

Background: Autophagy is essential for normal neuron maintenance but can also promote death.
Results: Brain-derived neurotrophic factor (BDNF) increases autophagosome formation but prevents excessive autophagic degradation by activating the mammalian target of rapamycin (mTOR).
Conclusion: BDNF-dependent neuron survival requires suppression of autophagic flux, which was impaired by both rapamycin and interleukin-1β (IL-1β).
Significance: mTOR inhibition subverts BDNF survival signals.
Keywords: Autophagy, Brain-derived Neurotrophic Factor (BDNF), Cell Death, Interleukin, Mammalian Target of Rapamycin (mTOR), Neuron, Protein Synthesis, Survival
Abstract
The mammalian target of rapamycin (mTOR) pathway has multiple important physiological functions, including regulation of protein synthesis, cell growth, autophagy, and synaptic plasticity. Activation of mTOR is necessary for the many beneficial effects of brain-derived neurotrophic factor (BDNF), including dendritic translation and memory formation in the hippocampus. At present, however, the role of mTOR in BDNF's support of survival is not clear. We report that mTOR activation is necessary for BDNF-dependent survival of primary rat hippocampal neurons, as either mTOR inhibition by rapamycin or genetic manipulation of the downstream molecule p70S6K specifically blocked BDNF rescue. Surprisingly, however, BDNF did not promote neuron survival by up-regulating mTOR-dependent protein synthesis or through mTOR-dependent suppression of caspase-3 activation. Instead, activated mTOR was responsible for BDNF's suppression of autophagic flux. shRNA against the autophagic machinery Atg7 or Atg5 prolonged the survival of neurons co-treated with BDNF and rapamycin, suggesting that suppression of mTOR in BDNF-treated cells resulted in excessive autophagy. Finally, acting as a physiological analog of rapamycin, IL-1β impaired BDNF signaling by way of inhibiting mTOR activation as follows: the cytokine induced caspase-independent neuronal death and accelerated autophagic flux in BDNF-treated cells. These findings reveal a novel mechanism of BDNF neuroprotection; BDNF not only prevents apoptosis through inhibiting caspase activation but also promotes neuron survival through modulation of autophagy. This protection mechanism is vulnerable under chronic inflammation, which deregulates autophagy through impairing mTOR signaling. These results may be relevant to age-related changes observed in neurodegenerative diseases.
Introduction
BDNF3 has shown promise as a therapy for brain injury or neurodegenerative diseases, as it has been found to improve cognitive function, reduce pathology (1, 2), and prevent neuron death (3). BDNF activation of PI3K/Akt is essential for neuron survival in vitro (4, 5). The PI3K/Akt pathway activates mTOR, which in turn promotes survival through control of protein synthesis, mitochondrial function, and autophagy (6–9). Activated mTOR signaling is reported in many cancer cells, and constitutively active mTOR mutants support survival in various cell types (10, 11). Furthermore, although BDNF activation of mTOR is important for the protein synthesis aspects of memory and long term potentiation consolidation (12–14), it is not known whether mTOR activation is essential for BDNF's promotion of neuron survival.
Although activation of mTOR is essential for many aspects of BDNF signaling, inhibiting mTOR can also be beneficial. Inhibiting mTOR with rapamycin can reduce pathology in a Parkinson disease model and extends life span of simple organisms and mice, perhaps through modulation of autophagy (15–18). The major form of autophagy, macroautophagy, is a constitutive form of self-digestion that is activated by nutrient starvation, accumulation of misfolded proteins, or mTOR inhibition. Autophagy is an essential component of the stress response of cells (19, 20); however, excessive autophagy can lead to cell death (21–24). There is evidence that autophagy is impaired in Alzheimer disease (25), and inhibitors of mTOR such as rapamycin are reported to induce autophagic clearance of pathogenic proteins in neurodegenerative diseases (26, 27). Considering the contrasting roles of activated mTOR on protein synthesis and autophagy, it was unclear which pathway would be more important for BDNF-dependent hippocampal neuron survival (28). We therefore determined the molecular signaling pathways and primary mechanism by which mTOR mediates BDNF protection against trophic factor deprivation-induced cell death.
We further explored the endogenous signals that may also regulate mTOR activation. We previously reported that the inflammatory cytokine IL-1β impaired BDNF-dependent cell survival and activation of Akt (5), suggesting that IL-1β can act as an endogenous inhibitor of the mTOR pathway. Here, we examined the effect of IL-1β on BDNF signaling through mTOR and suppression of autophagy-associated cell death. Our findings suggest that elevated levels of IL-1β impair neuronal function and also convert BDNF induction of autophagy from pro-survival to detrimental.
EXPERIMENTAL PROCEDURES
Cell Culture
Primary cultures of dissociated hippocampal neurons were prepared from E18 Sprague-Dawley rats as described previously (29). Cells were maintained in complete medium, defined as serum-free DMEM supplemented with B27, GlutaMAX, and penicillin/streptomycin (all culture reagents from Invitrogen). Unless otherwise specified, 50 ng/ml BDNF and 50 ng/ml IL-1β (PeproTech) were used to be consistent with previous reports from our laboratory (5), and rapamycin was 200 nm (Cell Signaling).
Survival Assay
At 5 days in vitro (DIV), cells were gently washed twice in withdrawal medium, defined as DMEM with GlutaMAX and penicillin/streptomycin but without B27 to mimic the conditions of published serum withdrawal experiments (4, 5). The treatment times used here were intended to mimic conditions of chronic inflammation, with 2 h of IL-1β pretreatment and 72 h of BDNF treatment. After switching to minimal medium, B27 and IL-1β (when applicable) were added immediately; inhibitors (when applicable) were added after 1.5 h, and BDNF was added after 2 h. Cells were maintained at 37 °C, 5% CO2 until 8 DIV, when cell survival was measured using the metabolic colorimetric 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, and absorbance was measured on a plate reader (Synergy-HT BioTek). To assess survival with immunofluorescence, cells were fixed in 4% paraformaldehyde, blocked/permeabilized in 5% goat serum in PBS with 0.1% Triton X-100 for 1 h, and stained for neuronal markers, including microtubule-associated protein 2 (MAP2), a dendrite-specific microtubule-associated protein that reveals cell body morphology, and for DNA using To-Pro3 or DAPI (30, 31). Because MAP2 staining completely labels neuronal soma, its presence can be used to identify living neurons or to mark the boundaries of the cell body using automated analysis. Antibodies used are listed in Table 1. MAP2-labeled cells with pale nuclear stain were counted as living. Dead cells were identified by strong To-Pro3 nuclear stain without MAP2 labeling. For quantification of cell survival/death, five separate fields in a grid pattern were quantified to produce a composite mean per treatment group.
TABLE 1.
Sources and dilutions of primary antibodies
The abbreviations used are as follows: WB, Western blotting; IFC, immunofluorescence.
| Target | Supplier and catalog no. | Method and dilution |
|---|---|---|
| Akt | Cell Signaling, 9272 | WB, 1:1000 |
| Atg5 | Sigma, A0731 | WB, 1:500 |
| Atg7 | Abcam, ab53255 | WB, 1:500 |
| Cleaved caspase-3 | Millipore, 9661 | WB, 1:1000 |
| HA | Cell Signaling, 2367 | IFC, 1:200 |
| LC3 | Cell Signaling, 3868 | WB, 1:1000; IFC, 1:200 |
| MAP2 | Abcam, ab5392 | IFC, 1:2000 |
| Pan-Neuronal Marker | Millipore, MAB2300 | IFC, 1:200 |
| mTOR | Cell Signaling, 2983 | WB, 1:1000 |
| p-Akt Ser-473 | Cell Signaling, 4060 | WB, 1:1000 |
| p-mTOR Ser-2448 | Cell Signaling, 5536 | WB, 1:1000 |
| p-p44/42 MAPK | Cell Signaling, 4370 | WB, 1:1000 |
| p-S6 Ser-240/244 | Cell Signaling, 5364 | WB, 1:1000; IFC, 1:1000 |
| p44/42 MAPK | Cell Signaling, 9102 | WB, 1:1000 |
| p62 | Cell Signaling, 5114 | WB, 1:1000 |
| S6 | Cell Signaling, 2317 | WB, 1:1000 |
| β-Actin | Sigma, A2066 | WB, 1:3000 |
For annexin V staining, cells were washed with warm PBS and then incubated in annexin V binding buffer with Alexa Fluor 488-conjugated annexin V (Invitrogen) for 15 min at 37 °C. Cells were washed with annexin V binding buffer, fixed in 4% paraformaldehyde in annexin V binding buffer, blocked/permeabilized, and stained with MAP2 and secondary antibody and coverslipped with DAPI. Healthy cells stain positive for MAP2, negative for annexin V, and show smooth, diffuse DAPI nuclear stain. Early apoptotic cells are outlined with annexin V as membrane inversion exposes phosphatidylserine, and their condensed nuclei stain strongly with DAPI. Dead neurons were identified by a nucleus that lacks MAP2 staining. The absence of annexin V stain on dead or dying neurons suggests that they are dying through a mechanism independent of apoptosis.
Western Blot Assay
After treatment, cells were washed in ice-cold PBS, then lysed in RIPA/Nonidet P-40 buffer containing protease and phosphatase inhibitor mixtures (Pierce, Thermo Fisher Scientific), and immediately frozen. Cells were harvested in Laemmli buffer, boiled, run on Criterion Tris-HCl gels, and then transferred to PVDF membranes according to the manufacturer's instructions (BD Biosciences). Antibodies used are listed in Table 1. ImageJ software was used for densitometry analysis. For LC3 analysis by Western blot, cells were incubated in 400 nm bafilomycin A1 (Baf A1) for the last 6 h of treatment to prevent complete degradation of autophagic targets.
Electron Microscopy
After 24 h of treatment, cells were fixed overnight at 4 °C in 2% paraformaldehyde, 2% glutaraldehyde in 0.1 m cacodylate buffer. Cells were rinsed with 0.1 m cacodylate buffer, postfixed with 1% osmium tetroxide in 0.1 m cacodylate buffer for 1 h, rinsed twice in double distilled H2O for 10 min, dehydrated in increasing serial dilutions of ethanol (70, 85, 95, and 100% two times) for 10 min each, put in propylene oxide (intermediate solvent) for 10 min twice, and incubated in propylene oxide/Spurr's resin (1:1 mix) for 1 h and in Spurr's resin overnight. For embedding, the bottoms were cut off of “Beem” capsules, and the capsules were filled with Spurr's resin and placed upside-down on the cells, and the resin was polymerized overnight at 60 °C. The Beem capsules were separated from the glass slides by immersion in liquid nitrogen. Subsequently, cells were sectioned at 60 nm using a Leica Ultracut UCT microtome. Sections were mounted on 150 mesh copper grids, stained with uranyl acetate and lead citrate, and viewed using a JE0L 1400 electron microscope. Images were captured using a Gatan digital camera.
Transfections
The enhanced YFP plasmid was obtained from Clontech. Plasmids containing p70S6K (32) in competent DH5a E. coli were as follows: constitutively active p70S6K (Addgene plasmid 8993 pRK7-HA-S6K1-E389-ΔCT), dominant negative p70S6K (Addgene plasmid 8986 pRK7-HA-S6K1-F5A), wild-type p70S6K (Addgene plasmid 8984 pRK7-HA-S6K1-WT), and the empty plasmid (Addgene 10883 pRK7) was used as a negative control. Insert DNA was purified using Invitrogen PureLink HiPure plasmid filter purification kit with precipitator module, resulting in an isolate with very low endotoxins. For p70S6K1 transfections, NanoJuice transfection reagent from Novagen was used (EMD Millipore). At 3 DIV, cells were transfected for 1 h in OptiMEM (Invitrogen) and then maintained in complete medium for 24 h until treatment. To measure survival, the first 15–20 transfected neurons identified from a consistent starting point within each well were imaged and later scored as described above.
Click Chemistry
Click chemistry is a metabolic pulse labeling method that measures protein synthesis with excellent linearity (33). At 5 DIV, cells were treated by replacement of complete medium with minimal medium containing rapamycin, anisomycin, BDNF, or vehicle. After 24 h, endogenous methionine was depleted by 30 min incubation in medium containing treatments but no methionine or cysteine. Metabolic labeling was performed for the last hour in medium without methionine but containing treatments and 200 μm l-azidohomoalanine. For cycloaddition reaction, cells previously fixed, permeabilized, and blocked were incubated in the Click-iT reaction buffer mix with 2 μm 488-alkyne for 1 h at room temperature (reagents from Invitrogen). Slides were coverslipped with Gold Prolonged Antifade reagent (Invitrogen) and imaged on an IX70 Olympus confocal microscope with a ×20/0.70 objective.
Immunocytochemistry
After treatment, cells were washed in cold PBS, fixed in 4% paraformaldehyde for 20 min, permeabilized for 20 min in 0.3% Triton X-100 in 0.2% BSA/PBS or 10 min in 100% methanol at −20 °C, and blocked in 5% goat serum in PBS with 0.1% Triton X-100 for 30 min at room temperature. Cells were incubated in primary antibody in 5% BSA overnight. Primary antibodies are listed in Table 1. Secondary antibodies conjugated to Alexa Fluor 405, 488, 546, or 647, annexin V conjugated to Alexa Fluor 488, and CM DiI (Invitrogen) were used. Slides were coverslipped with DAPI/antifade (Millipore) and imaged on an Olympus Fluoview FV1000 motorized inverted laser scanning confocal microscope with either a Plan-Apo ×40/1.3 NA or Plan-Apo ×60/1.42 NA, both oil immersion in which the laser power and confocal settings were kept constant within experiments. Unless otherwise noted, ImageJ software was used for analysis.
shRNA-mediated Knockdown
Thermo Fisher Scientific GIPZ lentiviral particles encoding GFP and either negative control or shRNA against Atg7 or Atg5 were used to knock down Atg7, 5′-AGCATCATCTTTGAAGTGA-3′, and Atg5, 5′-CGGTGGCTTCCTACTGTTA-3′. At 3 DIV, neurons were infected with either the negative control or shAtg7 or shAtg5. For Western blots, cells infected at 3 DIV were harvested in lysis buffer at 6 DIV and analyzed as described above. For survival experiments, neurons at 5 DIV were rinsed with withdrawal medium and incubated with survival treatments until 8 DIV. They were exposed to DiI for 30 min to stain living neurons and immediately fixed and stained for immunofluorescence analysis as described above (Cell Tracker CM-DiI, Invitrogen), including the pan-neuronal marker Millimark to identify cell morphology. Only living cells convert the colorless compound to a covalently bound fluorescent molecule, producing a specific label of surviving neurons, and only GFP-expressing cells were scored as living or dead.
LC3 Puncta Quantification
The program Volocity (PerkinElmer Life Sciences) was used for unbiased endogenous LC3 puncta analysis in fixed and stained neurons. Cells were identified by Volocity as a region with greater than 1 S.D. higher intensity MAP2 or HA stain than the mean intensity for that individual image. A puncta was identified as a region of LC3 stain that was more than 2 S.D. brighter than the individual image mean. All LC3 puncta larger than 0.1 μm within the MAP2 or HA stain were included in analysis, as any smaller puncta would be outside the reported size range of phagophores or autophagosomes of 0.2 to 10 μm (34). Autophagic flux, or the rate of protein degradation by autophagy, was defined as the increase in total LC3 area when the last stage of autophagy was blocked by incubation with an inhibitor of the vacuolar H+-ATPase, Baf A1. For immunofluorescence analysis, cells were incubated in 100 nm Baf A1 for the last 6 h of treatment. This lower dose of Baf A1 treatment was found in optimization experiments to preserve cell morphology better than the dose and time treatment used for Western blotting.
Statistical Analysis
Statistical analyses were performed with the GraphPad Prism 5 software. Unless otherwise indicated, a one-way or repeated ANOVA analysis was used to determine the overall effect of treatment, and Newman-Keuls post hoc was used for further analysis where a significant effect was observed in the ANOVA. A value of p < 0.05 was considered significant for all groups. All results are presented as means ± S.E., unless otherwise noted.
RESULTS
mTOR Activation Is Required for BDNF-dependent Neuron Survival
A classic protocol to measure trophic effects utilizes withdrawal of the cell media supplement serum or B27. Here, neurons grown in enriched medium supplemented with B27 were switched at 5 DIV to withdrawal medium (w/d, without B27) or withdrawal medium supplemented with BDNF. Counts of live/dead neurons in immunofluorescence images after 72 h revealed that the cell numbers in withdrawal medium were 40% of B27 controls, and in withdrawal medium supplemented with BDNF, the cell numbers were 110% of B27 controls, reflecting BDNF trophic support (Fig. 1, A and B). Addition of the mTOR inhibitor rapamycin (200 nm) attenuated BDNF rescue (49% versus 110% of B27 controls) but did not affect cell survival in cultures that were not treated with BDNF (48% versus 40% of B27 controls, Fig. 1, A and B). Furthermore, survival measurements by colorimetric metabolic MTT assay confirmed a dose-dependent inhibitory effect of rapamycin on BDNF-dependent rescue, IC50 = 0.2 nm, whereas no dose of rapamycin had an effect on cell survival in withdrawal medium (data not shown). Therefore, rapamycin specifically inhibits BDNF rescue without affecting neuron survival in withdrawal medium.
FIGURE 1.

BDNF activation of mTOR-p70S6K increases cell survival in a rapamycin-sensitive manner. A, immunofluorescent stain of neurons, red, MAP2; blue, To-Pro3. Insets, yellow arrows indicate living cells; arrowheads indicate dead cells, and scale bar indicates 50 μm. B, quantification of survival rates measured by immunofluorescence, two independent experiments, five replicates each. C, representative Western blots from cells treated for 1 h. D, quantification of p-mTOR levels normalized to control from Western blots in C, and data are from five separate experiments analyzed by repeated measures ANOVA. PD is 100 μm PD98059; Wort is 0.5 μm wortmannin, and Rap is 200 nm rapamycin. For all graphs, * is compared with control; # is compared with BDNF; ns, p > 0.05; *, p < 0.05; ** or ##, p < 0.01; *** or ###, p < 0.001.
BDNF signaling through the TrkB receptor activates mTOR through the PI3K-Akt pathway or through the Ras-Raf-MEK-MAPK pathway (35). To determine which arm of the signaling pathway was important for BDNF rescue, we used specific inhibitors of each pathway and assessed the effect of the inhibitors on BDNF-dependent phosphorylation of mTOR at Ser-2448, a site linked to the mTOR functional activation (36, 37). As illustrated in Fig. 1, C and D, 100 μm PD98059, an inhibitor of MEK that phosphorylates MAPK, did not block BDNF-dependent activation of mTOR, whereas 0.5 μm wortmannin, an inhibitor of PI3K, blocked mTOR activation as did 200 nm rapamycin (the positive control). These results indicate that BDNF activates mTOR primarily through PI3K-Akt and not significantly through MAPK.
Downstream of mTOR, p70S6K is reported to mediate insulin- and IGF-dependent cell survival and to prevent cell death after ischemia, through inactivation of the pro-apoptotic molecule Bad and by regulation of translation (7, 38, 39). Having determined that mTOR is essential for BDNF-dependent cell survival through pharmacological methods, we next examined the importance of p70S6K in survival by transfecting cells with a dominant negative form of p70S6K1.
We reasoned that if activation of mTOR and p70S6K is essential for BDNF rescue, then transfection with the dominant negative F5A-p70S6K should render neurons resistant to BDNF rescue. F5A-p70S6K neurons had lower survival levels in BDNF than did WT-p70S6K (survival, WT(BDNF) = 74.1% (n = 54) versus F5A(BDNF) = 44.7% (n = 38); WT versus F5A in BDNF Fisher's exact p = 0.005, 2 × 2 contingency table). F5A-p70S6K-transfected neurons survived in withdrawal medium at levels comparable with WT p70S6K-transfected neurons, suggesting that although p70S6K signaling was essential for BDNF's trophic effects, it was not essential for neuron survival in the absence of BDNF trophic signals (survival, WT(w/d) = 53.6% (n = 84) versus F5A(w/d) = 48.7% (n = 39); WT versus, F5A in w/d p = 0.699). This experiment provides genetic support for our conclusion from the rapamycin data, that activation of mTOR and p70S6K is essential for BDNF-dependent neuron survival. Next, we examined the molecular mechanism by which activated mTOR signaling mediates BDNF survival signals.
mTOR-dependent Protein Synthesis Is Essential for Survival but Is Not Selectively Up-regulated by BDNF
mTOR is reported to regulate cell survival through three main pathways as follows: 1) modulation of protein synthesis through phosphorylation of ribosomal protein S6 (38); 2) modulation of mitochondrial activity and suppression of caspase-3 (7, 40); and 3) modulation of autophagy (41). Because S6 is an evolutionarily conserved component of the 40 S ribosomal subunit, mTOR regulates assembly of the ribosomal complex and 5′ cap-dependent mRNA translation initiation (42, 43). In fibroblast and lymphoid cells, activated p70S6K induces production of anti-apoptotic proteins (44, 45). Thus, we hypothesized that BDNF may promote hippocampal neuron survival by mTOR-p70S6K-induced protein synthesis.
As a first step to test the essential role of mTOR-dependent protein synthesis, we compared the effects of rapamycin (which blocks mTOR) with anisomycin (which blocks all protein synthesis) on BDNF-dependent cell survival. For this, we used the MTT assay and established that anisomycin IC50 = 27.8 nm. At low doses (rapamycin 2 nm and anisomycin 25 nm), both inhibitors were equally effective in blocking BDNF-dependent survival (Fig. 2A) but importantly, treatment with both inhibitors produced an additive effect. In contrast, at high doses (rapamycin 200 nm and anisomycin 25 μm), there was occlusion such that the combination produced no more blockade than rapamycin alone (Fig. 2B). These results are consistent with the hypothesis that neuron survival requires mTOR-dependent protein synthesis.
FIGURE 2.

BDNF treatment in the context of mTOR inhibition induces caspase-independent cell death. A, low doses of rapamycin (rap) (2 nm) and anisomycin (ani) (25 nm) had an additive inhibitory effect on neuron survival in combination. B, high doses of rapamycin (200 nm) and anisomycin (2.5 μm) did not have an inhibitory effect on neuron survival in combination. Graphs in A and B each reflect three independent experiments, with each group measured in triplicate. C, Click chemistry metabolic pulse labeling of newly synthesized proteins, displayed with the ImageJ heatmap LUT-orangeHOT, scale shown, and boundaries from MAP2 outlined in white. D, quantification of newly synthesized proteins tagged during the last hour of a 24-h treatment normalized to MAP2. 2 nm rapamycin and 25 nm anisomycin were used in five independent experiments. E, Western blot of cell homogenates after 48 h of treatment. F, quantification of cleaved caspase 3 levels over time, as measured by Western blot, four independent experiments. G, after 48 h of treatment, living neurons were stained with annexin V (green), fixed, and labeled with MAP2 (red) and DAPI (blue). For all graphs, * is compared with control; # is compared with BDNF; ns, p > 0.05; * or #, p < 0.05; ** or ##, p < 0.01; *** or ###, p < 0.001; scale bars, 20 μm.
Next, we assessed the extent to which rapamycin versus anisomycin blocked protein synthesis. Assessment of total protein synthesis rates by pulse labeling after 24 h of treatment (33) revealed that cells in BDNF-containing medium averaged 1.4-fold higher rates of protein synthesis than cells in withdrawal medium (Fig. 2, C and D). Low doses of the inhibitors rapamycin and anisomycin were used to reduce the likelihood of off-target drug effects. Anisomycin (25 nm) completely blocked protein synthesis in neurons regardless of treatment (Fig. 2, C and D). Rapamycin (2 nm) reduced protein synthesis in BDNF-treated cells and, surprisingly, also in withdrawal medium by a similar amount. Because mTOR-dependent protein synthesis is not uniquely increased by BDNF, these data suggest that under deprived conditions, mTOR-dependent protein synthesis is not mainly driven by BDNF. Because rapamycin suppressed total protein synthesis (Fig. 2, C and D) but did not affect cell survival in withdrawal medium (Fig. 1B), increased mTOR-dependent translation is therefore unlikely to be the mechanism by which BDNF promotes neuron survival. Therefore, we examined the remaining putative mechanisms of mTOR-dependent cell survival, mitochondrial and autophagic regulation.
BDNF Suppresses Caspase-3 Activation in a Rapamycin-insensitive Manner
mTOR regulation of metabolism is essential for cell survival, as mTOR regulates mitochondrial function through the mitochondria transcriptional regulator PGC-1α, reducing accumulation of ROS and inactivating mitochondrial Bad (7, 39, 40). We reasoned that if mTOR inhibition during BDNF treatment caused cell death through mitochondrial dysfunction, the neurons would subsequently die through caspase-mediated type I cell death (22, 46). Therefore, we assessed markers of apoptosis to distinguish between cell death due to mitochondrial or autophagic dysregulation. We initially confirmed that BDNF maintained elevated p-S6 levels at least 48 h after treatment (Fig. 2E). Next, levels of cleaved (active) caspase-3 were found to be higher in withdrawal medium than in B27-treated cells at 48 and 72 h (Fig. 2, E and F), consistent with previous reports of growth factor withdrawal-induced apoptosis (10). Cells treated with BDNF showed consistently low caspase-3 levels at all time points, but importantly, addition of rapamycin did not affect cleaved caspase-3 levels (Fig. 2, E and F) even though it blocked BDNF-dependent cell survival (Fig. 1B). Furthermore, cells in withdrawal medium often stained positive for annexin V, whereas cells treated with BDNF and rapamycin rarely stained positive for annexin V, a marker of phosphatidylserine inversion that is characteristic of cells undergoing early apoptosis (Fig. 2G). These data suggest that cells co-treated with BDNF and rapamycin do not die through caspase-associated apoptosis. Furthermore, the data argue against a primary role of mitochondrial dysfunction inducing cell death in neurons treated with BDNF during mTOR inhibition. Having shown that neither mTOR-dependent protein synthesis nor mTOR mitochondrial regulation is likely to be the primary mechanism of BDNF-dependent survival, we next examined the role of autophagy in BDNF-mTOR survival signaling.
BDNF Limits Autophagic Flux through mTOR
During macroautophagy, hereafter autophagy, stages of autophagosome initiation are regulated separately from the final stages of autophagosome-lysosome fusion and subsequent degradation of autolysosome contents. The overall rate of conversion of proteins to metabolites is often described as autophagic flux. Because activated mTOR is known to suppress autophagic flux and rapamycin increases autophagic flux (47, 48), we hypothesized that in withdrawal conditions BDNF was suppressing excessive autophagy through activation of mTOR. To test this hypothesis, we measured changes in the levels of the microtubule-associated protein 1 light chain 3 (MAP1-LC3/Atg8/hereafter LC3). During autophagosome initiation, cytosolic LC3 (LC3-I) is cleaved to expose Gly-120 and subsequently lipidated to produce LC3-II, which binds to a nascent double-membrane segment called a phagophore (49). LC3 conversion is dependent on both Atg5 and Atg7, and completion of autophagy reduces endogenous levels of the degradation-tagging molecule, p62. LC3-I and LC3-II have different electrophoretic mobility that allows their identification by Western blot.
LC3-I and LC3-II levels were measured by Western blot at various time points during the treatments in withdrawal medium, i.e. BDNF, rapamycin, and BDNF with rapamycin. After 6 h of BDNF treatment, LC3-I and -II levels were higher than in withdrawal treatment, regardless of the addition of rapamycin (Fig. 3, A and B, light bars). This may suggest that the neurotrophin increases the biogenesis of the LC3 molecule. LC3-II levels can increase under conditions of increased autophagic initiation or decreased autophagic degradation or a combination of both. To parse out the contributions of autophagosome initiation versus degradation on LC3-II levels, an inhibitor of the last step of autophagy was employed. Baf A1 prevents lysosome acidification and thus the degradation of LC3-II (50). Addition of Baf A1 to neurons treated for 6 h resulted in significant accumulation of LC3-II only in withdrawal treatment and in BDNF with rapamycin treatment, reflecting high levels of autophagic flux (Fig. 3, A and B, dark bars).
FIGURE 3.

BDNF limits autophagic degradation through mTOR. A, Western blots for LC3 with actin for loading control after 6 h of treatment. Bafilomycin treatment (Baf A1, 400 nm, 6 h) was used to detect autophagic flux. rap, rapamycin. B, quantification of 6-h treatment blots: LC3-I (upper band) or LC3-II (lower band) compared with actin, normalized to 24-h withdrawal (w/d) levels. C, Western blots for LC3 after 24 h of treatment. D, quantification of LC3-I/actin and LC3-II/actin after 24 h of treatment, normalized to 24-h withdrawal levels. E, Western blot of p62 protein levels after 48 h of treatment. F, quantification of p62/actin and after 48 h of treatment. Graphs in A–F reflect four independent experiments. G, electron microscopic image of neurons treated with BDNF and rapamycin for 24 h. White arrowhead indicates autophagic vesicle. H, zoom of EM image in G, white arrows indicate electron-lucent cleft between double-membraned walls of vesicle. AV, autophagic vesicle; nuc, nucleus; lyso, lysosome; scale bars, 1 μm. For all graphs, * is compared with control unless otherwise specified; *, p < 0.05.
After 24-h survival treatments, there was no difference from withdrawal in LC3-I or LC3-II levels in the absence Baf A1 (Fig. 3, C and D, light bars). However, the addition of Baf A1 induced significant accumulation of both LC3-I and LC3-II levels in neurons treated for 24 h with BDNF and rapamycin, suggesting high autophagic flux (Fig. 3, C and D, dark bars). These LC3 measurements by Western blot suggest that neurons in withdrawal medium undergo a temporary increase in autophagic flux after 6 h of treatment, although neurons treated with BDNF and rapamycin maintain high levels of autophagic flux after both 6 and 24 h of treatment.
An additional readout of autophagic flux is the endogenous marker, p62, which is degraded by autophagy and accumulates in a delayed manner when autophagic degradation is inhibited (48). Interestingly, p62 levels were significantly lower in BDNF with rapamycin-treated cells than in BDNF-treated cells after 48 h of treatment (Fig. 3E). This supports the notion that the addition of rapamycin to BDNF-treated cells robustly enhanced autophagic flux within the first 48 h of treatment. Although mTOR suppression by rapamycin is known to accelerate autophagic flux in complete medium (48), the combined effect of BDNF treatment with rapamycin had not previously been reported. To document rapamycin induction of autophagy in the presence of BDNF, we examined neurons by electron microscopy after 24 h of treatment. Indeed, mTOR suppression by rapamycin induced the appearance of multiple double membrane-bound, electron-dense vacuoles containing partially degraded organelles consistent with autophagic vesicles (Fig. 3, G and H). Also observed were large, empty vesicles consistent with distended cathepsin-D-filled lysosomes described previously (51).
Endogenous LC3-associated autophagic structures were next probed by immunofluorescence and analyzed quantitatively to define how LC3 structures are regulated in the course of autophagy (48). Membrane-bound LC3-II appears as discrete puncta (shown in red) associated with large, empty vacuoles in autophagic neurons (Fig. 4A, left). Similar to LC3-II levels measured by immunoblot, an increase in LC3 puncta number after treatment may reflect either increased autophagic initiation or decreased degradation of LC3 through inhibition of autophagic flux (48, 52). Fig. 4B shows total LC3 puncta area per cell after 6 h of treatment in complete medium. Baf A1 (100 nm) increased total LC3 puncta area per cell (puncta load) compared with vehicle (Fig. 4B). A comparison of the puncta load between Baf A1-treated and vehicle-treated cells reveals the amount of LC3 that would have been degraded if autophagy was allowed to proceed unimpeded or flux (53, 54).
FIGURE 4.

Endogenous LC3 can be used to monitor autophagy. A, neuron treated with Baf A1 for 6 h in complete medium. The color raw image on the left is LC3 puncta (red) within MAP2 stain (green) and DAPI/nuclei (blue) and the black and white image to the right is the LC3 puncta identified by automated analysis using Volocity. B, total LC3 puncta area per cell after 6 h of Baf A1 (100 nm) or rapamycin (200 nm) treatment in complete medium. The dashed line reflects the total area of LC3 identified in untreated neurons. C, individual puncta size and number after Baf A1 treatment in complete medium. D, individual puncta size and number after rapamycin treatment in complete medium. Three independent experiments in which each group was sampled 10 times, so each group mean reflects values for 60 neurons; * is compared with control, *, p < 0.05.
Baf A1 treatment caused individual LC3 puncta to increase in size, possibly reflecting autophagosome-autophagosome fusion (48). In contrast, rapamycin (200 nm) accelerates autophagic flux (48) and decreased the total LC3 puncta load compared with control levels (Fig. 4B). Specifically, rapamycin significantly decreased the number of LC3 puncta, although it did not significantly affect individual puncta size (Fig. 4D). Rapamycin is reported to increase the proportion of larger, mature autophagolysosomes to smaller autophagosomes (51), so these data likely reflect an acceleration of autophagolysosome maturation and degradation that is triggered by rapamycin.
None of the above treatments significantly affected LC3 puncta size after 24 h (Fig. 5C); however, comparison of total LC3 puncta area per cell with versus without Baf A1 reveals that autophagic flux is low in BDNF-treated cells, but addition of rapamycin significantly elevates autophagic flux (Fig. 5, A and D). These data support our hypothesis that the presence of both rapamycin and BDNF causes neurons to die with elevated autophagy.
FIGURE 5.

BDNF increases LC3 puncta number through both mTOR-dependent and -independent pathways. A, immunofluorescence images of neurons after 24-h survival treatment with and without Baf A1. Green, MAP2; red, LC3; blue, DAPI. The black and white images were obtained by automated LC3 puncta identification and analysis. rap, rapamycin. B, quantification of LC3 puncta number after 24-h treatment. C, quantification of individual LC3 puncta size. D, LC3 puncta flux calculated by comparison of total puncta load with or without Baf A1. Baf A1 (100 nm) was used for the last 3 h of treatment. Data are from six slides, with 10 neurons analyzed per group per slide. E, neurons transfected with p70S6K mutants were stained for LC3 and analyzed by Volocity; red, LC3; green, HA; blue, DAPI. F, quantification of LC3 puncta number from four experiments for a total of 70 WT neurons, 38 ΔCT neurons, and 30 F5A neurons in withdrawal medium, and 67 WT neurons, 42 ΔCT neurons, and 33 F5A neurons in BDNF-containing medium. For all graphs,* is compared with control; # is compared with BDNF; *, or #, p < 0.05; **, p < 0.01; scale bars, 20 μm.
Furthermore, LC3 puncta number was lower in cells treated with rapamycin but higher in cells treated with BDNF (Fig. 4, A and B) compared with cells in withdrawal medium. Importantly, rapamycin addition to BDNF did not suppress LC3 puncta number to the level of rapamycin treatment alone. This highlights that BDNF acts on autophagy through both mTOR-dependent and mTOR-independent pathways (Fig. 5B and diagrammed in Fig. 8). To evaluate the specific contribution of the mTOR-dependent pathway of BDNF regulation of autophagy, we next examined the downstream target of mTOR, p70S6K, which is critically involved in autophagy regulation.
FIGURE 8.
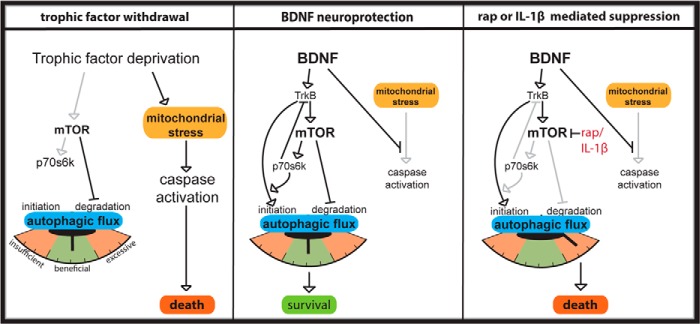
Hypothesized pathways for BDNF regulation of autophagy in primary neurons. In trophic factor withdrawal medium (left panel), there is low autophagic initiation and therefore low autophagic flux, but mitochondrial stress leads to apoptosis through caspase activation. If rapamycin is added to withdrawal medium, the mTOR inhibition of the final step of autophagy is released, but cells lack sufficient (mTOR-independent) autophagic initiation to provide substrates to maintain high levels of autophagic flux. In BDNF treatment (middle panel), caspase activation is suppressed and neurons experience a balance of mTOR-independent autophagic initiation and mTOR-dependent suppression of autophagosome degradation. Under conditions of mTOR suppression due to rapamycin or IL-1β (right panel), mTOR-independent autophagic initiation is not restrained by p70S6K feedback and rapamycin (rap) promotes autolysosome maturation, resulting in excessive autophagic flux and cell death.
p70S6K Moderates the Intensity of BDNF-dependent Autophagic Initiation
Having established the role of mTOR in mediating BDNF suppression of autophagy, we returned to examine the role of p70S6K on regulation of autophagy. Phosphorylation levels of p70S6K generally inversely correlate with autophagy levels and therefore are often reported as an indirect marker of autophagy (51). However, although the rest of the mTOR pathway suppresses autophagy, the molecule p70S6K is essential for induction of normal levels of starvation-induced autophagy through translational regulation of autophagic machinery (55). In contrast, p70s6k can also act as a negative feedback pathway through inhibitory phosphorylation of proteins upstream of mTOR (56, 57). Therefore, activation of p70S6K can act as a gatekeeper regulating the magnitude of autophagy activation during starvation-induced mTOR inactivation, as diagrammed in Fig. 8 (58–60).
To explore the role of p70S6K, we assessed the effect of p70S6K activation or inactivation on LC3 puncta number in withdrawal medium by transfecting constitutively active ΔCT-p70S6K. There was a significant effect of BDNF treatment overall, indicating that BDNF can increase LC3 puncta number (two-way ANOVA, medium versus BDNF, p < 0.0001, Fig. 5F). Interestingly, in neurons treated with BDNF, expression of ΔCT-p70S6K resulted in more LC3 puncta per cell than expression of WT-p70S6K.
Because ΔCT-p70S6K is constitutively active and does not rely on upstream signals for activation, it is resistant to the negative feedback effects of activated p70S6K. Therefore, the increase in LC3 puncta number in BDNF with constitutively active p70S6K suggests that activated p70S6K enhances BDNF autophagosome initiation. It may indicate that p70S6K limits the scope of autophagic induction through its endogenous negative feedback pathway, as modeled in Fig. 8. LC3 puncta number was similar between dominant negative F5A-transfected cells and controls, suggesting that p70S6K activation is not essential for LC3 puncta formation. These data are consistent with both a positive role of p70S6K in BDNF formation of autophagosomes and also an auto-inhibitory feedback mechanism.
Taken together, the LC3 measurements by Western blot, flux measurements by immunofluorescence, and levels of p62 suggest that 24 h after survival treatment in withdrawal medium, the addition of rapamycin maintains high autophagic flux compared with cells treated with BDNF alone. Furthermore, the lack of active caspase-3 and annexin V staining and high levels of autophagic flux in cells co-treated with BDNF and rapamycin also led us to hypothesize that neurons were dying through autophagy dysregulation.
Neuron Death Induced by BDNF with Rapamycin Treatment Is Prevented by Autophagy Gene Knockdown
If neurons treated with a combination of BDNF and rapamycin were dying due to excess autophagy, then reducing autophagy should rescue them (23, 61). A commonly used strategy is to target the autophagy-associated protein Atg7, which regulates autophagosome elongation through conjugation of LC3 to vesicle membranes, and knockdown that prevents conversion of LC3-I to LC3-II (23, 62, 63). We used lentiviral GIPZ particles to infect neurons with shRNA against Atg7 or Atg5, resulting in about 50% transfection efficiency. Infection with either the shAtg7- or shAtg5-encoding lentivirus reduced Atg7 or Atg5 protein levels by 50 and 10%, respectively, by Western blot, when compared with the negative control virus (Fig. 6A). Expression of either shAtg7 or shAtg5 lentivirus also reduced the conversion of LC3-I to LC3-II, validating the knockdown of autophagy (Fig. 6A).
FIGURE 6.
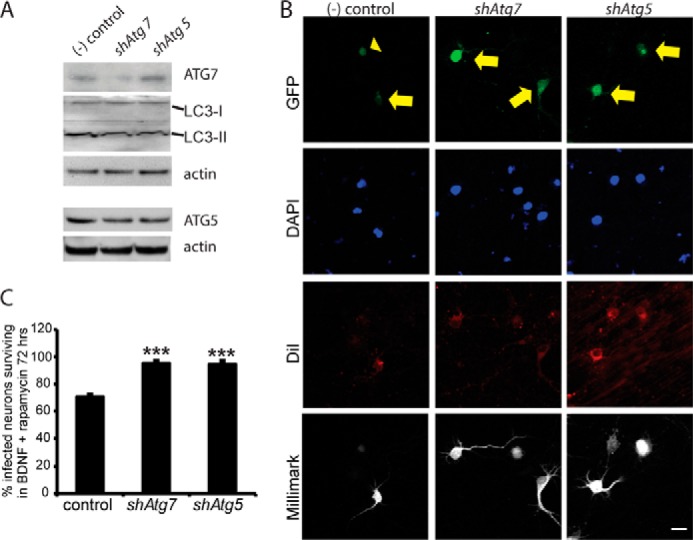
Autophagy reduction rescues neurons co-treated with BDNF and rapamycin. A, Western blots of lentivirus-infected cells show Atg7 and Atg5 knockdown by shAtg7 and shAtg5, respectively. Actin normalization revealed specific knockdown by each shRNA construct. Knockdown effects were more evident in survival experiments, in which only GFP+ neurons were analyzed, than in Western blot validation, probably reflecting low transfection efficiency. Functional reduction in autophagy is shown by reduced ratio of LC3-II to LC3-I proteins. B, infected cells express GFP and were scored as living if they stained positive for DiI and for the pan-neuronal marker Millimark. Infected cells were scored as dead if they expressed GFP but were DiI-negative and stained negative or at a low level with Millimark. C, quantification of B, three independent experiments for a total of at least 75 transfected neurons per group. Autophagy suppression increases neuron survival in co-treatment with BDNF and rapamycin. Scale bar, 20 μm, ***, p < 0.001.
Importantly, neurons with reduced autophagy survived better than controls in BDNF and rapamycin co-treatment (Fig. 6, B and C). Therefore, genetic reduction of autophagy blocked the harmful effects of rapamycin, consistent with the hypothesis that cells treated with BDNF in the context of mTOR inhibition are dying due to excessive autophagy.
IL-1β Prevents BDNF-dependent Neuron Survival through the mTOR-Autophagy Pathway
In addition to rapamycin, endogenous signals may also regulate mTOR activation. Previous reports from our laboratory showed that IL-1β impaired BDNF signaling and neuron rescue (5). IL-1β acts as a physiological inhibitor of trophic signaling by preventing transmission of ligand signals through insulin, IGF, and BDNF receptors (5, 64). Considering the previous data and the canonical BDNF pathway signaling, we hypothesized that IL-1β impairs BDNF survival signaling through inhibiting mTOR pathway activation. To test this, the effects of IL-1β and rapamycin on BDNF-dependent cell survival were compared using the MTT assay. IL-1β had no effect on cell survival in withdrawal medium, although IL-1β and rapamycin each inhibited BDNF-dependent cell survival (Fig. 7A). Importantly, addition of IL-1β did not significantly affect survival in cells treated with BDNF and rapamycin, producing no additive effect. These results are consistent with the hypothesis that IL-1β modulation of BDNF-dependent survival signaling is largely through the mTOR pathway. Furthermore, Fig. 7B shows that cells co-treated with BDNF and IL-1β die without elevated active caspase-3 levels, in agreement with the caspase-independent mode of cell death observed in rapamycin and BDNF co-treated cells.
FIGURE 7.
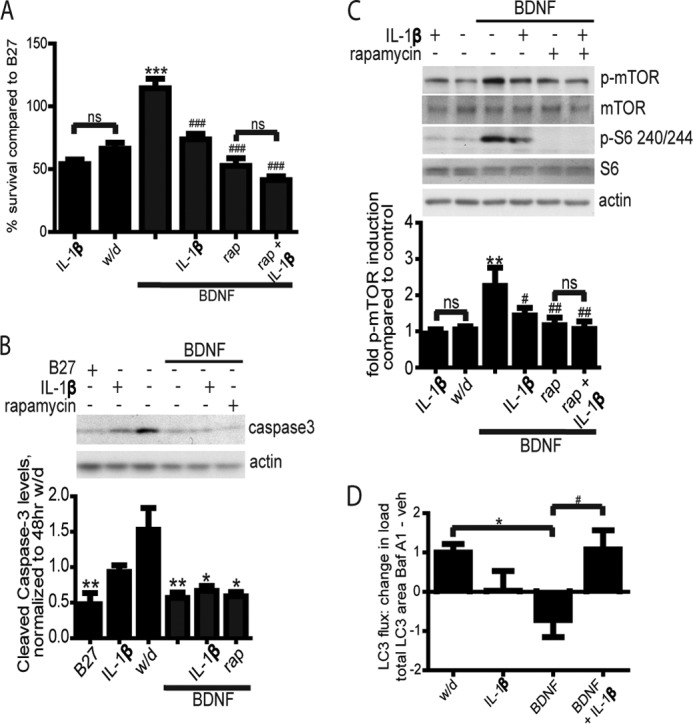
IL-1β inhibits BDNF-dependent neuron survival via the mTOR-autophagy pathway. A, cell survival by MTT, four independent experiments each performed in triplicate. rap, rapamycin. B, Western blot of active cleaved caspase-3 after 72 h of treatment and quantification relative to actin, normalized to withdrawal (w/d) levels, five independent experiments. C, Western blots and quantification of phospho-mTOR and p-S6, five independent experiments analyzed by repeated measures ANOVA. D, total LC3 puncta load per cell, Baf A1 minus vehicle (100 nm Baf for the last 6 h of treatment), five independent experiments. For all graphs, * is compared with control; # is compared with BDNF; * or #, p < 0.05; ** or ##, p < 0.01; ***, or ###, p < 0.001; ns, ns, p > 0.05.
IL-1β was previously found to inhibit BDNF induction of Akt phosphorylation, but it was not established whether mTOR signaling or autophagy was affected (5). Fig. 7C shows that 24 h of IL-1β treatment suppresses mTOR phosphorylation at Ser-2448 by BDNF but did not further suppress mTOR activation in cells treated with BDNF and rapamycin. This supports the hypothesis that IL-1β inhibits the same pathway targeted by rapamycin, namely mTOR. Furthermore, IL-1β regulation of BDNF signaling through mTOR is functionally significant, as BDNF activation of S6 was similarly modulated by these treatments; BDNF increased phosphorylation levels of S6, and this was inhibited by both rapamycin and IL-1β (Fig. 7C). Finally, examination of LC3 puncta load in cells treated with or without Baf A1 revealed that similar to rapamycin the addition of IL-1β to BDNF indeed elevated autophagic flux compared with BDNF treatment alone (Fig. 7D). Together, the survival, signaling, and autophagic flux data support the idea that IL-1β acts as an endogenous regulator of the BDNF-mTOR-autophagy survival pathway.
DISCUSSION
The present data show that BDNF signaling through mTOR supports neuron survival primarily through modulation of autophagy rather than through an increase in mTOR-dependent total protein synthesis or by blocking caspase-3-dependent cell death. Our data clearly show that the mTOR pathway activation is necessary for BDNF protection, as either rapamycin or p70S6K knockdown blocked BDNF rescue. Neurons in deprived conditions exhibited similar levels of mTOR-dependent protein synthesis after 24 h, regardless of BDNF treatment, suggesting that translation modulation is not the primary mechanism of BDNF-mTOR-dependent survival. Furthermore, neurons treated with BDNF and rapamycin died with low levels of active cleaved caspase-3 and high levels of autophagy. In contrast, multiple measurements of autophagy confirmed that addition of rapamycin elevated autophagic flux 24 h after treatment compared with BDNF treatment alone, and genetic knockdown of the autophagy gene Atg7 or Atg5 delayed cell death in the presence of BDNF with rapamycin. Finally, acting as an endogenous inhibitor of mTOR, IL-1β induced cell death associated with excess autophagy and low active caspase-3 levels in neurons treated with BDNF. These data suggest that a context of mTOR inhibition due to inflammatory cytokines or rapamycin treatment may switch the outcome of BDNF trophic signals from trophic to pro-death via excessive autophagy.
The protein synthesis experiments produced two results. First, rapamycin inhibited protein synthesis in both BDNF-treated and control neurons, suggesting that there was some low level of mTOR activation even in withdrawal conditions after 24 h. This may be because although mTOR is inactivated immediately upon trophic factor removal, after prolonged deprivation mTOR can be partially re-activated by metabolites released from the autolysosome (59). Second, because a low concentration of anisomycin blocked BDNF-dependent rescue without reducing survival in withdrawal medium, BDNF may also promote cell survival through induction of mTOR-independent protein synthesis. Translation of a subset of mRNAs can continue despite mTOR inhibition using cap-independent initiation if the mRNA has an internal ribosome entry site. In fact, some reports have found that translation of stress proteins such as heat shock proteins is enhanced during suppressed mTOR signaling (65, 66). We conclude that although protein synthesis may be generally essential for neuron survival, the primary role of activated mTOR in mediating BDNF rescue is through regulation of autophagy. These data also illustrate the multiple roles of BDNF/mTOR signaling in neurons and the importance of signaling compartmentalization. In dendrites, mTOR-dependent protein synthesis is essential to mediate BDNF's effects on plasticity (13), although precise control of mTOR-dependent autophagy is essential in the cell body to mediate BDNF-dependent survival. Local BDNF/mTOR modulation of autophagy, however, may also play a role in synaptic plasticity, as blocking mTOR activation in stimulated neurons induces autophagy-mediated AMPA receptor degradation in spines (63). Further research is needed to clarify the role of BDNF and mTOR in activity-dependent autophagy.
It was previously established that trophic factor withdrawal results in caspase-3-mediated apoptosis (10). Although rapamycin is known to increase autophagic flux (47, 48), in our experiments rapamycin exposure in withdrawal medium in the absence of BDNF did not switch the cell death phenotype away from caspase-mediated apoptosis to death characterized by low caspase-3 and high autophagic flux. Why then did addition of BDNF to rapamycin-treated cells switch cell death from caspase-associated to caspase-independent? One possibility is that in addition to suppressing active caspase-3 levels, BDNF primes neurons for degradative clearance through autophagic initiation while concomitantly suppressing autophagic flux through mTOR activation (diagrammed in Fig. 8). Rapamycin alone removes mTOR inhibition of autophagic flux but may be insufficient to maintain high levels of flux in cells without BDNF-dependent autophagic initiation. Consistent with this idea, BDNF increased LC3-I/II levels after 6 h of treatment and maintained an elevated LC3 puncta number during 24 h of treatment regardless of mTOR suppression. Indeed, the formation of new autophagosomes can occur independent of mTOR and requires activation of a complex of proteins containing the class III phosphatidylinositol 3-kinase (PI3K), Vps34, and Beclin (26, 67, 68). Therefore, treatment with BDNF during mTOR inhibition may have a dual effect of both accelerating autophagic induction and flux and removing the p70S6K feedback inhibition that normally dampens PI3K-dependent autophagic initiation.
The functional relationship between autophagy and cell death is complex (22, 69). Under certain conditions, especially when the apoptotic machinery is intact and active caspase levels are high, autophagy acts as a stress adaptation that prevents cell death. This is consistent with the beneficial effects of rapamycin for autophagy induction in transgenic animal models of Alzheimer disease, in which the activity levels of caspases are high (46, 70). Under other conditions, however, autophagy can contribute to cell death (21, 62, 71–73). The clearest evidence for autophagy as a cell death mechanism comes from cellular models deficient in apoptosis. For instance, autophagic cell death can be induced by etoposide (an inhibitor of topoisomerase II and a common apoptotic reagent) in cells from Bax−/−/Bak−/− double knock-out mice (74). Our results are consistent with these studies and suggest a role of BDNF in inhibition of both apoptosis and excessive autophagy. We further show that both rapamycin and IL-1β primarily activate mTOR-sensitive autophagy in neurons where apoptosis is inhibited by BDNF; therefore, inducing autophagy forces cells to die independent of caspase-3 activation (Fig. 7D).
The trophic factor deprivation paradigm shares some mechanisms with aging, because levels of trophic factors are known to decrease with age and aged brains have less activation of trophic factor signaling pathway molecules (80, 81). Furthermore, many endogenous features associated with brain aging can alter mTOR signaling, including elevated cytokine levels (56). Previously, our laboratory reported that IL-1β blocks BDNF promotion of both plasticity (82) and neuron survival (5) in a paradigm of neurotrophin resistance. This study extends the effect of IL-1β modulation to BDNF activation of mTOR and cell survival through suppression of autophagic flux. This is in agreement with other publications, which show that activation of the interleukin-1 receptor/Toll-like receptors induces autophagy (83, 84). In fact, chronic inflammation is reported to reduce the responsiveness of mTOR to trophic factor activation through inhibitory phosphorylation of receptor adaptor proteins and result in elevated autophagy in adipocytes (85). IL-1β has also been reported to modulate mTOR reactivity through IKKβ action immediately upstream of mTOR, at TSC1 (86), suggesting that IL-1β can resemble an endogenous rapamycin analog.
In all, these findings suggest that modulation of BDNF-induced mTOR activation controls autophagy and neurodegeneration. Thus, our data suggest that it may be advantageous to minimize chronic inflammation to preserve endogenous trophic function or promote positive outcomes during therapeutic treatments with trophic factors (1–3, 87–89). Also, the data suggest that pharmacologically inducing autophagy to clear pathological protein aggregates may further drive degeneration if the endogenous inhibitory feedback pathways are overridden. Perhaps a safer method to induce protein clearance through autophagy might be through fasting (90) or physical exercise (91). Overall, pharmaceutical and nonpharmaceutical strategies to regulate mTOR would be a valuable focus for therapeutic interventions.
Acknowledgments
We are indebted to Brittany Aguilar for assistance in performing the experiments. We are also grateful for helpful discussions with Joan Steffan, for the p70S6K plasmids from John Blenis, and for use of the confocal microscope and Volocity software in the Developmental Biology Center Optical Biology Core Facility at the University of California at Irvine.
This work was supported, in whole or in part, by National Institutes of Health Grant P01-AG000538 (to C. W. C.) and by NRSA Award MH14599-29 (to E. D. S.) from the NIMH.
- BDNF
- brain-derived neurotrophic factor
- mTOR
- mammalian target of rapamycin
- DIV
- days in vitro
- ANOVA
- analysis of variance
- Baf A1
- bafilomycin A1
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- IRS
- insulin receptor substrate
- w/d
- withdrawal
- CM
- chloromethyl
- DiI
- 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate.
REFERENCES
- 1. Blurton-Jones M., Kitazawa M., Martinez-Coria H., Castello N. A., Müller F.-J., Loring J. F., Yamasaki T. R., Poon W. W., Green K. N., LaFerla F. M. (2009) Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 106, 13594–13599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ploughman M., Windle V., MacLellan C. L., White N., Doré J. J., Corbett D. (2009) Brain-derived neurotrophic factor contributes to recovery of skilled reaching after focal ischemia in rats. Stroke 40, 1490–1495 [DOI] [PubMed] [Google Scholar]
- 3. Nagahara A. H., Merrill D. A., Coppola G., Tsukada S., Schroeder B. E., Shaked G. M., Wang L., Blesch A., Kim A., Conner J. M., Rockenstein E., Chao M. V., Koo E. H., Geschwind D., Masliah E., Chiba A. A., Tuszynski M. H. (2009) Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat. Med. 15, 331–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hetman M., Kanning K., Cavanaugh J. E., Xia Z. (1999) Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J. Biol. Chem. 274, 22569–22580 [DOI] [PubMed] [Google Scholar]
- 5. Tong L., Balazs R., Soiampornkul R., Thangnipon W., Cotman C. W. (2008) Interleukin-1β impairs brain derived neurotrophic factor-induced signal transduction. Neurobiol. Aging 29, 1380–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonni A., Brunet A., West A. E., Datta S. R., Takasu M. A., Greenberg M. E. (1999) Cell survival promoted by the ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286, 1358–1362 [DOI] [PubMed] [Google Scholar]
- 7. Pastor M. D., García-Yébenes I., Fradejas N., Pérez-Ortiz J. M., Mora-Lee S., Tranque P., Moro M. A., Pende M., Calvo S. (2009) mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J. Biol. Chem. 284, 22067–22078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shaw R. J., Cantley L. C. (2006) Ras, PI(3)K, and mTOR signalling controls tumour cell growth. Nature 441, 424–430 [DOI] [PubMed] [Google Scholar]
- 9. Wullschleger S., Loewith R., Hall M. N. (2006) TOR signaling in growth and metabolism. Cell 124, 471–484 [DOI] [PubMed] [Google Scholar]
- 10. Edinger A. L., Thompson C. B. (2004) An activated mTOR mutant supports growth factor-independent, nutrient-dependent cell survival. Oncogene 23, 5654–5663 [DOI] [PubMed] [Google Scholar]
- 11. Sato T., Nakashima A., Guo L., Coffman K., Tamanoi F. (2010) Single amino-acid changes that confer constitutive activation of mTOR are discovered in human cancer. Oncogene 29, 2746–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bekinschtein P., Cammarota M., Igaz L. M., Bevilaqua L. R., Izquierdo I., Medina J. H. (2007) Persistence of long-term memory storage requires a late protein synthesis-and BDNF-dependent phase in the hippocampus. Neuron 53, 261–277 [DOI] [PubMed] [Google Scholar]
- 13. Cammalleri M., Lütjens R., Berton F., King A. R., Simpson C., Francesconi W., Sanna P. P. (2003) Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc. Natl. Acad. Sci. U.S.A. 100, 14368–14373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Stoica L., Zhu P. J., Huang W., Zhou H., Kozma S. C., Costa-Mattioli M. (2011) Selective pharmacogenetic inhibition of mammalian target of rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc. Natl. Acad. Sci. U.S.A. 108, 3791–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Malagelada C., Jin Z. H., Jackson-Lewis V., Przedborski S., Greene L. A. (2010) Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J. Neurosci. 30, 1166–1175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kapahi P., Zid B. M., Harper T., Koslover D., Sapin V., Benzer S. (2004) Regulation of life span in Drosophila by modulation of genes in the TOR signaling pathway. Curr. Biol. 14, 885–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harrison D. E., Strong R., Sharp Z. D., Nelson J. F., Astle C. M., Flurkey K., Nadon N. L., Wilkinson J. E., Frenkel K., Carter C. S., Pahor M., Javors M. A., Fernandez E., Miller R. A. (2009) Rapamycin fed late in life extends life span in genetically heterogeneous mice. Nature 460, 392–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Miller R. A., Harrison D. E., Astle C. M., Baur J. A., Boyd A. R., de Cabo R., Fernandez E., Flurkey K., Javors M. A., Nelson J. F., Orihuela C. J., Pletcher S., Sharp Z. D., Sinclair D., Starnes J. W., Wilkinson J. E., Nadon N. L., Strong R. (2011) Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A Biol. Sci. Med. Sci. 66, 191–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H., Mizushima N. (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 [DOI] [PubMed] [Google Scholar]
- 20. Komatsu M., Waguri S., Chiba T., Murata S., Iwata J., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K. (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 [DOI] [PubMed] [Google Scholar]
- 21. Koike M., Shibata M., Tadakoshi M., Gotoh K., Komatsu M., Waguri S., Kawahara N., Kuida K., Nagata S., Kominami E., Tanaka K., Uchiyama Y. (2008) Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am. J. Pathol. 172, 454–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jaeger P. A., Wyss-Coray T. (2009) All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol. Neurodegener. 4, 16–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lee J.-A., Gao F.-B. (2009) Inhibition of autophagy induction delays neuronal cell loss caused by dysfunctional ESCRT-III in frontotemporal dementia. J. Neurosci. 29, 8506–8511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Higgins G. C., Devenish R. J., Beart P. M., Nagley P. (2011) Autophagic activity in cortical neurons under acute oxidative stress directly contributes to cell death. Cell. Mol. Life Sci. 68, 3725–3740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma T., Hoeffer C. A., Capetillo-Zarate E., Yu F., Wong H., Lin M. T., Tampellini D., Klann E., Blitzer R. D., Gouras G. K. (2010) Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer's disease. PLoS ONE 5, e12845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yamamoto A., Cremona M. L., Rothman J. E. (2006) Autophagy-mediated clearance of huntingtin aggregates triggered by the insulin-signaling pathway. J. Cell Biol. 172, 719–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitra S., Tsvetkov A. S., Finkbeiner S. (2009) Protein turnover differences between neurons and other cells. Autophagy 5, 1037–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hoeffer C. A., Klann E. (2010) mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci. 33, 67–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pike C. J., Burdick D., Walencewicz A. J., Glabe C. G., Cotman C. W. (1993) Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J. Neurosci. 13, 1676–1687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Caceres A., Binder L. I., Payne M. R., Bender P., Rebhun L., Steward O. (1984) Differential subcellular localization of tubulin and the microtubule-associated protein MAP2 in brain tissue as revealed by immunocytochemistry with monoclonal hybridoma antibodies. J. Neurosci. 4, 394–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Caceres A., Banker G., Steward O., Binder L., Payne M. (1984) MAP2 is localized to the dendrites of hippocampal neurons which develop in culture. Brain Res. 315, 314–318 [DOI] [PubMed] [Google Scholar]
- 32. Schalm S. S., Blenis J. (2002) Identification of a conserved motif required for mTOR signaling. Curr. Biol. 12, 632–639 [DOI] [PubMed] [Google Scholar]
- 33. Dieterich D. C., Lee J. J., Link A. J., Graumann J., Tirrell D. A., Schuman E. M. (2007) Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2, 532–540 [DOI] [PubMed] [Google Scholar]
- 34. Mizushima N. (2009) in Methods in Enzymology (Daniel J. Klionsky, ed) pp. 13–23, Academic Press, New York [Google Scholar]
- 35. Huang E. J., Reichardt L. F. (2001) Neurotrophins: roles in neuronal development and function. Annu. Rev. Neurosci. 24, 677–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Navé B. T., Ouwens M., Withers D. J., Alessi D. R., Shepherd P. R. (1999) Mammalian target of rapamycin is a direct target for protein kinase B: identification of a convergence point for opposing effects of insulin and amino-acid deficiency on protein translation. Biochem. J. 344, 427–431 [PMC free article] [PubMed] [Google Scholar]
- 37. Sekulić A., Hudson C. C., Homme J. L., Yin P., Otterness D. M., Karnitz L. M., Abraham R. T. (2000) A direct linkage between the phosphoinositide 3-kinase-AKT signaling pathway and the mammalian target of rapamycin in mitogen-stimulated and transformed cells. Cancer Res. 60, 3504–3513 [PubMed] [Google Scholar]
- 38. Wu X., Reiter C. E., Antonetti D. A., Kimball S. R., Jefferson L. S., Gardner T. W. (2004) Insulin promotes rat retinal neuronal cell survival in a p70S6K-dependent manner. J. Biol. Chem. 279, 9167–9175 [DOI] [PubMed] [Google Scholar]
- 39. Harada H., Andersen J. S., Mann M., Terada N., Korsmeyer S. J. (2001) p70S6 kinase signals cell survival as well as growth, inactivating the pro-apoptotic molecule BAD. Proc. Natl. Acad. Sci. U.S.A. 98, 9666–9670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cunningham J. T., Rodgers J. T., Arlow D. H., Vazquez F., Mootha V. K., Puigserver P. (2007) mTOR controls mitochondrial oxidative function through a YY1-PGC-1[agr] transcriptional complex. Nature 450, 736–740 [DOI] [PubMed] [Google Scholar]
- 41. Armour S. M., Baur J. A., Hsieh S. N., Land-Bracha A., Thomas S. M., Sinclair D. A. (2009) Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging 1, 515–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim D.-H., Sarbassov D. D., Ali S. M., King J. E., Latek R. R., Erdjument-Bromage H., Tempst P., Sabatini D. M. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175 [DOI] [PubMed] [Google Scholar]
- 43. Loewith R., Jacinto E., Wullschleger S., Lorberg A., Crespo J. L., Bonenfant D., Oppliger W., Jenoe P., Hall M. N. (2002) Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468 [DOI] [PubMed] [Google Scholar]
- 44. Zhao P., Meng Q., Liu L.-Z., You Y.-P., Liu N., Jiang B.-H. (2010) Regulation of survivin by PI3K/Akt/p70S6K1 pathway. Biochem. Biophys. Res. Commun. 395, 219–224 [DOI] [PubMed] [Google Scholar]
- 45. Vega F., Medeiros L. J., Leventaki V., Atwell C., Cho-Vega J. H., Tian L., Claret F.-X., Rassidakis G. Z. (2006) Activation of mammalian target of rapamycin signaling pathway contributes to tumor cell survival in anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Cancer Res. 66, 6589–6597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Snigdha S., Smith E. D., Prieto G. A., Cotman C. W. (2012) Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci. Bull. 28, 14–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Meijer A. J., Codogno P. (2004) Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 36, 2445–2462 [DOI] [PubMed] [Google Scholar]
- 48. Klionsky D. J., Abdalla F. C., Abeliovich H., Abraham R. T., Acevedo-Arozena A., Adeli K., Agholme L., Agnello M., Agostinis P., Aguirre-Ghiso J. A., Ahn H. J., Ait-Mohamed O., Ait-Si-Ali S., Akematsu T., Akira S., Al-Younes H. M., Al-Zeer M. A., Albert M. L., Albin R. L., Alegre-Abarrategui J., Aleo M. F., Alirezaei M., Almasan A., Almonte-Becerril M., Amano A., Amaravadi R., Amarnath S., Amer A. O., Andrieu-Abadie N., Anantharam V., Ann D. K., Anoopkumar-Dukie S., Aoki H., Apostolova N., Arancia G., Aris J. P., Asanuma K., Asare N. Y., Ashida H., Askanas V., Askew D. S., Auberger P., Baba M., Backues S. K., Baehrecke E. H., Bahr B. A., Bai X.-Y., Bailly Y., Baiocchi R., Baldini G., Balduini W., Ballabio A., Bamber B. A., Bampton E. T., Bánhegyi G., Bartholomew C. R., Bassham D. C., Bast R. C., Jr., Batoko H., Bay B.-H., Beau I., Béchet D. M., Begley T. J., Behl C., Behrends C., Bekri S., Bellaire B., Bendall L. J., Benetti L., Berliocchi L., Bernardi H., Bernassola F., Besteiro S., Bhatia-Kissova I., Bi X., Biard-Piechaczyk M., Blum J. S., Boise L. H., Bonaldo P., Boone D. L., Bornhauser B. C., Bortoluci K. R., Bossis I., Bost F., Bourquin J.-P., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N. R., Brancolini C., Brech A., Brenman J. E., Brennand A., Bresnick E. H., Brest P., Bridges D., Bristol M. L., Brookes P. S., Brown E. J., Brumell J. H., Brunetti-Pierri N., Brunk U. T., Bulman D. E., Bultman S. J., Bultynck G., Burbulla L. F., Bursch W., Butchar J. P., Buzgariu W., Bydlowski S. P., Cadwell K., Cahová M., Cai D., Cai J., Cai Q., Calabretta B., Calvo-Garrido J., Camougrand N., Campanella M., Campos-Salinas J., Candi E., Cao L., Caplan A. B., Carding S. R., Cardoso S. M., Carew J. S., Carlin C. R., Carmignac V., Carneiro L. A., Carra S., Caruso R. A., Casari G., Casas C., Castino R., Cebollero E., Cecconi F., Celli J., Chaachouay H., Chae H.-J., Chai C.-Y., Chan D. C., Chan E. Y., Chang R. C., Che C.-M., Chen C.-C., Chen G.-C., Chen G.-Q., Chen M., Chen Q., Chen S. S., Chen W., Chen X., Chen X., Chen X., Chen Y.-G., Chen Y., Chen Y., Chen Y.-J., Chen Z., Cheng A., Cheng C. H., Cheng Y., Cheong H., Cheong J.-H., Cherry S., Chess-Williams R., Cheung Z. H., Chevet E., Chiang H.-L., Chiarelli R., Chiba T., Chin L.-S., Chiou S.-H., Chisari F. V., Cho C. H., Cho D.-H., Choi A. M., Choi D., Choi K. S., Choi M. E., Chouaib S., Choubey D., Choubey V., Chu C. T., Chuang T.-H., Chueh S.-H., Chun T., Chwae Y.-J., Chye M.-L., Ciarcia R., Ciriolo M. R., Clague M. J., Clark R. S., Clarke P. G. H., Clarke R., Codogno P., Coller H. A., Colombo M. I., Comincini S., Condello M., Condorelli F., Cookson M. R., Coombs G. H., Coppens I., Corbalan R., Cossart P., Costelli P., Costes S., Coto-Montes A., Couve E., Coxon F. P., Cregg J. M., Crespo J. L., Cronjé M. J., Cuervo A. M., Cullen J. J., Czaja M. J., D'Amelio M., Darfeuille-Michaud A., Davids L. M., Davies F. E., De Felici M., de Groot J. F., de Haan C. A., De Martino L., De Milito A., De Tata V., Debnath J., Degterev A., Dehay B., Delbridge L. M., Demarchi F., Deng Y. Z., Dengjel J., Dent P., Denton D., Deretic V., Desai S. D., Devenish R. J., Di Gioacchino M., Di Paolo G., Di Pietro C., Díaz-Araya G., Díaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dikic I., Dinesh-Kumar S. P., Ding W.-X., Distelhorst C. W., Diwan A., Djavaheri-Mergny M., Dokudovskaya S., Dong Z., Dorsey F. C., Dosenko V., Dowling J. J., Doxsey S., Dreux M., Drew M. E., Duan Q., Duchosal M. A., Duff K., Dugail I., Durbeej M., Duszenko M., Edelstein C. L., Edinger A. L., Egea G., Eichinger L., Eissa N. T., Ekmekcioglu S., El-Deiry W. S., Elazar Z., Elgendy M., Ellerby L. M., Eng K. E., Engelbrecht A.-M., Engelender S., Erenpreisa J., Escalante R., Esclatine A., Eskelinen E.-L., Espert L., Espina V., Fan H., Fan J., Fan Q.-W., Fan Z., Fang S., Fang Y., Fanto M., Fanzani A., Farkas T., Farré J.-C., Faure M., Fechheimer M., Feng C. G., Feng J., Feng Q., Feng Y., Fésüs L., Feuer R., Figueiredo-Pereira M. E., Fimia G. M., Fingar D. C., Finkbeiner S., Finkel T., Finley K. D., Fiorito F., Fisher E. A., Fisher P. B., Flajolet M., Florez-McClure M. L., Florio S., Fon E. A., Fornai F., Fortunato F., Fotedar R., Fowler D. H., Fox H. S., Franco R., Frankel L. B., Fransen M., Fuentes J. M., Fueyo J., Fujii J., Fujisaki K., Fujita E., Fukuda M., Furukawa R. H., Gaestel M., Gailly P., Gajewska M., Galliot B., Galy V., Ganesh S., Ganetzky B., Ganley I. G., Gao F.-B., Gao G. F., Gao J., Garcia L., Garcia-Manero G., Garcia-Marcos M., Garmyn M., Gartel A. L., Gatti E., Gautel M., Gawriluk T. R., Gegg M. E., Geng J., Germain M., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghosh P., Giammarioli A. M., Giatromanolaki A. N., Gibson S. B., Gilkerson R. W., Ginger M. L., Ginsberg H. N., Golab J., Goligorsky M. S., Golstein P., Gomez-Manzano C., Goncu E., Gongora C., Gonzalez C. D., Gonzalez R., González-Estévez C., González-Polo R. A., Gonzalez-Rey E., Gorbunov N. V., Gorski S., Goruppi S., Gottlieb R. A., Gozuacik D., Granato G. E., Grant G. D., Green K. N., Gregorc A., Gros F., Grose C., Grunt T. W., Gual P., Guan J.-L., Guan K.-L., Guichard S. M., Gukovskaya A. S., Gukovsky I., Gunst J., Gustafsson A. B., Halayko A. J., Hale A. N., Halonen S. K., Hamasaki M., Han F., Han T., Hancock M. K., Hansen M., Harada H., Harada M., Hardt S. E., Harper J. W., Harris A. L., Harris J., Harris S. D., Hashimoto M., Haspel J. A., Hayashi S., Hazelhurst L. A., He C., He Y.-W., Hébert M.-J., Heidenreich K. A., Helfrich M. H., Helgason G. V., Henske E. P., Herman B., Herman P. K., Hetz C., Hilfiker S., Hill J. A., Hocking L. J., Hofman P., Hofmann T. G., Höhfeld J., Holyoake T. L., Hong M.-H., Hood D. A., Hotamisligil G. S., Houwerzijl E. J., Høyer-Hansen M., Hu B., Hu C.-A., Hu H.-M., Hua Y., Huang C., Huang J., Huang S., Huang W.-P., Huber T. B., Huh W.-K., Hung T.-H., Hupp T. R., Hur G. M., Hurley J. B., Hussain S. N., Hussey P. J., Hwang J. J., Hwang S., Ichihara A., Ilkhanizadeh S., Inoki K., Into T., Iovane V., Iovanna J. L., Ip N. Y., Isaka Y., Ishida H., Isidoro C., Isobe K., Iwasaki A., Izquierdo M., Izumi Y., Jaakkola P. M., Jäättelä M., Jackson G. R., Jackson W. T., Janji B., Jendrach M., Jeon J.-H., Jeung E.-B., Jiang H., Jiang H., Jiang J. X., Jiang M., Jiang Q., Jiang X., Jiang X., Jiménez A., Jin M., Jin S., Joe C. O., Johansen T., Johnson D. E., Johnson G. V. W., Jones N. L., Joseph B., Joseph S. K., Joubert A. M., Juhász G., Juillerat-Jeanneret L., Jung C. H., Jung Y.-K., Kaarniranta K., Kaasik A., Kabuta T., Kadowaki M., Kagedal K., Kamada Y., Kaminskyy V. O., Kampinga H. H., Kanamori H., Kang C., Kang K. B., Kang K. I., Kang R., Kang Y.-A., Kanki T., Kanneganti T.-D., Kanno H., Kanthasamy A. G., Kanthasamy A., Karantza V., Kaushal G. P., Kaushik S., Kawazoe Y., Ke P.-Y., Kehrl J. H., Kelekar A., Kerkhoff C., Kessel D. H., Khalil H., Kiel J. A., Kiger A. A., Kihara A., Kim D. R., Kim D.-H., Kim D.-H., Kim E.-K., Kim H.-R., Kim J.-S., Kim J. H., Kim J. C., Kim J. K., Kim P. K., Kim S. W., Kim Y.-S., Kim Y., Kimchi A., Kimmelman A. C., King J. S., Kinsella T. J., Kirkin V., Kirshenbaum L. A., Kitamoto K., Kitazato K., Klein L., Klimecki W. T., Klucken J., Knecht E., Ko B. C., Koch J. C., Koga H., Koh J.-Y., Koh Y. H., Koike M., Komatsu M., Kominami E., Kong H. J., Kong W.-J., Korolchuk V. I., Kotake Y., Koukourakis M. I., Kouri Flores J. B., Kovács A. L., Kraft C., Krainc D., Krämer H., Kretz-Remy C., Krichevsky A. M., Kroemer G., Krüger R., Krut O., Ktistakis N. T., Kuan C.-Y., Kucharczyk R., Kumar A., Kumar R., Kumar S., Kundu M., Kung H.-J., Kurz T., Kwon H. J., La Spada A. R., Lafont F., Lamark T., Landry J., Lane J. D., Lapaquette P., Laporte J. F., László L., Lavandero S., Lavoie J. N., Layfield R., Lazo P. A., Le W., Le Cam L., Ledbetter D. J., Lee A. J. X., Lee B.-W., Lee G. M., Lee J., Lee J.-H., Lee M., Lee M.-S., Lee S. H., Leeuwenburgh C., Legembre P., Legouis R., Lehmann M., Lei H.-Y., Lei Q.-Y., Leib D. A., Leiro J., Lemasters J. J., Lemoine A., Lesniak M. S., Lev D., Levenson V. V., Levine B., Levy E., Li F., Li J.-L., Li L., Li S., Li W., Li X.-J., Li Y., Li Y.-P., Liang C., Liang Q., Liao Y.-F., Liberski P. P., Lieberman A., Lim H. J., Lim K.-L., Lim K., Lin C.-F., Lin F.-C., Lin J., Lin J. D., Lin K., Lin W.-W., Lin W.-C., Lin Y.-L., Linden R., Lingor P., Lippincott-Schwartz J., Lisanti M. P., Liton P. B., Liu B., Liu C.-F., Liu K., Liu L., Liu Q. A., Liu W., Liu Y.-C., Liu Y., Lockshin R. A., Lok C.-N., Lonial S., Loos B., Lopez-Berestein G., López-Otín C., Lossi L., Lotze M. T., Lów P., Lu B., Lu B., Lu B., Lu Z., Luciano F., Lukacs N. W., Lund A. H., Lynch-Day M. A., Ma Y., Macian F., MacKeigan J. P., Macleod K. F., Madeo F., Maiuri L., Maiuri M. C., Malagoli D., Malicdan M. C., Malorni W., Man N., Mandelkow E.-M., Manon S., Manov I., Mao K., Mao X., Mao Z., Marambaud P., Marazziti D., Marcel Y. L., Marchbank K., Marchetti P., Marciniak S. J., Marcondes M., Mardi M., Marfe G., Mariño G., Markaki M., Marten M. R., Martin S. J., Martinand-Mari C., Martinet W., Martinez-Vicente M., Masini M., Matarrese P., Matsuo S., Matteoni R., Mayer A., Mazure N. M., McConkey D. J., McConnell M. J., McDermott C., McDonald C., McInerney G. M., McKenna S. L., McLaughlin B., McLean P. J., McMaster C. R., McQuibban G. A., Meijer A. J., Meisler M. H., Meléndez A., Melia T. J., Melino G., Mena M. A., Menendez J. A., Menna-Barreto R. F., Menon M. B., Menzies F. M., Mercer C. A., Merighi A., Merry D. E., Meschini S., Meyer C. G., Meyer T. F., Miao C.-Y., Miao J.-Y., Michels P. A., Michiels C., Mijaljica D., Milojkovic A., Minucci S., Miracco C., Miranti C. K., Mitroulis I., Miyazawa K., Mizushima N., Mograbi B., Mohseni S., Molero X., Mollereau B., Mollinedo F., Momoi T., Monastyrska I., Monick M. M., Monteiro M. J., Moore M. N., Mora R., Moreau K., Moreira P. I., Moriyasu Y., Moscat J., Mostowy S., Mottram J. C., Motyl T., Moussa C. E., Müller S., Muller S., Münger K., Münz C., Murphy L. O., Murphy M. E., Musarò A., Mysorekar I., Nagata E., Nagata K., Nahimana A., Nair U., Nakagawa T., Nakahira K., Nakano H., Nakatogawa H., Nanjundan M., Naqvi N. I., Narendra D. P., Narita M., Navarro M., Nawrocki S. T., Nazarko T. Y., Nemchenko A., Netea M. G., Neufeld T. P., Ney P. A., Nezis I. P., Nguyen H. P., Nie D., Nishino I., Nislow C., Nixon R. A., Noda T., Noegel A. A., Nogalska A., Noguchi S., Notterpek L., Novak I., Nozaki T., Nukina N., Nürnberger T., Nyfeler B., Obara K., Oberley T. D., Oddo S., Ogawa M., Ohashi T., Okamoto K., Oleinick N. L., Oliver F. J., Olsen L. J., Olsson S., Opota O., Osborne T. F., Ostrander G. K., Otsu K., Ou J. J., Ouimet M., Overholtzer M., Ozpolat B., Paganetti P., Pagnini U., Pallet N., Palmer G. E., Palumbo C., Pan T., Panaretakis T., Pandey U. B., Papackova Z., Papassideri I., Paris I., Park J., Park O. K., Parys J. B., Parzych K. R., Patschan S., Patterson C., Pattingre S., Pawelek J. M., Peng J., Perlmutter D. H., Perrotta I., Perry G., Pervaiz S., Peter M., Peters G. J., Petersen M., Petrovski G., Phang J. M., Piacentini M., Pierre P., Pierrefite-Carle V., Pierron G., Pinkas-Kramarski R., Piras A., Piri N., Platanias L. C., Pöggeler S., Poirot M., Poletti A., Poüs C., Pozuelo-Rubio M., Prætorius-Ibba M., Prasad A., Prescott M., Priault M., Produit-Zengaffinen N., Progulske-Fox A., Proikas-Cezanne T., Przedborski S., Przyklenk K., Puertollano R., Puyal J., Qian S.-B., Qin L., Qin Z.-H., Quaggin S. E., Raben N., Rabinowich H., Rabkin S. W., Rahman I., Rami A., Ramm G., Randall G., Randow F., Rao V. A., Rathmell J. C., Ravikumar B., Ray S. K., Reed B. H., Reed J. C., Reggiori F., Régnier-Vigouroux A., Reichert A. S., Reiners J. J., Jr., Reiter R. J., Ren J., Revuelta J. L., Rhodes C. J., Ritis K., Rizzo E., Robbins J., Roberge M., Roca H., Roccheri M. C., Rocchi S., Rodemann H. P., Rodríguez de Córdoba S., Rohrer B., Roninson I. B., Rosen K., Rost-Roszkowska M. M., Rouis M., Rouschop K. M. A., Rovetta F., Rubin B. P., Rubinsztein D. C., Ruckdeschel K., Rucker E. B., 3rd, Rudich A., Rudolf E., Ruiz-Opazo N., Russo R., Rusten T. E., Ryan K. M., Ryter S. W., Sabatini D. M., Sadoshima J., Saha T., Saitoh T., Sakagami H., Sakai Y., Salekdeh G. H., Salomoni P., Salvaterra P. M., Salvesen G., Salvioli R., Sanchez A. M., Sánchez-Alcázar J. A., Sánchez-Prieto R., Sandri M., Sankar U., Sansanwal P., Santambrogio L., Saran S., Sarkar S., Sarwal M., Sasakawa C., Sasnauskiene A., Sass M., Sato K., Sato M., Schapira A. H., Scharl M., Schätzl H. M., Scheper W., Schiaffino S., Schneider C., Schneider M. E., Schneider-Stock R., Schoenlein P. V., Schorderet D. F., Schüller C., Schwartz G. K., Scorrano L., Sealy L., Seglen P. O., Segura-Aguilar J., Seiliez I., Seleverstov O., Sell C., Seo J. B., Separovic D., Setaluri V., Setoguchi T., Settembre C., Shacka J. J., Shanmugam M., Shapiro I. M., Shaulian E., Shaw R. J., Shelhamer J. H., Shen H.-M., Shen W.-C., Sheng Z.-H., Shi Y., Shibuya K., Shidoji Y., Shieh J.-J., Shih C.-M., Shimada Y., Shimizu S., Shintani T., Shirihai O. S., Shore G. C., Sibirny A. A., Sidhu S. B., Sikorska B., Silva-Zacarin E. C., Simmons A., Simon A. K., Simon H.-U., Simone C., Simonsen A., Sinclair D. A., Singh R., Sinha D., Sinicrope F. A., Sirko A., Siu P. M., Sivridis E., Skop V., Skulachev V. P., Slack R. S., Smaili S. S., Smith D. R., Soengas M. S., Soldati T., Song X., Sood A. K., Soong T. W., Sotgia F., Spector S. A., Spies C. D., Springer W., Srinivasula S. M., Stefanis L., Steffan J. S., Stendel R., Stenmark H., Stephanou A., Stern S. T., Sternberg C., Stork B., Strålfors P., Subauste C. S., Sui X., Sulzer D., Sun J., Sun S.-Y., Sun Z.-J., Sung J. J., Suzuki K., Suzuki T., Swanson M. S., Swanton C., Sweeney S. T., Sy L.-K., Szabadkai G., Tabas I., Taegtmeyer H., Tafani M., Takács-Vellai K., Takano Y., Takegawa K., Takemura G., Takeshita F., Talbot N. J., Tan K. S., Tanaka K., Tanaka K., Tang D., Tang D., Tanida I., Tannous B. A., Tavernarakis N., Taylor G. S., Taylor G. A., Taylor J. P., Terada L. S., Terman A., Tettamanti G., Thevissen K., Thompson C. B., Thorburn A., Thumm M., Tian F., Tian Y., Tocchini-Valentini G., Tolkovsky A. M., Tomino Y., Tönges L., Tooze S. A., Tournier C., Tower J., Towns R., Trajkovic V., Travassos L. H., Tsai T.-F., Tschan M. P., Tsubata T., Tsung A., Turk B., Turner L. S., Tyagi S. C., Uchiyama Y., Ueno T., Umekawa M., Umemiya-Shirafuji R., Unni V. K., Vaccaro M. I., Valente E. M., Van den Berghe G., van der Klei I. J., van Doorn W., van Dyk L. F., van Egmond M., van Grunsven L. A., Vandenabeele P., Vandenberghe W. P., Vanhorebeek I., Vaquero E. C., Velasco G., Vellai T., Vicencio J. M., Vierstra R. D., Vila M., Vindis C., Viola G., Viscomi M. T., Voitsekhovskaja O. V., von Haefen C., Votruba M., Wada K., Wade-Martins R., Walker C. L., Walsh C. M., Walter J., Wan X.-B., Wang A., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H., Wang H.-G., Wang H.-D., Wang J., Wang K., Wang M., Wang R. C., Wang X., Wang X., Wang Y.-J., Wang Y., Wang Z., Wang Z. C., Wang Z., Wansink D. G., Ward D. M., Watada H., Waters S. L., Webster P., Wei L., Weihl C. C., Weiss W. A., Welford S. M., Wen L.-P., Whitehouse C. A., Whitton J. L., Whitworth A. J., Wileman T., Wiley J. W., Wilkinson S., Willbold D., Williams R. L., Williamson P. R., Wouters B. G., Wu C., Wu D.-C., Wu W. K., Wyttenbach A., Xavier R. J., Xi Z., Xia P., Xiao G., Xie Z., Xie Z., Xu D., Xu J., Xu L., Xu X., Yamamoto A., Yamamoto A., Yamashina S., Yamashita M., Yan X., Yanagida M., Yang D.-S., Yang E., Yang J.-M., Yang S. Y., Yang W., Yang W. Y., Yang Z., Yao M.-C., Yao T.-P., Yeganeh B., Yen W.-L., Yin J., Yin X.-M., Yoo O.-J., Yoon G., Yoon S.-Y., Yorimitsu T., Yoshikawa Y., Yoshimori T., Yoshimoto K., You H. J., Youle R. J., Younes A., Yu L., Yu L., Yu S.-W., Yu W. H., Yuan Z.-M., Yue Z., Yun C.-H., Yuzaki M., Zabirnyk O., Silva-Zacarin E., Zacks D., Zacksenhaus E., Zaffaroni N., Zakeri Z., Zeh H. J., 3rd, Zeitlin S. O., Zhang H., Zhang H.-L., Zhang J., Zhang J.-P., Zhang L., Zhang L., Zhang M.-Y., Zhang X. D., Zhao M., Zhao Y.-F., Zhao Y., Zhao Z. J., Zheng X., Zhivotovsky B., Zhong Q., Zhou C.-Z., Zhu C., Zhu W.-G., Zhu X.-F., Zhu X., Zhu Y., Zoladek T., Zong W.-X., Zorzano A., Zschocke J., Zuckerbraun B. (2012) Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 8, 445–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tanida I., Ueno T., Kominami E. (2004) Human light chain 3/MAP1LC3B is cleaved at its carboxyl-terminal Met-121 to expose Gly-120 for lipidation and targeting to autophagosomal membranes. J. Biol. Chem. 279, 47704–47710 [DOI] [PubMed] [Google Scholar]
- 50. Petralia R. S., Schwartz C. M., Wang Y.-X., Kawamoto E. M., Mattson M. P., Yao P. J. (2013) Sonic hedgehog promotes autophagy in hippocampal neurons. Biol. Open 2, 499–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Boland B., Kumar A., Lee S., Platt F. M., Wegiel J., Yu W. H., Nixon R. A. (2008) Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J. Neurosci. 28, 6926–6937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Mizushima N., Yoshimori T., Levine B. (2010) Methods in mammalian autophagy research. Cell 140, 313–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bains M., Florez-McClure M. L., Heidenreich K. A. (2009) Insulin-like growth factor-I prevents the accumulation of autophagic vesicles and cell death in Purkinje neurons by increasing the rate of autophagosome-to-lysosome fusion and degradation. J. Biol. Chem. 284, 20398–20407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Neely K. M., Green K. N., LaFerla F. M. (2011) Presenilin is necessary for efficient proteolysis through the autophagy-lysosome system in a γ-secretase-independent manner. J. Neurosci. 31, 2781–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Scott R. C., Schuldiner O., Neufeld T. P. (2004) Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7, 167–178 [DOI] [PubMed] [Google Scholar]
- 56. Tremblay F., Krebs M., Dombrowski L., Brehm A., Bernroider E., Roth E., Nowotny P., Waldhäusl W., Marette A., Roden M. (2005) Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes 54, 2674–2684 [DOI] [PubMed] [Google Scholar]
- 57. Zhang J., Gao Z., Yin J., Quon M. J., Ye J. (2008) S6K directly phosphorylates IRS-1 on Ser-270 to promote insulin resistance in response to TNF-α signaling through IKK2. J. Biol. Chem. 283, 35375–35382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chang Y.-Y., Juhász G., Goraksha-Hicks P., Arsham A. M., Mallin D. R., Muller L. K., Neufeld T. P. (2009) Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem. Soc. Trans 37, 232–326 [DOI] [PubMed] [Google Scholar]
- 59. Yu L., McPhee C. K., Zheng L., Mardones G. A., Rong Y., Peng J., Mi N., Zhao Y., Liu Z., Wan F., Hailey D. W., Oorschot V., Klumperman J., Baehrecke E. H., Lenardo M. J. (2010) Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 465, 942–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Klionsky D. J., Meijer A. J., Codogno P. (2005) Autophagy and p70S6 kinase. Autophagy 1, 59–60 [DOI] [PubMed] [Google Scholar]
- 61. Du L., Hickey R. W., Bayir H., Watkins S. C., Tyurin V. A., Guo F., Kochanek P. M., Jenkins L. W., Ren J., Gibson G., Chu C. T., Kagan V. E., Clark R. S. (2009) Starving neurons show sex difference in autophagy. J. Biol. Chem. 284, 2383–2396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Walls K. C., Ghosh A. P., Franklin A. V., Klocke B. J., Ballestas M., Shacka J. J., Zhang J., Roth K. A. (2010) Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J. Biol. Chem. 285, 10497–10507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shehata M., Matsumura H., Okubo-Suzuki R., Ohkawa N., Inokuchi K. (2012) Neuronal stimulation induces autophagy in hippocampal neurons that is involved in AMPA receptor degradation after chemical long-term depression. J. Neurosci. 32, 10413–10422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yamada M., Ohnishi H., Sano S. i, Nakatani A., Ikeuchi T., Hatanaka H. (1997) Insulin receptor substrate (IRS)-1 and IRS-2 are tyrosine-phosphorylated and associated with phosphatidylinositol 3-kinase in response to brain-derived neurotrophic factor in cultured cerebral cortical neurons. J. Biol. Chem. 272, 30334–30339 [DOI] [PubMed] [Google Scholar]
- 65. Sun J., Conn C. S., Han Y., Yeung V., Qian S.-B. (2011) PI3K-mTORC1 attenuates stress response by inhibiting cap-independent Hsp70 translation. J. Biol. Chem. 286, 6791–6800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Qin X., Sarnow P. (2004) Preferential translation of internal ribosome entry site-containing mRNAs during the mitotic cycle in mammalian cells. J. Biol. Chem. 279, 13721–13728 [DOI] [PubMed] [Google Scholar]
- 67. Jaeger P. A., Wyss-Coray T. (2010) Beclin 1 complex in autophagy and Alzheimer disease. Arch. Neurol. 67, 1181–1184 [DOI] [PubMed] [Google Scholar]
- 68. Sarkar S., Floto R. A., Berger Z., Imarisio S., Cordenier A., Pasco M., Cook L. J., Rubinsztein D. C. (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J. Cell Biol. 170, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Levine B., Yuan J. (2005) Autophagy in cell death: an innocent convict? J. Clin. Invest. 115, 2679–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Hyman B. T. (2011) Caspase activation without apoptosis: insight into Aβ initiation of neurodegeneration. Nat. Neurosci. 14, 5–6 [DOI] [PubMed] [Google Scholar]
- 71. Yu L., Alva A., Su H., Dutt P., Freundt E., Welsh S., Baehrecke E. H., Lenardo M. J. (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304, 1500–1502 [DOI] [PubMed] [Google Scholar]
- 72. Puyal J., Vaslin A., Mottier V., Clarke P. G. (2009) Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann. Neurol. 66, 378–389 [DOI] [PubMed] [Google Scholar]
- 73. Dziedzic S. A., Caplan A. B. (2012) Autophagy proteins play cytoprotective and cytocidal roles in leucine starvation-induced cell death in Saccharomyces cerevisiae. Autophagy 8, 731–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Shimizu S., Kanaseki T., Mizushima N., Mizuta T., Arakawa-Kobayashi S., Thompson C. B., Tsujimoto Y. (2004) Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat. Cell Biol. 6, 1221–1228 [DOI] [PubMed] [Google Scholar]
- 75. Deleted in proof.
- 76. Deleted in proof.
- 77. Deleted in proof.
- 78. Deleted in proof.
- 79. Deleted in proof.
- 80. Peng S., Wuu J., Mufson E. J., Fahnestock M. (2005) Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer's disease. J. Neurochem. 93, 1412–1421 [DOI] [PubMed] [Google Scholar]
- 81. Francis B. M., Kim J., Barakat M. E., Fraenkl S., Yücel Y. H., Peng S., Michalski B., Fahnestock M., McLaurin J., Mount H. T. (2012) Object recognition memory and BDNF expression are reduced in young TgCRND8 mice. Neurobiol. Aging 33, 555–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Tong L., Prieto G. A., Kramár E. A., Smith E. D., Cribbs D. H., Lynch G., Cotman C. W. (2012) Brain-derived neurotrophic factor-dependent synaptic plasticity is suppressed by interleukin-1β via p38 mitogen-activated protein kinase. J. Neurosci. 32, 17714–17724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Delgado M. A., Elmaoued R. A., Davis A. S., Kyei G., Deretic V. (2008) Toll-like receptors control autophagy. EMBO J. 27, 1110–1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Sanjuan M. A., Dillon C. P., Tait S. W., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J. L., Withoff S., Green D. R. (2007) Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257 [DOI] [PubMed] [Google Scholar]
- 85. Ost A., Svensson K., Ruishalme I., Brännmark C., Franck N., Krook H., Sandström P., Kjolhede P., Strålfors P. (2010) Attenuated mTOR signaling and enhanced autophagy in adipocytes from obese patients with type 2 diabetes. Mol. Med. 16, 235–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lee D.-F., Kuo H.-P., Chen C.-T., Hsu J.-M., Chou C.-K., Wei Y., Sun H.-L., Li L.-Y., Ping B., Huang W.-C., He X., Hung J.-Y., Lai C.-C., Ding Q., Su J.-L., Yang J.-Y., Sahin A. A., Hortobagyi G. N., Tsai F.-J., Tsai C.-H., Hung M.-C. (2007) IKKβ suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell 130, 440–455 [DOI] [PubMed] [Google Scholar]
- 87. Fischer W., Wictorin K., Björklund A., Williams L. R., Varon S., Gage F. H. (1987) Amelioration of cholinergic neuron atrophy and spatial memory impairment in aged rats by nerve growth factor. Nature 329, 65–68 [DOI] [PubMed] [Google Scholar]
- 88. Autry A. E., Monteggia L. M. (2012) Brain-derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 64, 238–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Park H.-Y., Kim J. H., Sun Kim H., Park C. K. (2012) Stem cell-based delivery of brain-derived neurotrophic factor gene in the rat retina. Brain Res. 1469, 10–23 [DOI] [PubMed] [Google Scholar]
- 90. Alirezaei M., Kemball C. C., Flynn C. T., Wood M. R., Whitton J. L., Kiosses W. B. (2010) Short-term fasting induces profound neuronal autophagy. Autophagy 6, 702–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. He C., Sumpter R., Jr., Levine B. (2012) Exercise induces autophagy in peripheral tissues and in the brain. Autophagy 8, 1548–1551 [DOI] [PMC free article] [PubMed] [Google Scholar]