

Background: Androgen receptor (AR) signaling is indispensable for the development of castration-resistant prostate cancer.
Results: Hydrogen sulfide (H2S) inhibits AR transactivation, and the expression of cystathionine γ-lyase (CSE) and H2S production in antiandrogen-resistant prostate cancer cells is lower.
Conclusion: Lower level of H2S contributes to antiandrogen-resistant status in prostate cancer cells.
Significance: CSE/H2S system can serve as a valuable prognosis indicator for treatment of prostate cancer.
Keywords: Adrenergic Receptor, Aging, Cancer Biology, Cell Growth, Signal Transduction, Sulfhydryl
Abstract
Androgen receptor (AR) signaling is indispensable for the development of prostate cancer from the initial androgen-dependent state to a later aggressive androgen-resistant state. This study examined the role of hydrogen sulfide (H2S), a novel gasotransmitter, in the regulation of AR signaling as well as its mediation in androgen-independent cell growth in prostate cancer cells. Here we found that H2S inhibits cell proliferation of both androgen-dependent (LNCaP) and antiandrogen-resistant prostate cancer cells (LNCaP-B), with more significance on the latter, which was established by long term treatment of parental LNCaP cells with bicalutamide. The expression of cystathionine γ-lyase (CSE), a major H2S-producing enzyme in prostate tissue, was reduced in both human prostate cancer tissues and LNCaP-B cells. LNCaP-B cells were resistant to bicalutamide-induced cell growth inhibition, and CSE overexpression could rebuild the sensitivity of LNCaP-B cells to bicalutamide. H2S significantly repressed the expression of prostate-specific antigen (PSA) and TMPRSS2, two AR-targeted genes. In addition, H2S inhibited AR binding with PSA promoter and androgen-responsive element (ARE) luciferase activity. We further found that AR is post-translationally modified by H2S through S-sulfhydration. Mutation of cysteine 611 and cysteine 614 in the second zinc finger module of AR-DNA binding domain diminished the effects of H2S on AR S-sulfhydration and AR dimerization. These data suggest that reduced CSE/H2S signaling contributes to antiandrogen-resistant status, and sufficient level of H2S is able to inhibit AR transactivation and treat castration-resistant prostate cancer.
Introduction
Prostate cancer is a hormonally regulated malignancy, and androgen receptor (AR)2 signaling is essential for the normal development and functions of prostate as well as the initiation and progression of prostate cancer (1). Upon binding with androgens, AR undergoes a conformational change that ultimately leads to its nuclear translocation and controls the transcriptions of a specific set of androgen-responsive genes, such as prostate-specific antigen (PSA), and concomitant tumor progression (2, 3). Androgen deprivation therapy has been used for the initial treatment of prostate cancer; however, there is inevitable disease progression from an androgen-dependent state to an aggressive androgen-resistant state, leading to castration-resistant prostate cancer (CRPC) (3, 4). The mechanisms underlying the development of CRPC are largely unclear, although AR signaling still plays important roles in CRPC (5).
The AR consists of four structurally and functionally distinct domains, a poorly conserved N-terminal domain, a highly conserved DNA binding domain (DBD), a short hinge region, and a moderately conserved C-terminal ligand binding domain (6, 7). The DBD of AR contains two cysteine 4-type zinc fingers, which enable the AR to specifically recognize androgen-responsive elements (AREs) (8). The second zinc finger is indispensable for the stabilization and specificity of receptor DNA binding (8, 9). The zinc finger is redox-sensitive and therefore may be a molecular target for post-translational modification (9, 10). Nitric oxide (NO) was shown to suppress androgen-induced AR binding activity by S-nitrosylating cysteine 601, located in the second zinc finger (9). Mutation of cysteine 619 in the second zinc finger also caused AR dysfunction (11).
Hydrogen sulfide (H2S) as a novel gasotransmitter has been shown to regulate cancer cell growth and tumor progression (12–14). We recently observed that H2S mediates the anticancer effect of sulforaphane in an androgen-independent prostate cancer cell line (PC-3) (14). Other slow-releasing H2S donors, including GYY413 and S-propargyl-cysteine, have also been shown to generate anticancer effects (15, 16). Cystathionine γ-lyase (CSE) and cystathionine β-synthase (CBS), two main H2S-generating enzymes, were demonstrated to be expressed in human prostate tissues and prostate cancer cells (17). The concentration of cysteine (one substrate for producing H2S) in plasma was significantly decreased as a result of prostate tumor progression (18). One main signaling mechanism underlying H2S regulation of cellular function is post-translational modification of proteins through S-sulfhydration (19, 20). Given the importance of H2S in cancer protection and AR signaling in CRPC, the regulation of AR signaling by H2S and its mediation in androgen-dependent and -independent AR transactivation in prostate cancer cells remain to be delineated.
In the current study, we explored the involvement of CSE/H2S system in antiandrogen-resistant prostate cancer cell growth. Post-translational modification of AR by H2S through S-sulfhydration was examined. H2S inhibition of AR transactivation via interruption of AR dimerization was further studied. Our study indicates an important role of endogenous H2S in AR signaling and antiandrogen-resistant cell growth in CRPC.
EXPERIMENTAL PROCEDURES
Mice and Cell Culture
CSE knock-out mice were generated as described previously (21). All animals were maintained on standard rodent chow and had free access to food and water. All animal experiments were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Lakehead University Animal Care Committee. After the mice were anesthetized, the prostate tissues were dissected and cleaned for protein extraction or H2S measurement.
The AR-positive human prostate cancer cell line (LNCaP) was purchased from the American Type Culture Collection (Manassas, VA) and cultured with RPMI 1640 medium (Sigma, Oakville, Ontario, Canada) supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C in a 5% CO2, 95% air atmosphere (22). Cos-1 cells (American Type Culture Collection) were cultured at 37 °C under 5% CO2 in minimal essential medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. Cos-1 cells were subjected to gene transfection when they had grown to 70–80% confluence. Charcoal/dextran-stripped FBS (5%) and phenol red-free medium were used for the experiments with androgen-dependent cell growth and AR transactivation. In one group of experiments, LNCaP cells were continually incubated with 2 μm bicalutamide in the steroid-free medium containing 5% charcoal/dextran-stripped FBS for 10 weeks, and formed colonies were cultured in steroid-free medium as antiandrogen-resistant LNCaP cell line (LNCaP-B) (22).
Determination of mRNA Level
Total RNA was isolated using TRI Reagent (Invitrogen, Burlington, Ontario, Canada). First strand cDNA was prepared by reverse transcription using M-MuLV reverse transcriptase and random hexamer primers according to the manufacturer's protocol (New England Biolabs, Ipswich, MA). Real-time PCR was performed in an iCycler iQ5 apparatus (Bio-Rad, Mississauga, Ontario, Canada) associated with the iCycler optical system software (version 3.1) using SYBR Green PCR master mix, as described previously (23). The primers of PSA were 5′-GCCCACCCAGGAGCCAGCACT-3′ (forward) and 5′-CCCCCAGAATCACCCGAGCAG-3′ (reverse). The primers of TMPRSS2 were 5′-GACGGCATTTGCGGGGATTTTG-3′ (forward) and 5′-CACCTTGGCAGCGTTCAGCACTTC-3′ (reverse) (24). The primers of β-actin were purchased from Ambion (Streetsville, Ontario, Canada).
ChIP Assay
The binding of AR with PSA promoter was determined by ChIP assay as described previously (25). The samples incubated with nonspecific IgG antibody acted as negative control. A fraction of the protein-DNA was not precipitated but set aside for the total chromatin examination (termed input). The aimed sequence containing ARE sites in the promoters of PSA and TMPSS2 was amplified by PCR. The primers for PSA were 5′-CTTGGAGTGCAAAGGATCTAG-3′ (forward) and reverse: 5′-CTGGGGAGCCTCCCCCAGGAGC-3′ (reverse). The primers for TMPRSS2 were 5′-CCAGAGCCTCCTCCAGGTTC-3′ (forward) and 5′-GTACTCAAGCGGATCCCAGTC-3′ (reverse). To exclude the unspecific AR binding, we also determined the binding of AR with the distant DNA region within PSA and TMPRSS2 promoter, which does not contain ARE sites predicted by bioinformatic analysis. Quantitative analyses of the AR and the promoter interaction were determined by real-time PCR, and binding intensity was normalized to the level of input by using the same primers.
Plasmid Preparation, Point Mutation, and Reporter Gene Assay
The plasmids pEGFP-AR was purchased from Addgene (Cambridge, MA) (26, 27). Single and/or double mutation of cysteine 595, 601, 611, 614, and/or 619 in AR to alanine were conducted using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) (23), respectively. The site-directed mutants were confirmed by DNA sequencing.
ARE reporter assay was determined by using the androgen receptor reporter kit from Qiagen (Toronto, Ontario, Canada). Briefly, LNCaP cells were transfected with a mixture of inducible androgen receptor-responsive firefly luciferase construct (400 ng) and constitutively expressing Renilla luciferase construct (10 ng). The AR-responsive luciferase construct encodes the firefly luciferase reporter gene under the control of a CMV promoter and tandem repeats of the AR transcriptional response element. After 48 h of transfection, luciferase activities were measured with a Dual-Luciferase reporter assay kit (Promega, Madison, WI) (25).
Western Blotting and Immunoprecipitation
After different treatments, cells or tissues were obtained and lysed. The protein lysates from human normal prostate tissue and prostate adenocarcinoma were obtained from OriGene Technologies Inc. (Rockville, MD). Equal amounts of proteins were boiled and separated by SDS-PAGE and electrophoretically transferred to nitrocellulose membrane as described previously (23). The dimer forms of AR were determined by using low temperature PAGE as described everywhere (28). To preserve dimer, all buffers and gels were prepared without 2-mercaptoethanol and SDS and pre-equilibrated to 4 °C prior to electrophoresis. All procedures, including electrophoresis and membrane transfer, were operated at 4 °C. The dilutions of primary antibodies were 1:2000 for CSE, CBS, and 3-mercaptopyruvate sulfurtransferase (Abnova), 1:200 for phosphorylated AR, AR, PSA, proliferative cell nuclear antigen (PCNA), cyclin D1, heat shock protein (HSP) 70 and 90 (Santa Cruz Biotechnology, Santa Cruz, CA), 1:2000 for GFP (Abcam, Toronto, Ontario, Canada), and 1:10000 for β-actin (Sigma).
For immunoprecipitation assay, soluble cell lysates were incubated with 1 μg of anti-HSP70 or anti-HSP90 antibodies for 4 h at 4 °C followed by incubation with protein A-agarose beads for 2 h at 4 °C. The beads were washed three times with lysis buffer, and bound proteins were eluted by boiling for 5 min with 2× SDS loading buffer and analyzed by Western blotting with anti-AR antibody (23).
Immunohistochemistry
The prostate tissues from CSE knock-out mice and wild-type mice were dissected, cleaned, and fixed by immersion in 4% paraformaldehyde for 18 h and then embedded in paraffin (21). Serial sections were cut at 4-μm thickness. After deparaffinizing and blocking the endogenous peroxidase with 0.5% hydrogen peroxide, the antigen retrieval was performed using a rice steamer. Sections were blocked with 5% normal goat serum for 10 min and then incubated in PCNA antibody diluted in the blocking serum for 1 h followed by biotinylated secondary antibody (Vector Laboratories, Burlingame, CA). Sections were incubated in streptavidin-peroxidase diluted 1/20 in Tris buffer for 10 min followed by development in the peroxidase substrate (Vector Laboratories) according to manufacturer's instructions. After counterstaining with hematoxylin, sections were dehydrated and mounted in Permount (Fisher Scientific). The PCNA-positive cells have dark brown nuclei, and the percentage of PCNA-positive cells was expressed as the ratio of PCNA-positive cells to total nucleated cells. At least two arbitrarily chosen fields per tissue cross section and three sections per tissue sample were examined.
RNA Interference and Adenovirus-modified CSE Expression
RNA interference was performed as described previously (23). Briefly, the cells were transfected with CSE gene-specific short interfering RNA (CSE-siRNA, Life Technologies, Burlington, Ontario, Canada) for 48 h using LipofectamineTM 2000 transfection agent in serum-free medium following manufacturer's protocol. The cells transfected with scrambled siRNA acted as a nonsilencing control. Recombinant CSE adenovirus vector (Ad-CSE) was constructed and transfected as described previously (29), and the recombinant adenovirus encoding bacterial β-galactosidase (Ad-LacZ) derived from the same vector was used as a control.
S-Sulfhydration Assay
H2S S-sulfhydration was performed as described previously (20, 23). Briefly, cells were homogenized in HEN buffer (250 mm Hepes-NaOH (pH 7.7), 1 mm EDTA, and 0.1 mm neocuproine) supplemented with 100 μm deferoxamine and centrifuged at 13,000 × g for 30 min at 4 °C. Cell lysates were added to blocking buffer (HEN buffer with 2.5% SDS and 20 mm methyl methanethiosulfonate) at 50 °C for 20 min with frequent vortexing. Methyl methanethiosulfonate was then removed by acetone, and the proteins were precipitated at −20 °C for 20 min. After acetone removal, the proteins were resuspended in HENS buffer (HEN buffer with 1% SDS). To the suspension was added 4 mm N-[6-(biotinamido)hexyl]-3′-(2′-pyridyldithio)propionamide (biotin-HPDP) in dimethyl sulfoxide without ascorbic acid. After incubation for 3 h at 25 °C, biotinylated proteins were precipitated by streptavidin-agarose beads, which were then washed with HENS buffer. The biotinylated proteins were eluted by SDS-PAGE gel and subjected to Western blotting analysis using antibodies against AR or GFP, glucocorticoid receptor (GR), estrogen receptor α (ERα), and GAPDH.
H2S Production Measurement
H2S production rate, reflecting CSE activity, was measured with a methyl blue method as described everywhere (23, 30). Briefly, the cells were lysed in 50 mm ice-cold potassium phosphate buffer (pH 6.8). The flasks containing reaction mixture (100 mm potassium phosphate buffer, 10 mm l-cysteine, 2 mm pyridoxal 5-phosphate, and 10% (w/v) cell/tissue homogenates) and center wells containing 0.5 ml of 1% zinc acetate and a piece of filter paper (2 × 2 cm) were flushed with N2 and incubated at 37 °C for 90 min. The reaction was stopped by adding 0.5 ml of 50% trichloroacetic acid, and the flasks were incubated at 37 °C for another 60 min. The contents of the center wells were transferred to test tubes each containing 3.5 ml of water. Then 0.5 ml of 20 mm N,N-dimethyl-p-phenylenediamine sulfate in 7.2 m HCl and 0.5 ml of 30 mm FeCl3 in 1.2 m HCl were added. The absorbance of the resulting solution at 670 nm was measured 20 min later with a spectrophotometer. The H2S concentration was calculated against the calibration curve of the standard H2S solutions.
Measurement of PSA
The concentration of PSA in culture medium was measured with a PSA enzyme-linked immunosorbent assay according to the manufacturer's instructions (Sigma).
Cell Growth and Viability Assays
To analyze cell growth, 5000 cells/well were plated in 96-well plates. LNCaP and LNCaP-B cells were maintained in charcoal/dextran-stripped FBS for 2 days before each experiment. Cell viabilities were determined using an MTS cell proliferation kit (Promega). The cells incubated with control medium were considered 100% viable (25).
Preparation of Nuclear and Cytosolic Extracts
Nuclear and cytosolic proteins were prepared with the CelLyticTM NuCLEARTM extraction kit (Sigma) as we described previously (23). In brief, the cells at 80% confluence were collected and lysed. Cell lysate was centrifuged at 500 × g for 10 min at 4 °C to separate nuclei and cytosol. Nuclei pellet was washed once with a lysis buffer and resuspended in an assay buffer. The nuclear and cytosolic fractions were then subjected to immunoblotting analysis using antibodies against AR, lamin A/C (nuclear marker), or tubulin (cytosolic marker), respectively.
Statistical Analysis
Data were presented as means ± S.E., and the data represented at least three independent experiments. Statistical comparisons were made using Student's t test or one-way analysis of variance followed by a post hoc analysis (Tukey test) where applicable, and significance level was set at p < 0.05.
RESULTS
Reduced CSE Expression and H2S Production in Human Prostate Cancer Tissues and Antiandrogen-resistant Prostate Cancer Cells
The expression of CSE, CBS, and 3-mercaptopyruvate sulfurtransferase, three major H2S-producing enzymes, was first determined and compared between human prostate adenocarcinoma and normal prostate tissues (31, 32). The expression of CSE but not CBS was significantly reduced in prostate cancer tissue when compared with normal prostate tissues. 3-Mercaptopyruvate sulfurtransferase was not detected in both tissues (Fig. 1A). To further elucidate the functional significance of H2S in the advanced progression of hormone-resistant prostate cancer, we established an in vitro model of antiandrogen-resistant prostate cancer cells (LNCaP-B) by long term treatment of AR-sensitive LNCaP cells with bicalutamide, a nonsteroidal antiandrogen agent (22). Interestingly, we observed that CSE expression is quite lower in LNCaP-B cells in comparison with their parental LNCaP cells, whereas CBS shows no difference between these two types of cells (Fig. 1B). As expected, H2S production rate in LNCaP-B cells was only 17.4% of that in LNCaP cells (Fig. 1C).
FIGURE 1.
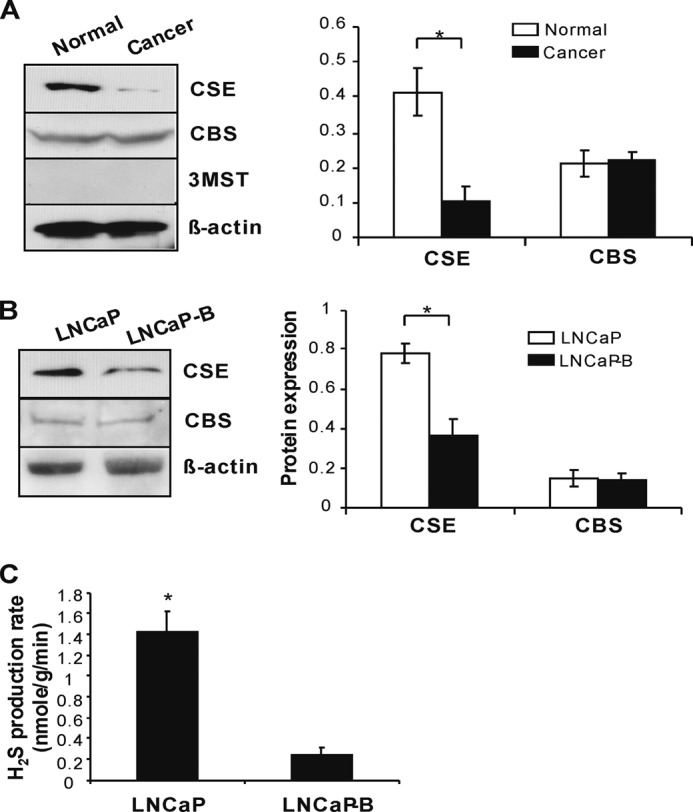
Reduced CSE expression and H2S production in human prostate cancer tissues and antiandrogen-resistant prostate cancer cells. A, lower CSE protein expression in human prostate adenocarcinoma. Human prostate adenocarcinoma and normal prostate protein lysates were obtained from OriGene Technologies Inc. and subjected to Western blotting using antibodies against CSE, CBS, and 3-mercaptopyruvate sulfurtransferase (3MST). The protein expression level of CSE or CBS was normalized with that of β-actin. *, p < 0.05. B, decreased CSE expression in antiandrogen-resistant prostate cancer cells (LNCaP-B). LNCaP-B cells were established by continually incubating LNCaP cells with 2 μm bicalutamide in the steroid-free medium containing 5% charcoal/dextran-stripped FBS for 10 weeks. *, p < 0.05. C, lower H2S production rate in LNCaP-B cells. H2S production rate was measured with a methylene blue method. *, p < 0.05. All experiments were repeated at least three times. Data are presented as means ± S.E.
Age is the greatest risk factor for prostate cancer (33). We found that CSE expression and H2S production in prostate tissue were reduced more than half in older wild-type mice (78 weeks old) when compared with young mice (12 weeks old) (Fig. 2, A and B). Age did not affect CBS expression in prostate tissue from both mice. We detected a very small proportion of H2S production in prostate tissues from CSE knock-out mice, which would be derived from the CBS-catalyzed reaction (Fig. 2B). In addition, the expression of PCNA and cyclin D1, two markers of cell proliferation, was significantly increased in prostate tissues from 78-week-old CSE knock-out mice (Fig. 2C). Immunohistochemistry data further demonstrated that there are more PCNA-positive cells in prostate tissue from 78-week-old CSE knock-out mice when compared with age-matched wild-type mice (Fig. 2D), pointing to the potential role of the CSE/H2S system in maintaining the balance of age-linked cell growth in prostate tissues.
FIGURE 2.

Increased cell proliferation in prostate tissues from aging CSE knock-out mice. A and B, down-regulated CSE expression and lower H2S production rate in prostate tissues from older wild-type mice. *, p < 0.05 versus all other groups. C, increased expression of PCNA and cyclin D1 in prostate tissues from older CSE knock-out mice. D, more PCNA-positive cells in prostate tissues from older CSE knock-out mice. Twelve- and 78-week-old CSE knock-out mice and age-matched wild-type littermates were used for these experiments. *, p < 0.05 versus all other three type of mice in the same group. +/+, wild-type; −/−, CSE knock-out. Eight to 10 mice of each type were used for C, and three mice of each type were used for D. Data are presented as means ± S.E.
Reduced CSE/H2S System Contributes to Castration-resistant Phenotype
After seeding the cells in new plates, LNCaP-B cells tended to adhere quickly to the bottom and grow in clusters when compared with parental LNCaP cells. Bicalutamide at 20 μm inhibited LNCaP cell viability by 38% but only 16% in LNCaP-B cells in medium containing 5% charcoal/dextran-stripped FBS, suggesting that LNCaP-B cells are resistant to bicalutamide-arrested cell growth. NaHS, a well known H2S donor, suppressed cell viability in both cell types but with more significance in LNCaP-B cells (37.5%) when compared with LNCaP cells (22%). The addition of NaHS rebuilt the sensitivity of LNCaP-B cells to bicalutamide-induced cell growth inhibition (Fig. 3A). We further found that LNCaP-B cells grow faster upon stimulation with low concentration of R1881, a synthetic androgen (1.97 times) when compared with LNCaP cells (1.36 times) (Fig. 3B). Exogenously applied NaHS significantly reversed R1881-induced cell growth in both cell types with more significance in LNCaP-B cells (Fig. 3B).
FIGURE 3.

Reduced CSE/H2S system contributes to antiandrogen-resistant phenotype. A, LNCaP-B cells were resistant to bicalutamide-induced but sensitive to H2S-induced cell growth inhibition. LNCaP and LNCaP-B cells were cultured in the steroid-free medium containing 5% charcoal/dextran-stripped FBS in the presence of bicalutamide (20 μm) and/or NaHS (30 μm) for 48 h. Cell viability was measured with an MTS cell proliferation kit. *, p < 0.05 versus all other groups in the same cell type. Bic, bicalutamide. B, H2S significantly reversed R1881-induced cell growth in LNCaP-B cells. LNCaP and LNCaP-B cells were cultured in the steroid-free medium containing 5% charcoal/dextran-stripped FBS in the presence of R1881 (10 nm) and/or NaHS (30 μm) for 48 h. Cell viability was measured with an MTS cell proliferation kit. *, p < 0.05 versus all other groups in the same cell type. C, RNA interference-mediated CSE knockdown. After LNCaP cells were transfected with CSE-siRNAs or negative siRNA (Neg-siRNA) for 48 h, the cells were collected for Western blotting analysis. D, CSE knockdown abolished bicalutamide-arrested cell growth. After LNCaP cells were transfected with CSE-siRNAs or negative siRNA (Neg-siRNA) for 48 h in the presence of bicalutamide (20 μm), cell viability was measured with an MTS cell proliferation kit. *, p < 0.05 versus all other groups. E, adenovirus-mediated CSE overexpression. After LNCaP-B cells were transfected with Ad-CSE or Ad-LacZ at 100 multiplicity of infection for 72 h, the cells were collected for Western blotting analysis. F, CSE overexpression rebuilt the sensitivity of LNCaP-B cells to bicalutamide. After LNCaP-B cells were transfected with Ad-CSE or Ad-LacZ at 100 multiplicity of infection for 72 h, cell viability was assessed with an MTS cell proliferation kit. *, p < 0.05 versus all other groups. All experiments were repeated at least three times. Data are presented as means ± S.E.
To investigate whether CSE expression contributes to antiandrogen-resistant cell growth in prostate cancer cells, we down-regulated CSE expression in LNCaP cells but up-regulated CSE expression in LNCaP-B cells (Fig. 3, C and E). Interestingly, we found that bicalutamide significantly reduces cell growth in negative control siRNA-transfected LNCaP cells, whereas bicalutamide has little effect on cell growth of LNCaP transfected with two different CSE siRNAs (Fig. 3D), respectively. Adenovirus-mediated CSE overexpression significantly reduced cell growth in LNCaP-B cells, which was further inhibited by bicalutamide. In contrast, bicalutamide had little effect on the cell growth of LNCaP-B cells transfected with adenovirus containing β-gal gene (Fig. 3F).
H2S Suppresses AR Transactivation
AR plays a central role in the progression of prostate cancer, which is tightly regulated by androgen. We first found that the expression of AR-targeted gene PSA but not AR is up-regulated in LNCaP-B cells when compared with LNCaP cells, and similarly, CSE knockdown significantly stimulates PSA but not AR expression in LNCaP cells (Fig. 4, A and B). R1881 induces protein expressions of both AR and PSA in LNCaP cells. The presence of NaHS did not affect AR protein expression, whereas NaHS significantly reduced both basal and R1881-stimulated PSA protein expression (Fig. 4C). Moreover, the basal or R1881-induced mRNA expression of PSA and TMPRSS2 (another AR target gene) was suppressed by NaHS (Fig. 4D) (24, 34). GYY4137, a slow-releasing H2S donor, also displayed inhibitory effect on the mRNA levels of PSA and TMPRSS2 (Fig. 4E) (15). We further measured medium PSA level in LNCaP cells after R1881 and NaHS treatment. R1881 stimulated medium PSA secretion by 458.3%, whereas NaHS inhibited PSA secretion by 37.5% (Fig. 4F). AR regulates gene transcriptions by binding the AREs located in the promoter/enhancer region (3). We next found that NaHS significantly suppresses ARE luciferase activity with or without R1881 stimulation (Fig. 4G). CSE overexpression displayed a similar effect as exogenously applied NaHS on inhibiting ARE luciferase activity (Fig. 4H). By using the ChIP assay, we further found that H2S lowers the binding of AR with the promoters of PSA- and TMPRSS2-containing ARE sites (Fig. 4I). No immunoprecipitation and amplification were seen with nonspecific IgG antibody. We further determined the binding of AR with the distant DNA region containing no ARE sites within PSA or TMPRSS2 promoter. As expected, no signaling was observed for the binding of AR with these distant DNA regions (Fig. 4I). All these data suggest that H2S suppresses AR transactivation by inhibiting AR binding with ARE. CSE expression and H2S production were not changed by either bicalutamide or R1881 in LNCaP cells (data not shown).
FIGURE 4.

H2S suppresses AR transactivation. A, PSA but not AR expression was up-regulated in LNCaP-B cells. *, p < 0.05 when compared with all groups. B, CSE knockdown induced PSA expression. After LNCaP cells were transfected with CSE-siRNAs or negative siRNA (Neg-siRNA) for 48 h, the cells were collected for Western blotting analysis. *, p < 0.05. C, H2S inhibited PSA protein expression. Twenty-four hours after LNCaP cells were treated with 10 nm R1881 in the presence or absence of NaHS (30 μm), the cells were collected for Western blotting analysis of AR and PSA expression. D, H2S inhibited the mRNA expression of PSA and TMPRSS2. Twenty-four hours after LNCaP cells were treated with 10 nm R1881 in the presence or absence of NaHS (30 μm), the cells were collected for real-time PCR analysis of AR and PSA mRNA expression. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. E, GYY4137 inhibited the expression of AR-targeted genes. Forty-eight hours after LNCaP cells were incubated with GYY4137 (100 μm), the cells were collected for analyzing the mRNA expression of PSA and TMPRSS2. *, p < 0.05. F, H2S inhibited basal and R1881-induced PSA secretion in LNCaP cells. Forty-eight hours after LNCaP cells were incubated with R1881 (10 nm) with or without NaHS (30 μm), the medium was collected for PSA measurement. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. G, H2S inhibited ARE luciferase activity. Forty-eight hours after LNCaP cells were transfected with inducible ARE luciferase construct in the presence or absence of R1881 (10 nm) and NaHS (30 μm), the cells were collected for measuring luciferase activity. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. H, CSE overexpression suppressed ARE luciferase activity. Twenty-four hours after LNCaP cells were transfected with Ad-CSE, the cells were transfected with inducible ARE luciferase construct in the presence of R1881 (10 nm) for another 48 h. *, p < 0.05 versus all other groups. I, H2S inhibited AR binding with ARE in AR-targeted genes. Forty-eight hours after LNCaP cells were treated with R1881 (10 nm) with or without NaHS (30 μm), the cells were subjected to ChIP assay to detect the binding of AR with ARE-containing promoter region in both PSA and TMPRSS2. The typical binding images are shown in the left panel, and quantitative analysis of AR and the promoter interaction measured by real-time PCR is shown in the right panel. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. All experiments were repeated at least three times. Data are presented as means ± S.E.
H2S Does Not Affect AR Nuclear Translocation and Phosphorylation
Next we investigated how H2S inhibits AR-mediated gene expression. In normal condition, AR interacts with HSP70 and HSP90 in cytoplasm and remains in an inactive state (3). Upon androgen stimulation, AR unbinds HSPs and translocates into the nucleus following increased AR phosphorylation and dimer formation, which will facilitate the binding of AR with ARE sequences and help transcription of AR-targeted genes (5). We first transfected Cos-1 cells with pAR-GFP plasmid either alone or with R1881. In the absence of R1881, GFP signals primarily localized in the cytosol. The addition of R1881 enhanced GFP signals and stimulated GFP signal translocation into the nucleus. Incubation of the cells with NaHS had little effect on GFP localization in the presence or absence of R1881 (Fig. 5A). We next found that the basal proportion of AR and the proportion of R1881-stimulated nuclear AR in LNCaP cells are not affected by NaHS treatment (Fig. 5B). The presence of NaHS did not affect the interaction of AR with HSP90 and HSP70, although we observed that R1881 significantly inhibits AR binding with HSP90 and HSP70 (Fig. 5C). We further demonstrated that NaHS has no effect on AR phosphorylation (Fig. 5D). All these data suggest that H2S-suppressed AR transactivation is independent of AR interaction with HSPs, nuclear translocation, and phosphorylation.
FIGURE 5.

H2S has no effect on AR nuclear translocation and phosphorylation. A, H2S did not affect cellular localization of AR. Seventy-two hours after LNCaP cells were incubated with R1881 (10 nm) with or without NaHS (30 μm), the cells were observed for GFP signal under an inverted Olympus IX70 microscope. B, H2S had no effect on AR nuclear localization. Forty-eight hours after LNCaP cells were incubated with R1881 (10 nm) with or without NaHS (30 μm), the cells were collected to extract nuclear and cytosolic protein. C, H2S did not change the binding of AR with HSPs. Forty-eight hours after LNCaP cells were incubated with R1881 (10 nm) with or without NaHS (30 μm), the cells were collected for immunoprecipitation (IP) assay. WB: Western blotting. D, H2S had little effect on AR phosphorylation (phos-AR). LNCaP cells were incubated with R1881 (10 nm) with or without NaHS (30 μm) for 2 h. The experiments were repeated at least three times.
H2S S-Sulfhydrates AR at the Second Cysteine-rich Zinc Finger Module
Post-translational modification of proteins by S-sulfhydrating their cysteine residue(s) is proposed to mediate most of the biological effects of H2S (19, 23). Here we found that NaHS at 30 μm enhanced AR S-sulfhydration in LNCaP cells (Fig. 6A). H2S suppression of AR transactivation even in the absence of androgen suggests us that H2S may directly act on AR-DBD to alter AR binding with ARE-containing genes. The DBD of AR includes 10 cysteine residues that form two cysteine 4-type zinc fingers with a Zn2+ in the center, which enables the AR to specifically recognize AREs (Fig. 6B) (7). To identify the S-sulfhydrated cysteine residue(s) in DBD, we individually mutated the 5 cysteine residues located in the second zinc finger, which is critical for AR dimerization and transactivation (9, 35). Single mutation of cysteine 611 or 614 but not other cysteine residues reduced AR S-sulfhydration in the presence of NaHS (Fig. 6C), and double mutation of both cysteine 611 and cysteine 614 completely abolished AR S-sulfhydration, pointing to the critical roles of cysteine 611 and cysteine 614 in AR S-sulfhydration (Fig. 6D). When both CSE and wild-type AR were overexpressed in Cos-1 cells, AR was clearly S-sulfhydrated. However, mutation of cysteine 611/614 abolished CSE overexpression-initiated AR S-sulfhydration, suggesting AR can also be S-sulfhydrated by overproduced endogenous H2S at cysteine 611/614 (Fig. 6E). Exogenously applied H2S had no effect on GR and ERα S-sulfhydration in LNCaP cells (Fig. 6F).
FIGURE 6.

H2S S-sulfhydrates AR at the second cysteine-rich zinc finger module. A, H2S S-sulfhydrated AR in LNCaP cells. The cells were incubated with R1881 (10 nm) with or without NaHS (30 μm) for 48 h. AR-load, samples were run on blots alongside total lysates and subjected to immunoblotting with antibody specific to AR. B, the structure domain of AR gene and its two cysteine 4-type zinc fingers in AR-DBD. C, single mutation of cysteine 611 (C611) or cysteine 614 (C614) but not other cysteine resides diminished H2S S-sulfhydration of AR. NTD, N-terminal domain; LBD, ligand-binding domain. D, double mutations of cysteine 611/614 completely eliminated H2S S-sulfhydration of AR. Cos-1 cells were transfected with the mutants for 48 h in the presence of NaHS (30 μm). E, CSE overexpression induced AR S-sulfhydration. Cos-l cells were transfected with Ad-LacZ or Ad-CSE with or without wild-type AR or AR cysteine 611/614 mutant for 48 h. F, H2S had no effect on GR and ERα S-sulfhydration. LNCaP cells were incubated NaHS (30 μm) for 48 h. GAPDH acted as a positive control. AR S-sulfhydration was detected by biotin switch assay with anti-AR antibody (A), anti-GFP antibody (C, D, and E), or anti-ERα and GR (F). The experiments were repeated at least three times.
Furthermore, we observed that the basal and R1881-induced AR dimer formation is inhibited by NaHS in LNCaP cells (Fig. 7A), which was validated in Cos-1 cells transfected with pAR-GFP vector (Fig. 7B). Interestingly, double mutation of cysteine 611/614 eliminates the inhibitory role of NaHS on AR dimmer formation, indicating that H2S suppresses AR transactivation via S-sulfhydrating both cysteine 611 and cysteine 614 in AR-DBD following reduced AR dimerization and AR binding with ARE (Fig. 7C). To investigate whether H2S-regulated prostate cancer cell growth is specific to AR functional change, we overexpressed AR in LNCaP cells (Fig. 7D). In control LNCaP cells, NaHS (30 μm) significantly inhibited cell growth, whereas AR overexpression induced cell growth and reversed the antigrowth effect of H2S (Fig. 7E), indicating H2S suppresses prostate cancer cell growth by at least partially targeting at AR.
FIGURE 7.

H2S inhibits AR dimerization. A, H2S attenuated AR dimer formation in LNCaP cells. The cells were incubated with or without R1881 (10 nm) and/or NaHS (30 μm) for 48 h. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. B, H2S inhibited AR dimerization in Cos-1 cells. Twenty-four hours after the cells were transfected with wild-type AR plasmid, R1881 (10 nm) and/or NaHS (30 μm) were added for another 48 h. *, p < 0.05 versus R1881 group and NaHS group; #, p < 0.05 versus R1881 group. C, mutation of C611/614 eliminated H2S-induced AR dimerization inhibition. Twenty-four hours after the cells were transfected with AR mutants, R1881 (10 nm) and/or NaHS (30 μm) were added for another 48 h. *, p < 0.05 versus R1881 group. D and E, AR overexpression abolished the antigrowth effect of H2S on prostate cancer cells. LNCaP cells were transfected with wild-type AR in the presence or absence of NaHS (30 μm) for 48 h. The experiments were repeated at least three times. Data are presented as means ± S.E.
DISCUSSION
Prostate cancer is one of the most commonly lethal malignancies occurring in western countries, and today, it is expected that 1 in 5 men will be diagnosed with this disease in their lifetime and that this number will rise to 1 in 4 within the next decade (33). Androgen ablation via surgical (orchiectomy) or medical (antiandrogens) castration as a first-line therapy has been recognized for a longer time (36). Despite all these therapies, prostate cancer cells invariably develop a variety of cellular pathways to survive and proliferate in an androgen-depleted environment, leading to eventually fatal outcome (4, 5). Although several hypotheses, including AR gene amplification, AR gene mutations, involvement of coregulators, and ligand-independent activation of AR, have been proposed to contribute to CRPC, further understanding of the molecular mechanisms triggered by AR signaling during CRPC is critical for designing new potential targets against advanced prostate cancer progression (3, 36).
In this study, we showed that CSE exhibits a significant decreased expression pattern in clinical advanced prostate cancer tissues and antiandrogen-resistant prostate cancer cells. The level of endogenous H2S, a novel gasotransmitter, was also lower in antiandrogen-resistant prostate cancer cells, suggesting that CSE/H2S signaling is involved in the development of CRPC (32). This seems to be the case as overexpression of CSE or supplement of exogenously applied H2S re-established the sensitivity of antiandrogen-resistant prostate cancer cells to bicalutamide-inhibited cell growth. In contrast, knockdown of CSE in LNCaP cells, both androgen-responsive and bicalutamide-sensitive prostate cells, endowed the cells to grow even in the presence of bicalutamide. Many factors have been reported to induce androgen-independent growth of prostate cancer cells (22, 37, 38). The production of NO, another well defined gasotransmitter, was higher in antiandrogen-resistant prostate cancer cells, and treatment of a nitrous oxide synthase inhibitor l-NG-nitroarginine methyl ester could resensitize the growth response to bicalutamide (22). Chronic inflammation induced by IL-1β or overexpression of c-Myc also conferred androgen-independent prostate cancer cell growth (37, 38). Although we demonstrated the reduced CSE expression and H2S production in human prostate cancer tissues and antiandrogen-resistant prostate cancer cells, further comparison of CSE/H2S change between human androgen-resistant and androgen-dependent prostate cancer tissues will definitely help the clinical application of H2S against prostate cancer.
To elucidate how decreased CSE/H2S system contributes to antiandrogen-resistant cell growth, we analyzed the regulation of AR signaling by H2S. Continued reliance on AR signaling is a hallmark of prostate cancer progression (1). It is known that the unbound AR resides predominantly in the cytoplasm in a complex containing HSP70 and HSP90; the presence of androgen initiates a cascade of events that leads to AR release from HSPs following increased AR phosphorylation, AR nuclear translocation, and dimerization (3, 10). We first confirmed that the presence of R1881, a synthetic androgen, induces AR protein expression, inhibits HSPs binding with AR, enhances AR phosphorylation, and stimulates AR nuclear translocation, pointing to the crucial role of androgen in activating AR signaling. In contrast, we observed that H2S has little effect on AR protein expression and AR phosphorylation. H2S also did not affect the interaction of AR with HSPs and AR subcellular distribution, even in the absence of R1881. Despite these findings, H2S did inhibit AR transactivation, as evidenced by decreased AR binding with AREs situated in the promoter region of AR target genes and lower ARE luciferase activity. Adenovirus-mediated CSE overexpression also significantly attenuated ARE luciferase activity independent of androgen. Consistent with these data, H2S decreased the mRNA expression of PSA and TRPPS2, two AR-responsive genes. All these data suggest that H2S eliminates AR transactivation possibly at the DNA binding level because the presence of R1881 does not affect the inhibitory role of H2S on AR transactivation.
The mechanisms underlying H2S-inhibited AR transactivation may involve the post-translational regulation of AR by S-sulfhydration. H2S, as a redox agent, can directly interact with sulfhydryl groups in the cysteine residue(s) of selective proteins, yielding a hydropersulfide moiety (–SSH), termed as S-sulfhydration (23). S-Sulfhydration leads to conformational change following altered protein function, which is a novel and important redox signaling mechanism in regulating different cellular functions (19, 32). In our study, we found that the full-length of AR can be S-sulfhydrated in the presence of H2S. H2S suppressing AR downstream gene expression without changing AR interaction with HSPs as well as nuclear localization suggests to us that H2S may directly target at AR-DBD. AR-DBD contains two cysteine-rich zinc finger modules (6). The first zinc finger is responsible for the recognition of AREs, whereas the second zinc finger is involved in DNA-dependent dimerization and the stabilization and specificity of DNA binding (8). We further identified that both cysteine 611 and cysteine 614 in the second zinc finger motif of DBD are responsible for AR S-sulfhydration because mutation of these two cysteine residues completely abrogated S-sulfhydration of AR. Other cysteine residues, including cysteines 595, 601, and 619 in the second zinc finger module of AR-DBD, do not appear to be responsible for AR S-sulfhydration. Most hormone receptors have a DNA binding domain, and the DNA binding domain of AR is highly identical to other hormone receptors, such as GR (76%) and ERα (56%). However, both GR and ERα were not S-sulfhydrated by H2S, excluding the involvement of these receptors in H2S-regulated prostate cancer cell growth. Protein S-sulfhydration on specific cysteine residue(s) is dependent on many factors, such as protein structure, the position of cysteine and the surrounding amino acids, local electric charge and redox status, etc. (19, 20). Thus our data elucidate a unique mechanism of H2S action on repressing AR transactivation through S-sulfhydrating the second zinc finger motif located in AR-DBD.
The interaction of H2S and AR through both cysteine 611 and cysteine 614 may destroy zinc-sulfur cluster and cause a structural change in AR-DBD, triggering abnormal AR dimerization and DNA binding ability (39). AR as a nuclear receptor is often activated as DBDs bind as dimers to two hexameric sequences orientated as direct or inverted repeats (35, 40). The second zinc finger module of AR provides DNA-dependent receptor dimerization necessary for most of the protein-DNA interaction (41). Indeed, the present study demonstrated that H2S inhibits AR dimerization in prostate cancer cells. Mutation studies showed that both cysteine 611 and cysteine 614 are involved in the inhibitory role of H2S on AR dimer formation. Lower AR dimerization would make AR hardly or inadequately bind with AREs, leading to reduced expression of AR-targeted genes. Future studies need to explore how H2S S-sulfhydration of cysteine 611/614 alters AR zinc finger structure and dimerization.
The risk of getting prostate cancer increases with age (3). About 60% of cases are diagnosed in men aged 65 or older, and it rarely occurs in men at age 40 or younger (33). In our studies, we found that CSE expression and H2S production in prostate tissues are significantly lower in mice when they became older, and CSE knock-out eliminates about 80% of H2S production in mouse prostate tissues. The aging CSE knock-out mice tend to have higher cell proliferation rate in prostate tissues, as evidenced by more PCNA-positive cells and higher expression of PCNA and cyclin D1.
In conclusion, this study demonstrated that reduced expression of CSE occurred in human prostate cancer tissues and antiandrogen-resistant prostate cancer cells, and maintenance of sufficient level of H2S could effectively inhibit antiandrogen-resistant growth of prostate cancer cells by suppressing AR transactivation through S-sulfhydrating the second zinc finger module located in DBD. Importantly, our studies suggest that the CSE/H2S system can serve not only as a valuable prognosis indicator but also as an effective therapeutic target for treatment of both the early state of prostate cancer and CRPC.
This study was supported by a grant-in-aid from the Heart and Stroke Foundation of Canada.
- AR
- androgen receptor
- ARE
- androgen responsive element
- PSA
- prostate-specific antigen
- CRPC
- castration-resistant prostate cancer
- CSE
- cystathionine γ-lyase
- Ad-CSE
- recombinant adenovirus vector encoding CSE gene
- Ad-LacZ
- recombinant adenovirus encoding β-galactosidase gene
- CBS
- cystathionine β-synthase
- DBD
- DNA binding domain
- ERα
- estrogen receptor α
- GR
- glucocorticoid receptor
- HSP
- heat shock protein
- PCNA
- proliferative cell nuclear antigen
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt
- NaHS
- sodium hydrosulfide.
REFERENCES
- 1. Gelmann E. P. (2002) Molecular biology of the androgen receptor. J. Clin. Oncol. 20, 3001–3015 [DOI] [PubMed] [Google Scholar]
- 2. Gioeli D., Paschal B. M. (2012) Post-translational modification of the androgen receptor. Mol. Cell. Endocrinol. 352, 70–78 [DOI] [PubMed] [Google Scholar]
- 3. Lonergan P. E., Tindall D. J. (2011) Androgen receptor signaling in prostate cancer development and progression. J. Carcinog. 10, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lallous N., Dalal K., Cherkasov A., Rennie P. S. (2013) Targeting alternative sites on the androgen receptor to treat castration-resistant prostate cancer. Int. J. Mol. Sci. 14, 12496–12519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heinlein C. A., Chang C. (2004) Androgen receptor in prostate cancer. Endocr. Rev. 25, 276–308 [DOI] [PubMed] [Google Scholar]
- 6. Helsen C., Dubois V., Verfaillie A., Young J., Trekels M., Vancraenenbroeck R., De Maeyer M., Claessens F. (2012) Evidence for DNA-binding domain-ligand-binding domain communications in the androgen receptor. Mol. Cell. Biol. 32, 3033–3043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirawat S., Budman D. R., Kreis W. (2003) The androgen receptor: structure, mutations, and antiandrogens. Cancer Invest. 21, 400–417 [DOI] [PubMed] [Google Scholar]
- 8. Schoenmakers E., Alen P., Verrijdt G., Peeters B., Verhoeven G., Rombauts W., Claessens F. (1999) Differential DNA binding by the androgen and glucocorticoid receptors involves the second Zn-finger and a C-terminal extension of the DNA-binding domains. Biochem. J. 341, 515–521 [PMC free article] [PubMed] [Google Scholar]
- 9. Qin Y., Dey A., Purayil H. T., Daaka Y. (2013) Maintenance of androgen receptor inactivation by S-nitrosylation. Cancer Res. 73, 6690–6699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. van der Steen T., Tindall D. J., Huang H. (2013) Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 14, 14833–14859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nazareth L. V., Stenoien D. L., Bingman W. E., 3rd, James A. J., Wu C., Zhang Y., Edwards D. P., Mancini M., Marcelli M., Lamb D. J., Weigel N. L. (1999) A C619Y mutation in the human androgen receptor causes inactivation and mislocalization of the receptor with concomitant sequestration of SRC-1 (steroid receptor coactivator 1). Mol. Endocrinol. 13, 2065–2075 [DOI] [PubMed] [Google Scholar]
- 12. Cao Q., Zhang L., Yang G., Xu C., Wang R. (2010) Butyrate-stimulated H2S production in colon cancer cells. Antioxid. Redox Signal. 12, 1101–1109 [DOI] [PubMed] [Google Scholar]
- 13. Kim S. H., Singh S. V. (2009) d,l-Sulforaphane causes transcriptional repression of androgen receptor in human prostate cancer cells. Mol. Cancer Ther. 8, 1946–1954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pei Y., Wu B., Cao Q., Wu L., Yang G. (2011) H2S mediates the anti-survival role of sulforaphane on human prostate cancer cells. Toxicol. Appl. Pharmacol. 257, 420–428 [DOI] [PubMed] [Google Scholar]
- 15. Lee Z. W., Zhou J., Chen C. S., Zhao Y., Tan C. H., Li L., Moore P. K., Deng L. W. (2011) The slow-releasing hydrogen sulfide donor, GYY4137, exhibits novel anti-cancer effects in vitro and in vivo. PLoS One 6, e21077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ma K., Liu Y., Zhu Q., Liu C. H., Duan J. L., Tan B. K., Zhu Y. Z. (2011) H2S donor, S-propargyl-cysteine, increases CSE in SGC-7901 and cancer-induced mice: evidence for a novel anti-cancer effect of endogenous H2S? PLoS One 6, e20525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo H., Gai J. W., Wang Y., Jin H. F., Du J. B., Jin J. (2012) Characterization of hydrogen sulfide and its synthases, cystathionine β-synthase and cystathionine γ-lyase, in human prostatic tissue and cells. Urology 79, 483e1–483e5 [DOI] [PubMed] [Google Scholar]
- 18. Al-Awadi F., Yang M., Tan Y., Han Q., Li S., Hoffman R. M. (2008) Human tumor growth in nude mice is associated with decreased plasma cysteine and homocysteine. Anticancer Res. 28, 2541–2544 [PubMed] [Google Scholar]
- 19. Gadalla M. M., Snyder S. H. (2010) Hydrogen sulfide as a gasotransmitter. J. Neurochem. 113, 14–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mustafa A. K., Gadalla M. M., Sen N., Kim S., Mu W., Gazi S. K., Barrow R. K., Yang G., Wang R., Snyder S. H. (2009) H2S signals through protein S-sulfhydration. Sci. Signal 2, ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang G., Wu L., Jiang B., Yang W., Qi J., Cao K., Meng Q., Mustafa A. K., Mu W., Zhang S., Snyder S. H., Wang R. (2008) H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ-lyase. Science 322, 587–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yu S., Jia L., Zhang Y., Wu D., Xu Z., Ng C. F., To K. K., Huang Y., Chan F. L. (2013) Increased expression of activated endothelial nitric oxide synthase contributes to antiandrogen resistance in prostate cancer cells by suppressing androgen receptor transactivation. Cancer Lett. 328, 83–94 [DOI] [PubMed] [Google Scholar]
- 23. Yang G., Zhao K., Ju Y., Mani S., Cao Q., Puukila S., Khaper N., Wu L., Wang R. (2013) Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid. Redox Signal 18, 1906–1919 [DOI] [PubMed] [Google Scholar]
- 24. Clinckemalie L., Spans L., Dubois V., Laurent M., Helsen C., Joniau S., Claessens F. (2013) Androgen regulation of the TMPRSS2 gene and the effect of a SNP in an androgen response element. Mol. Endocrinol. 27, 2028–2040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yang G., Pei Y., Teng H., Cao Q., Wang R. (2011) Specificity protein-1 as a critical regulator of human cystathionine γ-lyase expression during smooth muscle cell differentiation. J. Biol. Chem. 286, 26450–26460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kaczmarczyk S. J., Green J. E. (2003) Induction of cre recombinase activity using modified androgen receptor ligand binding domains: a sensitive assay for ligand-receptor interactions. Nucleic Acids Res. 31, e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Stenoien D. L., Cummings C. J., Adams H. P., Mancini M. G., Patel K., DeMartino G. N., Marcelli M., Weigel N. L., Mancini M. A. (1999) Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum. Mol. Genet. 8, 731–741 [DOI] [PubMed] [Google Scholar]
- 28. Klatt P., Schmidt K., Lehner D., Glatter O., Bächinger H. P., Mayer B. (1995) Structural analysis of porcine brain nitric oxide synthase reveals a role for tetrahydrobiopterin and l-arginine in the formation of an SDS-resistant dimer. EMBO J. 14, 3687–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang G., Wu L., Wang R. (2006) Pro-apoptotic effect of endogenous H2S on human aorta smooth muscle cells. FASEB J. 20, 553–555 [DOI] [PubMed] [Google Scholar]
- 30. Yang G., Tang G., Zhang L., Wu L., Wang R. (2011) Pathogenic role of cystathionine γ-lyase and hydrogen sulfide in type 1 diabetes mellitus. Am. J. Pathol. 179, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sen U., Sathnur P. B., Kundu S., Givvimani S., Coley D. M., Mishra P. K., Qipshidze N., Tyagi N., Metreveli N., Tyagi S. C. (2012) Increased endogenous H2S generation by CBS, CSE, and 3MST gene therapy improves ex vivo renovascular relaxation in hyperhomocysteinemia. Am. J. Physiol. Cell Physiol. 303, C41–C51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Edwards B. K., Noone A. M., Mariotto A. B., Simard E. P., Boscoe F. P., Henley S. J., Jemal A., Cho H., Anderson R. N., Kohler B. A., Eheman C. R., Ward E. M. (2014) Annual Report to the Nation on the status of cancer, 1975–2010, featuring prevalence of comorbidity and impact on survival among persons with lung, colorectal, breast, or prostate cancer. Cancer 120, 1290–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wissmann M., Yin N., Müller J. M., Greschik H., Fodor B. D., Jenuwein T., Vogler C., Schneider R., Günther T., Buettner R., Metzger E., Schüle R. (2007) Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat. Cell Biol. 9, 347–353 [DOI] [PubMed] [Google Scholar]
- 34. Wong C. I., Zhou Z. X., Sar M., Wilson E. M. (1993) Steroid requirement for androgen receptor dimerization and DNA binding: modulation by intramolecular interactions between the NH2-terminal and steroid-binding domains. J. Biol. Chem. 268, 19004–19012 [PubMed] [Google Scholar]
- 35. Pienta K. J., Bradley D. (2006) Mechanisms underlying the development of androgen-independent prostate cancer. Clin. Cancer Res. 12, 1665–1671 [DOI] [PubMed] [Google Scholar]
- 36. Wang R. (2012) Physiological implications of hydrogen sulfide: a whiff exploration that blossomed. Physiol. Rev. 92, 791–896 [DOI] [PubMed] [Google Scholar]
- 37. Bernard D., Pourtier-Manzanedo A., Gil J., Beach D. H. (2003) Myc confers androgen-independent prostate cancer cell growth. J. Clin. Invest. 112, 1724–1731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McCourt C., Maxwell P., Mazzucchelli R., Montironi R., Scarpelli M., Salto-Tellez M., O'Sullivan J. M., Longley D. B., Waugh D. J. (2012) Elevation of c-FLIP in castrate-resistant prostate cancer antagonizes therapeutic response to androgen receptor-targeted therapy. Clin. Cancer Res. 18, 3822–3833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Shaffer P. L., Jivan A., Dollins D. E., Claessens F., Gewirth D. T. (2004) Structural basis of androgen receptor binding to selective androgen response elements. Proc. Natl. Acad. Sci. U.S.A. 101, 4758–4763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Centenera M. M., Harris J. M., Tilley W. D., Butler L. M. (2008) The contribution of different androgen receptor domains to receptor dimerization and signaling. Mol. Endocrinol. 22, 2373–2382 [DOI] [PubMed] [Google Scholar]
- 41. van Royen M. E., van Cappellen W. A., de Vos C., Houtsmuller A. B., Trapman J. (2012) Stepwise androgen receptor dimerization. J. Cell Sci. 125, 1970–1979 [DOI] [PubMed] [Google Scholar]