

Background: The nucleolar protein PICT1 regulates p53 via ribosomal protein L11 and MDM2.
Results: Nucleolar stress induces PICT1 degradation that is mediated by the proteasome in a ubiquitination-independent manner.
Conclusion: PICT1 employs noncanonical proteasome-mediated degradation machinery to sense nucleolar stress.
Significance: This finding provides new insight into the molecular mechanisms underlying the nucleolar stress response.
Keywords: Cell Signaling, Nucleolus, Proteasome, Protein Degradation, Signal Transduction, Stress Response, Nucleolar Stress
Abstract
The nucleolar protein PICT1 regulates tumor suppressor p53 by tethering ribosomal protein L11 within the nucleolus to repress the binding of L11 to the E3 ligase MDM2. PICT1 depletion results in the release of L11 to the nucleoplasm to inhibit MDM2, leading to p53 activation. Here, we demonstrate that nucleolar stress induces proteasome-mediated degradation of PICT1 in a ubiquitin-independent manner. Treatment of H1299 cells with nucleolar stress inducers, such as actinomycin D, 5-fluorouridine, or doxorubicin, induced the degradation of PICT1 protein. The proteasome inhibitors MG132, lactacystin, and epoxomicin blocked PICT1 degradation, whereas the inhibition of E1 ubiquitin-activating enzyme by a specific inhibitor and genetic inactivation fail to repress PICT1 degradation. In addition, the 20 S proteasome was able to degrade purified PICT1 protein in vitro. We also found a PICT1 mutant showing nucleoplasmic localization did not undergo nucleolar stress-induced degradation, although the same mutant underwent in vitro degradation by the 20 S proteasome, suggesting that nucleolar localization is indispensable for the stress-induced PICT1 degradation. These results suggest that PICT1 employs atypical proteasome-mediated degradation machinery to sense nucleolar stress within the nucleolus.
Introduction
The nucleolus is a large nuclear domain that provides an environment for the synthesis of ribosomes; in the nucleolus, 28 S, 18 S, and 5.8 S ribosomal RNAs (rRNAs) are transcribed and, together with 5 S rRNA, are processed and assembled into the ribosomal subunits. Accumulating evidence suggests an important role for the nucleolus as a cellular stress sensor, in addition to the workshop for ribosome biogenesis (1, 2). Ribosome biogenesis is a process that must be tightly regulated to achieve proper cellular proliferation and growth. Perturbations of this process, known as nucleolar (or ribosomal) stress, induced by the inhibition of rRNA processing, synthesis, and ribosome assembly, cause cell cycle arrest and apoptosis (1, 2). Certain ribosomal proteins and the tumor suppressor p53 are known to play a pivotal role in response to such perturbation (2–9). Recent studies have unveiled that 5 S ribonucleoprotein (RNP)3 functions as a key module that regulates p53 in response to nucleolar stress (10–12). Depletion of 5 S RNP components (5 S rRNA and ribosomal proteins L5 and L11) blocks stress-induced p53 activation. Intriguingly, c-Myc, which regulates ribosome biogenesis through 5 S rRNA transcription, forms a feedback loop with L11 to regulate 5 S RNP; c-Myc transactivates L11, although L11 represses c-Myc at both the transcriptional and post-translational levels (10, 13, 14). This recent progress provides novel insight into how nucleolar stress induces p53 activation; however, the mechanism underlying the sensing of the stress by the ribosomal protein-p53 axis has remained elusive.
PICT1 was initially identified as a protein that binds the tumor suppressor PTEN (15). We also have demonstrated that PICT1 also functions as a regulator of the tumor suppressor p53 (16). PICT1 predominantly localizes to the nucleolus, where it may form part of a large protein complex consisting of a set of ribosomal proteins, including L11 (16). When PICT1 is depleted, L11 is released from the complex and translocates from the nucleolus to the nucleoplasm, where it binds the E3 ligase MDM2 and inhibits its function in the nucleoplasm. The inhibition of MDM2 E3 ligase activity eventually increases p53 protein levels and activates p53-mediated cellular responses. In fact, it has been shown that actinomycin D (ActD) treatment, which induces nucleolar stress by inhibiting nascent rRNA synthesis, dramatically reduces PICT1 protein and increases p53 protein in mouse embryonic stem cells (16). A recent study also showed that nucleolar stress (induced by ActD) and genotoxic stress (caused by DNA-damaging agents) induce translocation of PICT1 from the nucleolus to the nucleoplasm (17). In addition, it has been shown that PICT1 directly binds to 5 S rRNA and facilitates the integration of 5 S RNP into the ribosome (11, 12). Taken together, these observations suggest that PICT1 is a component of stress-sensing machinery in the nucleolus and that PICT1 degradation may be a crucial step for the subsequent p53-dependent stress response, although the mechanism underlying control of stress-induced PICT1 degradation remains unknown.
In this study, we demonstrate that stress-induced PICT1 degradation is sensitive to proteasome inhibitors, whereas the stress does not increase ubiquitination of PICT1. Furthermore, the inhibition of E1 ubiquitin-activating enzyme fails to repress PICT1 degradation, suggesting ubiquitin-independent proteasomal degradation of PICT1. We also demonstrate that nucleolar localization of PICT1 is required for PICT1 degradation.
EXPERIMENTAL PROCEDURES
Materials
Chemicals used in this study were as follows: 5-fluorouridine (FUrd) from Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan); ActD and doxorubicin hydrochloride from Sigma; MG132, lactacystin, epoxomicin, pepstatin A, and E64d from Peptide Institute Inc. (Osaka, Japan); CA-074Me from Calbiochem; and Pyr41 from Focus Biomolecules (Fort Washington, PA). Affinity-purified anti-PICT1 antibodies were described previously (16, 18, 19). Other antibodies used in the study were as follows: anti-p53 (FL-393) and anti-nucleolin/C23 (MS-3) from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA); anti-Myc 9B11 from Cell Signaling Technology Inc. (Danvers, MA); anti-Myc polyclonal antibody from Bethyl Laboratories, Inc. (Montgomery, TX); anti-cyclin D1 (556470) from Pharmingen; anti-ACTB from BioMatrix Research Inc. (Chiba, Japan); and anti-ubiquitin (Ubi-1) from EMD Millipore (Billerica, MA).
DNA Constructs
Plasmid vectors Myc-PICT1/pCAGGS and Myc-PICT1/pEF1 were described previously (16, 18, 19). To generate Myc-PICT1/pOSTet15T3, Myc-PICT1 cDNA was amplified by PCR using primers 5′-gaa ttc acc atg gaa caa aaa ctc atc tca gaa gaa-3′ and 5′-gc ggc cgc tta caa ctg gat ctc acg gaa cg-3′ and Myc-PICT1/pCAGGS as a template, digested by EcoRI and NotI, and cloned into the EcoRI-NotI site of pOSTet15T3. The pOSTet15T3 vector with Epstein-Barr virus-based episomal replication system was kindly provided from Dr. Yoshihiro Miwa (University of Tsukuba, Japan). To generate Myc-PICT1(d342–449)/pEF1 and Myc-PICT1(d342–449)/pOSTet15T3, PCR was performed with primers 5′-gag cag cag atg atc gag cct cga gag ag-3′ and 5′-ctc gat cat ctg ctg ctc cgt ctt ctt ctc-3′ and Myc-PICT1/pEF1 and Myc-PICT1/pOSTet15T3, respectively, as template. The PCR products were used to transform Escherichia coli XL10-Gold strain cells (Stratagene, La Jolla, CA). To generate FLAG-PTEN(T382A/T383A)/pCMV5, mutations (acc to gcc (codon 382); act to gct (codon 383)) were introduced by employing the same PCR-based strategy using FLAG-PTEN/pCMV5 (20) as template. Full-length enhanced GFP cDNA was amplified from pEGFP-N3 (Invitrogen) and tagged with a Myc sequence at the 5′-end by PCR, followed by cloning into the EcoRI-XbaI site of pCMV5 to generate Myc-EGFP/pCMV5. The pCMV5 vector was kindly provided from Dr. David W. Russell (University of Texas Southwestern Medical Center). Prime STAR max DNA polymerase (Takara, Shiga, Japan) was used for all PCR amplifications. Sequences of the constructed vectors were confirmed using the Big Dye Terminator method (Applied Biosystems, Carlsbad, CA). His-tagged ubiquitin expression construct pMT107 (21) was kindly provided by Dr. Dirk Bohmann (University of Rochester Medical Center).
Cell Culture
H1299 (human non-small cell lung carcinoma, ATCC CRL-5803), HeLa (human cervical carcinoma, ATCC CCL-2), U2OS (human osteosarcoma, ATCC HTB-96), MCF-7 (human breast adenocarcinoma, ATCC HTB-22) cells, HuH-7 cells (human hepatoma, JCRB 0403), and HEK293 (human embryonic kidney, ATCC CRL-1573) were cultured at 37 °C in DMEM supplemented with 10% FBS and penicillin/streptomycin. HL-60 (human acute promyelocytic leukemia, JCRB IFO-50022) and PC-3 (human prostate carcinoma, ATCC CRL-1435) were cultured at 37 °C in RPMI 1640 medium supplemented with 10% FBS and penicillin/streptomycin. Ts85 cells (mouse mammary carcinoma, RIKEN Cell Bank RCB0033) were cultured at 33 °C in RPMI 1640 medium supplemented with 10% FBS and penicillin/streptomycin. H1299 cells were kindly provided by Drs. Takehiko Kamijo and Akira Nakagawara (Chiba Cancer Center Research Institute). MCF-7 cells were provided by Cell Resource Center for Biomedical Research Institute of Development, Aging and Cancer, Tohoku University, Japan. HuH-7 cells were kindly provided by Drs. Tomoko Date and Takaji Wakita (National Institute of Infectious Diseases). To generate H1299/Myc-PICT1(d342–449) cells, in which expression of Myc-PICT1(d342–449) is under the control of the tetracycline-responsive element, H1299 cells were maintained for 2 weeks in the presence of 400 μg/ml G418 after transfection with Myc-PICT1(d342–449)/pOSTet15T3 using TurboFect (Pierce) according to the manufacturer's protocol. HeLa cells stably expressing Myc-PICT1 (WT or d342–449) were isolated using a cloning cylinder method and cultured in the presence of 800 μg/ml G418 after transfection with Myc-PICT1/pEF1 or Myc-PICT1(d342–449)/pEF1 using TurboFect according to the manufacturer's protocol. For drug treatment, an appropriate number of cells (e.g. 7 × 104 H1299 cells) were plated on 24-well plate(s) and cultured for 1 day. Degree of confluence (90–100%) and normal morphology of cells were confirmed prior to performing each experiment.
Immunoblot Analysis
Cells were directly lysed in an appropriate amount (50–70 μl per well for 24-well format) of urea lysis solution (7.2 m urea, 1.6% (w/v) Nonidet P-40, 2% (w/v) SDS, 50 mm DTT, 0.1% (w/v) bromphenol blue), followed by sonication to reduce viscosity. The concentration of protein in each sample was determined using the Bradford ultra kit (Expedeon, Inc., San Diego) and BSA (Nacalai Tesque, Kyoto, Japan) as a standard protein. Proteins (20 μg) were resolved on an SDS-polyacrylamide gel and transferred to polyvinylidene fluoride membrane filters (Pall, Port Washington, NY). Filters were probed with specified antibodies as described previously (19, 22). The relative intensity of immunoreactive bands (PICT1 and ACTB) was determined by LAS1000 (GE Healthcare). Representative images from at least three independent experiments are presented in each figure.
Immunopurification of Myc-PICT1 and Myc-EGFP Proteins
To prepare Myc-PICT1 proteins (WT and d342–449), HeLa cells were transfected with either Myc-PICT1/pEF1 or Myc-PICT1(d342–449)/pEF1 using TurboFect transfection reagent according to the manufacturer's protocol. After a 2-day incubation, nuclei were isolated as described on the Lamond Lab web site and in Refs. 23, 24. The nuclei (3 × 107) were then resuspended in 0.2 ml of high salt RIPA buffer (50 mm HEPES-NaOH, 500 mm NaCl, 1% (w/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, protease inhibitor mixture (Nacalai Tesque, Kyoto, Japan), 50 units of Benzonase (EMD Chemicals, Inc., Billerica, CA), pH 7.4) and incubated for 30 min at 4 °C. The suspension was sonicated to disrupt nuclei, and insoluble materials were precipitated by centrifugation (12,000 × g, 15 min, 4 °C). Cleared nuclear extract was added to 0.8 ml of no-salt RIPA buffer (50 mm HEPES-NaOH, 1% (w/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, protease inhibitor mixture, pH 7.4) and added to 1 μg of anti-Myc 9B11 antibody with 20 μl of a slurry of protein G-Sepharose beads (GE Healthcare). After incubation for 2 h at 4 °C with gentle rotation, the beads were collected by centrifugation, washed with RIPA-100 buffer (1 ml four times; 50 mm HEPES-NaOH, 100 mm NaCl, 1% (w/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, pH 7.4), and resuspended into 0.1 ml of RIPA-100 buffer.
To prepare Myc-EGFP protein, HeLa cells (6 × 106) were transfected with Myc-EGFP/pCMV5. After a 2-day incubation, the cells were lysed in 1 ml of RIPA buffer (50 mm HEPES-NaOH, 100 mm NaCl, 1% (w/v) Nonidet P-40, 0.5% (w/v) sodium deoxycholate, protease inhibitor mixture, pH 7.4), and insoluble materials were then precipitated by centrifugation (12,000 × g, 15 min, 4 °C). Anti-Myc 9B11 antibody (1 μg) with protein G-Sepharose beads (20 μl of a slurry) was added to the cleared extract. After incubation for 2 h at 4 °C with gentle rotation, the beads were collected by centrifugation, washed with RIPA-100 buffer (1 ml four times), and resuspended into 0.1 ml of RIPA-100 buffer.
20 S Proteasome-mediated in Vitro Degradation Assay
Two μl of a slurry of immunoprecipitate (Myc-PICT1 or Myc-EGFP) was incubated with 0.5 μg of the 20 S proteasome (Boston Biochem, Inc., Cambridge, MA) in 12 μl of reaction mix (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 10 mm MgCl2, 1 mm DTT). After incubation at 37 °C, the reaction was terminated by the addition of 4 μl of four times Laemmli sample buffer (200 mm Tris-HCl, 40% (v/v) glycerol, 2% (w/v) SDS, 20% (v/v) β-mercaptoethanol, 0.4% (w/v) bromphenol blue, pH 6.8). Proteins were analyzed by immunoblotting using anti-Myc polyclonal antibody (used at 1:3,000 dilution).
Immunofluorescence Assay
Cells grown on coverslips were subjected to drug treatment as described in each figure legend. All procedures described below were conducted at room temperature, unless otherwise stated. Cells were washed in PBS, fixed, and permeabilized in methanol for 5 min. After washing in PBS, cells were incubated in PBS containing 2% BSA for 30 min, followed by incubation for 12 h at 4 °C with affinity-purified anti-PICT1 antibody (used at 1:60 dilution) (18) and anti-nucleolin antibody (used at 1:100 dilution). After washing in PBS for 10 min, samples were probed with Alexa Fluor 488-conjugated anti-rabbit IgG (1:1,000 dilution, Invitrogen) and Alexa Fluor 594-conjugated anti-mouse IgG (1:1,000 dilution, Invitrogen) with DAPI for 30 min in PBS containing 2% BSA and washed with PBS twice. For the analysis of Myc-tagged PICT1, anti-Myc 9B11 antibody (used at 1:1,000 dilution) and either Alexa Fluor 488- or Alexa Fluor 647-conjugated anti-mouse IgG (1:1,000 dilution, Invitrogen) were used as primary and secondary antibodies, respectively. Fluorescence analysis was performed with a BioZero microscope (KEYENCE, Osaka, Japan) equipped with a Plan Fluor ELWD 20× objective (Nikon, Tokyo, Japan). Representative images from at least three independent experiments are presented in each figure.
Evaluation of PICT1 Ubiquitination
Cells (90% confluency) were transfected with pMT107 in combination with Myc-PICT1/pCAGGS, Myc-PICT1/pEF1, or FLAG-PTEN(T382A/T383A)/pCMV5 using TurboFect transfection reagent according to the manufacturer's protocol. After a 1–2-day incubation, the cells (on 6-well plate) were treated with FUrd (1 mm) and either epoxomicin (1 μm) or MG132 (10 μm) for 6–8 h, rinsed with PBS, and extracted in 1 ml per dish denaturing extraction buffer (8 m urea, 0.1 m HEPES-KOH, 0.1% Nonidet P-40, 0.5 m NaCl, 10 mm β-mercaptoethanol, 1× protease inhibitor mixture, pH 7.9). The lysate was sonicated to reduce viscosity, and 80 μl of a slurry of HIS-Select Nickel Affinity Gel (Sigma) was added. After incubation for 3 h at 4 °C with gentle rotation, the beads were collected by centrifugation and washed with denaturing extraction buffer (1 ml three times), and 45 μl of denaturing extraction buffer and 15 μl four times Laemmli sample buffer were added. Extracted proteins and the lysates were analyzed by immunoblotting.
RESULTS
Stress-induced PICT1 Degradation Is Mediated by the Proteasome
We previously demonstrated that, in mouse embryonic stem cells, nucleolar stress induces down-regulation of PICT1 protein and that depletion of PICT1 results in p53 induction as the result of MDM2 inhibition (16). Here, we first tested the effect of p53 status on down-regulation of PICT1 in response to nucleolar stress, using various cell lines. Treatment of cells with FUrd, which is converted to fluorouridine triphosphate, thereby inducing nucleolar stress by the incorporation of this nucleotide into nascent RNA, decreased PICT1 protein level in HeLa (p53 inactivated by HPV18-E6), MCF-7 (p53-WT), H1299 (p53-null), and U2OS (p53-WT) cells (Fig. 1, A and B). It is noteworthy that FUrd treatment did not affect PICT1 mRNA levels (data not shown), suggesting that down-regulation was the result of PICT1 protein degradation. We also tested HL-60 (p53-null), HuH-7 (p53-mutated), PC-3 (p53-null), and HEK293 (p53 stabilized by Ad5-E1B) cells and observed PICT1 degradation in these cells in response to nucleolar stress (Fig. 1, C–F), suggesting that p53 status does not affect PICT1 degradation. Although p53 status may not affect PICT1 degradation, to eliminate the potential effect of a p53-mediated cellular response, we used p53-null H1299 cells for further analyses, unless otherwise stated.
FIGURE 1.

Nucleolar stress induces PICT1 degradation in various cell lines. HeLa, MCF-7, H1299, U2OS, HL-60, HuH-7, PC-3, and HEK293 cells were treated with 1 mm FUrd for the indicated times. Cell lysates were prepared and subjected to immunoblot analysis as described under “Experimental Procedures” (A, C, and E). The calculated PICT1/ACTB value of each control (0 h) was set to 100%, and normalized values are presented in B, D, and F. A and B, HeLa, MCF-7, H1299, and U2OS; C and D, HL-60 and HuH-7; E and F, PC-3 and HEK293.
Protein degradation in cells is primarily mediated by either the proteasome or the lysosome. In H1299 cells, PICT1 degradation was induced by ActD (5 nm), an inhibitor of RNA polymerase I, and by doxorubicin (1 μm), a DNA-damaging anticancer agent, in addition to FUrd (Fig. 2A). Three distinct proteasome inhibitors (MG132, epoxomicin, and lactacystin) all blocked PICT1 degradation despite the presence of stress-inducing agents, and rather it seemed to increase PICT1 expression levels (Fig. 2A). In contrast, inhibitors of lysosomal proteases (pepstatin A for cathepsin D, CA-074Me for cathepsin B, and E64d for cathepsins B/H/L) did not block the degradation (Fig. 2B). It is noteworthy that neither these inhibitors by themselves nor the vehicle (DMSO, ≤0.2%) decreased PICT1 expression levels after 6 h of incubation (data not shown). In addition, treatment of cells with bafilomycin B1, a specific inhibitor of V-ATPase, that inhibits acidification and protein degradation in lysosomes, was also unable to repress the degradation (data not shown). These results suggest that the major degradation machinery for PICT1 protein is the proteasome.
FIGURE 2.

Proteasome inhibitors, but not lysosome inhibitors, block nucleolar stress-induced PICT1 degradation in H1299 cells. H1299 cells were treated with proteasome (A) or lysosome (B) inhibitors for 5 min, followed by the addition of the indicated stress inducers. After incubation for 6 h, cell lysates were prepared and subjected to immunoblot analysis as described under “Experimental Procedures.” The calculated PICT1/ACTB value of each control experiment was set to 100%, and normalized values are presented as average ± range of duplicate experiments (A). MG132, 10 μm MG132; Epoxo, 1 μm epoxomicin; Lacta, 1 μm lactacystin; CA-074, 20 μm CA-074Me; PepA, 25 μm pepstatin A; E64d, 30 μm E64d; ActD, 5 nm actinomycin D; Doxo, 1 μm doxorubicin; FUrd, 1 mm FUrd.
Stress-induced PICT1 Degradation Is Independent of Polyubiquitination
In the canonical proteasome-mediated protein degradation system, polyubiquitin covalently linked to the target protein is recognized as the degradation signal by the 26 S proteasome holo-complex. We hypothesized that PICT1 could be polyubiquitinated upon nucleolar stress to be degraded by the proteasome; thus, we assessed the alterations in ubiquitination levels of PICT1 protein during nucleolar stress. We expressed His-tagged ubiquitin in H1299 and HeLa cells and tried to detect His-ubiquitin-modified PICT1 by Ni2+-NTA-agarose pulldown and anti-PICT1 immunoblotting. Treatment of cells with epoxomicin or MG132 resulted in a robust increase in bulk ubiquitination in the cell (Fig. 3, A and B, and data not shown); however, we could not detect any ubiquitinated endogenous PICT1 after a 6-h treatment with FUrd, even in the presence of proteasome inhibitors (Fig. 3, A–C). Ubiquitinated PICT1 became detectable when PICT1 was overexpressed with His-ubiquitin (Fig. 3, A–C). However PICT1 ubiquitination was not altered even after the treatment with the proteasome inhibitor or FUrd, alone or together (Fig. 3, A–C). In contrast, MG132-induced increased ubiquitination of PTEN(T382A/T383A) mutant, which is known to be degraded by the proteasome in a ubiquitin-dependent manner (25, 26), was observed by the same pulldown assay system (Fig. 3D). These results imply that nucleolar stress does not induce polyubiquitination of PICT1 and that ubiquitination of PICT1 is not necessarily required for degradation.
FIGURE 3.

Nucleolar stress does not alter PICT1 ubiquitination. A and B, HeLa cells grown on 6-well plates were transfected with 1.5 μg of pMT107 (His-Ub, +) or pCMV5 (His-Ub, −) in combination with 3.5 μg of Myc-PICT1/pCAGGS (Myc-PICT1, +) or pCAGGS (Myc-PICT1, −). After incubation for 2 day, the cells were treated for 7 h with 10 μm MG132 or vehicle (DMSO). Ubiquitinated proteins were pulled down with Ni2+-NTA beads and subjected to immunoblot analysis. C, H1299 cells grown on 6-cm dishes were transfected with 4 μg each of pMT107 and pEF1 (His-Ub) or 4 μg each of pMT107 and Myc-PICT1/pEF1 (His-Ub+Myc-PICT1). After a 4-h incubation, the cells were trypsinized and plated onto four wells of 6-well plate, and incubated for 1 day. The cells were treated for 6 h with 1 mm FUrd and/or 1 μm epoxomicin (Epoxo). Ubiquitinated proteins were pulled down with Ni2+-NTA beads and subjected to immunoblot analysis as described under “Experimental Procedures.” D, HeLa cells grown on 6-cm dishes were transfected with 5 μg of pMT107 (His-Ub, +) or pcDNA3 (His-Ub, −) in combination with 5 μg of FLAG-PTEN(T382A/T383A)/pCMV5 (PTEN(TA/TA), +) or pCMV5 (PTEN(TA/TA), −). After incubation for 2 days, the cells were treated for 8 h with 10 μm MG132 or vehicle (DMSO). Ubiquitinated proteins were pulled down with Ni2+-NTA beads and subjected to immunoblot (IB) analysis.
To further explore this possibility, we next inhibited the E1 ubiquitin-activating enzyme and evaluated the effect on PICT1 degradation. The E1 enzyme activates ubiquitin by adenylating the C-terminal glycine carboxyl group, the first step in ubiquitin-protein isopeptide bond formation. We used ts85 cells, an FM3A-derived temperature-sensitive mutant cell line, in which the E1 enzyme is inactivated at restrictive temperature (39 °C), to evaluate the contribution of the E1 enzyme (and the E1 enzyme-mediated canonical ubiquitin-conjugation system) to nucleolar stress-induced PICT1 degradation. In ts85 cells, total ubiquitination in the cell lysate was dramatically decreased (by 90%, as judged by anti-ubiquitin immunoreactivity) when the cells were incubated at restrictive temperature for 18 h (Fig. 4A). As expected from the result shown in Fig. 3, comparable degradation of PICT1 was observed at permissive (33 °C) and restrictive temperatures in response to ActD treatment (Fig. 4B). In contrast, turnover of cyclin D1 mediated by the canonical ubiquitin-proteasome system was significantly retarded at restrictive temperature (Fig. 4C). To further confirm the ubiquitin-independent degradation of PICT1, we tested the effect of the E1 enzyme-specific inhibitor Pyr41 on stress-induced PICT1 degradation. As shown in Fig. 5A, either FUrd or ActD treatment decreased PICT1 protein in H1299 cells, consistent with results described above. Pyr41 treatment did not inhibit PICT1 degradation, although epoxomicin treatment did (Fig. 5A). In addition, knockdown of E1 enzyme UBE1 and/or potential E1 enzyme UBA6 (27, 28) did not affect nucleolar stress-induced PICT1 degradation (Fig. 5, B–D). To further test the possibility that PICT1 was directly degraded by the proteasome, we prepared Myc-tagged PICT1 protein using an immunopurification technique and subjected it to in vitro proteasome degradation assay. As shown in Fig. 6A, Myc-tagged PICT1 protein was degraded by uncapped 20 S proteasome within 60 min, whereas anti-Myc IgG heavy chain, which was used for PICT1 immunoprecipitation and remained with PICT1 protein in the reaction, was not. Immunopurified Myc-tagged enhanced GFP also showed resistance to the degradation (Fig. 6B), suggesting that PICT1 was selectively degraded by the 20 S proteasome in vitro. These results, taken together, suggest that nucleolar stress-induced PICT1 degradation is dependent on the proteasome but independent of polyubiquitination.
FIGURE 4.
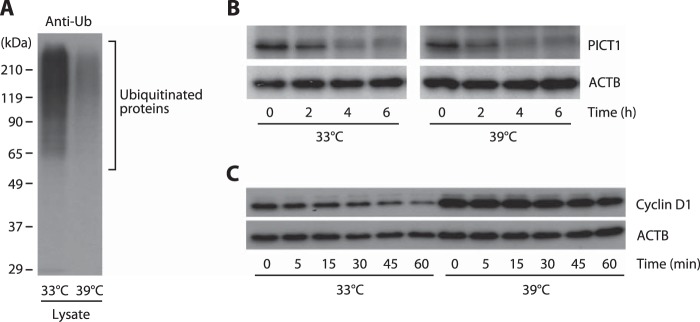
Inhibition of the E1 enzyme does not affect nucleolar stress-induced PICT1 degradation in ts85 cells. ts85 cells were incubated at either 33 or 39 °C for 18 h. Cell lysates were prepared, and ubiquitination (Ub) of cellular proteins was analyzed (A). After incubation at the indicated temperature, cells (3.5 × 105 cells per 0.9 ml) were further incubated with 5 nm ActD (B) or 50 μg/ml cycloheximide (C) for the indicated times. Cell lysates were prepared and subjected to immunoblot analysis as described under “Experimental Procedures.”
FIGURE 5.

Pharmacological and genetic inhibition of the E1 enzyme does not affect nucleolar stress-induced PICT1 degradation in H1299 cells. A, H1299 cells were treated with 1 μm epoxomicin (Epoxo), 10 μg/ml Pyr41, or vehicle (DMSO) for 5 min, followed by the addition of 1 mm FUrd, 5 nm ActD, DMSO, or no addition (−). After incubation for 6 h, cell lysates were prepared and subjected to immunoblot analysis as described under “Experimental Procedures.” B–D, HeLa cells were transfected with indicated siRNAs using Lipofectamine RNAi Max according to the manufacturer's protocol. B, synthesized siRNAs with target sequence. C, after a 72-h incubation, RNAs were extracted, and the expression of UBE1, UBA6, and ACTB mRNAs was analyzed by RT-PCR. Primers used are represented. D, cells were also treated for the indicated times with 5 ng/ml ActD. Cell lysates were then subjected to immunoblot analysis as described under “Experimental Procedures.”
FIGURE 6.
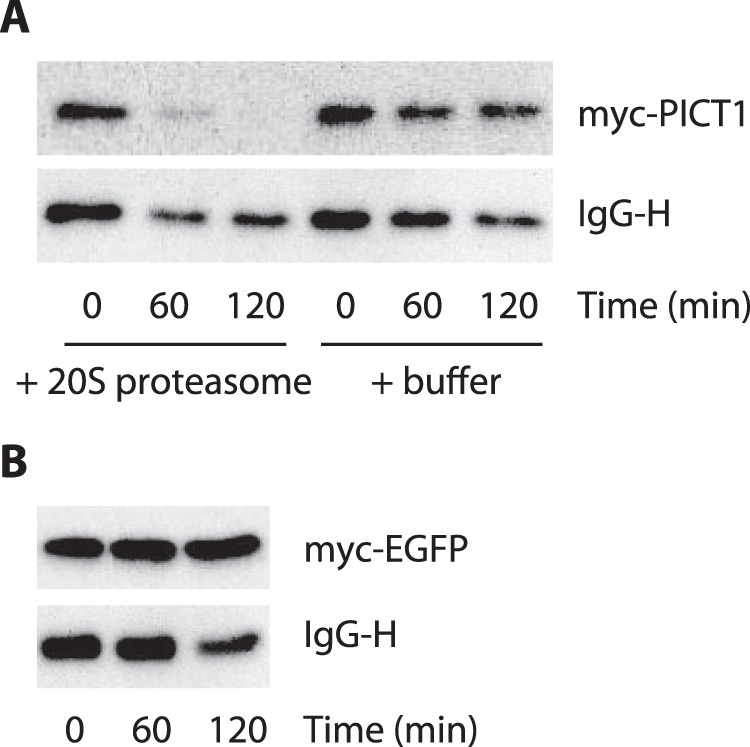
20 S proteasome degrades PICT1. Immunopurified Myc-PICT1 (A) and Myc-EGFP (B) were subjected to 20 S proteasome-mediated in vitro degradation assay. After incubation for the indicated times, Myc-tagged proteins and mouse IgG heavy chain (IgG-H) were detected by immunoblot analysis as described under “Experimental Procedures.”
Nucleolar Localization of PICT1 Is Required for Degradation
Next, we asked where PICT1 degradation occurred. PICT1 predominantly localized in the nucleolus under unstressed conditions, although quite a small subpopulation was found in the nucleoplasm and the cytosol (Fig. 7A). To test the importance of nucleolar localization of PICT1 for degradation, we made a mutant PICT1 in which the putative nucleolar localization signal was deleted. Although bioinformatic analyses show that PICT1 does not contain any known functional domains or motifs, previous studies have shown that the basic amino acid cluster within the region of amino acids 342–445 functions as a nucleolar localization signal (29, 30). Therefore, we made a deletion mutant (PICT1(d342–449)) and tested its degradation in response to nucleolar stress. First, we transiently expressed PICT1(d342–449) in H1299 cells and confirmed that the deletion mutant preferentially localized to the nucleoplasm rather than the nucleolus (Fig. 7A). Thus, we established that H1299 cells stably harbor the expression vector of the deletion mutant. The cells were treated with doxycycline to induce expression of the mutant and exposed to nucleolar stress induced by FUrd. As shown in Fig. 7B, endogenous PICT1 was degraded by exposure to the stress, whereas the deletion mutant PICT1(d342–449) was not. Intriguingly, under an in vitro setting, degradation of PICT1(d342–449) by 20 S proteasome was similar to that of wild-type PICT1 (Fig. 7C), indicating that the deletion mutant has the potential to be degraded by the proteasome. Thus, mislocalization prevented PICT1 from the degradation, suggesting that PICT1 is degraded within the nucleolus. However, several reports have indicated that proteasomes are present in the cytosol and the nucleoplasm but not in the nucleolus (31, 32), indicating PICT1 and the proteasome does not have any opportunity to meet in the nucleolus.
FIGURE 7.
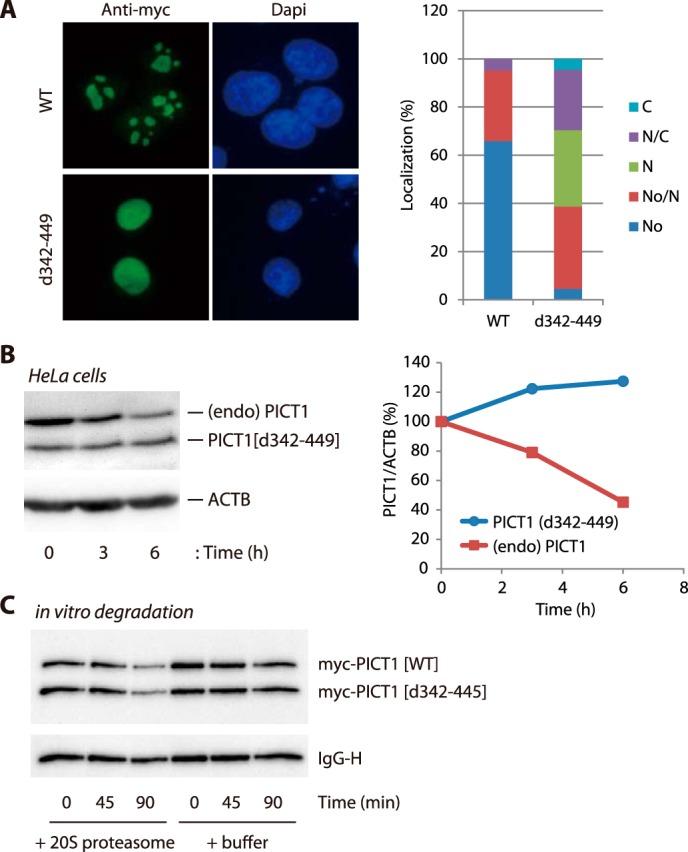
PICT1 deletion mutant (d342–449) is resistant to nucleolar stress-induced degradation and shows nuclear localization. A, H1299 cells were transfected with Myc-PICT1/pEF1 (WT) or Myc-PICT1(d342–449)/pEF1 (d342–449). After a 2-day incubation, intracellular localization of ectopically expressed Myc-tagged PICT1 protein was analyzed by immunofluorescence assay as described under “Experimental Procedures.” One hundred cells were analyzed for each sample, and localization patterns of Myc-tagged PICT1 are presented in the right panel. No, nucleolar; No/N, nucleolar and nuclear; N, nuclear; N/C, nuclear and cytosolic; C, cytosolic. B, H1299/Myc-PICT1(d342–449) cells were treated with 10 ng/ml doxycycline for 24 h to induce expression of PICT1(d342–449) mutant protein in cells. The cells were treated with 1 mm FUrd for the indicated times, and cell lysates were prepared and subjected to immunoblot analysis as described under “Experimental Procedures.” The calculated PICT1/ACTB value of each control (0 h) was set to 100%, and normalized values are presented in the right panel. PICT1(d342–449), blue; endogenous PICT1, red. C, comparable amount of immunopurified Myc-PICT1 proteins (WT and d342–449) were mixed and subjected to 20 S proteasome-mediated in vitro degradation assay. After incubation for the indicated times, Myc-tagged proteins and mouse IgG heavy chain (IgG-H) was detected by immunoblot analysis as described under “Experimental Procedures.”
Therefore we next tested translocation of PICT1 in response to nucleolar stress. As shown in Fig. 8, PICT1 as well as nucleolin, another nucleolar resident, predominantly localized to the nucleolus in unstressed cells. After ActD or FUrd treatment, nucleolin was dispersed entirely to the nucleoplasm, consistent with previous observations (33, 34), and most PICT1 was degraded and barely detectable (Fig. 8). In the presence of epoxomicin, PICT1 degradation was blocked, and PICT1 stayed in the nucleolus, even after a 6-h treatment with these drugs (Fig. 8). Taken together, these results raise the possibility that PICT1 shuttles from the nucleolus to the cytosol where degradation of PICT1 by the proteasome occurs. In addition, the PICT1 shuttling would be sensitive to proteasome inhibitors. Therefore, we next tested intracellular localization of PICT1(d342–449), a degradation-resistant mutant, under nucleolar stress. In accordance with the above observations, PICT1(d342–449) preferentially localized in the nucleoplasm, whereas wild-type PICT1 showed nucleolar localization (Fig. 9). Under nucleolar stress, wild-type PICT1 underwent degradation in the absence of epoxomicin; however, PICT1(d342–449) mutant stayed in the nucleoplasm despite the presence of epoxomicin (Fig. 9), further suggesting that translocation of PICT1 from the nucleolus to the cytosol is critical for the degradation.
FIGURE 8.
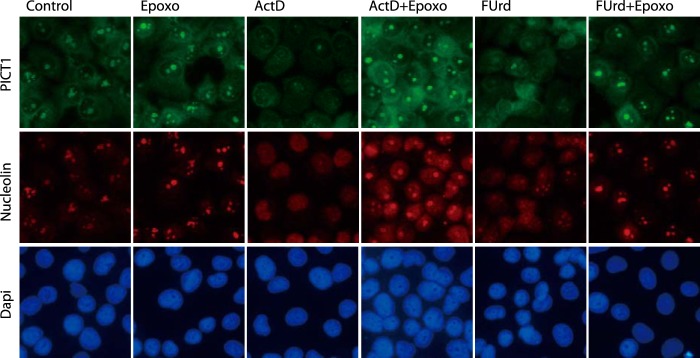
Nucleolar stress does not alter intracellular localization of PICT1. H1299 cells were treated for 6 h with 1 μm epoxomicin (Epoxo), 5 nm ActD, 1 mm FUrd, alone or together as indicated. Intracellular localization of endogenous PICT1 and nucleolin was analyzed by immunofluorescence assay as described under “Experimental Procedures.”
FIGURE 9.
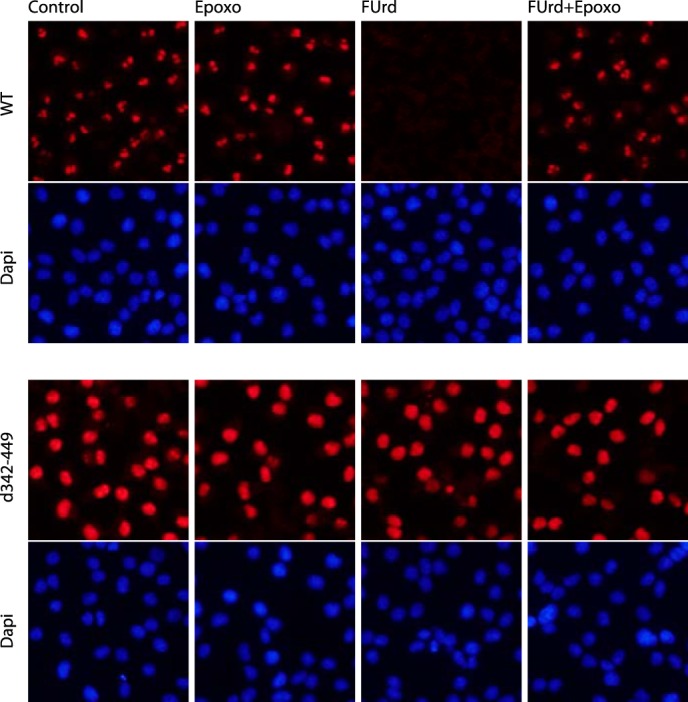
Nucleolar stress does not affect intracellular localization of PICT1 deletion mutant (d342–449). HeLa cells stably expressing Myc-PICT1/pEF1 (WT) or Myc-PICT1(d342–449)/pEF1 (d342–449) were treated for 6 h with 1 μm epoxomicin (Epoxo), 1 mm FUrd, alone or together as indicated. Intracellular localization of ectopically expressed Myc-tagged PICT1 protein was analyzed by immunofluorescence assay as described under “Experimental Procedures.”
DISCUSSION
In this study, we have shown that nucleolar stress-induced PICT1 degradation is mediated by the proteasome in a ubiquitination-independent manner. This finding provides new insight into the molecular mechanisms underlying the nucleolar stress response and participation of the PICT1-p53 axis in tumorigenesis. Ubiquitination-independent, proteasome-mediated degradation of proteins has been reported by several research groups, and one group has demonstrated that 20 S and 26 S proteasomes potentially cleave more than 20% of all cellular proteins (35). Recent work suggests that intrinsically disordered regions (IDRs) in the protein, which lack stable tertiary and/or secondary structures under physiological conditions, become target sites for ubiquitination-independent degradation by the proteasome (36–38). IDRs are also known to be involved in one-to-many signaling processes as signal integrators via binding to multiple partners (39, 40). IDR prediction programs (DisEMBL, PONDR-FIT, and DISOPRED2, etc.) indicate that PICT1 contains multiple IDRs. Fig. 10 shows the results of PONDR-FIT (41) and DISOPRED2 (42) analyses, suggesting three large IDRs (1–46, 131–188, and 285–347) in PICT1. Although further study will be required to address the significance of IDRs in PICT1 degradation, these predictions, together with results of in vitro PICT1 degradation by 20 S proteasome (Fig. 6), raise the possibility that a PICT1 IDR(s) plays a critical role in PICT1 degradation, as described in the following model. (i) A PICT1 IDR mediates binding to multiple partners, such as ribosomal proteins and RNA-processing enzymes, to form a large protein complex in the nucleolus. (ii) Nucleolar stress in some way decreases the integrity of this protein complex and induces dissociation of proteins, including PICT1, from the complex. (iii) Released PICT1, in which the IDR is no longer masked by the binding partner(s), translocates into the cytosol and is targeted by the proteasome to be degraded.
FIGURE 10.
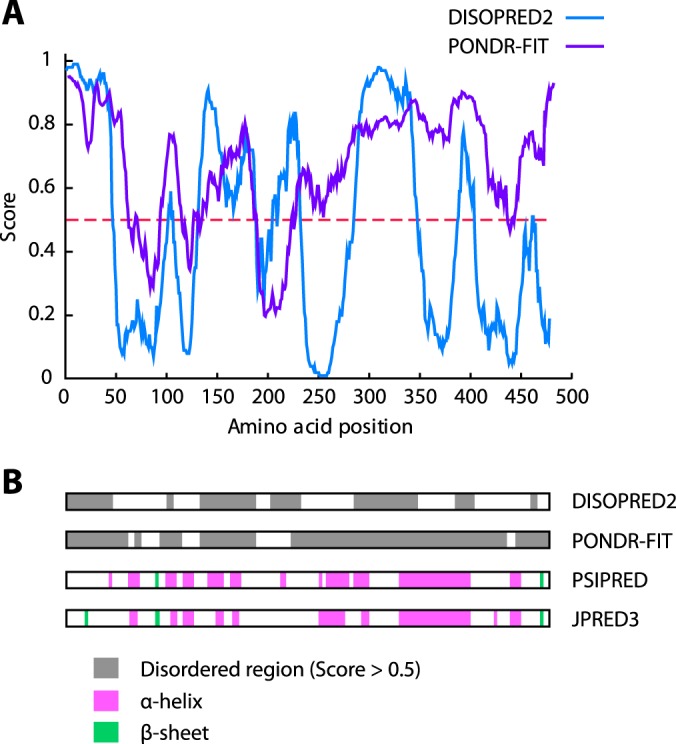
Bioinformatic analyses of PICT1 protein for disorder and secondary structure predictions. Regions of disorder were predicted with DISOPRED2 and PONDR-FIT. A, results are represented by blue (DISOPRED2) and purple (PONDR-FIT) lines. B, disordered regions (gray box) were represented with results of secondary structure prediction with PSIPRED (46) and JPRED3 (47). Pink and green boxes represent α-helix and β-sheet, respectively.
The next question that arises here is the nature of the trigger for PICT1 degradation. Specifically, what triggers the loss of integrity of the PICT1 complex? A decreased amount of rRNA, experimentally induced by inhibition of RNA polymerase I, is one of the major causal factors of nucleolar stress. PICT1 encompasses multiple stretches of basic amino acids, which potentially bind to nucleic acids. In support of this notion, a recent study shows the binding of PICT1 to 5 S rRNA and the importance of PICT1 for the integration of the 5 S RNP particle into the ribosome (12). It is plausible that PICT1 binds directly to specific rRNA species and that a stoichiometric imbalance in the PICT1-rRNA interaction leads to PICT1 destabilization and degradation.
The requirement of nucleolar localization of PICT1 for the degradation (Fig. 7) raises another question of how the PICT1 deletion mutant (d342–449) maintains the stability in the nucleoplasm even under nucleolar stress. To address this question, it should be noted that deletion of the region (342–449) did not completely block nucleolar localization of PICT1; there remained nucleolar (4.5%) and nucleolar/nuclear (25%) localized PICT1 populations (Fig. 7A). This suggests that the region (342–449) not only functions as a nucleolar localization signal but also has other critical functions (binding to other nuclear molecules, providing sites for post-translational modifications, etc.) for regulating the fate of PICT1 protein in cells. Intriguingly, there are some reports that show the accumulation of proteasome components in the nucleolus when cells are treated with proteasome inhibitors (43, 44). These imply that a certain population of proteasomes transiently localizes within the nucleolus. Thus, it is another possibility that naked PICT1 as a result of nucleolar stress is targeted by the proteasome to be degraded, although whether nucleolar stress increases nucleolar proteasome activity should be addressed.
Nucleolar stress influences diverse types of nucleolar proteins. As shown in Fig. 8, nucleolin was redistributed to the nucleoplasm upon the induction of stress, and this effect was blocked by epoxomicin. Intriguingly, Pyr41 did not block the redistribution of nucleolin (data not shown), suggesting that a proteasome-dependent, but ubiquitin-independent, process also exists upstream of nucleolin redistribution. Nucleostemin, another nucleolar resident and positive regulator of cell proliferation, was recently reported to be degraded in response to nucleolar stress, again in a proteasome-dependent but ubiquitin-independent manner (45). Although these results imply that PICT1 functions as an upstream signaling molecule for nucleolin and nucleostemin, depletion of PICT1 affected neither nucleolin nor nucleostemin (16). There might be complex signaling networks that employ a ubiquitin-independent proteasomal degradation system as a tool for nucleolar stress response. Further study will be required to identify the role of PICT1 and draw a more detailed picture of the nucleolar stress response system.
Acknowledgments
We are grateful to Dr. Dirk Bohmann (University of Rochester Medical Center), Dr. Kenji Tago (Jichi Medical University), Dr. Yoshihiro Miwa (University of Tsukuba), and Dr. David W. Russell (University of Texas Southwestern Medical Center) for providing plasmid vectors. We are also grateful to Drs. Takehiko Kamijo and Akira Nakagawara (Chiba Cancer Center Research Institute) for providing H1299 cells and to Drs. Tomoko Date and Takaji Wakita (National Institute of Infectious Diseases) for providing HuH-7 cells. We thank Dr. Fumiaki Okahara for contributing to this work at the initial stage.
This work was supported by grants-in-aid for scientific research (KAKENHI) from the Ministry of Education, Culture, Sports, and Technology of Japan.
- RNP
- ribonucleoprotein
- ActD
- actinomycin D
- FUrd
- 5-fluorouridine
- IDR
- intrinsically disordered region
- Ni2+-NTA
- nickel-nitrilotriacetic acid
- ACTB
- β-actin.
REFERENCES
- 1. Boulon S., Westman B. J., Hutten S., Boisvert F. M., Lamond A. I. (2010) The nucleolus under stress. Mol. Cell 40, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Deisenroth C., Zhang Y. (2010) Ribosome biogenesis surveillance: probing the ribosomal protein-Mdm2-p53 pathway. Oncogene 29, 4253–4260 [DOI] [PubMed] [Google Scholar]
- 3. Bursać S., Brdovčak M. C., Pfannkuchen M., Orsolić I., Golomb L., Zhu Y., Katz C., Daftuar L., Grabušić K., Vukelić I., Filić V., Oren M., Prives C., Volarevic S. (2012) Mutual protection of ribosomal proteins L5 and L11 from degradation is essential for p53 activation upon ribosomal biogenesis stress. Proc. Natl. Acad. Sci. U.S.A. 109, 20467–20472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fumagalli S., Ivanenkov V. V., Teng T., Thomas G. (2012) Suprainduction of p53 by disruption of 40S and 60S ribosome biogenesis leads to the activation of a novel G2/M checkpoint. Genes Dev. 26, 1028–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Horn H. F., Vousden K. H. (2008) Cooperation between the ribosomal proteins L5 and L11 in the p53 pathway. Oncogene 27, 5774–5784 [DOI] [PubMed] [Google Scholar]
- 6. Sun X. X., Dai M. S., Lu H. (2007) 5-Fluorouracil activation of p53 involves an MDM2-ribosomal protein interaction. J. Biol. Chem. 282, 8052–8059 [DOI] [PubMed] [Google Scholar]
- 7. Sun X. X., Dai M. S., Lu H. (2008) Mycophenolic acid activation of p53 requires ribosomal proteins L5 and L11. J. Biol. Chem. 283, 12387–12392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sun X. X., Wang Y. G., Xirodimas D. P., Dai M. S. (2010) Perturbation of 60 S ribosomal biogenesis results in ribosomal protein L5- and L11-dependent p53 activation. J. Biol. Chem. 285, 25812–25821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y., Lu H. (2009) Signaling to p53: ribosomal proteins find their way. Cancer Cell 16, 369–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dai M. S., Sun X. X., Lu H. (2010) Ribosomal protein L11 associates with c-Myc at 5 S rRNA and tRNA genes and regulates their expression. J. Biol. Chem. 285, 12587–12594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Donati G., Peddigari S., Mercer C. A., Thomas G. (2013) 5 S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 4, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sloan K. E., Bohnsack M. T., Watkins N. J. (2013) The 5 S RNP couples p53 homeostasis to ribosome biogenesis and nucleolar stress. Cell Rep. 5, 237–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Challagundla K. B., Sun X. X., Zhang X., DeVine T., Zhang Q., Sears R. C., Dai M. S. (2011) Ribosomal protein L11 recruits miR-24/miRISC to repress c-Myc expression in response to ribosomal stress. Mol. Cell. Biol. 31, 4007–4021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dai M. S., Arnold H., Sun X. X., Sears R., Lu H. (2007) Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 26, 3332–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Okahara F., Ikawa H., Kanaho Y., Maehama T. (2004) Regulation of PTEN phosphorylation and stability by a tumor suppressor candidate protein. J. Biol. Chem. 279, 45300–45303 [DOI] [PubMed] [Google Scholar]
- 16. Sasaki M., Kawahara K., Nishio M., Mimori K., Kogo R., Hamada K., Itoh B., Wang J., Komatsu Y., Yang Y. R., Hikasa H., Horie Y., Yamashita T., Kamijo T., Zhang Y., Zhu Y., Prives C., Nakano T., Mak T. W., Sasaki T., Maehama T., Mori M., Suzuki A. (2011) Regulation of the MDM2-P53 pathway and tumor growth by PICT1 via nucleolar RPL11. Nat. Med. 17, 944–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lee S., Kim J. Y., Kim Y. J., Seok K. O., Kim J. H., Chang Y. J., Kang H. Y., Park J. H. (2012) Nucleolar protein GLTSCR2 stabilizes p53 in response to ribosomal stresses. Cell Death Differ. 19, 1613–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okahara F., Itoh K., Ebihara M., Kobayashi M., Maruyama H., Kanaho Y., Maehama T. (2005) Production of research-grade antibody by in vivo electroporation of DNA-encoding target protein. Anal. Biochem. 336, 138–140 [DOI] [PubMed] [Google Scholar]
- 19. Okahara F., Itoh K., Nakagawara A., Murakami M., Kanaho Y., Maehama T. (2006) Critical role of PICT-1, a tumor suppressor candidate, in phosphatidylinositol 3,4,5-trisphosphate signals and tumorigenic transformation. Mol. Biol. Cell 17, 4888–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Maehama T., Dixon J. E. (1998) The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 273, 13375–13378 [DOI] [PubMed] [Google Scholar]
- 21. Treier M., Staszewski L. M., Bohmann D. (1994) Ubiquitin-dependent c-Jun degradation in vivo is mediated by the δ domain. Cell 78, 787–798 [DOI] [PubMed] [Google Scholar]
- 22. Maehama T., Tanaka M., Nishina H., Murakami M., Kanaho Y., Hanada K. (2008) RalA functions as an indispensable signal mediator for the nutrient-sensing system. J. Biol. Chem. 283, 35053–35059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Andersen J. S., Lam Y. W., Leung A. K., Ong S. E., Lyon C. E., Lamond A. I., Mann M. (2005) Nucleolar proteome dynamics. Nature 433, 77–83 [DOI] [PubMed] [Google Scholar]
- 24. Andersen J. S., Lyon C. E., Fox A. H., Leung A. K., Lam Y. W., Steen H., Mann M., Lamond A. I. (2002) Directed proteomic analysis of the human nucleolus. Curr. Biol. 12, 1–11 [DOI] [PubMed] [Google Scholar]
- 25. Torres J., Pulido R. (2001) The tumor suppressor PTEN is phosphorylated by the protein kinase CK2 at its C terminus. Implications for PTEN stability to proteasome-mediated degradation. J. Biol. Chem. 276, 993–998 [DOI] [PubMed] [Google Scholar]
- 26. Vazquez F., Ramaswamy S., Nakamura N., Sellers W. R. (2000) Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 20, 5010–5018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jin J., Li X., Gygi S. P., Harper J. W. (2007) Dual E1 activation systems for ubiquitin differentially regulate E2 enzyme charging. Nature 447, 1135–1138 [DOI] [PubMed] [Google Scholar]
- 28. Pelzer C., Kassner I., Matentzoglu K., Singh R. K., Wollscheid H. P., Scheffner M., Schmidtke G., Groettrup M. (2007) UBE1L2, a novel E1 enzyme specific for ubiquitin. J. Biol. Chem. 282, 23010–23014 [DOI] [PubMed] [Google Scholar]
- 29. Kalt I., Borodianskiy-Shteinberg T., Schachor A., Sarid R. (2010) GLTSCR2/PICT-1, a putative tumor suppressor gene product, induces the nucleolar targeting of the Kaposi's sarcoma-associated herpesvirus KS-Bcl-2 protein. J. Virol. 84, 2935–2945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kalt I., Levy A., Borodianskiy-Shteinberg T., Sarid R. (2012) Nucleolar localization of GLTSCR2/PICT-1 is mediated by multiple unique nucleolar localization sequences. PLoS One 7, e30825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. von Mikecz A. (2006) The nuclear ubiquitin-proteasome system. J. Cell Sci. 119, 1977–1984 [DOI] [PubMed] [Google Scholar]
- 32. Wójcik C., DeMartino G. N. (2003) Intracellular localization of proteasomes. Int. J. Biochem. Cell Biol. 35, 579–589 [DOI] [PubMed] [Google Scholar]
- 33. Huang M., Itahana K., Zhang Y., Mitchell B. S. (2009) Depletion of guanine nucleotides leads to the Mdm2-dependent proteasomal degradation of nucleostemin. Cancer Res. 69, 3004–3012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yamauchi T., Keough R. A., Gonda T. J., Ishii S. (2008) Ribosomal stress induces processing of Mybbp1a and its translocation from the nucleolus to the nucleoplasm. Genes Cells 13, 27–39 [DOI] [PubMed] [Google Scholar]
- 35. Baugh J. M., Viktorova E. G., Pilipenko E. V. (2009) Proteasomes can degrade a significant proportion of cellular proteins independent of ubiquitination. J. Mol. Biol. 386, 814–827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Melo S. P., Barbour K. W., Berger F. G. (2011) Cooperation between an intrinsically disordered region and a helical segment is required for ubiquitin-independent degradation by the proteasome. J. Biol. Chem. 286, 36559–36567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Peña M. M., Melo S. P., Xing Y. Y., White K., Barbour K. W., Berger F. G. (2009) The intrinsically disordered N-terminal domain of thymidylate synthase targets the enzyme to the ubiquitin-independent proteasomal degradation pathway. J. Biol. Chem. 284, 31597–31607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Singh Gautam A. K., Balakrishnan S., Venkatraman P. (2012) Direct ubiquitin independent recognition and degradation of a folded protein by the eukaryotic proteasomes–origin of intrinsic degradation signals. PLoS One 7, e34864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tompa P. (2012) Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 37, 509–516 [DOI] [PubMed] [Google Scholar]
- 40. Wright P. E., Dyson H. J. (1999) Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 293, 321–331 [DOI] [PubMed] [Google Scholar]
- 41. Xue B., Dunbrack R. L., Williams R. W., Dunker A. K., Uversky V. N. (2010) PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 1804, 996–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ward J. J., Sodhi J. S., McGuffin L. J., Buxton B. F., Jones D. T. (2004) Prediction and functional analysis of native disorder in proteins from the three kingdoms of life. J. Mol. Biol. 337, 635–645 [DOI] [PubMed] [Google Scholar]
- 43. Arabi A., Rustum C., Hallberg E., Wright A. P. (2003) Accumulation of c-Myc and proteasomes at the nucleoli of cells containing elevated c-Myc protein levels. J. Cell Sci. 116, 1707–1717 [DOI] [PubMed] [Google Scholar]
- 44. Mattsson K., Pokrovskaja K., Kiss C., Klein G., Szekely L. (2001) Proteins associated with the promyelocytic leukemia gene product (PML)-containing nuclear body move to the nucleolus upon inhibition of proteasome-dependent protein degradation. Proc. Natl. Acad. Sci. U.S.A. 98, 1012–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lo D., Dai M. S., Sun X. X., Zeng S. X., Lu H. (2012) Ubiquitin- and MDM2 E3 ligase-independent proteasomal turnover of nucleostemin in response to GTP depletion. J. Biol. Chem. 287, 10013–10020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jones D. T. (1999) Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 [DOI] [PubMed] [Google Scholar]
- 47. Cole C., Barber J. D., Barton G. J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201 [DOI] [PMC free article] [PubMed] [Google Scholar]