

Background: Branched-chain amino acids (BCAA) are elevated in maple syrup urine disease, obesity, and type 2 diabetes.
Results: We show that benzothiophene carboxylate derivatives are allosteric inhibitors of branched-chain α-ketoacid dehydrogenase kinase (BDK).
Conclusion: These BDK inhibitors robustly augment BCAA oxidation in mice, resulting in lower plasma BCAA.
Significance: The BDK inhibitors are potentially useful for treatment of the above disorders.
Keywords: Diabetes, Drug Design, Genetic Disease, Obesity, Protein Kinase, Allosteric Kinase Inhibitors, Branched-chain alpha-Ketoacid Dehydrogenase Complex, Branched-chain alpha-Ketoacids, Branched-chain Amino Acids, Maple Syrup Urine Disease
Abstract
The mitochondrial branched-chain α-ketoacid dehydrogenase complex (BCKDC) is negatively regulated by reversible phosphorylation. BCKDC kinase (BDK) inhibitors that augment BCKDC flux have been shown to reduce branched-chain amino acid (BCAA) concentrations in vivo. In the present study, we employed high-throughput screens to identify compound 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid (BT2) as a novel BDK inhibitor (IC50 = 3.19 μm). BT2 binds to the same site in BDK as other known allosteric BDK inhibitors, including (S)-α-cholorophenylproprionate ((S)-CPP). BT2 binding to BDK triggers helix movements in the N-terminal domain, resulting in the dissociation of BDK from the BCKDC accompanied by accelerated degradation of the released kinase in vivo. BT2 shows excellent pharmacokinetics (terminal T½ = 730 min) and metabolic stability (no degradation in 240 min), which are significantly better than those of (S)-CPP. BT2, its analog 3-chloro-6-fluorobenzo[b]thiophene-2-carboxylic acid (BT2F), and a prodrug of BT2 (i.e. N-(4-acetamido-1,2,5-oxadiazol-3-yl)-3,6-dichlorobenzo[b]thiophene-2-carboxamide (BT3)) significantly increase residual BCKDC activity in cultured cells and primary hepatocytes from patients and a mouse model of maple syrup urine disease. Administration of BT2 at 20 mg/kg/day to wild-type mice for 1 week leads to nearly complete dephosphorylation and maximal activation of BCKDC in heart, muscle, kidneys, and liver with reduction in plasma BCAA concentrations. The availability of benzothiophene carboxylate derivatives as stable BDK inhibitors may prove useful for the treatment of metabolic disease caused by elevated BCAA concentrations.
Introduction
The degradation of branched-chain amino acids (BCAA)4 leucine, isoleucine, and valine begins with transamination to produce the corresponding branch-chain α-ketoacids (BCKA). The second common step, the irreversible oxidative decarboxylation of BCKA, is catalyzed by the single branched-chain α-ketoacid dehydrogenase complex (BCKDC) in mitochondria. The homeostasis of BCAA is critical in health and disease. BCAA supplementation reduces oxidative stress, which in turn promotes survival in rats with advanced liver cirrhosis (1) and supports mitochondrial biogenesis in cardiac and skeletal muscle (2). On the other hand, in patients with inherited maple syrup urine disease (MSUD), the accumulation of BCAA and BCKA caused by the dysfunction of BCKDC leads to sometimes fatal acidosis, neurological derangement, and mental retardation (3, 4). High blood BCAA concentrations are linked to the development of insulin resistance and are useful metabolic markers in type 2 diabetes risk assessment (5, 6). Thus, impaired BCAA catabolism plays a pivotal role in the pathogenesis of metabolic and neurological diseases.
The 4.5-million dalton mammalian BCKDC consists of multiple copies of three catalytic components (i.e. branched-chain α-ketoacid decarboxylase (E1), dihydrolipoyl transacylase (E2), and dihydrolipoamide dehydrogenase (E3)) as well as two regulatory enzymes: BCKD kinase (BDK) and BCKD phosphatase (BDP). BDK and BDP tightly regulate the activity of BCKDC through the phosphorylation (inactivation)/dephosphorylation (activation), respectively, of the E1α subunits (7, 8). The BCKDC is organized around the cubic 24-meric E2 core, to which other enzyme components are attached through non-covalent interactions (9). The docking of BDK and BDP to the E2 core of BCKDC via the lipoyl-bearing domain (LBD) of each E2 subunit co-localizes them to the E1 substrate and augments respective kinase and phosphatase activities through conformational changes (10–12).
The BCKDC catalyzes the rate-limiting step in the disposal of BCAA (13, 14). Therefore, modulation of BDK activity constitutes a major mechanism for BCAA homeostasis in vivo (15), and BDK offers a therapeutic target to increase BCKDC flux and ameliorate accumulated BCAA and BCKA in disease states. BDK is inhibited by α-ketoisocaproate (KIC) from leucine, resulting in the activation of BCKDC in perfused rat hearts (16). The inhibition of BDK by small molecules, such as KIC, prompted the development and identification of a series of KIC analogs that function as BDK inhibitors (16, 17). These include α-chloroisocaproate (CIC) (18), phenylpyruvate (17), clofibric acid (19), and phenylbutyrate (20). However, these BDK inhibitors are either nonspecific (e.g. phenylbutyrate) or less than robust as BDK inhibitors, with reported I40 (concentration for 40% inhibition) in the submillimolar range (e.g. CIC, phenylpyruvate, and clofibric acid).
Our laboratory has focused on the development of novel BDK inhibitors for therapeutic approaches to reducing BCAA/BCKA concentrations in MSUD and obesity as well as type 2 diabetes. We previously reported the structure-based design and synthesis of (S)-α-cholorophenylproprionate ((S)-CPP) as a BDK inhibitor (21). We showed that (S)-CPP robustly inhibits BDK activity by binding to the allosteric site in the N-terminal domain of BDK. Treatment of wild-type mice with (S)-CPP resulted in increased BCKDC activity in various tissues with reduced plasma BCAA concentrations. However, (S)-CPP required high doses for the treatments due to its relatively short half-life in vivo. In the present study, we conducted high-throughput screens of a small chemical library and identified a new class of BDK inhibitors represented by 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid (BT2). BT2 binds to the same allosteric site as (S)-CPP, with both compounds sharing the same inhibitory mechanism. Nevertheless, BT2 exhibits significantly higher stability than (S)-CPP in vivo and requires a far lower dose than (S)-CPP for long term treatments. The novel BDK inhibitor BT2 has potential for clinical applications to lower BCAA/BCKA concentration in the above disease states.
EXPERIMENTAL PROCEDURES
Sources and Synthesis of Small Molecule Compounds
Compound 476-I16 (N,N′-(1,2,5-oxadiazole-3,4-diyl)bis(3,6-dichlorobenzo[b]thiophene-2-carboxamide)) and BT1 (N-(4-amino-1,2,5-oxadiazol-3-yl)-3,6-dichlorobenzo[b]thiophene-2-carboxamide) were purchased from TimTec (Newark, DE); BT2 (3,6-dichlorobenzo[b]thiophene-2-carboxylic acid) and BT2F (3-chloro-6-fluorobenzo[b]thiophene-2-carboxylic acid) were obtained from Sigma-Aldrich.
BT3 (N-(4-acetamido-1,2,5-oxadiazol-3-yl)-3,6-dichlorobenzo[b]thiophene-2-carboxamide) was synthesized as follows. In a 25-ml round-bottomed flask equipped with a magnetic stirrer, 247 mg (1 mmol, 1 eq) of BT2, 155 mg (1 mmol, 1 eq) of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, and 135 mg (1 mmol, 1 eq) of hydroxybenzotriazole were dissolved in 5 ml of dimethylformamide at room temperature. The resulting mixture was stirred at room temperature under an atmosphere of N2 for 10 min before 142 mg (1 mmol, 1 eq) of N-(4-amino-1,2,5-oxadiazol-3-yl)acetamide was added dropwise (in 1 ml of dimethylformamide solution). The reaction mixture was stirred at room temperature for 12 h and quenched with 10 ml of ethyl acetate and aqueous saturated NaHCO3 (5 ml). The organic layer was separated, washed sequentially with 0.1 n aqueous HCl solution (10 ml), aqueous saturated NaHCO3 (10 ml), and brine (10 ml). The organic layer was dried over MgSO4 and evaporated under reduced pressure. The resulting residue was subjected to flash column chromatography (silica gel; ethyl acetate/hexane, 1:5–1:2) to give pure BT3 (181 mg, 49% yield) as a white solid.
Proteins
N-terminal maltose-binding protein-tagged rat BDK was expressed in pGroESL-cotransformed BL-21 cells and purified as described previously (10). For crystallization purposes, C-terminal His6-tagged rat BDK was expressed and purified also as described previously (12, 22).
The construct LBD-Php is a fusion protein containing a 14-residue phosphorylation peptide (Php) that corresponds to residues 286–300 of the E1α subunit, 287RIGHHSTSDDSSAY300, cloned into the C-terminal end of the C-terminal His-tagged LBD (residues 1–99 of E2 subunit) with a 9-residue-long linker, LENLYFQGT, between them. The fusion protein was expressed, purified, and lipoylated as the His-tagged LBD, as described previously (23, 24). When used in BDK assays, the LBD portion in the fusion protein binds and activates BDK, whereas the bound BDK is brought juxtaposed to the peptide substrate (Php) to facilitate phosphorylation.
Mouse Hepatocytes and Embryonic Fibroblasts and Human Lymphoblasts
Primary hepatocytes were isolated from wild-type and intermediate MSUD (iMSUD) mice as described previously (25). Wild-type and BDP−/− (PP2Cm−/−) mouse embryonic fibroblasts (MEF) were kindly supplied by Dr. Yibin Wang of UCLA Medical Center (26). Wild-type and iMSUD lymphoblasts were prepared from blood samples of normal subjects and patients carrying the homozygous R252H E1α or R240C E2 mutations (4).
Crystallization, Structure Determination, and Refinements of the BDK and BT2 Complexes
Purified C-terminal His6-tagged BDK protein was crystallized as described previously (21). The crystals were incubated with 0.2 mm BT2 and ADP or AMPPNP at 20 °C overnight before being transferred to a cryosolution containing 20% glycerol and snap-frozen in liquid nitrogen. X-ray diffraction data collection, processing, and refinements were as described previously (21). The statistics for data collection and refinement are shown in Table 1.
TABLE 1.
Data collection and refinement statistics
| BT2 + ADP | BT2 + AMPPNP | |
|---|---|---|
| Protein Data Bank code | 4E00 | 4E01 |
| Data collection | ||
| Space groupa | P42212 | P42212 |
| Cell dimensions | ||
| a, b (Å) | 128.06 | 127.28 |
| c (Å) | 73.78 | 73.99 |
| α, β, γ (degrees) | α = β = γ = 90° | α = β = γ = 90° |
| Resolution (Å) | 50-2.15 (2.19-2.15)b | 50-1.97 (2.00-1.97) |
| Rmerge | 6.4 (81.9) | 7.7 (93.7) |
| I/σI | 28.6 (3.2) | 20.5 (2.1) |
| Completeness (%) | 99.7 (100.0) | 99.6 (97.7) |
| Redundancy | 9.6 (9.4) | 9.8 (8.0) |
| Refinement | ||
| Resolution (Å) | 2.15 | 1.97 |
| No. of reflections | 34021 | 43743 |
| Rwork/Rfree (%) | 20.7/23.4 | 19.7/22.0 |
| No. of atoms | ||
| Protein | 2493 | 2486 |
| BT2 | 14 | 14 |
| Nucleotide | 27 | 31 |
| Mg2+ | 1 | 1 |
| K+ | 1 | 1 |
| Water | 71 | 118 |
| B-Factors | ||
| Protein | 59.2 | 51.1 |
| BT2 | 43.3 | 37.2 |
| Nucleotide | 49.8 | 34.8 |
| Mg2+ | 59.7 | 41.2 |
| K+ | 50.7 | 37.0 |
| Water | 50.8 | 48.5 |
| Root mean square deviations | ||
| Bond lengths (Å) | 0.008 | 0.007 |
| Bond angles (degrees) | 1.17 | 1.11 |
a One molecule/asymmetric unit, ∼70% solvent content.
b Values in parentheses are for the highest resolution shell.
High-throughput Screens for BDK Inhibitors
The phosphorylation reaction product ADP is detected using the ADP Hunter assay kit (DiscoveRx, Fremont, CA). This assay kit contains the coupling enzymes pyruvate kinase and pyruvate oxidase, which function in sequence to produce hydrogen peroxide from ADP. The product hydrogen peroxide, when catalyzed by the third enzyme peroxidase, converts the dye precursor Amplex to fluorescent resorufin. The final product, upon excitation at 530 nm, generates a fluorescence emission at 590 nm. The assay is robust with relatively low background signal; the Z′ factor is in the excellent range of 0.79–0.86. The Z′ factor for each plate is calculated according to the equation (27), Z′ = 1 − (3σm + 3σ0)/|μm − μ0|; σm and μm are the S.D. and average, respectively, of the maximal signals in wells where the BDK reaction occurs minus inhibitor; σ0 and μ0 are the S.D. and average, respectively, of background signals from in wells where BDK is omitted. A compound (12 μm per assay) is considered a “hit” when its signal is less than 3 S.D. values from the mean fluorescence value of the no-inhibition control (100% signal). The hits correspond to >30–40% inhibition of BDK activity. In a secondary screening, all hits from the primary screens were cherry-picked and assayed again in triplicate for validation. An assay control containing ADP and no BDK was also instituted to rule out the inhibition of the coupling enzymes, instead of BDK, by false-positive compounds.
Assay for Inhibition of BDK Activity
To determine the IC50 for BDK inhibitors, a mixture containing 0.2 μm BDK, 6 μm E1, 0.5 μm of E2, and various amounts of inhibitor was incubated at 25 °C for 10 min in a buffer of 20 mm Tris-Cl (pH 7.5), 100 mm KCl, 5 mm MgCl2, 2 mm dithiothreitol (DTT), 0.02% (v/v) Tween 20, and 0.1 mg/ml bovine serum albumin before the reaction. All inhibition titrations were performed at eight dose points ranging from 100 nm to 316 μm in a 3.162-fold dilution series, with each inhibitor concentration tested in duplicate. The remaining steps were described previously (28).
Metabolic Stability, Protein Binding, and Pharmacokinetics Studies
BT2 and BT3 were monitored by LC-MS/MS with the mass spectrometer in MRM (multiple reaction monitoring) mode by following the precursor to fragment ion transition 246.9–202.9 (negative mode) and 373.0–230.9 (positive mode), respectively. S9 studies of BT2 and BT3 were performed as described previously with the addition of standard curves to calculate absolute concentrations of BT2 and BT3 (21). Pharmacokinetic studies with BT2 in 5% DMSO, 10% cremophor EL, and 85% 0.1 m sodium bicarbonate, pH 9.0, were performed in CD-1 female mice from Charles River Laboratories (Wilmington, MA) also as reported previously (21). The fraction of compound bound to plasma protein was determined by a mass balance ultrafiltration method as described previously (29).
Mouse Studies with BDK Inhibitor BT2
8–10-week-old C57BL/6J male mice were randomized into two groups: vehicle- and BT2-treated. A total of 4 mice were included in each group. Mice were weighed on the day of the treatment and used to determine the administered dosage. BT2 was dissolved in DMSO and diluted into 5% DMSO, 10% cremophor EL, and 85% 0.1 m sodium bicarbonate, pH 9.0, for delivery. Animals were dosed daily in the morning for 7 days by intraperitoneal injection in a volume of 0.2 ml at 20 mg/kg/day using a 1-ml syringe with a 30-gauge needle. At 60 min after the last injection, animals were euthanized using carbon dioxide asphyxiation followed by cervical dislocation and then dissected. Blood was harvested by cardiac puncture and stored on ice. Acidified citrate dextrose was used as an anticoagulant. Immediately after blood collection, heart, liver, kidneys, and both hind leg quadriceps muscles were removed and snap frozen in liquid nitrogen. Average ischemia time before organ harvest was 1.75 min. Blood was centrifuged in an Eppendorf 5415R refrigerated microcentrifuge at 9,300 × g to isolate plasma, which was subsequently stored at −80 °C. All animal studies were approved by the University of Texas Southwestern Medical Center Institutional Animal Care and Use Committee.
Assays for BCKDC Activity in Cells and Mouse Tissues
Whole-cell assays for the decarboxylation of α-keto[1-14C] isovalerate and assays for reconstituted BCKDC activity using cell homogenates were carried out as prescribed previously (30). To measure BCKDC activity in tissues, individual kidneys (100–150 mg), hearts (120–200 mg), muscle (100–200 mg), and livers (250–400 mg) harvested from vehicle- and BT2-treated mice were flash-frozen in liquid nitrogen, thawed, and then manually homogenized in an ice-cold glass homogenizer containing 1 ml of a homogenizing buffer. The homogenizing buffer contained 30 mm KPi, pH 7.5, 3 mm EDTA, 5 mm DTT, 1 mm KIC, 3% fetal bovine serum, 5% Triton X-100, and 1 μm leupeptin. Samples were transferred to ice-cold 10-ml polycarbonate tubes and spun in an ultracentrifuge at 25,000 × g for 10 min to pellet cell and tissue debris. Supernatants were removed and stored on ice until diluted (1:3 for muscle, 1:6 for kidneys and heart, and 1:20 for liver) with a dilution buffer containing 50 mm HEPES, pH 7.5, 0.5 mm DTT, 0.1% Triton X-100, 3% fetal bovine serum, and 1 μm leupeptin. Diluted samples (50 μl) were placed in 24-well assay plates containing 295 μl of assay buffer per well. Assay buffer contained 30 mm KPi, pH 7.5, 0.4 mm CoA, 3 mm NAD+, 5% fetal bovine serum, 2 mm thiamine diphosphate, 2 mm MgCl2, and 65 μg of human E3. Twenty-five μl of α-keto[1-14C]isovalerate substrate was added to each well to begin the activity assay then sealed with a clear mylar adhesive film. Assay plates were incubated at 37 °C for 30 min and then placed back on ice and allowed to cool for 10 min. 50 μl of a 20% TCA solution was added to each well in order to stop the reactions. Assay plates were incubated further at 37 °C for 45 min. 14CO2 trapped on 2 m NaOH soaked filter wicks was counted in a liquid scintillation counter.
Western Blotting
Antibody against an E1 peptide containing phosphorylated Ser-292 (pE1) was a gift from Christopher Lynch (Pennsylvania State University School of Medicine) (31). Antibody against the E1 protein was prepared in the antibody core facility at the University of Texas Southwestern Medical Center and affinity-purified. Antibodies against GAPDH and BDK were obtained from MitoSciences/Abcam (Cambridge, MA) and LSBio (Seattle, WA), respectively. The chemiluminescence immunoassay was carried out as described previously (21). One ml of Luminata Forte Western HRP (Millipore Corp., Billerica, MA) substrate reagent was pipetted across the membrane for signal detection in a FluorChem E system (Cell Biosciences, Santa Clara, CA). The blots of four different tissues probed with the same antibodies were exposed and processed concurrently. Densitometry analysis was performed using the software AlphaVIEW SA provided by Cell Biosciences, Inc.
Determination of BCAA Concentrations
Plasma valine and leucine/isoleucine concentrations were determined by LC-MS/MS in a 4000 Qtrap mass spectrometer coupled to a Shimadzu Prominence LC as described previously (21). In the present analysis, valine and leucine/isoleucine standard curves were prepared by spiking known concentrations of each amino acid into murine plasma. The endogenous signal from blank plasma was subtracted from each point on the standard curve, and the data were plotted in GraphPad Prism. BCAA concentrations in treated samples were determined based on this standard curve.
Quantitative PCR
Total RNA from tissues was isolated with the RNeasy minikit (Qiagen) and converted into cDNA using the QuantiTect SYBR Green RT-PCR kit (Qiagene). PCR was performed on an AB 7900HT fast real-time PCR system (Applied Biosystems) using a BDK-specific primer pair (5′-CTCCCACCATGATGCTCTATTC-3′ (forward) and 5′-GAGCAATCCTCACTGGTAACTC-3′ (reverse)) or a GAPDH-specific primer pair (5′-CAAGGTCATCCATGACAACTTTG-3′ (forward) and 5′-GAGCAATCCTCACTGGTAACTC-3′ (reverse)). GAPDH was used as an internal control to normalize RNA input.
Statistical Analysis
Prism version 6.0 (GraphPad Inc.) was used to perform the two-tailed unpaired t test for comparison between groups. S.D. values are depicted as error bars in the plots.
RESULTS
High-throughput Screening Identified Benzothiophene Carboxylate Derivatives as Novel BDK Inhibitors
To carry out the high-throughput screens of BDK inhibitors, we developed a robust fluorescence-based assay. The assay employed a novel fusion peptide that links the LBD of E2 to a 14-mer peptide containing phosphorylation site 1 (Ser-292) and the flanking amino acid residues of the E1α subunit. The LBD binds and activates BDK, whereas the tethered BDK is brought juxtaposed to the peptide substrate through the linker. The phosphorylation reaction product ADP is detected using the ADP Hunter assay kit (DiscoveRx, Fremont, CA). This assay kit contains the coupling enzymes pyruvate kinase and pyruvate oxidase, which function in sequence to produce hydrogen peroxide from ADP. The product hydrogen peroxide, when catalyzed by the third enzyme peroxidase, converts the dye precursor Amplex to fluorescent resorufin. The final product, upon excitation at 530 nm, generates a fluorescence emission at 590 nm. The assay is robust, with relatively low background signal; the Z′ factor is in the excellent range of 0.79–0.86 (see “Experimental Procedures”).
In the high-throughput screen core facility, we screened a 2,000-compound pilot library. Among the four initially confirmed hits, compound 476-I16 (Fig. 1A) shows the best IC50 of ∼10 μm. However, LC-MS/MS analysis disclosed an m/z value drastically smaller than predicted from its chemical formula (data not shown). An HPLC elution profile subsequently indicated that >95% of compound 476-I16 had been degraded into two smaller fragments, BT1 and BT2 (Fig. 1B). The BT1 fraction is a very weak inhibitor for BDK in vitro (IC50 > 400 μm) (Fig. 1C), which is at variance with BT2, which inactivates BDK efficiently (IC50 = 3.4 μm) (Fig. 1D). In cell-based assays, the inhibition of BDK is reflected by increased BCKDC activity in MEF. Paradoxically, the BT1 faction shows significantly more enhancement of BCKDC activity than BT2, with parental compound 467-I16 (a mixture of BT1 and BT2) showing intermediate activation (Fig. 1E). We interpreted the data as indicating that BT1 is converted to BT2, with the latter being the actual BDK inhibitor. It follows that BT1 is more efficient than BT2 with respect to delivery into the cell resulting in the more profound augmentation of BCKDC activity; therefore, by definition, BT1 is a prodrug of BT2. The interconversion between 476-I16 (m/z = 559.5), BT1 (m/z = 329.8), and BT2 (m/z = 245.9) was confirmed by LC-MS analysis (Fig. 1F). An analog of BT1 (i.e. BT3 (m/z = 371.8)) was synthesized, which was also converted to BT2 (Fig. 1F) and used as another prodrug of BT2 in subsequent cell-based studies. The novel BDK inhibitor BT2 is similar to the known inhibitor (S)-CPP (Fig. 1G) that we reported earlier (21) in that both compounds possess the carboxylate moiety. However, BT2 contains a benzothiophene derivative instead of a phenol ring as in (S)-CPP. Another benzothiophene carboxylate derivative, BT2F (Fig. 1G), was found to also be a BDK inhibitor and used in cell culture studies (see below).
FIGURE 1.

Identification of benzothiophene carboxylate derivatives as novel BDK inhibitors. A, candidate BDK inhibitor 476-I16 identified by high-throughput screening of a 2,000-compound chemical library. B, separation of compound 476-I16 into smaller compound fractions BT1 and BT2 by reversed-phase HPLC. C, absence of BDK inhibition by BT1. D, inhibition of BDK as a function of BT2 concentrations. E, activation of BCKDC in MEF cells by BT1, BT2, and a mixture of both from compound 476-I16, compared with the DMSO control. F, chemical structures, degradation schemes, and molecular masses (m/z) of benzothiophene carboxylate derivatives. BT3 is an analog of BT1. G, chemical structures of (S)-CPP, BT2, and BT2F. Error bars, S.D.
BT2 and (S)-CPP Bind to the Same Allosteric Site in BDK
The crystal structure of BDK containing BT2 was determined. Fig. 2A shows that BT2 binds to an allosteric site in the N-terminal domain of each monomer in homodimeric BDK, similar to (S)-CPP, as we reported previously (21). The allosteric site is 20–30 Å away from the ATP/ADP-binding pocket located in the C-terminal domain. A detailed structure (Fig. 2B) shows that the carboxylate oxygen atoms contact side chains of Tyr-99, His-132, Arg-167, and Arg-171, and the benzothiophene moiety is accommodated in a hydrophobic pocket formed by side chains of Leu-68, Ile-72, Leu-106, Val-125, Leu-128, Leu-129, and Ile-170. The configuration of bound BT2 (Fig. 2C) is similar to that of bound (S)-CPP (Fig. 2D), reported previously (21). However, there are subtle differences between the two configurations of bound ligands. The top view of the surface map (in magenta mesh), contoured by side chains of the surrounding amino acids, shows that the entire bound BT2 is in a strict planar conformation (Fig. 2E). Both the front and side views illustrate that bound BT2 fits snugly in the pocket defined by the surface map (Fig. 2E). In contrast, the alkylphenyl moiety of bound (S)-CPP is out of the planar conformation, as depicted in the top view (Fig. 2F). The front and side views show more unoccupied spaces in the pocket harboring (S)-CPP than BT2 (Fig. 2F). The results indicate that the allosteric site in BDK can be contoured by the bound ligands into different conformations.
FIGURE 2.
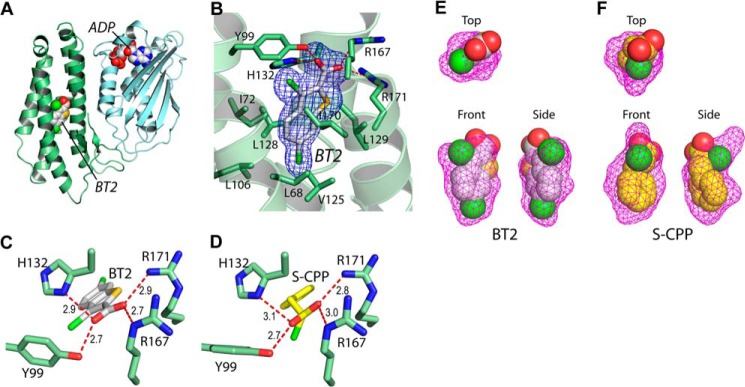
Crystal structures of rat BDK in complex with BDK inhibitors BT2 and (S)-CPP. A, bound BT2 and ADP (in the space-filling model) in the N-terminal domain (green) and C-terminal domain (cyan), respectively, of a BDK monomer. B, the Fo − Fc omit density map of bound BT2 in the allosteric site. C and D, hydrogen bonding networks of bound BT2 and (S)-CPP, respectively, in the allosteric site. E and F, surface maps of the allosteric pocket harboring BT2 and (S)-CPP, respectively.
BT2 and Its Prodrug BT3 Markedly Augment BCKDC Activity in Cultured Cells and Primary Hepatocytes
When compared with the DMSO-treated control, prodrug BT3 stimulates BCKDC activity far more robustly than BT2 in wild-type MEF cells cultured in the presence of a 40 μm concentration of either compound for 48 h (Fig. 3A, left). In BDP−/− MEF cells, BCKDC is largely phosphorylated due to the absence of the phosphatase. When cultured in the presence of either 20 μm BT2 or BT3 for 96 h, de novo synthesized BCKDC, in part, becomes dephosphorylated and active; again, more active BCKDC is present in BT3-treated than in BT2-treated cells (Fig. 3A, right). These results suggest the presence of the oxydiazol ring-containing promoiety in BT3 results in more efficient delivery of the compound than BT2 into MEF cells, as reflected by a greater -fold activation of BCKDC activity than BT2.
FIGURE 3.
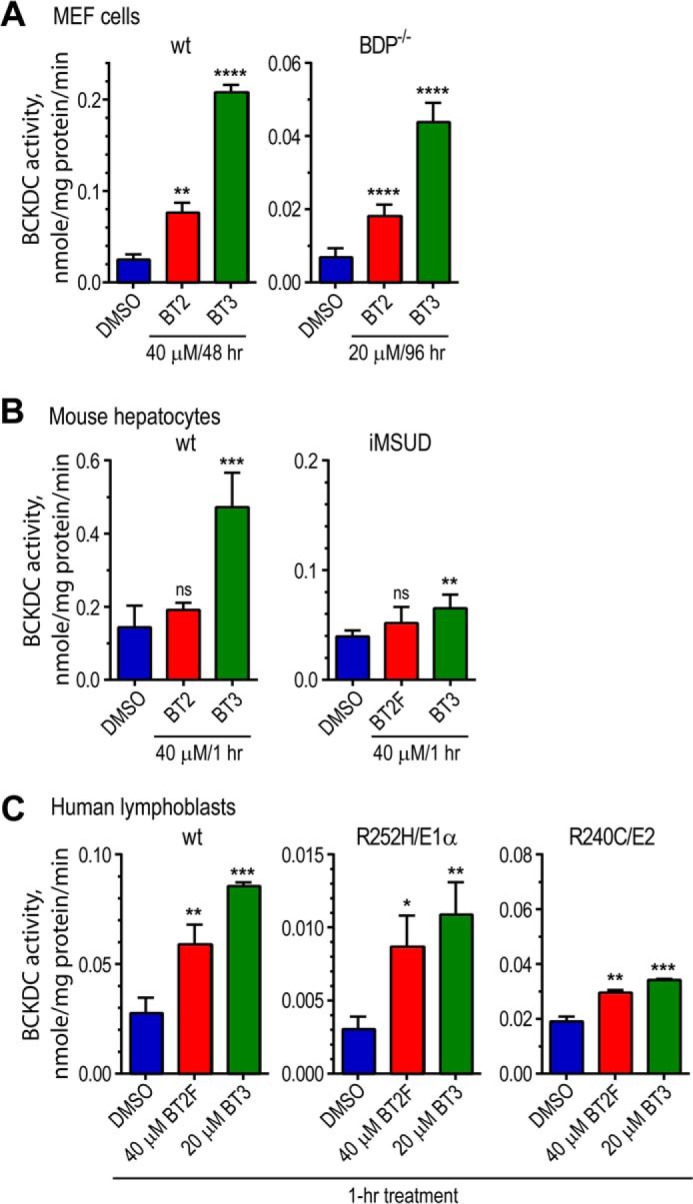
BT2, BT2F, and BT3 significantly increase BCKDC activity in wild-type and MSUD cells. A, wild-type MEF cells were cultured with 40 μm BT2, BT2F, or BT3 for 48 h with DMSO as a control. BDP−/− cells manifesting intermediate MSUD were grown in the presence of 20 μm BT2 or BT3 for 96 h. BCKDC activity was measured by the intact cell assay using α-keto[1-14C] isovalerate as a substrate. B, primary hepatocytes prepared from wild-type MSUD or iMSUD mice were incubated in the Krebs buffer with BT2 or BT3 (40 μm) for 1 h and assayed for BCKDC activity in a reconstituted reaction mixture. C, harvested lymphoblasts from control subjects and intermediate MSUD patients carrying R252H/E1α or R240C/E2 (both mature sequences) were incubated in the Krebs buffer for 1 h with 40 μm BT2 or BT2F or 20 μm BT3. Wild-type and mutant BCKDC activity was measured by the intact cell assay. Error bars, S.D.
Primary hepatocytes were prepared from wild-type and intermediate MSUD mice. The iMSUD mice harbor a hepatic-specific trimeric E2 (instead of wild-type 24-meric E2) in an E2-null background, resulting in only 5% hepatic BCKDC activity relative to wild type (32). Primary hepatocytes from wild-type mice treated for 1 h with 40 μm BT3, but not BT2, show significant augmentation of BCKDC activity (Fig. 3B, left). Similarly, primary hepatocytes from iMSUD mice exhibit a significant increase in mutant BCKDC activity when treated for 1 h with 40 μm BT3 but not BT2 (Fig. 3B, right). The increased BCKDC activity in wild-type and iMSUD hepatocytes coincides with significantly reduced phosphorylation in the E1α subunit of wild-type and mutant BCKDC (data not shown). These results again indicate that BT3 as a prodrug of BT2 is more efficient than BT2 itself in enhancing BCKDC activity at the cellular level. The smaller -fold increase in BCKDC activity in wild-type primary hepatocytes compared with wild-type MEF cells is explained by the fact that the BCKDC in primary hepatocytes is markedly less phosphorylated than that in MEF cells.
The efficacy of the BDK inhibitors in restoring BCKDC activity in lymphoblast cell lines from MSUD patients was also investigated. Treatment of wild-type lymphoblasts with 20 μm BT3 in the culture medium for 1 h results in markedly higher BCKDC activity than the DMSO control (Fig. 3C, left). Treatment with a BT2 analog, BT2F, in which the chlorine in the 6-position of BT2 is replaced with a fluorine, shows smaller increases in wild-type BCKDC activity compared with BT3-treated lymphoblast. Incubations of MSUD lymphoblasts carrying the homozygous R252H mutation in the E1α subunit with 20 μm BT3 and 40 μm BT2F led to significantly higher mutant BCKDC activity than the DMSO control, with BT3-treated cells showing higher activity than the BT2F-treated ones (Fig. 3C, middle). Increased mutant BCKDC activity also occurs, when lymphoblasts from an MSUD patient carrying the homozygous R240C mutation in the E2 subunit were treated similarly with BT3 and BT2F, with higher activation observed with the former compound (Fig. 3C, right).
BT2 Shows Good in Vitro Metabolic Stability That Translates into a Long Half-life and High Plasma Levels in Vivo
When BT3 was incubated with liver S9 fractions comprising the cytosolic and microsomal portion of hepatocytes, the compound rapidly disappeared from the solution (T½ = 40 min) with a rapid rise in the BT2 concentration (Fig. 4A). The result obtained with these liver fractions corroborates the conversion of BT1 or BT3 to BT2 inside the cell, as deduced from the above physical and cellular studies (Fig. 1, A–F). The incubation of BT2 with the S9 fraction shows that compound concentration remains unchanged in 240 min (Fig. 4B), suggesting that BT2 is highly stable. Not surprisingly, when BT2 is administered in vivo by the intraperitoneal route, it is slowly cleared from the plasma as well, with a terminal T½ = 730 min (Fig. 4C). Interestingly, BT2 also has an extremely low volume of distribution, close to only twice the blood volume of a mouse. In vitro studies revealed that a high percentage (99.3%) of BT2 is bound to plasma protein (Table 2). In addition, the carboxylic acid moiety on the molecule is probably charged at physiological pH, making tissue distribution inefficient. The slow clearance of BT2 from plasma can probably be explained by the high metabolic stability (Fig. 4B), binding of the vast majority of the compound to the plasma protein fraction, and limited tissue distribution (Table 2). Details of the pharmacological properties of BT2 are summarized in Table 2.
FIGURE 4.

Metabolic stability and pharmacokinetics of BT2 and BT3. A, metabolic stability of BT3 in S9 fractions. BT3 (2 μm) was incubated with S9 factions; concentrations of BT3 and BT2 in aliquots taken at different time points were determined by LC-MS/MS. B, metabolic stability of BT2 in S9 factions. The remaining BT2 levels at different time points were determined by LC-MS/MS and expressed as ln(percentage of initial BT2 concentration). C, pharmacokinetics of plasma BT2 depletion. BT2 (10 mg/kg) was administered to CD-1 male mice by intraperitoneal injection. Plasma was collected from different time points (n = 3). BT2 concentrations were determined by LC-MS/MS. ns, not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 when compared with the group treated with DMSO. Error bars, S.D.
TABLE 2.
Pharmacokinetic parameters for (S)-CPP and BT2
Terminal T½, half-life of the terminal phase; Cmax, observed maximum plasma concentration; Tmax, time to reach Cmax; AUClast, area under the concentration-time curve from 0 to the last measured point; Vz/F, apparent volume of distribution during terminal phase; CL/F, volume of plasma cleared of the drug per unit time, where F is the fraction bioavailable as compared with an intravenous dose, which is not known.
| BT2 | (S)-CPPa | |
|---|---|---|
| Plasma pharmacokinetics | ||
| Dose | 10 mg/kg IP | 40 mg/kg IP |
| Terminal T½ | 730 min | 127 min |
| Cmax | 71,600 ng/ml | 88,067 ng/ml |
| Tmax | 30 min | 10 min |
| AUClast | 69,834,000 min·ng/ml | 8,354,759 min·ng/ml |
| Vz/F | 3.80 ml | 20.8 ml |
| CL/F | 0.00361 ml/min | 0.113 ml/min |
| Stability in S9 fraction | ||
| T½ | ≫ 240 min | 187 min |
| Plasma protein binding | ||
| Fraction bound | 99.3% | 72.8% |
a The (S)-CPP data, except that for plasma protein binding, were taken from Ref. 21 for the purpose of comparison.
BT2 Administration Markedly Augments BCKDC Activity in Different Tissues
Based on the above pharmacokinetics of BT2, wild-type mice fed a standard chow diet were dosed with 5% DMSO, 10% cremophor EL, and 85% of 0.1 m sodium bicarbonate, pH 9.0 (vehicle) or 20 mg/kg/day BT2 for 5 days. At the end of the treatment, the mice were sacrificed at 9 a.m., when the animals were in the fed state. Fig. 5A (top) shows that BCKDC activity was robustly (12.3-fold) enhanced in the heart compared with the vehicle-treated animals. Less activation was obtained in muscle and kidney at 3.6- and 3.8-fold, respectively. Essentially no increase in BCKDC activity was observed in liver, where the enzyme complex is essentially dephosphorylated before the BT2 treatment. The -fold activation of BCKDC activity in the above tissues correlates with decreased phosphorylation in heart, muscle, and kidney after the long term BT2 treatment (Fig. 5A, bottom). Plasma BCAA concentrations in vehicle- and BT2-treated mice were determined by LC-MS/MS. The long term treatment with BT2 results in reduction in plasma valine (p = 0.14) and leucine + isoleucine concentrations (p = 0.12) (Fig. 5B).
FIGURE 5.

Robust simulation of BDKDC activity in mouse tissues with decreased plasma BCAA concentrations. A, BCKDC activity in heart, muscle, kidney, and liver from wild-type C57BL/6J mice treated with vehicle (V) or BT2 (T) for 1 week. Shaded bars represent the mean of BCKDC activities in tissues from an individual mouse (n = 4); *, p < 0.05; ****, p < 0.0001. B, relative protein levels of phosphorylated E1 (pE1) and total E1 (E1) in tissues from vehicle- and BT2-treated mice. GAPDH served as a loading control. The amounts of tissue homogenate applied were 15 μg for liver, 30 μg for heart and kidney, and 60 μg for muscle. C, decreased plasma BCAA concentrations in BT2-treated mice compared with vehicle-treated (n = 4). Leu + Ile, sum of the two amino acids that are indistinguishable in LC-MS/MS. Error bars, S.D.
BT2 Treatment Reduces BDK Protein Levels in Vivo
Tissues from the above vehicle- and BT2-treated wild-type mice were analyzed for the BDK protein and mRNA levels by Western blotting and quantitative PCR measurements. The protein levels of BDK in kidneys and heart were reduced to averages of 39 and 24%, respectively, in the BT2-treated tissues compared with the vehicle-treated ones (Fig. 6A). BDK protein levels in liver and muscle were too low to be detected (data not shown). The mRNA levels in BT2-treated kidneys are unchanged relative to those in the vehicle-treated kidneys (Fig. 6B), which shows that the BT2 treatment does not alter the expression of the BDK. These results, taken together, indicate that the dissociation of BDK from the BCKDC in BT2-treated tissues accelerates the degradation of BDK, resulting in reduced levels of the kinase.
FIGURE 6.
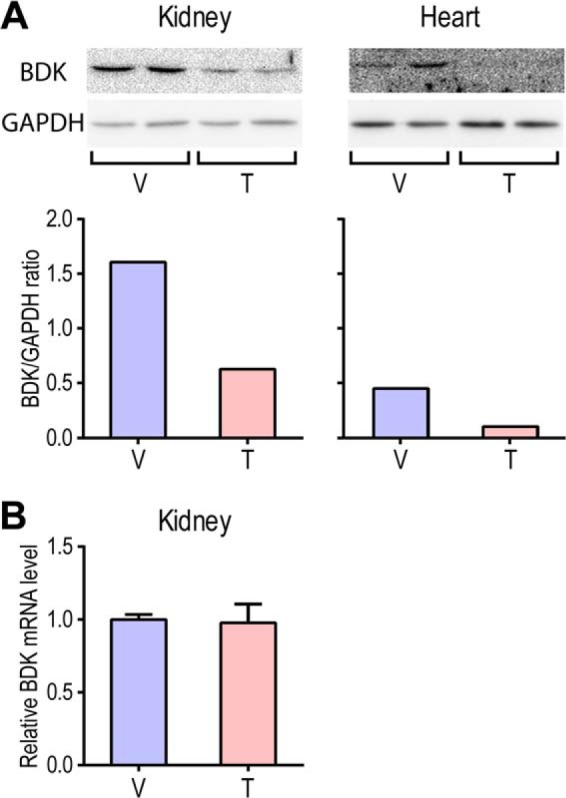
Protein and mRNA levels of BDK in kidneys and heart from vehicle- and BT2-treated wild-type mice. A, Western blots (top) of kidney and heart homogenates from vehicle-treated (V) and BT2-treated (T) mice with GAPDH as loading controls. The quantification of band densities shows that BDK protein levels in BT2-treated kidney and heart are reduced to 39 and 24%, respectively, compared with the vehicle-treated ones (bottom). B, BDK mRNA levels determined by quantitative PCR in kidneys from BT2-treated mice relative to the vehicle-treated ones (set as 1.0). The GAPDH mRNA level was used as an internal control. Error bars, S.D.
DISCUSSION
BDK inhibitors that augment BCKDC flux and reduce BCAA concentrations provide a plausible approach to the treatment of disorders in BCAA catabolism. As described above, there are presently only a handful of known BDK inhibitors. However, these compounds are unsatisfactory in that they have either high IC50 values or low metabolic stability in vivo. These properties render the known BDK inhibitors less than ideal for long term studies in animal models for disorders in BCAA metabolism. In the present study, we employed high-throughput screening to identify what turned out to be a prodrug of a BDK inhibitor in BT2. BT2 belongs to a family of benzothiophene derivatives, several of which are already reported to be enzyme inhibitors. Specifically, benzothiophene carboxylates, including BT2, are moieties of potent Mcl-1 inhibitors developed by a fragment-based method for the treatment of a wide range of cancers (33). Benzothiophene carboxamide derivatives shown to be inhibitors of enoyl-acyl carrier protein reductase from Plasmodium falciparum are important potential anti-malarial drugs (34). Moreover, BT2-related benzothiophene hydroxamic acids are botulinum nerotoxin serotype A protease inhibitors (35). These results along with the present study, which shows that BT2 is a BDK inhibitor, establish benzothiophene derivatives as important pharmaceutical compounds for treatment of human disease.
We reported earlier that the known BDK inhibitors, such as (S)-CPP, (R)-CPP, phenylbutyrate, (S)-CIC, and (R)-CIC, bind to the same allosteric site in the N-terminal domain of the BCK subunit (21). Here, we show that BT2 also occupies this allosteric site in BDK, similar to (S)-CPP. In the BDK-BT2 structure, the benzothiophene ring is housed in a hydrophobic niche, which accommodates the phenol ring in the BDK-(S)-CPP structure, with the carboxylate oxygen atoms from both ligands interacting similarly with side chains of the same hydrophilic residues (Fig. 2, C and D). The better fit of BT2 in the binding pocket than (S)-CPP (Fig. 2, E and F) may explain the slightly better IC50 of the former (3.19 μm) compared with the latter (6.3 μm) (21). Analogous to (S)-CPP (21), the binding of BT2 to the allosteric pocket triggers helix movement that communicates to the lipoyl-bearing domain, resulting in the dissociation of BDK from the BCKDC scaffold. The release of BDK from the BCKDC was previously observed with other BDK inhibitors and BCKA that bind to the same allosteric site (12, 36). In this work, we provide the first evidence indicating that the free BDK is less stable than the bound form, which results in the reduced level of BDK protein in kidney and heart from the BT2-treated versus the vehicle-treated wild-type mice (Fig. 6, A and B). Thus, allosteric BDK inhibitors, such as BT2 and (S)-CPP, have a dual function of reducing both kinase activity and the protein level, with both mechanisms contributing to the reduced phosphorylation and activation of BCKDC in vivo (Fig. 5, A and B).
The size limit of the allosteric site precludes the binding of the significantly larger BT2 prodrug BT1, as indicated by the absence of BDK inhibition by the latter compound (Fig. 1C). Therefore, it is serendipitous that during the high-throughput screens, compound 476-I16 in the chemical library was partially decomposed into daughter compounds BT1 and BT2 (Fig. 1B). The presence of BT2 in the decomposed 476-I16 compound mixture enabled us to eventually identify BT2 as a novel allosteric inhibitor for BDK. On the other hand, the presence of the oxadiazole moiety in the prodrug of BT2 (i.e. BT3) masks the charged carboxylate group in BT2 and probably promotes the more efficient import of BT3 than BT2 into cells. This feature explains the higher -fold increases in BCKDC activity by BT3 than BT2 in cell culture and primary hepatocytes (Fig. 3). Although BT3 should be theoretically more efficacious in animal studies than BT2, we were unable to find an acceptable formulation to administer BT3 for preclinical animal trials despite extensive efforts. Moreover, BT3 was found to be nearly 100% cleared from the plasma within 30 min following intraperitoneal administration in mice using 100% DMSO (data not shown). The combined results indicate that BT3 is not likely to provide much of an advantage over BT2 in animal studies. Therefore, BT2 was utilized for all in vivo studies.
In addition to showing a slightly better fit into the binding pocket of BDK, BT2 also possesses better pharmacokinetic properties than (S)-CPP (Table 2). BT2 is more stable than (S)-CPP, as indicated by the lower plasma clearance and longer terminal T½ for BT2 versus (S)-CPP in vivo as well as better in vitro metabolic stability, as shown in S9 assays (T½ > 240 min for BT2 versus T½ = 178 min for (S)-CPP). The higher percentage of binding to plasma proteins for BT2 (99.3%) versus (S)-CPP (72.8%) probably also results in slower rates of elimination for the former compound. The improved pharmacological properties of BT2 over (S)-CPP allow for the use of a significantly lower dose of BT2 than (S)-CPP for in vivo studies. We showed previously that a short term dosing of (S)-CPP at 160 mg/kg intraperitoneally to wild-type mice resulted in complete dephosphorylation of BCKDC in all tissues with concomitant reduction in plasma BCAA concentrations (21). In the long term study described here, we treated wild-type mice with BT2 at 20 mg/kg/day intraperitoneally for 1 week and also achieved nearly complete dephosphorylation and maximal activation of BCKDC (Fig. 5). In a parallel long term study with (S)-CPP, similar results can be obtained only with a 2 dose/day regimen of 80 mg/kg in the morning and 160 mg/kg in the afternoon by intraperitoneal injections (data not shown). Therefore, the more stable and potent BDK inhibitor in BT2, compared with (S)-CPP, offers an efficient tool to modulate BCAA catabolism in vivo.
BCAA and related metabolites are associated with insulin resistance, diabetes, and coronary artery disease (37–39) and are predictive of incident type 2 diabetes (5). These metabolites are prognostic of obesity intervention outcomes (40) and show favorable responses to therapeutic procedures such as gastric bypass (41). These strong associations of BCAA with human disease argue for a better understanding of the mechanisms that drive changes in their circulating levels and for testing the therapeutic impact of new methods for altering BCAA metabolism (42). These recent findings advance the BCAA field beyond the historical observations of Cahill and others linking BCAA and obesity/insulin resistance (43, 44). The results have renewed interest in the possible cause-effect relationships between elevated BCAA and insulin resistance in type 2 diabetes. Modulation of BCAA catabolism in vivo by BDK inhibitors like BT2 may eventually shed light on the cause-effect relationship of elevated BCAA in animal models for insulin-resistant type 2 diabetes.
Acknowledgments
We are indebted to Drs. Shuquang Wei and Bruce Posner for assistance in the high-throughput screens of BDK inhibitors, Dr. Changguang Wang for conducting measurements of protein binding for BT2 and (S)-CPP, and Dr. Laszlo Kurti for help in the HPLC separation of compounds BT1 and BT2. We are grateful to Dr. Yibin Wang for the generous supply of wild-type and BDP−/− MEF cells, Crystal structures presented in this report are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source, operated under Department of Energy Contract DE-AC02-06CH11357.
This work was supported, in whole or in part, by National Institutes of Health Grants DK62306, DK26758, and DK92921. This work was also supported by Welch Foundation Grants I-1286 and I-1748 and the Sloan Research Fellowship.
The atomic coordinates and structure factors (codes 4E00 and 4E01) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BCAA
- branched-chain amino acid(s)
- BCKA
- branched-chain α-ketoacid(s)
- BCKD
- branched-chain α-ketoacid dehydrogenase
- BCKDC
- branched-chain α-ketoacid dehydrogenase complex
- BDK
- branched-chain α-ketoacid dehydrogenase kinase
- E1
- branched-chain α-ketoacid decarboxylase
- E2
- dihydrolipoyl transacylase
- E3
- dihydrolipoamide dehydrogenase
- LBD
- lipoyl-bearing domain
- KIC
- α-ketoisocaproate
- BT2
- 3,6-dichlorobenzo[b]thiophene-2-carboxylic acid
- CPP
- α-cholorophenylproprionate
- MSUD
- maple syrup urine disease
- iMSUD
- intermediate MSUD
- BDP
- BCKD phosphatase
- CIC
- α-chloroisocaproate
- AMPPNP
- 5′-adenylyl-β,γ-imidodiphosphate
- MEF
- mouse embryo fibroblast(s).
REFERENCES
- 1. Iwasa M., Kobayashi Y., Mifuji-Moroka R., Hara N., Miyachi H., Sugimoto R., Tanaka H., Fujita N., Gabazza E. C., Takei Y. (2013) Branched-chain amino acid supplementation reduces oxidative stress and prolongs survival in rats with advanced liver cirrhosis. PloS One 8, e70309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. D'Antona G., Ragni M., Cardile A., Tedesco L., Dossena M., Bruttini F., Caliaro F., Corsetti G., Bottinelli R., Carruba M. O., Valerio A., Nisoli E. (2010) Branched-chain amino acid supplementation promotes survival and supports cardiac and skeletal muscle mitochondrial biogenesis in middle-aged mice. Cell Metab. 12, 362–372 [DOI] [PubMed] [Google Scholar]
- 3. Chuang D. T., Shih V. E. (2001) Maple syrup urine disease (branched-chain ketoaciduria). in The Metabolic and Molecular Basis of Inherited Disease, 8th Ed (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Vogelstein B., Childs B., eds) pp. 1971–2006, McGraw-Hill, New York [Google Scholar]
- 4. Chuang D. T., Wynn R. M., Shih V. E. (2008) Maple syrup urine disease (branched-chain ketoaciduria): an update. in The Metabolic and Molecular Basis of Inherited Disease, Online Ed (Scriver C. R., Beaudet A. L., Sly W. S., Valle D., Vogelstein B., Childs B., eds) pp. 1–42, McGraw-Hill, New York [Google Scholar]
- 5. Wang T. J., Larson M. G., Vasan R. S., Cheng S., Rhee E. P., McCabe E., Lewis G. D., Fox C. S., Jacques P. F., Fernandez C., O'Donnell C. J., Carr S. A., Mootha V. K., Florez J. C., Souza A., Melander O., Clish C. B., Gerszten R. E. (2011) Metabolite profiles and the risk of developing diabetes. Nat. Med. 17, 448–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Würtz P., Soininen P., Kangas A. J., Rönnemaa T., Lehtimäki T., Kähönen M., Viikari J. S., Raitakari O. T., Ala-Korpela M. (2013) Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care 36, 648–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao Y., Hawes J., Popov K. M., Jaskiewicz J., Shimomura Y., Crabb D. W., Harris R. A. (1994) Site-directed mutagenesis of phosphorylation sites of the branched chain α-ketoacid dehydrogenase complex. J. Biol. Chem. 269, 18583–18587 [PubMed] [Google Scholar]
- 8. Wynn R. M., Kato M., Machius M., Chuang J. L., Li J., Tomchick D. R., Chuang D. T. (2004) Molecular mechanism for regulation of the human mitochondrial branched-chain α-ketoacid dehydrogenase complex by phosphorylation. Structure 12, 2185–2196 [DOI] [PubMed] [Google Scholar]
- 9. Aevarsson A., Seger K., Turley S., Sokatch J. R., Hol W. G. (1999) Crystal structure of 2-oxoisovalerate and dehydrogenase and the architecture of 2-oxo acid dehydrogenase multienzyme complexes. Nat. Struct. Biol. 6, 785–792 [DOI] [PubMed] [Google Scholar]
- 10. Davie J. R., Wynn R. M., Meng M., Huang Y. S., Aalund G., Chuang D. T., Lau K. S. (1995) Expression and characterization of branched-chain α-ketoacid dehydrogenase kinase from the rat. Is it a histidine-protein kinase? J. Biol. Chem. 270, 19861–19867 [DOI] [PubMed] [Google Scholar]
- 11. Wynn R. M., Li J., Brautigam C. A., Chuang J. L., Chuang D. T. (2012) Structural and biochemical characterization of human mitochondrial branched-chain α-ketoacid dehydrogenase phosphatase. J. Biol. Chem. 287, 9178–9192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou M., Lu G., Gao C., Wang Y., Sun H. (2012) Tissue-specific and nutrient regulation of the branched-chain α-keto acid dehydrogenase phosphatase, protein phosphatase 2Cm (PP2Cm). J. Biol. Chem. 287, 23397–23406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Harper A. E., Miller R. H., Block K. P. (1984) Branched-chain amino acid metabolism. Annu. Rev. Nutr. 4, 409–454 [DOI] [PubMed] [Google Scholar]
- 14. Harris R. A., Popov K. M., Zhao Y., Shimomura Y. (1994) Regulation of branched-chain amino acid catabolism. J. Nutr. 124, 1499S–1502S [DOI] [PubMed] [Google Scholar]
- 15. Harris R. A., Joshi M., Jeoung N. H. (2004) Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem. Biophys. Res. Commun. 313, 391–396 [DOI] [PubMed] [Google Scholar]
- 16. Paxton R., Harris R. A. (1984) Regulation of branched-chain α-ketoacid dehydrogenase kinase. Arch. Biochem. Biophys. 231, 48–57 [DOI] [PubMed] [Google Scholar]
- 17. Paxton R., Harris R. A. (1984) Clofibric acid, phenylpyruvate, and dichloroacetate inhibition of branched-chain α-ketoacid dehydrogenase kinase in vitro and in perfused rat heart. Arch. Biochem. Biophys. 231, 58–66 [DOI] [PubMed] [Google Scholar]
- 18. Harris R. A., Paxton R., DePaoli-Roach A. A. (1982) Inhibition of branched chain α-ketoacid dehydrogenase kinase activity by α-chloroisocaproate. J. Biol. Chem. 257, 13915–13918 [PubMed] [Google Scholar]
- 19. Kobayashi R., Murakami T., Obayashi M., Nakai N., Jaskiewicz J., Fujiwara Y., Shimomura Y., Harris R. A. (2002) Clofibric acid stimulates branched-chain amino acid catabolism by three mechanisms. Arch. Biochem. Biophys. 407, 231–240 [DOI] [PubMed] [Google Scholar]
- 20. Brunetti-Pierri N., Lanpher B., Erez A., Ananieva E. A., Islam M., Marini J. C., Sun Q., Yu C., Hegde M., Li J., Wynn R. M., Chuang D. T., Hutson S., Lee B. (2011) Phenylbutyrate therapy for maple syrup urine disease. Hum. Mol. Genet. 20, 631–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tso S. C., Qi X., Gui W. J., Chuang J. L., Morlock L. K., Wallace A. L., Ahmed K., Laxman S., Campeau P. M., Lee B. H., Hutson S. M., Tu B. P., Williams N. S., Tambar U. K., Wynn R. M., Chuang D. T. (2013) Structure-based design and mechanisms of allosteric inhibitors for mitochondrial branched-chain α-ketoacid dehydrogenase kinase. Proc. Natl. Acad. Sci. U.S.A. 110, 9728–9733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Machius M., Chuang J. L., Wynn R. M., Tomchick D. R., Chuang D. T. (2001) Structure of rat BCKD kinase: nucleotide-induced domain communication in a mitochondrial protein kinase. Proc. Natl. Acad. Sci. U.S.A. 98, 11218–11223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chuang J. L., Wynn R. M., Chuang D. T. (2002) The C-terminal hinge region of lipoic acid-bearing domain of E2b is essential for domain interaction with branched-chain α-keto acid dehydrogenase kinase. J. Biol. Chem. 277, 36905–36908 [DOI] [PubMed] [Google Scholar]
- 24. Chuang J. L., Davie J. R., Wynn R. M., Chuang D. T. (2000) Production of recombinant mammalian holo-E2 and E3 and reconstitution of functional branched-chain α-keto acid dehydrogenase complex with recombinant E1. Methods Enzymol. 324, 192–200 [DOI] [PubMed] [Google Scholar]
- 25. Skvorak K. J., Paul H. S., Dorko K., Marongiu F., Ellis E., Chace D., Ferguson C., Gibson K. M., Homanics G. E., Strom S. C. (2009) Hepatocyte transplantation improves phenotype and extends survival in a murine model of intermediate maple syrup urine disease. Mol. Ther. 17, 1266–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lu G., Sun H., She P., Youn J. Y., Warburton S., Ping P., Vondriska T. M., Cai H., Lynch C. J., Wang Y. (2009) Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J. Clin. Invest. 119, 1678–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 28. Wynn R. M., Kato M., Chuang J. L., Tso S. C., Li J., Chuang D. T. (2008) Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J. Biol. Chem. 283, 25305–25315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang C., Williams N. S. (2013) A mass balance approach for calculation of recovery and binding enables the use of ultrafiltration as a rapid method for measurement of plasma protein binding for even highly lipophilic compounds. J. Pharm. Biomed. Anal. 75, 112–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chuang J. L., Chuang D. T. (2000) Diagnosis and mutational analysis of maple syrup urine disease using cell cultures. Methods Enzymol. 324, 413–423 [DOI] [PubMed] [Google Scholar]
- 31. She P., Van Horn C., Reid T., Hutson S. M., Cooney R. N., Lynch C. J. (2007) Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 293, E1552–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Homanics G. E., Skvorak K., Ferguson C., Watkins S., Paul H. S. (2006) Production and characterization of murine models of classic and intermediate maple syrup urine disease. BMC Med. Genet. 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friberg A., Vigil D., Zhao B., Daniels R. N., Burke J. P., Garcia-Barrantes P. M., Camper D., Chauder B. A., Lee T., Olejniczak E. T., Fesik S. W. (2013) Discovery of potent myeloid cell leukemia 1 (Mcl-1) inhibitors using fragment-based methods and structure-based design. J. Med. Chem. 56, 15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Banerjee T., Sharma S. K., Kapoor N., Dwivedi V., Surolia N., Surolia A. (2011) Benzothiophene carboxamide derivatives as inhibitors of Plasmodium falciparum enoyl-ACP reductase. IUBMB life 63, 1101–1110 [DOI] [PubMed] [Google Scholar]
- 35. Capek P., Zhang Y., Barlow D. J., Houseknecht K. L., Smith G. R., Dickerson T. J. (2011) Enhancing the pharmacokinetic properties of botulinum neurotoxin serotype A protease inhibitors through rational design. ACS Chem. Neurosci. 2, 288–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Murakami T., Matsuo M., Shimizu A., Shimomura Y. (2005) Dissociation of branched-chain α-keto acid dehydrogenase kinase (BDK) from branched-chain α-keto acid dehydrogenase complex (BCKDC) by BDK inhibitors. J. Nutr. Sci. Vitaminol. 51, 48–50 [DOI] [PubMed] [Google Scholar]
- 37. Newgard C. B., An J., Bain J. R., Muehlbauer M. J., Stevens R. D., Lien L. F., Haqq A. M., Shah S. H., Arlotto M., Slentz C. A., Rochon J., Gallup D., Ilkayeva O., Wenner B. R., Yancy W. S., Jr., Eisenson H., Musante G., Surwit R. S., Millington D. S., Butler M. D., Svetkey L. P. (2009) A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 9, 311–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Huffman K. M., Shah S. H., Stevens R. D., Bain J. R., Muehlbauer M., Slentz C. A., Tanner C. J., Kuchibhatla M., Houmard J. A., Newgard C. B., Kraus W. E. (2009) Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care 32, 1678–1683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shah S. H., Bain J. R., Muehlbauer M. J., Stevens R. D., Crosslin D. R., Haynes C., Dungan J., Newby L. K., Hauser E. R., Ginsburg G. S., Newgard C. B., Kraus W. E. (2010) Association of a peripheral blood metabolic profile with coronary artery disease and risk of subsequent cardiovascular events. Circ. Cardiovasc. Genet. 3, 207–214 [DOI] [PubMed] [Google Scholar]
- 40. Shah S. H., Crosslin D. R., Haynes C. S., Nelson S., Turer C. B., Stevens R. D., Muehlbauer M. J., Wenner B. R., Bain J. R., Laferrère B., Gorroochurn P., Teixeira J., Brantley P. J., Stevens V. J., Hollis J. F., Appel L. J., Lien L. F., Batch B., Newgard C. B., Svetkey L. P. (2012) Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia 55, 321–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Laferrère B., Reilly D., Arias S., Swerdlow N., Gorroochurn P., Bawa B., Bose M., Teixeira J., Stevens R. D., Wenner B. R., Bain J. R., Muehlbauer M. J., Haqq A., Lien L., Shah S. H., Svetkey L. P., Newgard C. B. (2011) Differential metabolic impact of gastric bypass surgery versus dietary intervention in obese diabetic subjects despite identical weight loss. Sci. Transl. Med. 3, 80re2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Newgard C. B. (2012) Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 15, 606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Felig P. (1975) Amino acid metabolism in man. Annu. Rev. Biochem. 44, 933–955 [DOI] [PubMed] [Google Scholar]
- 44. Felig P., Marliss E., Cahill G. F., Jr. (1969) Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med. 281, 811–816 [DOI] [PubMed] [Google Scholar]