

Background: SEC24 is responsible for selectively recruiting cargo proteins into COPII vesicles.
Results: Mice completely lacking one of the four SEC24 paralogs (SEC24C) die early in post-implantation embryonic development.
Conclusion: SEC24C is essential for embryonic development but is dispensable in a number of cell types.
Significance: These findings provide new insight into the function of SEC24C in vivo.
Keywords: Development, Endoplasmic Reticulum (ER), Gene Knockout, Intracellular Trafficking, Mouse Genetics, COPII
Abstract
COPII-coated vesicles mediate the transport of newly synthesized proteins from the endoplasmic reticulum to the Golgi. SEC24 is the COPII component primarily responsible for recruitment of protein cargoes into nascent vesicles. There are four Sec24 paralogs in mammals, with mice deficient in SEC24A, -B, and -D exhibiting a wide range of phenotypes. We now report the characterization of mice with deficiency in the fourth Sec24 paralog, SEC24C. Although mice haploinsufficient for Sec24c exhibit no apparent abnormalities, homozygous deficiency results in embryonic lethality at approximately embryonic day 7. Tissue-specific deletion of Sec24c in hepatocytes, pancreatic cells, smooth muscle cells, and intestinal epithelial cells results in phenotypically normal mice. Thus, SEC24C is required in early mammalian development but is dispensable in a number of tissues, likely as a result of compensation by other Sec24 paralogs. The embryonic lethality resulting from loss of SEC24C occurs considerably later than the lethality previously observed in SEC24D deficiency; it is clearly distinct from the restricted neural tube phenotype of Sec24b null embryos and the mild hypocholesterolemic phenotype of adult Sec24a null mice. Taken together, these results demonstrate that the four Sec24 paralogs have developed unique functions over the course of vertebrate evolution.
Introduction
Approximately one-third of all mammalian proteins traverse the secretory pathway en route to their final destinations, including cell surface membranes and intracellular compartments, as well as the extracellular space (1, 2). These proteins begin their journey in the endoplasmic reticulum (ER),3 where they are recruited into newly forming COPII vesicles located at ribosome-free ER exit sites (3, 4). The COPII vesicles containing these cargoes are then trafficked from the ER to the ER-Golgi intermediate compartment.
The mechanism of COPII coat assembly has been elegantly dissected in Saccharomyces cerevisiae, and the fundamental mechanisms appear to be conserved from yeast to humans (5). Vesicle formation is initiated with the activation of the small GTPase Sar1p by the GEF Sec12p, which is localized on the ER membrane. GTP binding to Sar1p induces a conformational change and insertion of a small amphipathic helix of Sar1p into the ER membrane and begins the process of vesicle formation (6, 7). Activated Sar1p recruits the Sec23p-Sec24p heterodimer to the ER, forming the inner layer of the COPII coat (8, 9). Polymerization of Sec13p and Sec31p heterotetramers to form the outer layer promotes further curvature and budding of the nascent COPII vesicle from the ER (10).
SEC24 is the COPII component thought to be primarily responsible for cargo recruitment, with ER exit motifs on protein cargoes interacting with specific binding sites on SEC24 (11, 12). SEC24-cargo interactions are either direct, in the case of transmembrane proteins, or require a transmembrane adaptor to mediate the interaction of ER luminal cargo with the COPII coat located on the cytoplasmic face of the ER membrane. Examples include the well characterized interaction between the adaptor component LMAN1/MCFD2 and its ER luminal cargoes Factor V and Factor VIII (13, 14).
In S. cerevisiae, loss of Sec24p is lethal, although deficiency of either of two nonessential Sec24p paralogs, Iss1p or Lst1p, results in specific cargo-transport defects (15). The mammalian genome encodes four paralogs of Sec24 (Sec24a–d), which can be classified into two subgroups, SEC24A/B and SEC24C/D, based on protein sequence identity, with the A/B subgroup more closely related to the ancestral yeast paralog Sec24p (16, 17). All four paralogs contain a highly conserved C terminus and a variable N-terminal region, with previous reports suggesting differences between the two SEC24 subgroups in their affinity for cargo-sorting signals (18, 19). All four Sec24 paralogs appear to be ubiquitously expressed in the adult and in the developing mouse (20, 21).
Mutations in several Sec24 paralogs in zebrafish suggest a critical role in the secretion of extracellular matrix collagens (22–24). Although there have been no diseases attributed to mutations in any of the human SEC24 paralogs, mutations in SEC23A result in the human disorder cranio-lenticulo-sutral dysplasia (25), and mutations in human SEC23B cause congenital dyserythropoietic anemia type II (26). In contrast to humans, Sec23-deficient mice exhibit perinatal lethality due to pancreatic destruction (27). Mice deficient in SEC24A exhibit low plasma cholesterol levels, attributed to a reduction in secretion of the regulatory protein PCSK9 (28). SEC24B-deficient mice have a specific block in the secretion of VANGL2, leading to a neural tube closure defect (29). SEC24D-deficient mice die very early in embryonic development (before E3.5) (20). We now report that the loss of murine SEC24C also results in embryonic lethality, although at a later time point (∼E7 to E8). However, ablation of SEC24C in multiple tissues is surprisingly well tolerated.
EXPERIMENTAL PROCEDURES
Generation of SEC24C-deficient Mice
ES cell clone EPD0241-2-A11 was obtained from the European Mouse Mutagenesis program (EUCOMM). This clone is heterozygous for the allele Sec24ctm1a(EUCOMM)Wtsi, a Sec24c allele with conditional potential, which will be referred to as Sec24cGT. ES cells were cultured and expanded for microinjection and genomic DNA isolation as described previously (30). ES cell mouse chimeras were generated as described (31) and subsequently bred to B6(Cg)-Tyrc-2J/J (JAX stock 000058) to achieve germ line transmission. ES cell-derived F1 black progeny were genotyped using primers to detect the presence (Sec24cGT) or absence (Sec24cWT) of the targeted allele (primers Ex and E). All primers used in this study are listed in Table 1. The Sec24cGT allele was maintained by continuous backcrosses to C57BL/6J mice. Genotyping was performed with mouse tail clip DNA using Go-Taq Green Master Mix (Promega, Madison, WI), and the resulting PCR products were resolved by 2% agarose gel electrophoresis.
TABLE 1.
List of primers used in this study
All sequences are listed 5′ to 3′.
| Primer name | Sequence (5′ to 3′) |
|---|---|
| Genotyping and LR-PCR primers | |
| A | AAGGCGCATAACGATACCA |
| B | CACAACGGGTTCTTCTGTTAGTCCC |
| C | CTTGAGGCAAGAATGCAAAACAAGAATC |
| Ex | GTACTAGGTGAGCCTGAAATCAATG |
| E | ACTAAGATGGGTCCACAAAAGAGC |
| FLP1 | GGTCCAACTGCAGCCCAAGCTTCC |
| FLP2 | GTGGATCGATCCTACCCCTTGCG |
| Cre-fwd | TTACCGGTCGATGCAACGAGT |
| Cre-rev | TTCCATCAGTGAACGAACCTGG |
| GF3 | GCTGATACTGATACTAGGATCCACGGACAG |
| GR4 | GCACTGCTAACAGTTCGCTATTCCTTCCG |
| G | CACACCTCCCCCTGAACCTGAAAC |
| Tg-F | AAATCTGTGCGGAGCCGAAATCTG |
| Tg-R | GCATGAACATGGTTAGCAGAGGCT |
| RT-PCR primers | |
| GAPDH-fwd | TGTGATGGGTGTGAACCACGAGAA |
| GAPDH-rev | ACCAGTGGATGCAGGGATGATGTT |
| 24c-F | GTGTGACCGGCGGCTCTA |
| 24c-R | GGGGCTGCAGGTCCTGGTTG |
| 24d-F | TTGGCAAAGAATTCCTCGACCTG |
| 24d-R | ATCTTTGAGGAGGATGGCCTGGA |
| Primer V | GCCCTCAACCTAATTATGAGAGCCCA |
| Primer W | CCCGTACACCAGTAACAAATGGCT |
| Primer X | TCTCAGCAGTTTGGTCCTCCATTG |
| Primer Y | TTTGCTGCAGCTGATAACCAGG |
| Primers for transgene construction | |
| P1 | GAATACTCAAGCTTGCATGCCTGCAGGTCG |
| P2 | AGAGCATGCATGAGATCACCGGTCATCACCGTGGCGGCCGCTCATAATCAGCCATAC |
| P3 | ACGTACACCGGTGCTCTCATAATGAATG |
| P4 | CTAGTCTGCGGCCGCTTAGCTCAGTAGCTGCCGGATC |
| P5 | ACGTACACCGGTATTTTCATCATGAGCC |
| P6 | TCGCTAGGCGGCCGCTCAGTTAAGCAGTTG |
Long Range PCR to Confirm the Insertion Site of the Sec24c Gene Trap Allele
Genomic DNA isolated from the ES cell clone EPD0241-2-A11 and genomic DNA from a wild type C57BL/6J mouse tail clip were amplified by long range PCR to confirm correct targeting of the Sec24cGT allele with primers GF3 and primer B (5′ end) and GR4 and primer G (3′ end) using Phusion Hot Start II DNA Polymerase (Thermo Scientific), with products resolved on a 0.8% agarose gel.
Generation of a Sec24c Conditional Allele
To generate mice carrying the conditional Sec24ctm1c(EUCOMM)Wtsi allele (referred to here as Sec24cFL allele), Sec24c+/GT mice were crossed to mice transgenic for Flpe recombinase driven by an actin promoter (C57BL/6J background, JAX 005703). Mice were genotyped with primers Ex and E to detect both the wild type (308 bp) allele and the presence of the LoxP site (278 bp), primers A, B, and C to distinguish between the Sec24cGT (204 bp) and Sec24cFL (534 bp) alleles, and with primers FLP1 and FLP2 to detect the FLPE transgene (750 bp). Sec24c+/FL mice were backcrossed to C57BL/6J mice to remove the FLPe transgene.
Generation of a Sec24c Null Allele
Sec24c+/FL mice were crossed to EIIA-driven Cre recombinase transgenic mice (C57/BL6 background, JAX 003724), and offspring were genotyped with primers A, Ex, and E to distinguish the Sec24c+, Sec24cFL, and Sec24ctm1d(EUCOMM)Wtsi (referred to as the Sec24c−) alleles. The Cre transgene was detected with primers Cre-fwd and Cre-rev. Sec24c+/− mice were backcrossed to C57BL/6J mice to remove the Cre transgene.
Generation of Tissue-specific Knock-out Mice
Sec24cFL/FL mice were crossed to Sec24c+/−Cre+ using the following tissue-specific transgenes: P48-Cre (pancreas-specific, a generous gift from Christopher V. E. Wright (32)), villin-Cre (intestinal-epithelial specific, JAX 004586) (33), albumin-Cre (liver hepatocyte-specific, JAX 003574) (34), SM22-Cre mice (smooth muscle-specific, JAX 004746) (35), and Meox2-Cre transgene (ubiquitous expression beginning at embryonic day 5, JAX 003755) (36). Progeny were genotyped at the Sec24c locus with primers A, Ex, and E and with primers Cre-fwd and Cre-rev to detect the presence of the Cre transgene. The level of Cre-mediated excision was assessed by isolation of genomic DNA from tissue samples of either Sec24c+/FL Cre+ or Sec24cFL/− Cre+ mice and PCR using primers A, Ex, and E (Table 1) at the Sec24c locus.
Phenotypic Analysis of Sec24c+/−, Sec24c+/−, Sec24d+/GT, and Tissue-specific Knock-out Mice
Whole blood was collected, and complete blood counts were carried out as described previously (20). For histology, tissues were fixed in 4% paraformaldehyde in PBS overnight at 4 °C and then transferred to 30% EtOH, 50% EtOH, and 70% EtOH, three times each for 10 min. Processing, embedding, sectioning, and H&E staining were performed at the Microscopy and Image Analysis Laboratory, University of Michigan. Body weights were measured weekly from weaning up to 12 weeks of age. A high fat diet (45% of calories from fat) was purchased from Research Diets (New Brunswick, NJ), and animals were fed ad libitum. For insulin and blood lipid analysis, mice were fasted 16 h before blood collection. Blood was collected using heparin-coated collection tubes (Fisher) by retro-orbital bleeding from mice anesthetized with isoflurane. Plasma samples were then collected by centrifugation of heparinized blood samples at 3000 × g for 5 min at 4 °C. Plasma cholesterol and triglyceride levels were measured in 5 μl of plasma samples with colorimetric assays using the LiquiColor cholesterol test kit (Stanbio, Boerne, TX) or serum triglyceride determination kit (Sigma) according to the manufacturer's instructions. Insulin levels were measured using the ultrasensitive mouse insulin ELISA kit (Crystal Chem, Downers Grove, IL), per manufacturer's instructions.
Timed Matings
Timed matings were carried out by intercrossing Sec24c+/− mice. Embryos were harvested at multiple time points, including day E7.5–11.5 for genotyping and histological analysis. Genotyping was performed on genomic DNA isolated from embryonic yolk sacs. Blastocyst collection and genotyping were performed as described previously (37) using super-ovulated Sec24c+/− females and Sec24c+/− males.
Construction of Sec24c and Sec24d Transgenes and Generation of Transgenic Mice
Transgenes carrying either Sec24c or Sec24d were designed using the previously reported pCAG3Z vector (38). The CAG promoter contains the CMV intermediate-early enhancer and the chicken β-actin promoter to drive early, ubiquitous expression (39, 40) of either Sec24c or Sec24d. AgeI and NotI sites were added upstream of an SV40 early termination signal and a poly(A) signal isolated from Tg2.33 (38), and this was placed downstream of the CAG promoter in the pCAG3Z vector to create pCAG3zS. Sec24c (cDNA −9 to 3290) or Sec24d (cDNA −9 to 3098) was inserted between the CAG promoter and the SV40 signal using AgeI and NotI. DNA sequencing was performed on all constructs prior to microinjection to verify the integrity of the transgenes. All primers used to create these constructs (P1–P6) are listed in Table 1. pCAG3zS-Sec24c and pCAGzS-Sec24d were liberated from the vector backbone with a double digest with BamHI-HF/SphI-HF or SacI/HindIII, respectively (New England Biolabs), and transgenic mice were generated by the University of Michigan Transgenic Animal Model Core as described previously (41). Transgenic founders (C57BL/6J × SJL F2) for both lines were detected by PCR using Tg-F and Tg-R located in the promoter region of the transgenes. Mice transgenic for either pCAG3zS-Sec24c or pCAG3zS-Sec24d were crossed with Sec24c+/− mice to generate Sec24c+/− Tg+ mice, which were then crossed to Sec24c+/− mice to generate potential Sec24c−/− Tg+ mice. All progeny were genotyped at the Sec24c locus and for the presence of the transgene. Similar crosses were also performed with Sec24d+/GT (20) mice. Five founders for pCAG3zS-Sec24c were used to generate transgenic lines to test the ability of pCAG3zS-Sec24c to rescue the loss of Sec24c. Four pCAG3zS-Sec24d founders were used to generate transgenic lines.
Reverse Transcription-PCR
Total RNA was isolated from a panel of frozen tissues from transgenic mice from each of the founder lines using an RNeasy kit (Qiagen) as per manufacturers instructions. cDNA synthesis and PCR were carried out in one reaction using SuperScript® III one-step RT-PCR system with Platinum®Taq (Invitrogen) per manufacturer's instructions. Primers V, W, X, and located in Sec24c exons 4–7 were used to amplify signal from Sec24c cDNA. Primers 24c-F or 24d-F and primers 24c-R or 24d-R were used to detect cDNA specific for the Sec24c or Sec24d transgenes. GAPDH amplification with primers GAPDH-fwd and GAPDH-rev was carried out in parallel for each sample.
Statistical Analysis
p value for progeny genotypes were calculated by a χ2 test comparing the expected ratio of genotypes to those observed. For intercrosses of Sec24c+/GT, Sec24c+/FL, or Sec24c+/− mice, the expected ratio of homozygous mice to all other expected genotypes (25:75) was used. Complete blood count parameters were evaluated for significance using Student's t test. The α levels were adjusted for multiple observations according to the Bonferroni correction.
RESULTS
Sec24c mRNA expression was detected by RT-PCR in all 15 adult mouse tissues tested, consistent with previously reported human expression patterns (16, 42). RT-PCR analysis also detected an alternative in-frame exon (exon 6*, encoding 23 amino acids), present in a subset of Sec24c mRNAs (Fig. 1). The transcript containing this additional exon (Sec24c-2) was detected only in brown adipose tissue, skeletal muscle, brain (Fig. 1C), and the heart, with only low levels of the transcript skipping exon 6* (Sec24c-1) detected in these tissues (Fig. 1C). Sec24c-1 is the only splice form detected in lung, kidney, stomach, large intestine, and small intestine (Fig. 1C) as well as the adrenal gland, liver, salivary gland, testis, spleen, and white adipose tissue (data not shown).
FIGURE 1.

Identification of an alternative splice form of Sec24c. A, schematic of the two Sec24c splice forms observed in mice. Arrows indicate primers used to detect the presence of the alternative exon (6*). Shaded regions in exons 4 and 5 represent exonic sequence present in Sec24c-2 that is absent from another RefSeq splice variant annotated as containing exon 6* (NM_001168273.1), which was not detected in our RT-PCR. B, protein sequence alignment beginning at Thr-306 of SEC24C-1 and SEC24C-2 showing the additional 23 amino acids encoded by exon 6*. C, RT-PCR using primers V and W, which detects a 249-bp product from Sec24c-2 and a 180-bp product from Sec24c-1.
Correct targeting of the Sec24cGT allele (Fig. 2A) was confirmed by long range PCR (Fig. 2B), with PCR genotyping primers (A, Ex, and E) differentiating between each Sec24c allele used in this study (Fig. 2C). Genotypes of 2-week-old progeny from Sec24c+/GT intercrosses revealed the expected number of wild type and heterozygous offspring but none of the expected ¼ Sec24cGT/GT mice (p < 2.3 × 10−6, Table 2A). Intercrosses of Sec24c+/FL (obtained by excision of the β-geo cassette from Sec24cGT, Fig. 2A) generated the expected number of Sec24cFL/FL mice (Table 2B) demonstrating that loss of Sec24cGT/GT mice results from the presence of the gene trap cassette, rather than a passenger gene effect (43). EIIA-Cre-mediated excision of Sec24cFL generated the Sec24c− allele (Fig. 2A), which lacks exon 3 and results in a frameshift and early termination codon. Intercrosses of Sec24c+/− mice confirm uniform loss of Sec24c−/− mice by 2 weeks of age (Table 2C). At embryonic day 10.5–11, no intact Sec24c−/− embryos were observed. However, empty yolk sacs were present and could be separated from maternal tissue, with genotyping identifying the missing embryos as Sec24c−/−. Genotypes were also assessed at E9.5; again, for null embryos, only empty yolk sacs were detected, although at E8.5, one Sec24c−/− embryo appeared to have been dead for ∼12 h (blinded observation prior to genotyping). Histological analysis of E7.5 embryos and decidual swellings from a Sec24c+/− intercross revealed a series of morphologies ranging in severity from embryos that appeared to be gastrulating and developing normally (Fig. 3, A, B, and D), those that had not yet begun gastrulation and exhibited a thinned embryonic ectoderm (Fig. 3C), those in which the embryonic ectoderm was disorganized (Fig. 3E), and implantation sites containing remnants of the egg cylinder and membranes (Fig. 3F). Although no genotypes were assigned to the sectioned implantation sites (Fig. 3, D–F), Sec24c+/+ (Fig. 3A) and Sec24c+/− (Fig. 3B) embryos appear to develop normally, in contrast to the Sec24c−/− embryo depicted in Fig. 3C. Taken together, these data suggest that SEC24C deficiency is lethal between E7.0 and E8.5. Although the cause of death remains unknown, it appears that SEC24C is required in the embryonic ectoderm just prior to gastrulation. Analysis of progeny from Sec24c+/− intercrosses and backcrosses to wild type mice indicates that heterozygous mice are present in the expected ratios (Table 2C).
FIGURE 2.

Generation of Sec24c conditional and null alleles. A, schematic of the original Sec24cGT allele. FLPe-mediated excision yields the conditional Sec24cFL allele, and subsequent Cre-mediated excision gives rise to the null Sec24c− allele. Primers used for genotyping are indicated. B, long range PCR confirms correct targeting for the original Sec24cGT allele. Primers GF3 and GR4 are located outside of the homology arms. Primers GF3 and B amplify a 6234-bp product from the 5′ end of the Sec24cGT allele, and primers G and GR4 amplify a 6648-bp product from the 3′ end. As expected, neither set of primers yields a product from the Sec24cWT allele. C, genotyping with primers A, Ex, and E distinguishes between the wild type (308 bp), gene trap or floxed (278 bp), and null alleles (225 bp).
TABLE 2.
Sec24c allelic series genotype distributions
A, results of Sec24c+/GT intercrosses and backcrosses are shown. B, results of Sec24c+/FL intercrosses are shown. C, results of Sec24c+/− intercrosses and backcrosses are shown.
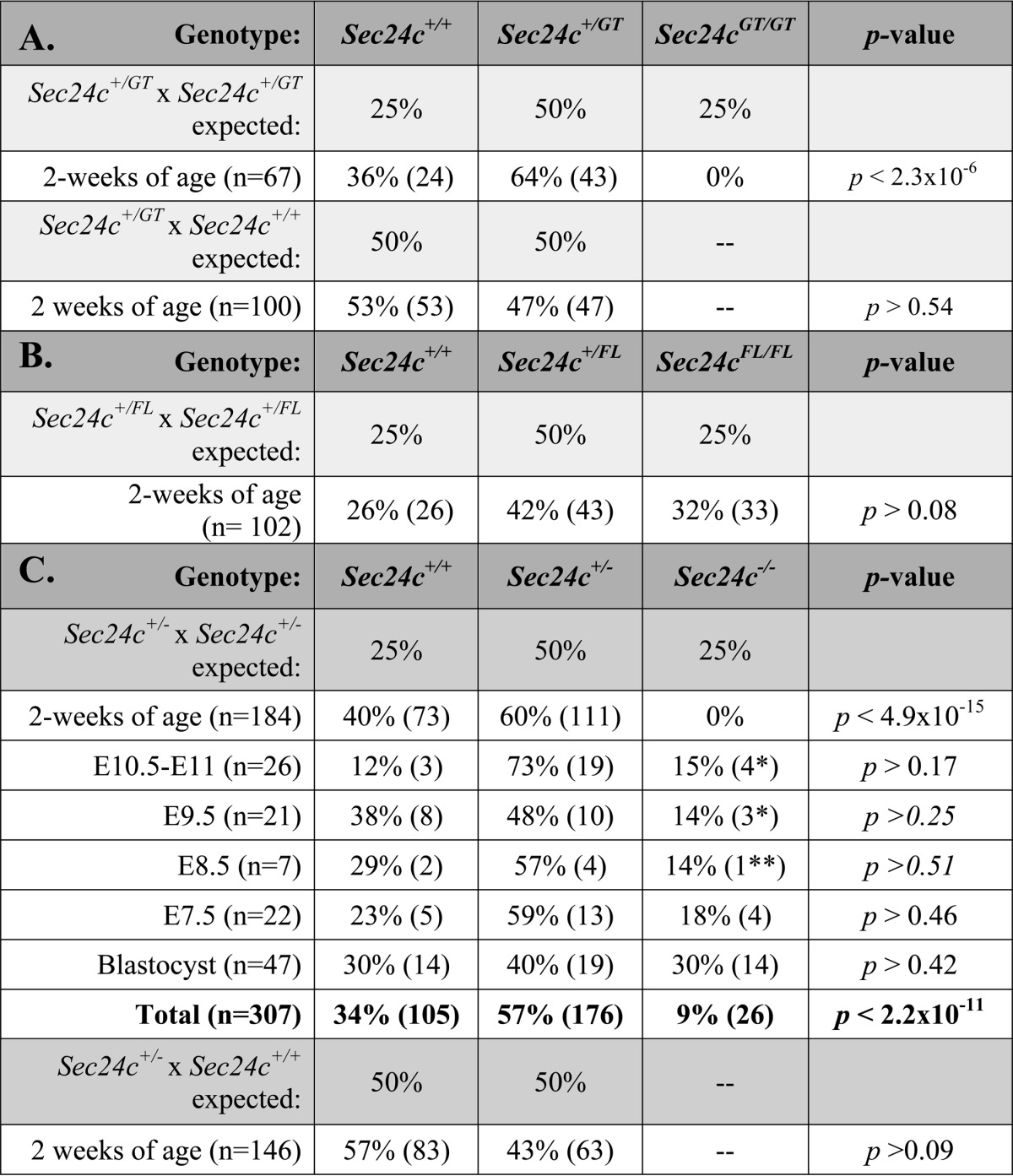
* All Sec24c−/− embryos from E9.5 to E11 were absent, but yolk sacs were able to be isolated for genotyping.
** Embryo was noted to have been dead for ∼12 h. All embryonic observations were made prior to genotyping analysis.
FIGURE 3.
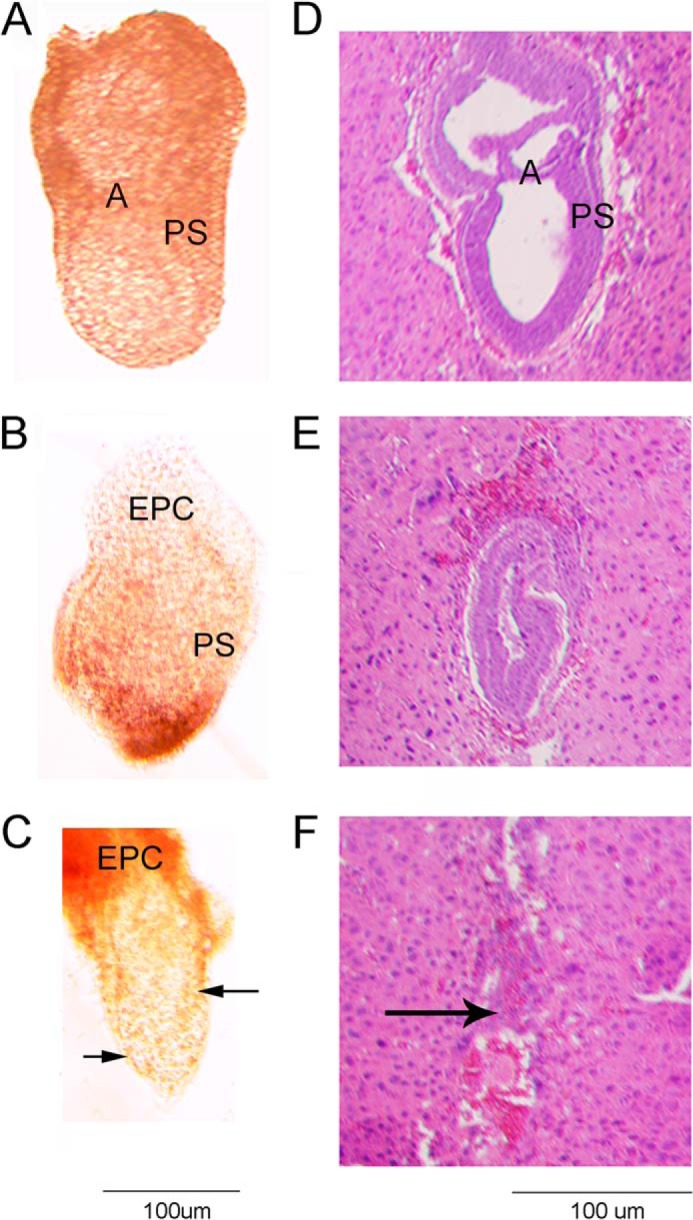
Phase contrast (A–C) and H&E staining (D–F) of E7.5 embryos resulting from Sec24c+/− intercrosses. E7.5 embryos were removed from the decidua and photographed prior to fixation (A–C), or whole decidual swellings were fixed and sectioned (D–F). Images are representative of the types of embryonic development/loss observed in 22 embryos. A, B, and D illustrate normal gastrulation. Genotypes were obtained for A–C (A, Sec24c+/+; B, Sec24c+/−; C, Sec24c−/−). The embryo in C exhibits thinning of the embryonic ectoderm. E illustrates a disorganized embryo, and F shows remnants of the egg cylinder. Anterior is to the left of each embryo; A = amniotic fold; EPC = ectoplacental cone; PS = primitive streak. Arrow in F indicates remnants of the embryo.
As noted above, the expected numbers of Sec24c+/GT and Sec24c+/− mice are observed at 2 weeks of age. Growth and complete blood counts of heterozygous mice were also indistinguishable from their wild type littermates (Fig. 4 and Table 3). Adult Sec24c+/− mice are fertile and exhibit no gross or microscopic abnormalities on standard autopsy examination (Fig. 4C). Although only a small number of wild type littermates were followed beyond 100 days, no significant difference was observed in the life span of Sec24c+/− mice (n = 21) compared with controls.
FIGURE 4.

Sec24c+/− mice are phenotypically normal. Growth curves of female (A) and male (B) mice indicate no difference in body weights between heterozygous mice and wild type littermate controls, with p > 0.05 at all time points. Error bars represent standard deviation. C, H&E staining of Sec24c+/− tissues reveals no abnormalities in a panel of tissues, including adrenal gland (panel i), intestine (panel ii), heart (panel iii), pancreas (panel iv), kidney (panel v), liver (panel vi), lung (panel vii), and spleen (panel viii). Scale bar, 0.25 μm.
TABLE 3.
Complete blood count analysis of mice from Sec24c+/− × Sec24d+GT intercrosses
Tail bleeds were carried out at 8–12 weeks of age. Data from males and females were pooled after confirming no significant difference between males and females of the same genotypes. The following abbreviations are used: HGB, hemoglobin; HCT, hematocrit; MCV, mean corpuscular volume; MCH, mean corpuscular hemoglobin; MCHC, MCH concentration; RDW, red cell distribution width; PLT, platelet count; MPV, mean platelet volume.
| Parameter | Genotype |
|||
|---|---|---|---|---|
| Sec24c+/+ | Sec24c+/− | Sec24d+/GT | Sec24c+/− Sec24d+/GT | |
| WBC (× 103 cells/μl) | 7.4 ± 0.9 | 8.2 ± 1.3 | 9.9 ± 1.4 | 9.0 ± 1.1 |
| RBC (× 106 cells/μl) | 8.7 ± 0.8 | 9.5 ± 0.3 | 9.7 ± 0.2 | 9.9 ± 0.3 |
| HGB (g/dl) | 12.2 ± 1.0 | 12.2 ± 0.5 | 13.3 ± 0.2 | 13.0 ± 0.5 |
| HCT (%) | 45.2 ± 2.6 | 45.7 ± 1.5 | 48.5 ± 0.7 | 48.4 ± 1.3 |
| MCV (fl) | 48.8 ± 0.6 | 48.1 ± 0.5 | 49.8 ± 0.4 | 49.1 ± 0.4 |
| MCH (pg) | 13.1 ± 0.3 | 12.6 ± 0.2 | 13.6 ± 0.1 | 13.1 ± 0.1 |
| MCHC (g/dl) | 26.9 ± 0.7 | 26.3 ± 0.5 | 27.2 ± 0.4 | 26.8 ± 0.4 |
| RDW (%) | 13.5 ± 0.3 | 13.5 ± 0.2 | 13.9 ± 0.5 | 14.0 ± 0.6 |
| PLT (× 103 cells/μl) | 976.0 ± 171.7 | 1221.7 ± 53.3 | 1186.7 ± 80.9 | 1076.3 ± 73.0 |
| MPV (fl) | 5.3 ± 0.6 | 5.0 ± 0.2 | 4.8 ± 0.2 | 5.1 ± 0.2 |
To test the potential overlap in function between SEC24C and SEC24D (16), we performed intercrosses between Sec24c+/− and Sec24d+/GT mice (Table 4) (20). At 2 weeks of age, double heterozygous mice are present at the expected Mendelian ratio (p > 0.12). No differences were observed in growth to 12 weeks (Fig. 5, A and B) or complete blood count analyses (Table 3), when compared with WT or single heterozygous littermates. Although only a small number of wild type littermates were followed beyond 100 days, no significant difference was observed in the life span of Sec24c+/− Sec24d+/GT mice (n = 27) compared with controls. Routine autopsy and histological survey were also unremarkable (Fig. 5C).
TABLE 4.
Results of Sec24c+/−Sec24d+/GT intercross
Mice are genotyped at 2 weeks of age. Sec24d+/GT mice are on C57BL/6J background (>n18).
| Genotype | Sec24c+/+ Sec24d+/+ | Sec24c+/− Sec24d+/+ | Sec24c+/+ Sec24d+/GT | Sec24c+/− Sec24d+/GT | p value |
|---|---|---|---|---|---|
| Expected ratio of F1 progeny | 25% | 25% | 25% | 25% | |
| Observed F1 progeny (n = 94) | 32% (30) | 26% (24) | 24% (23) | 18% (17) | p > 0.12 |
FIGURE 5.

Sec24c+/−Sec24d+/GT mice are phenotypically normal. Growth curves of female (A) and male (B) mice indicate no difference in body weights between heterozygous mice and littermate controls, with p > 0.05 at all time points. Error bars represent standard deviation. “Double het” = Sec24c+/− Sec24d+/GT. C, H&E staining of Sec24c+/− Sec24d+/GT tissues does not reveal any abnormalities in a panel of tissues, including adrenal gland (panel i), intestine (panel ii), heart (panel iii), pancreas (panel iv), kidney (panel v), liver (panel vi), lung (panel vii), and spleen (panel viii). Scale bar, 0.25 μm.
Transgenes designed to express SEC24C or SEC24D from the ubiquitous chick β-actin promoter (Fig. 6A) were generated and tested for their ability to rescue the lethal Sec24c−/− phenotype. No Sec24c−/− Tg+ mice survived to weaning for either the SEC24C (1 line, n = 34) or SEC24D (1 line, n = 49) transgene. Similarly, crosses to the Sec24dGT mice failed to demonstrate rescue of the Sec24dGT/GT lethal phenotype by either transgene; for the SEC24D transgene, no rescues were observed out of 149 transgenic mice from five different founder lines, with n for each line ranging from 9 to 43 transgenic progeny. For the SEC24C transgene, no rescues were observed out of 130 transgenic mice from four founder lines, with n for each line ranging from 11 to 57 transgenic progeny. RT-PCR analysis indicates that mRNA from the transgenes is being expressed in all tissues tested (Fig. 6B).
FIGURE 6.

Ubiquitous transgenic expression of Sec24c and Sec24d. A, diagram of the pCAG3zS-Sec24c or -Sec24d transgenic construct, adapted from Ref. 38, including transcription start site, initial ATG, and polyadenylation signal. CMV IE = cytomegalovirus immediate-early enhancer; UTR = untranslated region; SV40 poly(A) = simian virus 40 polyadenylation signal. Primers 24c-F, 24c-R, 24d-F, and 24d-R were used for RT-PCR of transgenes expressing Sec24c or Sec24d, respectively. B, transgene expression in a panel of tissues for founder lines 24c-line1, 24d-line1, 24d-line2, and 24d-line3. Heart and brain RT samples were carried out in the absence of reverse transcriptase.
To test the requirement for SEC24C in specific adult tissues, Sec24cFL/FL mice were crossed with mice carrying Cre transgenes specific for the pancreas (p48-Cre), hepatocytes (albumin-Cre), smooth muscle cells (SM22-Cre), and intestinal epithelial cells (villin-Cre) (Table 5). Sec24cFL/− Cre+ mice were observed in the expected numbers for p48-Cre (p > 0.65), SM22-Cre (p > 0.46), villin-Cre (p > 0.46), and albumin-Cre (p > 0.10). Although Cre-mediated excision of the Sec24cFL allele was nearly complete in the pancreas of p48Cre+ mice (Fig. 7A), pancreatic acinar and islets appear entirely normal by routine histology (Fig. 7B). Sec24cFL/−p48-Cre+ also exhibit normal weight gain through 12 weeks of age compared with their littermate controls on either normal chow (Fig. 8, A and B) or high fat diet (HFD) (Fig. 9, A and B). Similar results were observed for the albumin-Cre and villin-Cre transgenes, although the levels of excision were not as complete in these tissues as in the pancreas of p48-Cre+ mice (Fig. 7). There were also no differences in plasma cholesterol or triglyceride levels in tissue-specific knockouts versus littermate controls on HFD for p48-Cre, albumin-Cre, and villin-Cre transgenes (Figs. 9–11), and insulin levels between Sec24cFL/−p48-Cre+ and corresponding littermate controls on HFD were also indistinguishable (Fig. 9E). Sec24cFL/− SM22-Cre+ mice exhibited no gross abnormalities and were able to carry litters to term, and although excision of Sec24c in smooth muscle cells in SM22-Cre+ mice was not measured directly, it is expected to be complete, based on previous reports (44). Sec24cFL/− Meox2-Cre+ mice were not observed at 2 weeks of age (p < 1.7 × 10−6, see Table 5).
TABLE 5.
Results of tissue-specific deletion of Sec24c
Mice are genotyped at 2 weeks of age. p48-Cre = pancreatic cells; SM22-Cre = smooth muscle cells; villin-Cre = intestinal epithelial cells; albumin-Cre = liver hepatocytes; Meox2-Cre = ubiquitous Cre expressed beginning around embryonic day 5.
| Genotype | Sec24c+/FL | Sec24c+/FL Cre+ | Sec24cFL/− | Sec24cFL/−Cre+ | p value |
|---|---|---|---|---|---|
| Expected ratio of test cross-progeny | 25% | 25% | 25% | 25% | |
| Cre line | |||||
| p48-Cre (n = 148) | 20.9% (31) | 25.7% (38) | 27.7% (41) | 25.7% (38) | p > 0.65 |
| SM22-Cre (n = 40) | 32.5% (13) | 15.0% (58) | 22.5% (9) | 30% (12) | p > 0.46 |
| Villin-Cre (n = 143) | 28.7% (41) | 22.4% (32) | 26.5% (38) | 22.4% (32) | p > 0.46 |
| Albumin-Cre (n = 123) | 23.6% (29) | 31.7% (39) | 26.0% (32) | 18.7% (23) | p > 0.10 |
| Meox2-Cre (n = 69) | 37.7% (26) | 20.3% (14) | 42.0% (29) | 0% (0) | p < 1.7 × 10−6 |
FIGURE 7.
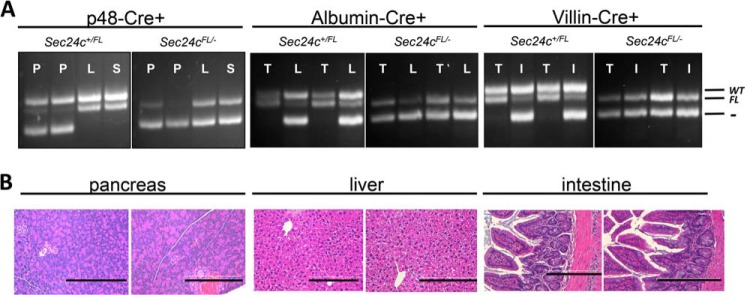
Tissue-specific Cre-mediated excision of Sec24cFL allele. A, genotyping for Cre-mediated excision in the pancreas (P), liver (L), spleen (S), intestine (I), and tail (T) in Sec24c+/FL and Sec24cFL/− mice carrying the p48-Cre, albumin-Cre, or villin-Cre transgenes. The floxed allele is excised to generate the null allele in the pancreas, liver, or intestine, respectively, but is preserved in other tissues. The sizes of the expected products for wild type (WT), flox (FL), and null (−) alleles are indicated. B, H&E staining of pancreatic tissue from p48-Cre+ mice, liver tissue from albumin-Cre+ mice, and intestine from villin-Cre+ mice show no abnormalities. Scale bar, 0.25 μm.
FIGURE 8.

Loss of SEC24C in the pancreas, liver, or intestine has no effect on growth. Body weight analysis over 12 weeks of Sec24cFL/−p48-Cre+ mice (A and B), Sec24cFL/− albumin-Cre+ mice (C and D), and Sec24cFL/−villin-Cre+ mice (E and F) with corresponding littermate controls in females and males. Insets show individual body weights at 12 weeks. p > 0.05 at all time points. Error bars represent standard deviation.
FIGURE 9.

Sec24cFL/−p48-Cre+ mice display expected diet-induced obesity. Sec24cFL/−p48-Cre+ and littermate controls show no difference in weight gain over 12 weeks on HFD for females (A) and males (B), with p > 0.05 at all time points. Cholesterol levels (C), triglyceride levels (D), and insulin levels (E) are indistinguishable from littermate controls after 12 weeks on HFD. All p values >0.05 and error bars represent standard deviation.
FIGURE 10.

Sec24cFL/−albumin-Cre+ mice display expected diet-induced obesity. Sec24cFL/−albumin-Cre+ and littermate controls show no difference in weight gain over 12 weeks on HFD for females (A) and males (B), with p > 0.05 at all time points. Cholesterol levels (C) and triglyceride levels (D) are indistinguishable from littermate controls after 12 weeks on HFD. All p values >0.05 and error bars represent standard deviation.
FIGURE 11.

Sec24cFL/−villin-Cre+ mice display expected diet-induced obesity. Sec24cFL/−villin-Cre+ and littermate controls show no difference in weight gain over 12 weeks on HFD for females (A) and males (B), with p > 0.05 at all time points. Cholesterol levels (C) and triglyceride levels (D) are indistinguishable from littermate controls after 12 weeks on HFD. All p values >0.05 and error bars represent standard deviation.
DISCUSSION
Our results demonstrate that SEC24C is required for post-implantation embryonic development in the mouse, with SEC24C-deficient embryos lost in the developmental interval between implantation and E8.5. The loss of Sec24cFL/− Meox2-Cre+ mice is consistent with these data, as Meox2-Cre is expressed ubiquitously beginning around embryonic day 5 (36). Data from tissue-specific knockouts reveal that SEC24C is dispensable in a number of cell types, including pancreatic acinar cells (involved in the secretion of digestive enzymes), islet cells (responsible for the production and secretion of a number of hormones, including insulin), hepatocytes (the primary cell type in the liver), intestinal epithelial cells (which line the intestine), and smooth muscle cells (found in the aorta, uterus, intestine, and in vascular walls), suggesting that the three remaining Sec24 paralogs can compensate for the loss of SEC24C in these tissues. These observations, together with the surprisingly limited but very distinct phenotypes of SEC23B (27), SEC24A (28), and SEC24B (29) deficiencies in the mouse and SAR1B (45), SEC23A (25), and SEC23B (26) deficiencies in humans, suggest at least partial overlap in function among the sets of mammalian SEC23, SEC24, and SAR1 paralogs, with unique requirements of a limited subset of cargoes likely accounting for the above phenotypes.
It has been reported that the serotonin transporter (SERT) requires SEC24C for efficient ER export (46). Given that SERT-deficient mice are viable and fertile (47), it is unlikely that the embryonic lethality observed in the SEC24C-deficient mice can be explained by defective trafficking of SERT to the plasma membrane. In addition, SERT does not become transcriptionally active until embryonic day 10 (47), well after the lethality we observed in the Sec24c−/− embryos. Thus, this embryonic lethality is likely due to a trafficking defect in another as yet unidentified secretory protein(s).
The apparently normal phenotype of Sec24c+/− and Sec24c+/− Sec24d+/GT mice demonstrates a tolerance for moderate quantitative changes in the levels of these two COPII components, consistent with previous reports of mice doubly heterozygous for Sec24a and Sec24d or Sec24b (28). Although an overlap in function between SEC24A and -B was observed in Sec24a−/− Sec24b+/− mice, similar experiments were precluded for SEC24C and -D, given the early embryonic lethality observed with both single homozygotes. The more severe phenotype of deficiencies in SEC24C/D compared with SEC24A/B is surprising, in light of the higher level of sequence identity between the SEC24A/B subfamily and the ancestral yeast paralog, Sec24p, and of SEC24C/D with the nonessential yeast homolog Lst1p (16). The later survival and unique phenotypes of Sec24a−/− and Sec24b−/− mice facilitated the identification of specific cargoes for each paralog (PCSK9 and VANGL2, respectively) largely explaining the observed phenotypes (28, 29). Given very early embryonic loss of mice deficient in either SEC24C or SEC24D, identification of a precise cause of death and the specific cargo(es) represents an even greater challenge. Nonetheless, our data suggest that SEC24C, like SEC24D, is required for the recruitment of critical cargo(es) at an early stage of development. The survival of SEC24C-deficient embryos well beyond the time point at which Sec24dGT/GT embryos are lost suggests a difference in SEC24C/D expression programs in early development and/or key cargo(es) uniquely dependent on SEC24D.
To date, no human disorders attributable to mutations in SEC24C have been identified. Analysis of data from the human Exome Variant Server (NHLBI GO Exome Sequencing Project, Seattle, WA, accessed January, 2014) identifies an allele frequency of 1% for predicted “damaging” variants in SEC24C in both African and European populations. This allele frequency would predict a population prevalence of 1.13 × 10−4 for individuals homozygous or compound heterozygous for damaging mutations in SEC24C. The absence of previous reports of human SEC24C deficiency could be due to early embryonic lethality, similar to the mouse phenotype. However, a much milder phenotype in SEC24C-deficient humans cannot be excluded. Of note, striking differences in phenotypes across species for deficiency of specific COPII paralogs have been reported previously, including very early embryonic lethality in SEC24D-deficient mice (20) compared with isolated skeletal defects in zebrafish (24) and congenital anemia in humans with mutations in SEC23B (26) compared with a pancreatic phenotype in mice (27).
Our analysis identified two alternatively spliced forms of mouse Sec24c mRNA, Sec24c-1 (RefSeq (49) accession NM_172596) and Sec24c-2 (not annotated in RefSeq). The latter contains an additional in-frame exon between exons 6 and 7 encoding 23 amino acids (exon 6* in Fig. 1A). Although another RefSeq splice variant of Sec24c containing a truncated exon 4 and exon 5, as well as exon 6*, is annotated (NM_001168273.1), it was undetectable in our RT-PCR analysis.
Sec24c-2 expression is restricted to brown fat, brain, and skeletal muscle (Fig. 1, B and C) and heart, with Sec24c-1 markedly reduced or absent in these tissues. Exon 6* and the flanking splice signals appear to be conserved in the human genome. A small number of human transcripts containing the corresponding sequence have been detected and annotated in GenBankTM (50), but it is unclear if the precise tissue-specific splice pattern is conserved. It has been reported that ∼94% of human transcripts undergo alternative splicing (51). Of those transcripts, 10–30% exhibit tissue-specific alternative spicing patterns (52), with these events occurring particularly frequently in the brain, but also in skeletal muscle and the heart (53). Of note, brown fat and skeletal muscle are also thought to arise from a common progenitor (54).
The addition of in-frame exon 6* results in the insertion of 23 amino acids at the N terminus of SEC24C, just upstream of the conserved zinc finger domain. This insertion appears to be a repeat of an upstream motif, (I/L)DPD(A/S)IPSP. These residues could provide a unique binding site for a subset of cargoes expressed in these tissues. The proximity of these additional SEC24C residues to the binding site of IXM (the t-SNARE ER exit signal for syntaxin 5 and membrin) could suggest a role in regulating interactions with these or another subset of cargoes, especially given that key residues within the hypervariable region of SEC24B have been shown to compete with t-SNARE binding (18).
Our efforts to rescue the lethal Sec24dGT/GT or Sec24c−/− phenotype with a ubiquitous Sec24c or Sec24d transgene were unsuccessful. Although transgene mRNA expression could be detected in all or most adult tissues examined (Fig. 6B), the level and/or developmental timing of this expression appears to be insufficient to replace that of the endogenous Sec24c and Sec24d genes. These results are in contrast to the previously reported rescue of SEC24D-deficient mice by BAC transgenes spanning the Sec24d locus (20), suggesting critical regulatory element(s) present in the BAC transgenes that are absent in the chicken β-actin promoter-driven constructs. As a result, the BAC transgene likely more faithfully recapitulates the endogenous expression pattern of SEC24D (48). Despite the failure of our transgenic constructs to rescue the lethality associated with SEC24C deficiency, the health and viability of Sec24cFL/FL mice (Table 2) demonstrates that the loss of Sec24cGT/GT and Sec24c−/− mice results from the disruption of Sec24c expression, either by the β-geo cassette (in Sec24cGT/GT mice) or the removal of exon 3 (in the case of the Sec24c−/− mice), rather than an effect on another locus (passenger gene) (43).
Taken together with previous reports, our data suggest a unique function for each SEC24 paralog. Although there may be overlap in cargo specificity, a subset of cargoes may require a particular mammalian SEC24 for efficient export from the ER. This specificity may be the result of intrinsic cargo selectivity among the SEC24 proteins, or alternatively, it may be a function of the particular spatio- and/or temporal expression pattern for each Sec24 gene.
Acknowledgments
We thank Dr. Jonathan Goldberg for helpful discussions regarding the Sec24c splice forms, Marta Dzaman for help with E7.5 histology, and Anya Kiseleva and Gengeng Yu for their technical support. We acknowledge Elizabeth Hughes, Keith Childs, Galina Gavrilina, and Debora VanHeyningen for preparation of ES cell mouse chimeras from gene-trapped ES clone EPD0241-2-A11 and the Transgenic Animal Model Core of the University of Michigan's Biomedical Research Core Facilities (core support was provided by the University of Michigan Multipurpose Arthritis Center, National Institutes of Health Grant AR20557, University of Michigan Cancer Center, and National Institutes of Health Grant CA46592). All histology work was performed in the Microscopy and Image Analysis Laboratory at the University of Michigan, Biomedical Research Core Facilities, with the assistance of Judy Poore. The Microscopy and Image Analysis Laboratory is a multiuser imaging facility supported by NCI, National Institutes of Health, O'Brien Renal Center, University of Michigan Medical School, and Endowment for the Basic Sciences, Department of Cell and Developmental Biology, and the University of Michigan.
This work was supported, in whole or in part, by National Institutes of Health Grants PO1HL057346 and RO1HL039693.
- ER
- endoplasmic reticulum
- HFD
- high fat diet
- SERT
- serotonin transporter.
REFERENCES
- 1. Bonifacino J. S., Glick B. S. (2004) The mechanisms of vesicle budding and fusion. Cell 116, 153–166 [DOI] [PubMed] [Google Scholar]
- 2. Palade G. (1975) Intracellular aspects of the process of protein synthesis. Science 189, 867–867 [DOI] [PubMed] [Google Scholar]
- 3. Budnik A., Stephens D. J. (2009) ER exit sites–localization and control of COPII vesicle formation. FEBS Lett. 583, 3796–3803 [DOI] [PubMed] [Google Scholar]
- 4. Lee M. C., Miller E. A. (2007) Molecular mechanisms of COPII vesicle formation. Semin. Cell Dev. Biol. 18, 424–434 [DOI] [PubMed] [Google Scholar]
- 5. Zanetti G., Pahuja K. B., Studer S., Shim S., Schekman R. (2012) COPII and the regulation of protein sorting in mammals. Nat. Cell Biol. 14, 20–28 [DOI] [PubMed] [Google Scholar]
- 6. Bielli A., Haney C. J., Gabreski G., Watkins S. C., Bannykh S. I., Aridor M. (2005) Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J. Cell Biol. 171, 919–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee M. C., Orci L., Hamamoto S., Futai E., Ravazzola M., Schekman R. (2005) Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122, 605–617 [DOI] [PubMed] [Google Scholar]
- 8. Aridor M., Weissman J., Bannykh S., Nuoffer C., Balch W. E. (1998) Cargo selection by the COPII budding machinery during export from the ER. J. Cell Biol. 141, 61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuehn M. J., Herrmann J. M., Schekman R. (1998) COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature 391, 187–190 [DOI] [PubMed] [Google Scholar]
- 10. Stagg S. M., Gürkan C., Fowler D. M., LaPointe P., Foss T. R., Potter C. S., Carragher B., Balch W. E. (2006) Structure of the Sec13/31 COPII coat cage. Nature 439, 234–238 [DOI] [PubMed] [Google Scholar]
- 11. Miller E., Antonny B., Hamamoto S., Schekman R. (2002) Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J. 21, 6105–6113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Miller E. A., Beilharz T. H., Malkus P. N., Lee M. C., Hamamoto S., Orci L., Schekman R. (2003) Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114, 497–509 [DOI] [PubMed] [Google Scholar]
- 13. Nichols W. C., Seligsohn U., Zivelin A., Terry V. H., Hertel C. E., Wheatley M. A., Moussalli M. J., Hauri H. P., Ciavarella N., Kaufman R. J., Ginsburg D. (1998) Mutations in the ER-Golgi intermediate compartment protein ERGIC-53 cause combined deficiency of coagulation factors V and VIII. Cell 93, 61–70 [DOI] [PubMed] [Google Scholar]
- 14. Zhang B., Cunningham M. A., Nichols W. C., Bernat J. A., Seligsohn U., Pipe S. W., McVey J. H., Schulte-Overberg U., de Bosch N. B., Ruiz-Saez A., White G. C., Tuddenham E. G., Kaufman R. J., Ginsburg D. (2003) Bleeding due to disruption of a cargo-specific ER to Golgi transport complex. Nat. Genet. 34, 220–225 [DOI] [PubMed] [Google Scholar]
- 15. Peng R., De Antoni A., Gallwitz D. (2000) Evidence for overlapping and distinct functions in protein transport of coat protein Sec24p family members. J. Biol. Chem. 275, 11521–11528 [DOI] [PubMed] [Google Scholar]
- 16. Tang B. L., Kausalya J., Low D. Y., Lock M. L., Hong W. (1999) A family of mammalian proteins homologous to yeast Sec24p. Biochem. Biophys. Res. Commun. 258, 679–684 [DOI] [PubMed] [Google Scholar]
- 17. Pagano A., Letourneur F., Garcia-Estefania D., Carpentier J. L., Orci L., Paccaud J. P. (1999) Sec24 proteins and sorting at the endoplasmic reticulum. J. Biol. Chem. 274, 7833–7840 [DOI] [PubMed] [Google Scholar]
- 18. Mancias J. D., Goldberg J. (2008) Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 27, 2918–2928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wendeler M. W., Paccaud J. P., Hauri H. P. (2007) Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep. 8, 258–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baines A. C., Adams E. J., Zhang B., Ginsburg D. (2013) Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS ONE 8, e61114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Richardson L., Venkataraman S., Stevenson P., Yang Y., Burton N., Rao J., Fisher M., Baldock R. A., Davidson D. R., Christiansen J. H. (2010) EMAGE mouse embryo spatial gene expression database: 2010 update. Nucleic Acids Res. 38, D703–D709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Neuhauss S. C., Solnica-Krezel L., Schier A. F., Zwartkruis F., Stemple D. L., Malicki J., Abdelilah S., Stainier D. Y., Driever W. (1996) Mutations affecting craniofacial development in zebrafish. Development 123, 357–367 [DOI] [PubMed] [Google Scholar]
- 23. Driever W., Solnica-Krezel L., Schier A. F., Neuhauss S. C., Malicki J., Stemple D. L., Stainier D. Y., Zwartkruis F., Abdelilah S., Rangini Z., Belak J., Boggs C. (1996) A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46 [DOI] [PubMed] [Google Scholar]
- 24. Sarmah S., Barrallo-Gimeno A., Melville D. B., Topczewski J., Solnica-Krezel L., Knapik E. W. (2010) Sec24D-dependent transport of extracellular matrix proteins is required for Zebrafish skeletal morphogenesis. PLoS ONE 5, e10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Boyadjiev S. A., Fromme J. C., Ben J., Chong S. S., Nauta C., Hur D. J., Zhang G., Hamamoto S., Schekman R., Ravazzola M., Orci L., Eyaid W. (2006) Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat. Genet. 38, 1192–1197 [DOI] [PubMed] [Google Scholar]
- 26. Schwarz K., Iolascon A., Verissimo F., Trede N. S., Horsley W., Chen W., Paw B. H., Hopfner K.-P., Holzmann K., Russo R., Esposito M. R., Spano D., De Falco L., Heinrich K., Joggerst B., Rojewski M. T., Perrotta S., Denecke J., Pannicke U., Delaunay J., Pepperkok R., Heimpel H. (2009) Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat. Genet. 41, 936–940 [DOI] [PubMed] [Google Scholar]
- 27. Tao J., Zhu M., Wang H., Afelik S., Vasievich M. P., Chen X.-W., Zhu G., Jensen J., Ginsburg D., Zhang B. (2012) SEC23B is required for the maintenance of murine professional secretory tissues. Proc. Natl. Acad. Sci. U.S.A. 109, E2001–E2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen X.-W., Wang H., Bajaj K., Zhang P., Meng Z.-X., Ma D., Bai Y., Liu H.-H., Adams E., Baines A., Yu G., Sartor M. A., Zhang B., Yi Z., Lin J., Young S. G., Schekman R., Ginsburg D. (2013) SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife 2, e00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Merte J., Jensen D., Wright K., Sarsfield S., Wang Y., Schekman R., Ginty D. D. (2010) Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat. Cell Biol. 12, 41–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kendall S. K., Samuelson L. C., Saunders T. L., Wood R. I., Camper S. A. (1995) Targeted disruption of the pituitary glycoprotein hormone α-subunit produces hypogonadal and hypothyroid mice. Genes Dev. 9, 2007–2019 [DOI] [PubMed] [Google Scholar]
- 31. Bahou W. F., Bowie E. J., Fass D. N., Ginsburg D. (1988) Molecular genetic analysis of porcine von Willebrand disease: tight linkage to the von Willebrand factor locus. Blood 72, 308–313 [PubMed] [Google Scholar]
- 32. Kawaguchi Y., Cooper B., Gannon M., Ray M., MacDonald R. J., Wright C. V. (2002) The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 32, 128–134 [DOI] [PubMed] [Google Scholar]
- 33. Madison B. B., Dunbar L., Qiao X. T., Braunstein K., Braunstein E., Gumucio D. L. (2002) cis elements of the Villin gene control expression in restricted domains of the vertical (Crypt) and horizontal (Duodenum, Cecum) axes of the intestine. J. Biol. Chem. 277, 33275–33283 [DOI] [PubMed] [Google Scholar]
- 34. Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. (1999) Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic β cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315 [DOI] [PubMed] [Google Scholar]
- 35. Boucher P., Gotthardt M., Li W.-P., Anderson R. G., Herz J. (2003) LRP: role in vascular wall integrity and protection from atherosclerosis. Science 300, 329–332 [DOI] [PubMed] [Google Scholar]
- 36. Tallquist M. D., Soriano P. (2000) Epiblast-restricted Cre expression in MORE mice: a tool to distinguish embryonic vs. extra-embryonic gene function. Genesis 26, 113–115 [DOI] [PubMed] [Google Scholar]
- 37. Van Keuren M. L., Gavrilina G. B., Filipiak W. E., Zeidler M. G., Saunders T. L. (2009) Generating transgenic mice from bacterial artificial chromosomes: transgenesis efficiency, integration and expression outcomes. Transgenic Res. 18, 769–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fahim A. T., Wang H., Feng J., Ginsburg D. (2009) Transgenic overexpression of a stable plasminogen activator inhibitor-1 variant. Thromb. Res. 123, 785–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Alexopoulou A. N. (2008) The CMV early enhancer/chicken β actin (CAG) promoter can be used to drive transgene expression during the differentiation of murine embryonic stem cells into vascular progenitors. BMC Cell Biol. 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hitoshi N., Kenichi Y., Junichi M. (1991) Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108, 193–199 [DOI] [PubMed] [Google Scholar]
- 41. Nagy A., Gertsenstein M., Vintersten K., Behringer R. (2003) Manipulating the Mouse Embyro: A Laboratory Manual, pp. 237–319, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 42. Krupp M., Marquardt J. U., Sahin U., Galle P. R., Castle J., Teufel A. (2012) RNA-Seq Atlas, a reference database for gene expression profiling in normal tissue by next-generation sequencing. Bioinformatics 28, 1184–1185 [DOI] [PubMed] [Google Scholar]
- 43. Westrick R. J., Mohlke K. L., Korepta L. M., Yang A. Y., Zhu G., Manning S. L., Winn M. E., Dougherty K. M., Ginsburg D. (2010) A spontaneous Irs1 passenger mutation linked to a gene targeted SerpinB2 allele. Proc. Natl. Acad. Sci. U.S.A. 107, 16904–16909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Frutkin A. D., Shi H., Otsuka G., Levéen P., Karlsson S., Dichek D. A. (2006) A critical developmental role for tgfbr2 in myogenic cell lineages is revealed in mice expressing SM22-Cre, not SMMHC-Cre. J. Mol. Cell. Cardiol. 41, 724–731 [DOI] [PubMed] [Google Scholar]
- 45. Jones B., Jones E. L., Bonney S. A., Patel H. N., Mensenkamp A. R., Eichenbaum-Voline S., Rudling M., Myrdal U., Annesi G., Naik S., Meadows N., Quattrone A., Islam S. A., Naoumova R. P., Angelin B., Infante R., Levy E., Roy C. C., Freemont P. S., Scott J., Shoulders C. C. (2003) Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat. Genet. 34, 29–31 [DOI] [PubMed] [Google Scholar]
- 46. Sucic S., El-Kasaby A., Kudlacek O., Sarker S., Sitte H. H., Marin P., Freissmuth M. (2011) The serotonin transporter is an exclusive client of the coat protein complex II (COPII) component SEC24C. J. Biol. Chem. 286, 16482–16490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bengel D., Murphy D. L., Andrews A. M., Wichems C. H., Feltner D., Heils A., Mössner R., Westphal H., Lesch K.-P. (1998) Altered brain serotonin homeostasis and locomotor insensitivity to 3,4-methylenedioxymethamphetamine (“Ecstasy”) in serotonin transporter-deficient mice. Mol. Pharmacol. 53, 649–655 [DOI] [PubMed] [Google Scholar]
- 48. Yang X. W., Gong S. (2005) An overview on the generation of BAC transgenic mice for neuroscience research. Curr. Protoc. Neurosci. 31, 5.20.1–5.20/11 [DOI] [PubMed] [Google Scholar]
- 49. Pruitt K. D., Tatusova T., Maglott D. R. (2005) NCBI Reference Sequence (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 33, D501–D504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Benson D. A., Cavanaugh M., Clark K., Karsch-Mizrachi I., Lipman D. J., Ostell J., Sayers E. W. (2013) GenBank. Nucleic Acids Res. 41, D36–D42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang E. T., Sandberg R., Luo S., Khrebtukova I., Zhang L., Mayr C., Kingsmore S. F., Schroth G. P., Burge C. B. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature 456, 470–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xu Q., Modrek B., Lee C. (2002) Genome-wide detection of tissue-specific alternative splicing in the human transcriptome. Nucleic Acids Res. 30, 3754–3766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chen M., Manley J. L. (2009) Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat. Rev. Mol. Cell Biol. 10, 741–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Seale P., Bjork B., Yang W., Kajimura S., Chin S., Kuang S., Scimè A., Devarakonda S., Conroe H. M., Erdjument-Bromage H., Tempst P., Rudnicki M. A., Beier D. R., Spiegelman B. M. (2008) PRDM16 controls brown fat/skeletal muscle switch. Nature 454, 961–967 [DOI] [PMC free article] [PubMed] [Google Scholar]