. 9, 779–790 [DOI] [PubMed] [Google Scholar]
- 6. Gerdes K. (ed) (2013) Prokaryotic Toxin-Antitoxins, pp. 69–92, Springer, Berlin/Heidelberg [Google Scholar]
- 7. Chan W. T., Nieto C., Harikrishna J. A., Khoo S. K., Othman R. Y., Espinosa M., Yeo C. C. (2011) Genetic regulation of the yefM-yoeB toxin-antitoxin locus of Streptococcus pneumoniae. J. Bacteriol. 193, 4612–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gotfredsen M., Gerdes K. (1998) The Escherichia coli relBE genes belong to a new toxin-antitoxin gene family. Mol. Microbiol. 29, 1065–1076 [DOI] [PubMed] [Google Scholar]
- 9. Hu Y., Benedik M. J., Wood T. K. (2012) Antitoxin DinJ influences the general stress response through transcript stabilizer CspE. Environ. Microbiol. 14, 669–679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kedzierska B., Lian L. Y., Hayes F. (2007) Toxin-antitoxin regulation: bimodal interaction of YefM-YoeB with paired DNA palindromes exerts transcriptional autorepression. Nucleic Acids Res. 35, 325–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim Y., Wang X., Zhang X. S., Grigoriu S., Page R., Peti W., Wood T. K. (2010) Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ. Microbiol. 12, 1105–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markiewicz P., Kleina L. G., Cruz C., Ehret S., Miller J. H. (1994) Genetic studies of the lac repressor. XIV. Analysis of 4000 altered Escherichia coli lac repressors reveals essential and non-essential residues, as well as “spacers” which do not require a specific sequence. J. Mol. Biol. 240, 421–433 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Kim Y., Hong S. H., Ma Q., Brown B. L., Pu M., Tarone A. M., Benedik M. J., Peti W., Page R., Wood T. K. (2011) Antitoxin MqsA helps mediate the bacterial general stress response. Nat. Chem. Biol. 7, 359–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dalton K. M., Crosson S. (2010) A conserved mode of protein recognition and binding in a ParD-ParE toxin-antitoxin complex. Biochemistry 49, 2205–2215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grady R., Hayes F. (2003) Axe-Txe, a broad-spectrum proteic toxin-antitoxin system specified by a multidrug-resistant, clinical isolate of Enterococcus faecium. Mol. Microbiol. 47, 1419–1432 [DOI] [PubMed] [Google Scholar]
- 16. Li G. Y., Zhang Y., Inouye M., Ikura M. (2009) Inhibitory mechanism of Escherichia coli RelE-RelB toxin-antitoxin module involves a helix displacement near an mRNA interferase active site. J. Biol. Chem. 284, 14628–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Roberts R. C., Ström A. R., Helinski D. R. (1994) The parDE operon of the broad-host-range plasmid RK2 specifies growth inhibition associated with plasmid loss. J. Mol. Biol. 237, 35–51 [DOI] [PubMed] [Google Scholar]
- 18. Christensen S. K., Maenhaut-Michel G., Mine N., Gottesman S., Gerdes K., Van Melderen L. (2004) Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM-yoeB toxin-antitoxin system. Mol. Microbiol. 51, 1705–1717 [DOI] [PubMed] [Google Scholar]
- 19. Diago-Navarro E., Hernández-Arriaga A. M., Kubik S., Konieczny I., Díaz-Orejas R. (2013) Cleavage of the antitoxin of the parD toxin-antitoxin system is determined by the ClpAP protease and is modulated by the relative ratio of the toxin and the antitoxin. Plasmid 70, 78–85 [DOI] [PubMed] [Google Scholar]
- 20. Lehnherr H., Yarmolinsky M. B. (1995) Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 92, 3274–3277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Van Melderen L., Bernard P., Couturier M. (1994) Lon-dependent proteolysis of CcdA is the key control for activation of CcdB in plasmid-free segregant bacteria. Mol. Microbiol. 11, 1151–1157 [DOI] [PubMed] [Google Scholar]
- 22. Gerdes K., Christensen S. K., Løbner-Olesen A. (2005) Prokaryotic toxin-antitoxin stress response loci. Nat. Rev. Microbiol. 3, 371–382 [DOI] [PubMed] [Google Scholar]
- 23. Christensen S. K., Mikkelsen M., Pedersen K., Gerdes K. (2001) RelE, a global inhibitor of translation, is activated during nutritional stress. Proc. Natl. Acad. Sci. U.S.A. 98, 14328–14333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pedersen K., Christensen S. K., Gerdes K. (2002) Rapid induction and reversal of a bacteriostatic condition by controlled expression of toxins and antitoxins. Mol. Microbiol. 45, 501–510 [DOI] [PubMed] [Google Scholar]
- 25. Overgaard M., Borch J., Jørgensen M. G., Gerdes K. (2008) Messenger RNA interferase RelE controls relBE transcription by conditional cooperativity. Mol. Microbiol. 69, 841–857 [DOI] [PubMed] [Google Scholar]
- 26. Garcia-Pino A., Balasubramanian S., Wyns L., Gazit E., De Greve H., Magnuson R. D., Charlier D., van Nuland N. A., Loris R. (2010) Allostery and intrinsic disorder mediate transcription regulation by conditional cooperativity. Cell 142, 101–111 [DOI] [PubMed] [Google Scholar]
- 27. Magnuson R., Yarmolinsky M. B. (1998) Corepression of the P1 addiction operon by Phd and Doc. J. Bacteriol. 180, 6342–6351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Johnson E. P., Strom A. R., Helinski D. R. (1996) Plasmid RK2 toxin protein ParE: purification and interaction with the ParD antitoxin protein. J. Bacteriol. 178, 1420–1429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Afif H., Allali N., Couturier M., Van Melderen L. (2001) The ratio between CcdA and CcdB modulates the transcriptional repression of the ccd poison-antidote system. Mol. Microbiol. 41, 73–82 [DOI] [PubMed] [Google Scholar]
- 30. Armalyte J., Jurenaite M., Beinoraviciūte G., Teiserskas J., Suziedeliene E. (2012) Characterization of Escherichia coli dinJ-yafQ toxin-antitoxin system using insights from mutagenesis data. J. Bacteriol. 194, 1523–1532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Prysak M. H., Mozdzierz C. J., Cook A. M., Zhu L., Zhang Y., Inouye M., Woychik N. A. (2009) Bacterial toxin YafQ is an endoribonuclease that associates with the ribosome and blocks translation elongation through sequence-specific and frame-dependent mRNA cleavage. Mol. Microbiol. 71, 1071–1087 [DOI] [PubMed] [Google Scholar]
- 32. Park S. J., Son W. S., Lee B. J. (2013) Structural overview of toxin-antitoxin systems in infectious bacteria: a target for developing antimicrobial agents. Biochim. Biophys. Acta 1834, 1155–1167 [DOI] [PubMed] [Google Scholar]
- 33. Doublié S. (1997) Preparation of selenomethionyl proteins for phase determination. Methods Enzymol. 276, 523–530 [PubMed] [Google Scholar]
- 34. Van Duyne G. D., Standaert R. F., Karplus P. A., Schreiber S. L., Clardy J. (1993) Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105–124 [DOI] [PubMed] [Google Scholar]
- 35. Otwinowski Z., Minor W. (1997) in Methods in Enzymology (Carter C. W., Jr., Sweet R. M., eds) pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 36. Sheldrick G. M. (2010) Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pape T., Schneider T. R. (2004) HKL2MAP: a graphical user interface for phasing with the SHELX programs. J. Appl. Crystallogr. 37, 843–844 [Google Scholar]
- 38. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., Richardson J. S., Terwilliger T. C., Zwart P. H. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen V. B., Arendall W. B., 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.2r3pre Ed., Schrödinger, LLC, New York [Google Scholar]
- 42. Farinha M. A., Kropinski A. M. (1990) Construction of broad-host-range plasmid vectors for easy visible selection and analysis of promoters. J. Bacteriol 172, 3496–3499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miller J. H. (1972) Experiments in Molecular Genetics, pp. 352–355, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 44. Mattison K., Wilbur J. S., So M., Brennan R. G. (2006) Structure of FitAB from Neisseria gonorrhoeae bound to DNA reveals a tetramer of toxin-antitoxin heterodimers containing pin domains and ribbon-helix-helix motifs. J. Biol. Chem. 281, 37942–37951 [DOI] [PubMed] [Google Scholar]
- 45. Zhou Y., Larson J. D., Bottoms C. A., Arturo E. C., Henzl M. T., Jenkins J. L., Nix J. C., Becker D. F., Tanner J. J. (2008) Structural basis of the transcriptional regulation of the proline utilization regulon by multifunctional PutA. J. Mol. Biol. 381, 174–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hasegawa H., Holm L. (2009) Advances and pitfalls of protein structural alignment. Curr. Opin. Struct. Biol. 19, 341–348 [DOI] [PubMed] [Google Scholar]
- 47. Raumann B. E., Rould M. A., Pabo C. O., Sauer R. T. (1994) DNA recognition by β-sheets in the Arc repressor-operator crystal structure. Nature 367, 754–757 [DOI] [PubMed] [Google Scholar]
- 48. Zegers I., Haikal A. F., Palmer R., Wyns L. (1994) Crystal structure of RNase T1 with 3′-guanylic acid and guanosine. J. Biol. Chem. 269, 127–133 [PubMed] [Google Scholar]
- 49. Sevcik J., Dodson E. J., Dodson G. G. (1991) Determination and restrained least-squares refinement of the structures of ribonuclease Sa and its complex with 3′-guanylic acid at 1.8 Å resolution. Acta Crystallogr. Sect. B Struct. Sci. 47, 240–253 [PubMed] [Google Scholar]
- 50. Bøggild A., Sofos N., Andersen K. R., Feddersen A., Easter A. D., Passmore L. A., Brodersen D. E. (2012) The crystal structure of the intact E. coli RelBE toxin-antitoxin complex provides the structural basis for conditional cooperativity. Structure 20, 1641–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kamada K., Hanaoka F. (2005) Conformational change in the catalytic site of the ribonuclease YoeB toxin by YefM antitoxin. Mol. Cell 19, 497–509 [DOI] [PubMed] [Google Scholar]
- 52. Brown B. L., Grigoriu S., Kim Y., Arruda J. M., Davenport A., Wood T. K., Peti W., Page R. (2009) Three-dimensional structure of the MqsR:MqsA complex: a novel TA pair comprised of a toxin homologous to RelE and an antitoxin with unique properties. PLoS Pathog. 5, e1000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Holm L., Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Takagi H., Kakuta Y., Okada T., Yao M., Tanaka I., Kimura M. (2005) Crystal structure of archaeal toxin-antitoxin RelE-RelB complex with implications for toxin activity and antitoxin effects. Nat. Struct. Mol. Biol. 12, 327–331 [DOI] [PubMed] [Google Scholar]
- 56. Schureck M. A., Maehigashi T., Miles S. J., Marquez J., Cho S. E., Erdman R., Dunham C. M. (2014) Structure of the P. vulgaris HigB-(HigA)2-HigB toxin-antitoxin complex. J. Biol. Chem. 289, 1060–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Schreiter E. R., Drennan C. L. (2007) Ribbon-helix-helix transcription factors: variations on a theme. Nat. Rev. Microbiol. 5, 710–720 [DOI] [PubMed] [Google Scholar]
- 58. Waldburger C. D., Schildbach J. F., Sauer R. T. (1995) Are buried salt bridges important for protein stability and conformational specificity? Nat. Struct. Biol. 2, 122–128 [DOI] [PubMed] [Google Scholar]
- 59. Griffin M. A., Davis J. H., Strobel S. A. (2013) Bacterial toxin RelE: a highly efficient ribonuclease with exquisite substrate specificity using atypical catalytic residues. Biochemistry 52, 8633–8642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Miallau L., Jain P., Arbing M. A., Cascio D., Phan T., Ahn C. J., Chan S., Chernishof I., Maxson M., Chiang J., Jacobs W. R., Jr., Eisenberg D. S. (2013) Comparative proteomics identifies the cell-associated lethality of M. tuberculosis RelBE-like toxin-antitoxin complexes. Structure 21, 627–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shinagawa H. (1996) SOS response as an adaptive response to DNA damage in prokaryotes. EXS 77, 221–235 [DOI] [PubMed] [Google Scholar]
- 62. Maisonneuve E., Castro-Camargo M., Gerdes K. (2013) (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 154, 1140–1150 [DOI] [PubMed] [Google Scholar]
- 63. Winther K. S., Gerdes K. (2012) Regulation of enteric vapBC transcription: induction by VapC toxin dimer-breaking. Nucleic Acids Res. 40, 4347–4357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Van Melderen L., Thi M. H., Lecchi P., Gottesman S., Couturier M., Maurizi M. R. (1996) ATP-dependent degradation of CcdA by Lon protease. Effects of secondary structure and heterologous subunit interactions. J. Biol. Chem. 271, 27730–27738 [DOI] [PubMed] [Google Scholar]
- 65. Potrykus K., Cashel M. (2008) (p)ppGpp: still magical? Annu. Rev. Microbiol. 62, 35–51 [DOI] [PubMed] [Google Scholar]
- 66. Durfee T., Hansen A. M., Zhi H., Blattner F. R., Jin D. J. (2008) Transcription profiling of the stringent response in Escherichia coli. J. Bacteriol. 190, 1084–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Paul B. J., Barker M. M., Ross W., Schneider D. A., Webb C., Foster J. W., Gourse R. L. (2004) DksA: a critical component of the transcription initiation machinery that potentiates the regulation of rRNA promoters by (p)ppGpp and the initiating NTP. Cell 118, 311–322 [DOI] [PubMed] [Google Scholar]
- 68. Ashkenazy H., Erez E., Martz E., Pupko T., Ben-Tal N. (2010) ConSurf 2010: calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 38, W529–W533 [DOI] [PMC free article] [PubMed] [Google Scholar]