

Background: Vesicular Na+/H+ exchanger NHE5 is implicated in neuronal pH homeostasis, but its role is poorly understood.
Results: Activation of AMP-activated protein kinase (AMPK) increases plasmalemmal NHE5 abundance and activity in response to metabolic stress-induced acidosis.
Conclusion: Interaction of NHE5 and AMPK represents a novel mechanism for coupling energy metabolism to pH regulation in nervous tissue.
Significance: These findings provide new insight into nervous system pH homeostasis.
Keywords: AMP-activated Kinase (AMPK), Energy Metabolism, Neuron, pH Regulation, Sodium-proton Exchange
Abstract
Strict regulation of intra- and extracellular pH is an important determinant of nervous system function as many voltage-, ligand-, and H+-gated cationic channels are exquisitely sensitive to transient fluctuations in pH elicited by neural activity and pathophysiologic events such as hypoxia-ischemia and seizures. Multiple Na+/H+ exchangers (NHEs) are implicated in maintenance of neural pH homeostasis. However, aside from the ubiquitous NHE1 isoform, their relative contributions are poorly understood. NHE5 is of particular interest as it is preferentially expressed in brain relative to other tissues. In hippocampal neurons, NHE5 regulates steady-state cytoplasmic pH, but intriguingly the bulk of the transporter is stored in intracellular vesicles. Here, we show that NHE5 is a direct target for phosphorylation by the AMP-activated protein kinase (AMPK), a key sensor and regulator of cellular energy homeostasis in response to metabolic stresses. In NHE5-transfected non-neuronal cells, activation of AMPK by the AMP mimetic AICAR or by antimycin A, which blocks aerobic respiration and causes acidification, increased cell surface accumulation and activity of NHE5, and elevated intracellular pH. These effects were effectively blocked by the AMPK antagonist compound C, the NHE inhibitor HOE694, and mutation of a predicted AMPK recognition motif in the NHE5 C terminus. This regulatory pathway was also functional in primary hippocampal neurons, where AMPK activation of NHE5 protected the cells from sustained antimycin A-induced acidification. These data reveal a unique role for AMPK and NHE5 in regulating the pH homeostasis of hippocampal neurons during metabolic stress.
Introduction
Neuronal membrane excitability and neurotransmission are metabolically intensive activities that elicit transient fluctuations in intra- and extracellular pH (pHi and pHe, respectively) (1). These pH oscillations further modulate electrical activity by regulating the conductance of various pH-sensitive neurotransmitter-, voltage-, and proton-gated ion channels (2–5). Indeed, such pH transients are thought to serve as a physiologically relevant feedback mechanism to control nervous system function (6–8). Conversely, extreme perturbations of pH homeostasis have been linked to certain pathophysiologies, including tissue damage arising from hypoxia and ischemia (9) and seizures (10, 11).
Like other organ systems, neural pH homeostasis is maintained by a variety of membrane solute carriers, including Na+/H+ exchangers (NHEs)2 and assorted bicarbonate transporters (12, 13). Much of our understanding of the contributions of the NHE gene family to nervous system function is derived from studies of the ubiquitous amiloride-sensitive NHE1 isoform, which fulfills not only a general role in maintaining cytoplasmic pH near neutral, but can also act specifically at nerve terminals to modulate synaptic transmission (14, 15). Its functional importance is further highlighted by studies of Nhe1 null mice that exhibit ataxia, seizures, and selective loss of neurons in the cerebellum and brainstem (16–18). However, few deleterious changes were detected in other regions of the brain, implicating compensatory or unique contributions by other NHEs. NHE5 is of particular interest as its expression is largely confined to nervous tissue (19, 20). In particular, NHE5 is thought to play a prominent role in controlling the pHi of hippocampal neurons. Unlike NHE1, NHE5 is more resistant to inhibition by amiloride derivatives and benzyolguanidinium-based (e.g. HOE694) compounds (21, 22) and resides predominantly in intracellular vesicles (23, 24), although the significance of this distribution is uncertain. However, recent studies (25) found that NHE5 is rapidly recruited to the cell surface of hippocampal neurons in response to NMDA receptor-induced neural activity where it elevates pHi and suppresses growth of postsynaptic dendritic spines. This suggests that vesicular NHE5 may serve as a reservoir of functional transporters that are recruited to the cell surface in response to appropriate cues.
To better understand NHE5 regulation, we screened a human brain cDNA library for NHE5-interacting proteins and identified the α2 catalytic subunit of AMP-activated protein kinase (AMPK) as a putative partner. AMPK is an evolutionarily conserved serine/threonine protein kinase that functions as a key sensor and master regulator of energy homeostasis at the cellular and organismal levels (26–29). AMPK assembles as a heterotrimeric complex composed of a catalytic α subunit and two regulatory β and γ subunits, each of which is encoded by two or three distinct genes (α1, α2; β1, β2; γ1, γ2, γ3); thus there is a potential to form 12 distinct holoenzymes. The subunit isoforms are widely expressed in peripheral tissues, whereas the brain shows a more restricted pattern, containing predominantly α2, β1, and γ1, and to a lesser extent α1 and β2 (30).
Our results show that NHE5 forms a complex with both AMPKα1 and -α2 oligomers and that activation of AMPK regulates hippocampal neuronal pHi in response to metabolic stress-induced acidosis by promoting cell surface accumulation of NHE5. This interaction represents a potentially novel mechanism for coupling energy metabolism to pHi homeostasis in nervous tissue.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
Chemicals and reagents used for AP-1 cell culture were obtained from either BioShop Canada or Fisher Scientific, with the exception of α-minimum essential medium (αMEM), fetal bovine serum (FBS), penicillin/streptomycin, and trypsin-EDTA, all of which were purchased from Invitrogen. All products used for neuronal primary cell culture were purchased from Invitrogen, unless otherwise indicated. Protein localization studies using immunofluorescence and immunoblotting were performed using the following commercial antibodies: mouse monoclonal anti-hemagglutinin (HA) antibody (HA.11 clone 16B12) (Covance Inc., Berkeley, CA), rabbit polyclonal anti-HA (Abcam Inc., Cambridge, MA), mouse monoclonal anti-myc (EMD Millipore), rabbit polyclonal anti-AMPKα1, -α2, -γ1, and -γ2 and mouse monoclonal anti-AMPKα1/2 (Upstate Cell Signaling), rabbit polyclonal anti-AMPKβ (BD Transduction Laboratories), and rabbit polyclonal anti-phospho-AMPKα-pT172 antibody (Upstate Cell Signaling). Rabbit polyclonal anti-NHE5 was generated as previously described (24). Horseradish peroxidase-conjugated secondary IgG antibodies were purchased from Jackson ImmunoResearch Laboratories (West Grove, PA) and Alexa Fluor 647-conjugated goat anti-rabbit IgG was purchased from Invitrogen. The enhanced chemiluminescence system, protein G-Sepharose 4B, glutathione-Sepharose 4B, and pGEX-2T bacterial expression vector were purchased from GE Healthcare. Commercially available drugs used in this study include: 2-deoxyglucose (Sigma), antimycin A (Sigma), 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR) (Upstate Cell Signaling), 6-[4-(2-piperidin-1-yl-ethoxy)-phenyl)]-3-pyridin-4-yl-pyrrazolo[1,5-a]-pyrimidine (compound C) (EMD Millipore), and ethylisopropylamiloride (EIPA) (Sigma). (3-Methanesulfonyl-4-piperidinobenzoyl)guanidine hydrochloride (HOE694) was kindly provided by Dr. Hans-J. Lang (Aventis Pharma Deutschland GmbH, Frankfurt, Germany). All other reagents were obtained from Fisher Scientific or Sigma.
Yeast Two-hybrid Screening
The human NHE5 cDNA corresponding to residues Gly544–Leu896 (cytoplasmic C terminus) was amplified by PCR using two oligonucleotide primers (forward, 5′-ATG GCC ATG GCC ATT GGC CAC GTC TTG TCT TCC-3′; reverse, 5′-TAG CCCGGG CTA CAG CCG GCT GCC TCT GTT G-3′) and subcloned into the NcoI and XmaI sites of the pAS2–1 vector (pAS2–1/NHE5(544–896)) (Clontech, Palo Alto, CA) in-frame with the GAL4 1–147 DNA binding domain. S. cerevisiae strain AH109 (MATa, trp1–901, leu2–3, 112, ura3–52, his 3–200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, ura3::MEL1UAS-MEL1TATA-lacZ) was co-transformed with pAS2–1/NHE5(544–896), carrying the Trp gene and the GAL4 DNA binding domain and a human brain cDNA library cloned into the pACT2 vector (Clontech), carrying the LEU2 gene and the GAL4(768–881) activation domain using the modified lithium acetate method (97). Over 2 × 106 clones were screened on the synthetic complete (SC) media containing 0.67% bacto-yeast nitrogen base without amino acids (Difco Laboratories), 2% glucose, 1.5% bacto-agar, and 0.2% -Leu-Trp-His-Ade drop-out mixture and subjected to the β-galactosidase filter assay. For the β-galactosidase filter assay, a replica was taken from each plate onto filter paper (VWR Scientific Products, West Chester, PA), frozen rapidly in liquid nitrogen, and incubated with 0.35 mg/ml of 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) in buffer containing 6 mm Na2HPO4, 40 mm NaH2PO4, 10 mm KCl, 1 mm MgSO4 (pH 7.0) at room temperature for up to 4 h. The resulting Leu+, Trp+, His+, Ade+, LacZ+ colonies were streaked on SC-Leu-Trp-His-Ade plates two additional times and single colonies were inoculated into liquid SC, Leu−, Trp+ His−, Ade− media for 3–4 days culture to release the pAS2-1/NHE5(544–896) plasmid. Library-derived cDNA clones in pAC2-1 coding for putative NHE5 interacting proteins were rescued from yeast cells and directly transformed into DH5α cells. The identities of the clones were determined by DNA sequencing and compared against the National Cancer Center for Biotechnology Information database using the BLAST search program. One cDNA clone encoding the AMPKα2 subunit lacking the first 6 amino acids was identified by the two-hybrid screen. To isolate the full-length cDNA, PCR was carried out with the following set of oligonucleotide primers: forward, 5′-CCC AAG CTT GCC ACC ATG GCT GAG AAG CAG AAG CAC GAC GGG CGG GTG AAG ATC GGA C-; reverse, 5′-CGG AAT TCT TAA CGG GCT AAA GTA GTA CTA ATC AGA CTG G-3′ (HindIII and EcoRI sites were underlined). The amplified fragment was subcloned into the pCMV mammalian expression vector at the HindIII and EcoRI sites and the sequence was verified subsequently.
Construction of Epitope-tagged Gene Constructs
To examine NHE1 and NHE5 expression in heterologous cell systems, we used a mammalian expression plasmid (pCMV) containing the rat NHE1 cDNA containing a single influenza virus hemagglutinin (HA) epitope (-YPYDVPDYA-) at its extreme C terminus (NHE1HA) (31) and the human NHE5 cDNA tagged with a triple HA-epitope in its first exomembranous loop (NHE5HA3) (23). The full-length AMPKα2 cDNA was modified to contain a Myc epitope (-EQKLISEEDL-) at its extreme C terminus (AMPKα2myc). AMPKα2myc was constructed by PCR using the sense oligonucleotide 5′-CCC AAG CTT GCC ACC ATG GCT GAG AAG CAG AAG CAC GAC GGG CGG GTG AAG ATC GGA C-3′ containing a HindIII at the 5′-end and an antisense oligonucleotide containing the coding sequence for the Myc epitope and an XbaI site (5′-GCT CTA GAC TAG TTC AGG TCC TCC TCG CTA ATT AGC TTC TGT TCA ACG GGC TAA AGT AGT AAT CAG ACT GG-3′). The amplified fragment was subcloned into pCMV at the HindIII and XbaI sites, and the fidelity of the construct was verified by DNA sequencing. In addition, both NHE5 and AMPKα2 cDNAs were fused to enhanced green fluorescent protein at their respective C termini (NHE5GFP and AMPKα2GFP) using HindIII and EcoRI restriction sites in mammalian expression vector pAcGFP-N1 vector (BD Bioscience). Furthermore, the pAcGFP-N1 vector was used as a base to construct a pAcCherry-N1 vector, whereby GFP was replaced with monomeric cherry fluorescent protein (ChFP) using restriction sites BamHI and NotI. This cloning procedure leaves the multicloning site unaltered, thereby allowing for the analogous construction of both NHE5ChFP and AMPKα2ChFP using HindIII and EcoRI restriction sites.
Glutathione S-Transferase (GST) Pulldown Assays
For generating GST fusion proteins, different regions of the NHE5 cDNA were amplified by PCR, excised with BamHI and EcoRI, and inserted into the same restriction sites of the pGEX-2T bacterial expression vector (GE Healthcare) in-frame with the amino-terminal GST tag. Transformed BL21 Escherichia coli cells derived from single colonies were inoculated in 5 ml of 2YT + Amp (100 μg/ml) media overnight and protein expression was induced by further incubation in 50 ml of the same media containing 0.4 mm isopropyl 1-thio-d-galactopyranoside at 30 °C for 3 h. E. coli cells were collected by centrifugation, resuspended in 1 ml of lysis buffer containing 0.5% Nonidet P-40, 1 mm EDTA, 0.1 mg/ml of lysozyme and proteinase inhibitor mixtures (Roche Diagnostics) in PBS, incubated on ice for 15 min, and sonicated 4 times for 30 s on ice. Cell debris was removed by centrifugation at 16,000 × g for 20 min and an equal amount of the GST fusion proteins (∼10 μg) were purified by incubation with glutathione-Sepharose beads (GE Healthcare) at 4 °C for 2 h. Following four washes in lysis buffer without lysozyme, the beads were incubated with 35S-labeled in vitro translated protein generated by the TnT reticulocyte lysate system (Promega, Madison, WI). After overnight incubation at 4 °C the beads were washed six times in the same buffer and 35S-labeled TnT protein bound to the GST fusion protein was detected by SDS-PAGE followed by autoradiography. Loading of the proteins was confirmed by Coomassie Blue dye staining of the SDS-PAGE gel. For producing in vitro translated AMPKα2, the full-length AMPKα2 cDNA was ligated into the pCMV vector. The T7 promoter in the vector enabled in vitro transcription translation coupling reaction using rabbit reticulocyte lysates (Promega).
In Vitro Phosphorylation Assays
GST-NHE5 fusion proteins were purified by glutathione-Sepharose beads and a phosphorylation assay was performed by incubation with AMPK purified from rat liver (Upstate Biotechnology, Lake Placid, NY) according to the manufacturer's instruction with some modifications. Briefly, 10 μg of GST fusion proteins on the glutathione-Sepharose solid support were incubated with 10 milliunits of AMPK in a final volume of 25 μl containing 40 mm HEPES (pH 7.0), 400 μm AMP, 80 mm NaCl, 5 mm MgCl2, 0.8 mm EDTA, 0.4 mm dithiothreitol, and 10 μCi of [γ-32P]ATP (PerkinElmer Life Science). After incubation at 30 °C for 7 min, beads were sedimented by a quick centrifugation and then washed six times with ice-cold PBS containing 0.5% Nonidet P-40 and 1 mm EDTA. GST fusion proteins were eluted in SDS sample buffer containing 50 mm Tris-Cl (pH 6.8), 2% SDS, 0.1% bromphenol blue, and 10% glycerol and resolved by SDS-PAGE. The gel was stained with Coomassie Blue, dried, and exposed to x-ray film (Eastman Kodak, Rochester, NY).
Cell Culture
AP-1 cells (mutagenized Chinese hamster ovary cells devoid of plasma membrane Na+/H+ exchange activity) (32) were maintained in αMEM supplemented with 10% fetal bovine serum, penicillin (100 units/ml), streptomycin (100 μg/ml), and 25 mm NaHCO3 (pH 7.4). AP-1 cells stably expressing NHE1 and NHE5 (AP-1/NHE1 and AP-5/NHE5, respectively) were generated using an acid challenging selection process as previously described (22, 31). All cells were incubated in a humidified atmosphere of 95% air, 5% CO2 at 37 °C.
Primary cultures of hippocampal neurons were prepared from postnatal day 0–2 mice. Briefly, animals were killed by decapitation, their brains removed, and hippocampi was dissected out. Dissected hippocampi were maintained in chilled 100 μm HEPES-buffered saline solution supplemented with 0.6% glucose, then digested with 165 units of papain for 30 min at 37 °C. Recovered neurons and glial cells were dissociated by trituration and suspended in DMEM supplemented with 1% penicillin/streptomycin, 10% FBS, and 0.6% glucose. The suspension was passed through a BD Biosciences Falcon 70-μm cell strainer and placed into a 15-ml conical tube on top of an Opti-Prep density gradient and centrifuged at ∼800 × g as described previously (33). Fractions enriched in neurons were collected and washed with DMEM suspension media. The cells were then plated onto poly-d-lysine-coated 12-mm coverslips at an approximate density of 5000 cells/cm2 and placed in an incubator at 37 °C. After 24 h, plating media was replaced with neuronal growth media consisting of Neurobasal-A, 2% B-27 supplement, 1% GlutaMAX, 1% penicillin/streptomycin, and for only the first feeding, 20 μm arabinofuranosyl cytidine to inhibit glial cell proliferation. Neurons were fed every 3–4 days subsequently without arabinofuranosyl cytidine and maintained +14 days in vitro in a humidified atmosphere of 95% air, 5% CO2 at 37 °C.
Coimmunoprecipitations and Immunoblotting
Cell and tissue lysates were prepared in phosphate-buffered saline (PBS) containing 0.5% Nonidet P-40, protease inhibitor mixture (Roche Diagnostics), and 0.25% sodium deoxycholic acid. Depending on the experiment, cell lysates (1 ml) were incubated with rabbit polyclonal antibodies that recognize the HA-epitope, AMPKα1, -α2, -γ1, or -γ2 subunits (5 μg each). Brain tissue lysates were incubated with either mouse IgG or mouse monoclonal anti-AMPKα1/2 antibody (5 μg each). All lysates were incubated at 4 °C overnight on a tilt rocker platform, followed by 2 h of IgG binding to 25 μl of a 50% slurry of protein G-Sepharose beads on a tilt rocker. The beads were washed 4 times for 5 min each in lysis buffer. The proteins in the immunoprecipitates were fractionated by SDS-PAGE and detected by immunoblotting. Membranes containing immunoprecipitates were blocked for 1 h with 5% skim milk and PBS-T, and then probed with various primary antibodies as follows. NHE5HA3 and AMPKα2myc were detected using mouse monoclonal anti-HA and anti-myc antibodies, respectively. Native AMPKβ was detected with a rabbit pan polyclonal antibody that recognizes both AMPKβ1 and -β2. Native NHE5 was detected with an isoform-specific rabbit polyclonal antibody as described previously (24). All antibodies were diluted in 5% nonfat skim milk, and PBS-T (phosphate-buffered saline and 0.1% Tween 20). The signals were visualized with either 0.8 μg/ml of HRP-conjugated, goat anti-rabbit or anti-mouse secondary antibodies after a 1-h incubation. Immunoreactive bands were detected with ECL detection reagents (Western Lighting Ultra PerkinElmer kit) and Kodak x-ray film.
ATP Determination
Cell lysates were prepared in PBS containing 0.5% Nonidet P-40 and the ATP concentrations were determined with the Invitrogen ATP Determination Kit. The luciferase activity was measured with a Turner Designs TD-20/20 luminometer.
Confocal Microscopy
NHE5ChFP and AMPKα2GFP (1.5 μg each) were simultaneously transfected into AP-1 cells grown in 35-mm culture dishes with 25-mm glass coverslip bottoms (MatTek Corporation) using Lipofectamine2000 (Invitrogen) and their intracellular localizations were analyzed 48 h after transfection. Cells were assayed under live conditions in phenol red-free Iscove's MEM (Multicell) at 37 °C and 5% CO2 inside a Live Cell Instruments' environment chamber fitted to a Zeiss LSM710 confocal microscope. The fluorescence signals were analyzed using Zen (Zeiss) and Metamorph (Molecular Devices) software.
Förster Resonance Energy Transfer Assay
Förster or fluorescence resonance energy transfer (FRET) using the acceptor photobleaching method (34) was used to assess the interaction between NHE5 and AMPKα2 in intact cells. To this end, NHE5ChFP and AMPKα2GFP were simultaneously transfected into AP-1 cells seeded on 25-mm #1.5 coverslips (Thermo Fisher Scientific) and fixed with a 4% formaldehyde solution for 15 min at room temperature after 48 h. The coverslips were subsequently mounted onto glass slides with Aqua-Mount (Thermo Fisher Scientific). Cells co-expressing NHE5ChFP and AMPKα2GFP were excited with a 488-nm laser and their emission was collected from 500 to 700 nm with a spectral detector and binned every 10 nm. The ChFP and GFP signals were unmixed linearly using control NHE5ChFP and AMPKα2GFP expressed alone.
Excitation of NHE5ChFP with a 488-nm laser light showed low or no signal (data not shown). Regions of overlapping signal were measured for the average intensity per pixel. Subsequently, a 30-milliwatt, 561-nm laser at full power was used to photobleach NHE5ChFP that resulted in an increase in the GFP signal that was previously transferred to ChFP during FRET. Images were acquired and analyzed with Zeiss Zen software and corrected for background intensities and field non-uniformity.
Measurement of Na+/H+ Exchanger Activity
AP-1 cells alone or stably expressing human NHE5 were used for these studies. To measure basal plasmalemmal NHE5 activity, the 22Na+ influx assay was conducted as described previously (22) except that acid loading prior to 22Na+ uptake was omitted. Briefly, cells were grown to confluence in 24-well polystyrene tissue culture dishes, washed with isotonic choline chloride solution (130 mm choline chloride, 1 mm MgCl2, 2 mm CaCl2, 20 mm HEPES-Tris, pH 7.4) and incubated with 2 μCi/ml of 22Na+ in the same solution containing 1 mm ouabain (to inhibit Na+ efflux by the Na+/K+-ATPase) in the absence or presence of the specific NHE antagonist EIPA (200 μm) at room temperature for 5 min. This concentration of EIPA is sufficient to completely block NHE5 activity (IC50 ∼ 0.42 μm) (22). EIPA and its related analogues inhibit NHE activity by competitively blocking Na+ influx. The assay was terminated by rapidly washing the cells three times with 4 volumes of ice-cold NaCl stop solution (130 mm NaCl, 1 mm MgCl2, 2 mm CaCl2, 20 mm HEPES-NaOH, pH 7.4). Cell monolayers were subsequently solubilized by 0.25 ml of 0.5 m NaOH followed by neutralization with the same volume of 0.5 m HCl. Radioactivity was measured by liquid scintillation spectroscopy. Measurements of 22Na+ influx specific to the Na+/H+ exchanger were determined as the difference between the initial rates of 22Na+ influx in the absence and presence of EIPA and expressed as EIPA-inhibitable 22Na+ influx.
In experiments examining the kinetics of NHE5 activity as a function of the extracellular Na+ concentration, cells were acid-loaded using the NH4+-prepulse technique prior to measuring the influx of 22Na+ as described previously (22). To measure the activity of NHE5 as a function of the intracellular H+ (H+i) concentration, intracellular pH (pHi) was clamped over the range of 5.4 to 7.4 using the K+-nigericin method (35) as previously described (36). Data analysis and fitting was carried out using Origin 8 software.
Measurement of Intracellular pH
Cells grown on 12-mm glass coverslips are washed with a standard HEPES-buffered artificial cerebrospinal fluid (ACSF) (37) and incubated for 15–60 min at 37 °C with the pH-sensitive ratiometric dye BCECF-AM (10 μm) diluted into 10 mm HEPES-ACSF or 22 mm sodium bicarbonate-buffered ACSF. The cells were then washed with ACSF to remove the residual dye. Coverslips were placed into a Live Cell Instruments (LCI) magnetic chamber and filled with ACSF. The cells were examined at 37 °C in a LCI environment chamber fitted to a Zeiss LSM710 confocal microscope. Solutions were perfused onto the cells at ∼2 ml/min with a Gilson Minipuls 2 peristaltic pump. The cells were perfused with ACSF for ∼1 min prior to any treatments or manipulations. The BCECF dye was excited sequentially with a 20 MHz pulsed 440-nm laser and a 20 MHz pulsed 473-nm laser. The emission spectrum was collected between 510 and 650 nm for each excitation and a fluorescence ratio of the emission intensities from each excitation wavelength was calculated after background subtraction (473/440). The ratio was converted to pH using a calibration curve generated using the K+-nigericin clamp method. The images were analyzed with Zen (Zeiss) or Metamorph/Metaexpress (Molecular Devices) software.
Detection of Cell Surface NHE5 Expression
Plasma membrane proteins were isolated using a cell surface biotinylation assay as described previously (38, 39). Briefly, confluent AP-1/NHE5HA3 cells were incubated in serum-free αMEM for up to 60 min at 37 °C. Cells were rinsed twice with ice-cold PBS-CM (PBS containing 0.1 mm CaCl2 and 1 mm MgCl2) and then plasmalemmal proteins were labeled indiscriminately with membrane-impermeable N-hydroxysulfosuccinimydyl-SS-biotin (0.5 mg/ml, sulfo-NHS-SS-biotin; Pierce) for 30 min at 4 °C. The solution was then discarded and unreacted biotin was quenched three times with ice-cold PBS-CM containing 20 mm glycine. The cells were then lysed in PBS buffer containing 0.2% deoxycholic acid, 0.5% Triton X-100 and proteinase inhibitor mixture for 30 min on ice, and then centrifuged at 12,000 × g for 30 min at 4 °C to remove insoluble cellular debris. A portion of the resulting supernatant was removed and represents the “total fraction.” The remaining supernatant was incubated overnight with neutravidin-agarose beads to extract biotinylated “surface membrane proteins” according to the manufacturer's instructions. The proteins were then resolved by SDS-PAGE followed by immunoblotting. The intensities of the bands were quantified by densitometry of films exposed in the linear range, and then digitally imaged and analyzed with the FluorChemTM system (Alpha Innotech Corporation, CA).
Statistical Analysis
Unless otherwise stated, error bars represent the mean ± S.E. and statistical analysis was performed by using the one-way ANOVA Bonferroni post hoc test or the Student's t test.
RESULTS
Identification of AMPKα2 as an NHE5 Interacting Protein
Na+/H+ exchangers are predicted to contain 12 transmembrane helical segments within their amino-terminal region that form the cation permeation pathway, whereas the distal C-terminal portion of the exchanger faces the cytoplasm and is responsible for regulating transport activity in response to various stimuli (illustrated in Fig. 1A). To gain insight into the function and regulation of NHE5, a fragment spanning the bulk of the C-terminal cytoplasmic tail of NHE5 (amino acids Gly544–Leu896) was used as a molecular probe to screen a human brain cDNA library for potential binding proteins using the yeast two-hybrid system (40). Over 2 × 106 clones were screened from which 9 positive clones were obtained. One clone encoded almost the entire length of the catalytic α2 subunit of AMP-activated protein kinase (AMPKα2), with the exception of the first six amino acids. The yeast two-hybrid interaction between NHE5 and AMPKα2 was validated in mating assays by co-transforming yeast strains MATα strain Y187 and AH109 with pAS2–1/NHE5(544–896) and AMPKα2 obtained from the yeast two-hybrid screen. The NHE5(544–896) bait did not interact with the GAL4 activation domain encoded by pGAD10, and AMPKα2 did not interact with the GAL4 DNA binding domains encoded by pAS2-1, further supporting the possibility that these two proteins might be genuine interacting partners.
FIGURE 1.
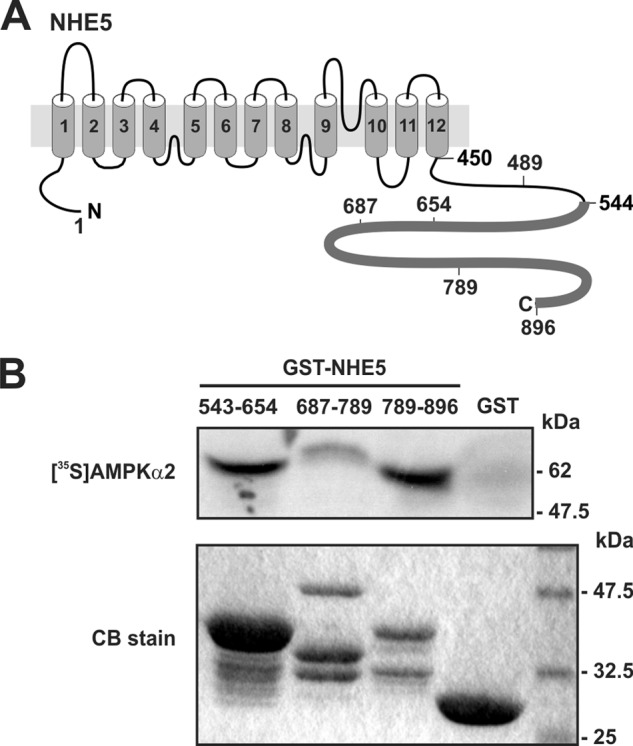
The cytoplasmic C terminus of NHE5 binds to the AMPKα2 catalytic subunit in vitro. A, schematic representation of the predicted transmembrane organization of NHE5. B, purified GST and GST-NHE5 fusion proteins encoding peptide segments that span the cytoplasmic C terminus of NHE5 were incubated with in vitro translated full-length 35S-labeled AMPKα2 protein. The proteins in the reaction solution were resolved by SDS-PAGE. Radioactivity was detected using a PhosphorImager and protein loading was visualized by staining with Coomassie Blue (CB) dye. Data are representative of at least three independent experiments.
Mapping the AMPKα-binding Site of NHE5
To validate the direct interaction between NHE5 and AMPKα2, in vitro GST fusion protein pulldown assays were used. Three GST-linked segments that spanned the C-terminal fragment of NHE5 used as bait in the yeast two-hybrid screen were incubated with in vitro synthesized full-length [35S]AMPKα2 and the resulting precipitates were evaluated for binding by SDS-PAGE and PhosphorImager analysis. The GST-NHE5 fusion proteins for each of the constructs migrated in the SDS-PAGE gel as multiple bands as detected by Coomassie Blue staining (Fig. 1B, lower panel). The higher molecular mass band for each construct represents the full-length fusion protein, whereas the faster migrating bands most likely represent GST-NHE5 proteolytic products. [35S]AMPKα2 bound mainly to two segments of NHE5 encompassing amino acids 543–654 and 789–896 (Fig. 1B, upper panel). Significantly, both segments were robustly phosphorylated by purified rat liver AMPK (containing mainly the α1 subunit, which shares 90% amino acid identity with α2 in its catalytic core) in the presence of added AMP (0.3 mm) (Fig. 2A). More extensive mapping analyses delimited the major phosphorylated sites to segments 571–601 and 845–870, and to a much lesser degree 602–624 (Fig. 2B and data not shown). Significantly, each of these segments possesses a putative consensus recognition sequence for AMPK, Φ-(X,β)-X-X-(S/T)-X-X-X-Φ (where Φ represents a hydrophobic residue Met, Leu, Phe, Ile, or Val, and β represents a positively charged residue Arg, Lys, or His) (41). Simultaneous Ala substitutions of putative target residues Ser575 and Ser577 (S575A/S577A), Ser618 and Ser620 (S618A/S620A), and Ser851, Ser853, Ser855, and Ser856 (S851A/S853A/S855A/S856A) abolished in vitro phosphorylation by AMPK (Fig. 2B). These data indicate that the C terminus of NHE5 is a direct substrate of AMPK, at least in vitro.
FIGURE 2.
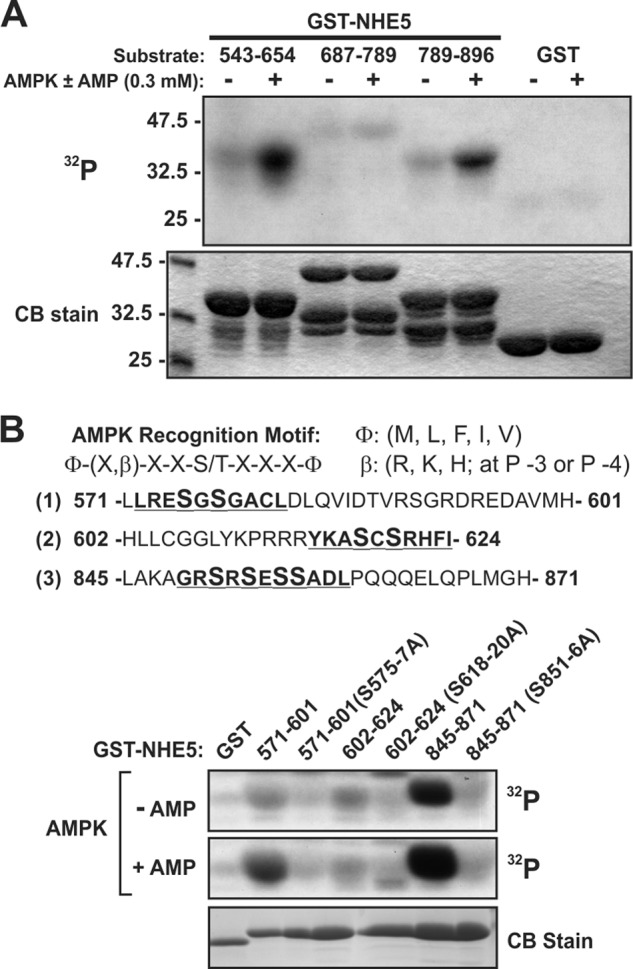
NHE5 is phosphorylated in vitro by AMPK. A, GST-NHE5 fusion proteins were incubated in vitro with [γ-32P]ATP and purified, partially activated, rat liver AMPK in the presence or absence of AMP (0.3 mm), subjected to SDS-PAGE, and analyzed by autoradiography. Protein loading was visualized by staining the gel with Coomassie Blue (CB) dye. B, GST-NHE5 fusion proteins (571–601, 602–624, and 845–871) containing simultaneous Ala substitutions of potential target Ser residues (highlighted in enlarged bold font size) in putative AMPK recognition motifs (upper panel) were incubated with purified rat liver AMPK and [γ-32P]ATP in vitro with or without AMP, subjected to SDS-PAGE and analyzed by autoradiography (lower panel). Data are representative of at least three independent experiments. S575–7A, S575A/S577A; S618–20A, S618A/S620A; S851–6A, S851A/S853A/S855A/S856A.
Interaction of NHE5 and AMPK in Whole Cells and Tissue
To verify the interaction of full-length NHE5 and AMPKα2 in mammalian cells, co-immunoprecipitation experiments were performed by transiently expressing a C-terminal Myc-tagged AMPKα2 construct (AMPKα2myc) in a mutagenized Chinese hamster ovary cell line (AP-1) that lacks endogenous plasmalemmal NHEs, but stably expresses a NHE5 construct engineered to contain a triple HA-epitope in its first extracellular loop (AP-1/NHE5HA3) as previously described (23). As shown in Fig. 3A, NHE5HA3 (∼99 kDa) and AMPKα2myc (∼60 kDa) formed an immunoprecipitable complex from co-transfected cells.
FIGURE 3.

NHE5 forms a macromolecular complex with AMPK subunits in heterologous and native tissue. A, AP-1 cells stably expressing triple HA-tagged NHE5 (NHE5HE3) were transiently transfected (24 h) with empty vector or full-length Myc-tagged AMPKα2 (AMPKα2myc). Cell lysates were prepared and incubated with a rabbit polyclonal anti-HA antibody (α-HAp) to immunoprecipitate NHE5HA3. The cell lysates and immunoprecipitates (IP) were analyzed by SDS-PAGE and immunoblotted (IB) with either a monoclonal anti-HA antibody (α-HAm) or anti-Myc (α-mycm) to detect NHE5HA3 or AMPKα2myc, respectively. B, to detect association of endogenous AMPK subunits with NHE5HA3, cell lysates were prepared from untransfected AP-1 cells or AP-1 cells stably expressing NHE5HA3 and then incubated with rabbit polyclonal antibodies against endogenous AMPKα1, -α2, -γ1, and -γ2 subunits. To detect a complex between NHE5 and the native AMPK β1/2 subunits, cell lysates were incubated with a rabbit polyclonal anti-HA antibody. The immunoprecipitates were analyzed by SDS-PAGE and immunoblotting with either mouse monoclonal anti-HA antibody to detect NHE5HA3 or with a commercially available pan anti-AMPKβ antibody that recognizes both the β1 and β2 subunits. C, to detect association of native AMPKα subunits with NHE5 in brain tissue, cell lysates were prepared from mouse brain (postnatal day 3) and then incubated with a control mouse IgG antibody or a mouse monoclonal antibody that recognizes both AMPKα1 and -α2 (α-AMPKα1/2m). The immunoprecipitates and cell lysates were analyzed by SDS-PAGE and immunoblotting. The IP blot was probed with a rabbit polyclonal antibody that specifically detects NHE5 (α-NHE5p). The lysate blots were probed with polyclonal antibodies that recognize NHE5 or AMPKα2. Data are representative of three independent experiments.
Using commercially available AMPK antibodies, we found by immunoblot analyses that AP-1 cells endogenously express α1, α2, β1/2, γ1, and γ2. To assess whether NHE5HA3 could associate with each of these native subunits, we performed another series of co-immunoprecipitation experiments using the stable AP-1/NHE5HA3 cell line. As shown in Fig. 3B, NHE5HA3 was able to form an immunoprecipitable complex with each native subunit, indicating that NHE5 can potentially associate with different heterotrimeric arrangements of AMPK. Importantly, native NHE5 was detected in an AMPK-containing protein complex immunoprecipitated from whole mouse brain lysates using a monoclonal antibody that recognizes both AMPKα1 and -α2 (Fig. 3C).
The association of NHE5 with AMPKα2 was also confirmed by immunofluorescence confocal microscopy in intact cells. AP-1 cells were co-transfected with NHE5 linked to monomeric cherry fluorescent protein (NHE5ChFP) and AMPKα2 fused to enhanced green fluorescence protein (AMPKα2GFP). Similar to previous observations of exogenous HA-tagged NHE5 and native NHE5 in nervous tissue and cells (23–25), a minor fraction of NHE5ChFP localized close to or at the plasma membrane, whereas the bulk resided in vesicles that accumulated in a juxtanuclear region (Fig. 4A). By comparison, most of the AMPKα2GFP signal was diffusely distributed throughout the nucleus and the cytoplasm, consistent with earlier findings (42), although a significant fraction was detected in punctate vesicles that also colocalized with NHE5ChFP. Although these results suggest a close association, this technique is hampered by the limited optical spatial resolution of the microscope, which is ∼200 nm. To determine the proximity of these two proteins more precisely, we applied FRET using the acceptor photobleaching method (34, 43). When applied to optical microscopy, it allows one to determine when two molecules are within several nanometers of each other, distances sufficiently close for bimolecular interactions to occur. The analyses (Fig. 4B) show that upon modest photobleaching of the acceptor molecule NHE5ChFP (∼7–8% quenching to minimize phototoxicity and ensure that the donor is not also being bleached), there was a corresponding, albeit larger, increase (∼27%, p < 0.05, Student's t test) in the fluorescence intensity of the excited donor AMPKα2GFP, energy that previously had been transferred to NHE5ChFP and indicative of a close physical interaction between these two proteins. Collectively, these results indicate that a fraction of AMPK can form a stable complex with NHE5 in heterologous cell systems as well as natively in whole brain.
FIGURE 4.
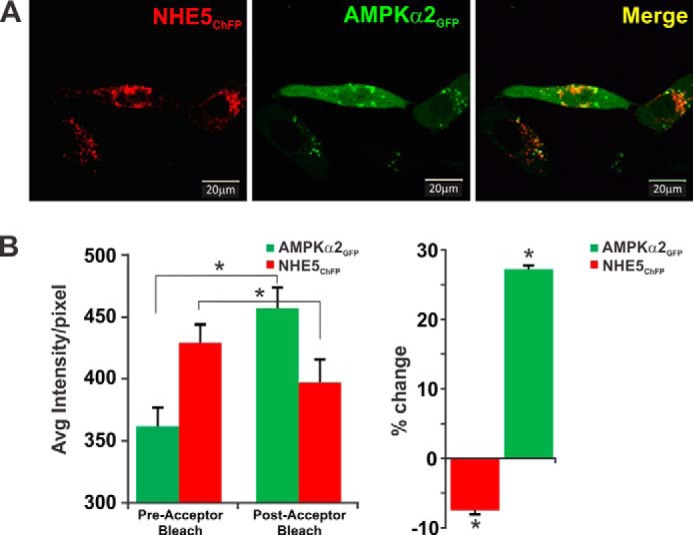
NHE5 and AMPKα2 colocalize in transfected AP-1 cells. A, immunofluorescence confocal microscopy images of AP-1 cells transiently co-transfected (48 h) with monomeric cherry fluorescent protein-tagged NHE5 (NHE5ChFP, red) and enhanced green fluorescent protein-tagged AMPKα2 (AMPKα2GFP, green). Overlapping signals in merged image are yellow. B, fluorescence (or Forster) FRET analysis of NHE5ChFP and AMPKα2GFP interactions were determined by the acceptor photobleaching method (see ”Experimental Procedures“). Photobleaching of NHE5ChFP resulted in an increase in the GFP signal previously transferred to ChFP during FRET (left panel). Percent change in average fluorescence per pixel, relative to pre-bleaching of NHE5ChFP is shown in the right panel. Data are representative of at least three independent experiments. Asterisks indicate p < 0.05, Student's paired t test.
A recent study indicated that chronic activation of AMPK with the membrane-permeant AMP mimetic AICAR (1 mm, 24-h) significantly enhanced NHE1 mRNA, protein, and activity in human embryonic kidney HEK293 cells (44). This observation left open the possibility that AMPK may interact physically with other NHEs. To investigate this question, NHE1HA was transiently expressed in AP-1 cells and assessed for its ability to form a complex with endogenous AMPKα1 or -α2. As shown in Fig. 5, NHE1 did not associate with either catalytic α subunit, indicative of an indirect relationship between AMPK and NHE1 in HEK293 cells. Hence, our data suggest that the association of AMPK with NHE5 is selective, at least within the confines of this comparison.
FIGURE 5.
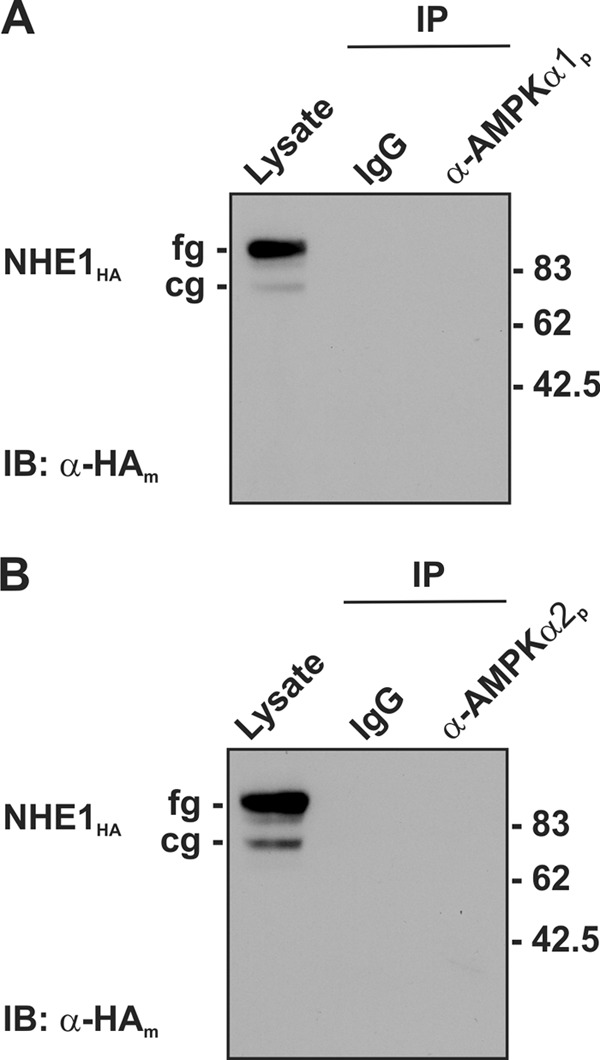
AMPK does not form a macromolecular complex with the Na+/H+ exchanger NHE1 isoform. To detect whether AMPK can associate with NHE1, cell lysates were prepared from AP-1 cells stably expressing NHE1HA and then incubated with rabbit IgG or polyclonal antibodies against the endogenous AMPKα1 and -α2 subunits. The immunoprecipitates (IP) were analyzed by SDS-PAGE and immunoblotting (IB) with mouse monoclonal anti-HA antibody to detect the core (cg) and fully (fg) glycosylated forms of NHE1HA. Data are representative of two independent experiments.
AMPK Regulation of NHE5
To examine the functional significance of the association between NHE5 and AMPK, we first examined their relationship in AP-1 cells. AP-1/NHE5HA3 cells were treated with AICAR to activate AMPK and plasmalemmal NHE5 activity was measured using an EIPA-inhibitable 22Na+ flux assay as described previously (22). As mentioned earlier, although NHE5 is more resistant to inhibition by EIPA (IC50 ∼ 0.42 μm) compared with NHE1 (IC50 ∼ 15 nm), it can be completely blocked at higher drug concentrations (i.e. 200 μm), thereby allowing discrimination of NHE5-mediated 22Na+ flux from nominal radioisotope uptake in this cell line under the imposed assay conditions.
As shown in Fig. 6, A and B, basal NHE5 activity increased (∼1.5–2-fold; p < 0.01, one-way ANOVA) in a dose- and time-dependent manner that correlated with increases in the active, phosphorylated form of AMPK (AMPKα-pThr172) (Fig. 6C). EIPA-inhibitable 22Na+ uptake in control untransfected AP-1 cells was negligible and unaffected by AICAR treatment, indicating that the enhanced 22Na+ influx of AICAR-treated AP-1/NHE5HA3 cells is attributable exclusively to NHE5.
FIGURE 6.
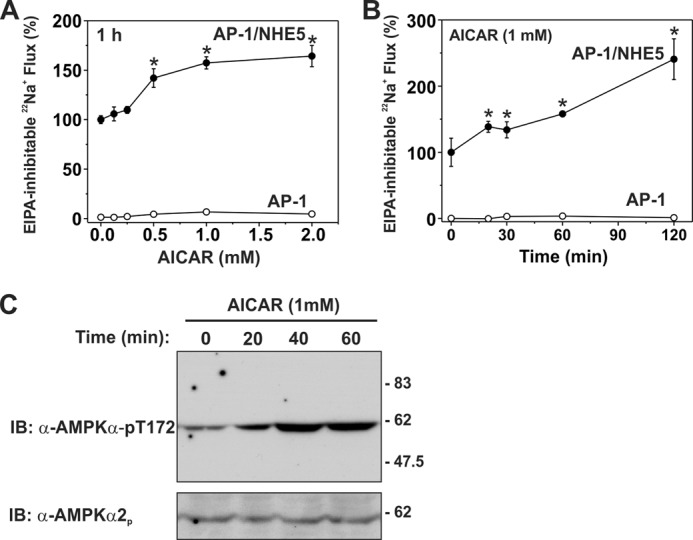
Activation of endogenous AMPK by AICAR increases NHE5 activity in a dose- and time-dependent manner. Untransfected and NHE5HA3-stably transfected AP-1 cells were pretreated with (A) increasing concentrations of AICAR (0–2.0 mm) for 60 min or (B) as a function of time (0–120 min) in the presence of 1 mm AICAR. Na+/H+ exchange activity in each cell line was measured as EIPA-inhibitable 22Na+ influx (nmol/min/mg of protein) and expressed as a percentage of the basal transport rate for each condition, normalized to NHE5 activity at 0 mm AICAR (A) or 0 min (B). C, AICAR-dependent activation of AMPK in AP-1/NHE5HA3 cells as a function of time was detected using a mouse monoclonal antibody that recognizes the phosphoactivated form of the AMPKα catalytic subunit (α-AMPKα-pT172). Loading was controlled by stripping and reprobing the blot with a rabbit polyclonal for AMPKα2 (α-AMPKα2p). Data are representative of at least three independent experiments. Asterisks indicate p < 0.01, one-way ANOVA. IB, immunoblot.
To further evaluate the acute effect of AICAR on NHE5 activity in relationship to pHi, stably expressing AP-1/NHE5HA3 cells were initially incubated with diluent or AICAR (1 mm) for 1 h. The cells were then perfused with HEPES-buffered ACSF (containing 126 mm NaCl) under nominally HCO3−/CO2-free conditions (to exclude potential contributions from bicarbonate transporters) in the continuing presence of either diluent or AICAR, followed by acidification using the NH4+ prepulse technique (45) and subsequent pHi recovery in Na+-rich ACSF. The pHi was monitored using the ratiometric pH-sensitive fluorescent dye BCECF and microscopy. For comparative purposes, AP-1/NHE1HA cells were also examined. Consistent with the radioisotopic measurements, acute pretreatment of AP-1/NHE5HA3 cells with AICAR significantly stimulated the pHi recovery rate relative to untreated controls, and also raised the steady-state pHi from 6.98 ± 0.05 to 7.34 ± 0.09 (p < 0.05, Student's t test) (Fig. 7A). Conversely, AICAR had no significant effect on pHi recovery in AP-1/NHE1HA cells or on resting pHi (resting pHi: control, 7.38 ± 0.08; AICAR, 7.24 ± 0.08) (p > 0.05, Student's t test) (Fig. 7B). To validate that the Na+-dependent pHi recoveries in the stable cell lines were due to the respective transfected NHE isoforms, we took advantage of their differential sensitivity to NHE antagonist HOE694, which is even more highly selective (∼2 orders of magnitude) for NHE1 relative to NHE5 than amiloride or EIPA (22). As shown in Fig. 7D, 1 μm HOE694 completely blocked Na+-dependent pHi recovery from an NH4+-imposed acid load in AP-1/NHE1HA cells. This effect was equivalent to that observed using solutions where Na+ was substituted with choline+, a non-transportable cation, thereby confirming that this flux is Na+ dependent. Conversely, substantially higher concentrations of HOE694 (500–1000 μm) were required to block pHi recovery in AP-1/NHE5HA3 cells (Fig. 7C), concentrations consistent with previous studies (22).
FIGURE 7.

Acute activation of AMPK by AICAR selectively enhanced intracellular pH recovery in AP-1 cells stably expressing NHE5. AP-1 cells stably expressing NHE5HA3 (A) and NHE1HA (B) were grown on 12-mm coverslips and loaded with the pH-sensitive fluorescent dye BCECF-AM (50 μm) in αMEM (supplemented with 10% FBS) in the absence or presence of the AMPK agonist AICAR (1 mm) for 1 h. The cells were then placed in a LCI environment chamber maintained at 37 °C and fitted to a Zeiss LSM710 confocal microscope. The cells were initially perfused with HEPES-buffered ACSF (contains 126 mm NaCl) in the continued absence or presence of AICAR and then acidified using the NH4+ prepulse technique, followed by recovery in Na+-rich ACSF. Intracellular pH was measured as described under ”Experimental Procedures.“ To assess the drug sensitivity of NHE5 (C) and NHE1 (D), following NH4Cl treatment the cells were allowed to recover in Na+-rich ACSF medium containing increasing concentrations (1–1000 μm) of the NHE-specific antagonist HOE694 as indicated, or in some cases ACSF medium where the NaCl was substituted with molar equivalents of choline chloride (i.e. 0 mm NaCl). The rate of pHi recovery following acidification was monitored as a function of time. The data represent the mean ± S.E. of three independent experiments, with 10–30 cells per experiment.
Activation of AMPK can also occur under pathophysiological conditions such as ischemia or nutrient deprivation. To induce changes in cellular metabolism that mimic ischemia and stimulate AMPK, mitochondrial inhibitors such as antimycin A (AA; 1 μg/ml) can be used to block complex III in the electron transport chain, thereby inhibiting aerobic energy production (46). In addition, generation of ATP by glycolysis can be blocked by 2-deoxyglucose (5 mm) (47, 48). As shown in Fig. 8A, 2-deoxyglucose drastically reduced ATP levels in AP-1 cells to ∼15% of steady-state levels within 20 min, whereas AA showed a lesser reduction (∼50%). Both treatments also increased the phosphoactivated form of AMPK (i.e. AMPKα-pT172), although the extent of phosphorylation was more pronounced (2–3-fold) for AA, presumably due to the greater supply of cellular ATP needed for optimal phosphorylation of AMPK by upstream kinases such as LKB1 (Fig. 8B). It was also noted that prolonged 2-deoxyglucose treatment caused the AP-1 cells to roundup and detach from the culture dishes. Hence, subsequent analyses of metabolic stress on NHE5 activity were confined to the use of AA. As shown in Fig. 9, AA treatment caused a highly reproducible biphasic increase in basal NHE5 activity over a 60-min period that reached levels comparable with that achieved with AICAR (see Fig. 6). The AMPK antagonist compound C (CC) (49) effectively blocked the AA-induced increase in NHE5 activity to levels measured in resting cells. Treatment with CC alone also caused a gradual decrease in NHE5 activity. The inhibitory effect of CC alone on NHE5 activity suggests that the transporter is tonically regulated by low levels of active AMPK-pThr172 in resting cells (see Fig. 8B).
FIGURE 8.
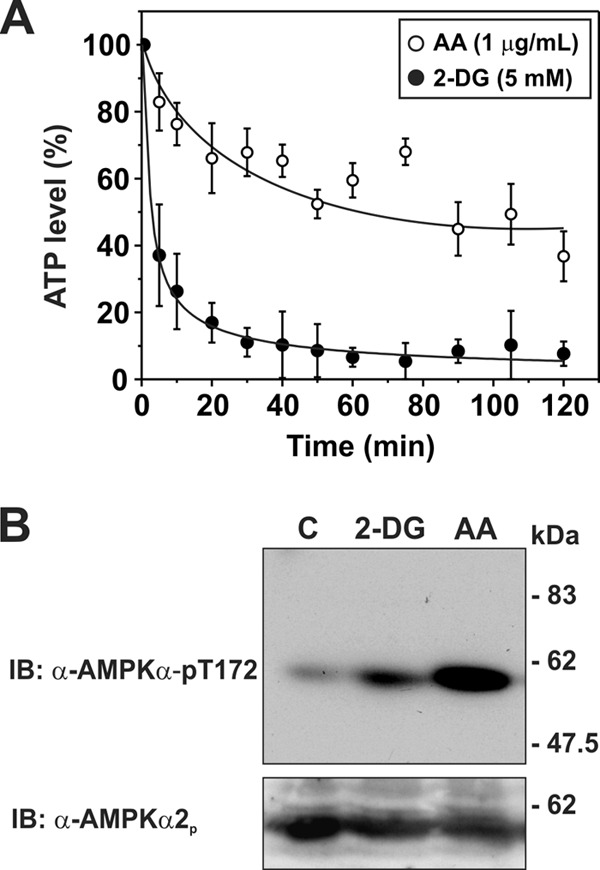
Effect of metabolic inhibitors on cellular ATP levels and activation of AMPK in AP-1/NHE5 cells. AP1/NHE5 cells were treated over a 120-min time course with inhibitors of either oxidative phosphorylation, antimycin A (1 μg/ml), or glycolysis, 2-deoxyglucose (5 mm). A, cytosolic ATP levels were measured from the cellular lysates with a chemiluminescent ATP determination kit. Data shown are representative of at least four independent experiments. B, activation of AMPK in AP-1/NHE5 cells was detected at 60 min using an antibody that recognizes the phospho-activated form of the AMPKα catalytic subunit (AMPKα-pT172). Loading was controlled by stripping and reprobing the blot with a rabbit polyclonal for AMPKα2. Data are representative of at least three independent experiments. IB, immunoblot.
FIGURE 9.
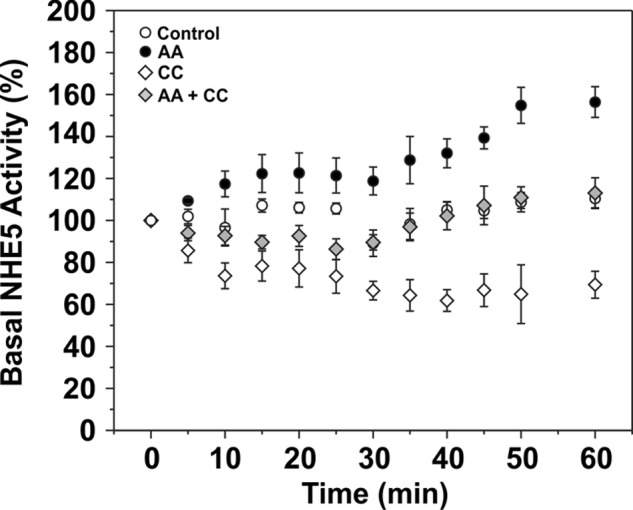
Activation of NHE5 in response to acute metabolic stress is dependent on AMPK. AP-1/NHE5HA3 cells were treated over a 60-min period either separately or together with AA (1 μg/ml) to activate AMPK and the specific AMPK inhibitor CC (20 μm). NHE5 activity was measured as EIPA-inhibitable 22Na+ influx (nmol/min/mg protein) and normalized as a percentage of the basal rate at the start of the experiment (0 min). Data were generated from quadruplicate samples in each of three independent experiments and presented as the mean ± S.E.
The resulting biphasic increase in plasmalemmal NHE5 activity elicited by AA treatment could result from one or more of the following possibilities: 1) AA-induced cytoplasmic acidification due to the greater dependence on glycolysis for metabolic energy with a corresponding increase in lactic acid accumulation and/or 2) an AMPK-mediated change in the transport kinetics of NHE5 (i.e. increased substrate affinities and/or transport velocity Vmax). Indeed we confirmed that, under HEPES-buffered media conditions, AA produced a gradual acidification (pHi 7.2 → 6.4) of untransfected AP-1 cells over a 20-min period, and remained at this level thereafter (Fig. 10A). In AP-1/NHE5HA3 cells, AA caused a less pronounced acidification that reached a maximum level of pHi ∼ 6.8 between 10 and 30 min exposure, and then pHi gradually recovered to pH ∼ 7.0 over the next 30 min although it did not yet reach resting control levels (pHi ∼ 7.2) (Fig. 10B). Significantly, this latter pHi recovery was prevented by inhibiting AMPK with CC or blocking NHE5 with high concentrations of the NHE antagonist HOE694 (1000 μm), thereby linking these two proteins to a common pathway. Hence, the acute AA-induced acidification could account, at least in part, for the initial increased catalytic rate of existing plasma membrane-resident NHE5, but does not fully explain the late-phase enhancement of NHE5 activity.
FIGURE 10.
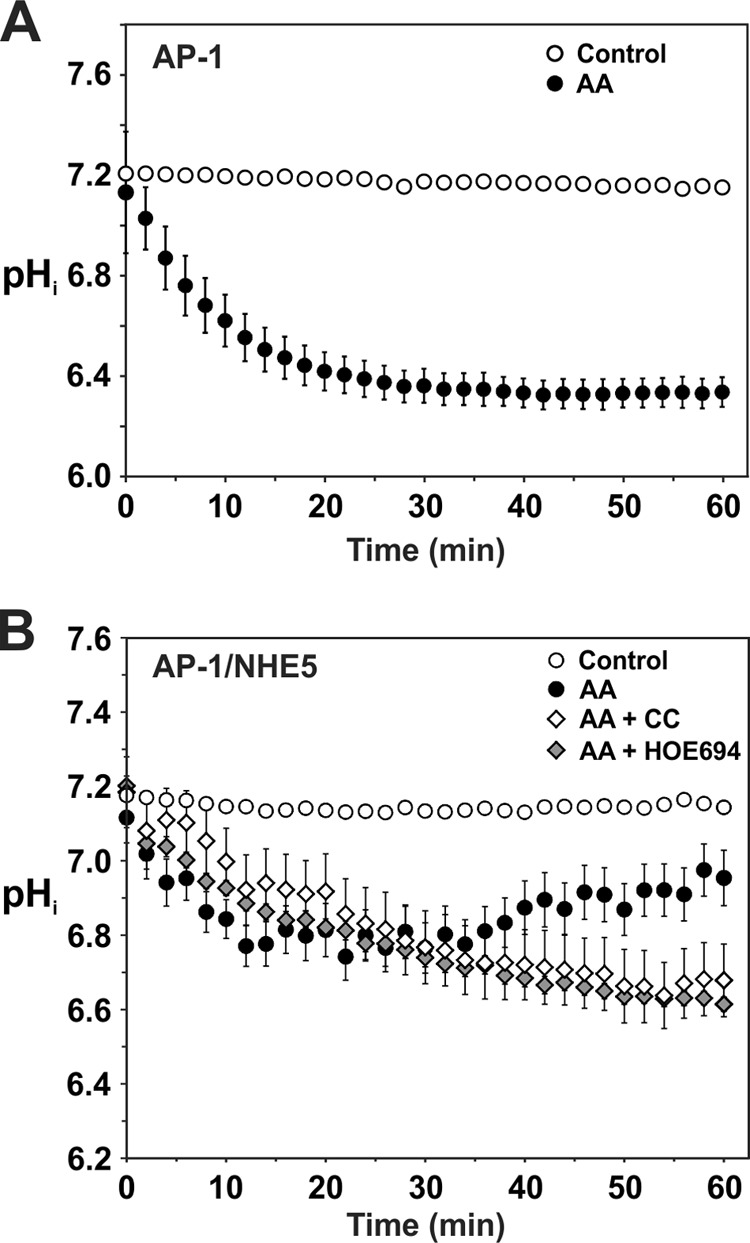
NHE5 regulates intracellular pH in AP-1 cells in response to metabolic stress-induced acidosis. A, untransfected AP-1 cells were treated with diluent or AA (1 μg/ml) over a 60-min period to inhibit oxidative phosphorylation and the effects on intracellular pH were measured using the ratiometric pH-sensitive fluorescent dye BCECF. B, AP-1/NHE5HA3 cells were treated with diluent, AA (1 mg/ml), AA plus CC (20 μm), or AA plus HOE694 (1000 μm) over a 60-min period and the effects on intracellular pH were measured. The data are presented as the mean ± S.E. of at least 3 independent experiments.
To further examine the late-phase activation of NHE5 activity, we measured its kinetic properties. As shown in Fig. 11, A and B, AA treatment (60 min) did not significantly alter the apparent affinities of NHE5 for Na+ (control KNa+ 16.9 ± 5.2 mm; AA KNa+ 14.9 ± 4.4 mm; p > 0.05, Student's t test) or H+ (control KH+ 0.39 ± 0.06 μm; AA KH+ 0.42 ± 0.09 μm; p > 0.05, Student's t test), respectively. However, the Vmax of NHE5 noticeably increased by ∼26% from a value of 85.9 ± 8.6 to 108.2 ± 11.0 nmol/min/mg of protein (p < 0.05, Student's t-test) (Fig. 11A), suggesting either an enhanced rate of turnover and/or increased transporters at the plasma membrane. To explore this latter possibility, plasmalemmal NHE5 abundance was measured as a function of time using a cell surface biotinylation assay (39). As illustrated in Fig. 12A and quantified in Fig. 12B, NHE5 cell surface expression was significantly increased (∼30–40%; p < 0.05, Student's t test) in response to AA, with the earliest increase detected at 30 min of exposure. Similar increases were also confirmed using a cell-based enzyme-linked immunosorbent assay to detect surface levels of the externally exposed triple HA-epitope present in NHE5 (data not shown). Significantly, the membrane redistribution of NHE5 was effectively blocked by CC (Fig. 12, C and D). Furthermore, examination of NHE5 constructs containing Ser to Ala substitutions of the two major AMPK phosphoacceptor sites identified in vitro (cf. Fig. 2) showed that simultaneous mutation of both S575A and S577A, but not the individual mutations or the combined S851A/S853A/S855A/S856A mutant, effectively abolished the AA-induced increase in NHE5 activity (Fig. 13A) and cell surface abundance (Fig. 13, B and C). This identifies AMPK phosphorylation of the proximal Ser575 and Ser577 residues as important in the membrane trafficking of NHE5. Conversely, the functional significance of AMPK phosphorylation of the more distal AMPK motif is unclear. The biochemical detection of the AA-induced increase in NHE5 surface abundance was also confirmed visually by fluorescence imaging where wild-type but not mutant (S575A/S577A) GFP-tagged NHE5 (NHE5GFP) increased its abundance at or near the plasma membrane as a function of time (i.e. after ∼30 min treatment) (Fig. 13D). Notably, this enhanced surface expression is coincident with the second phase of the biphasic increase in AA-mediated stimulation of NHE5 activity.
FIGURE 11.
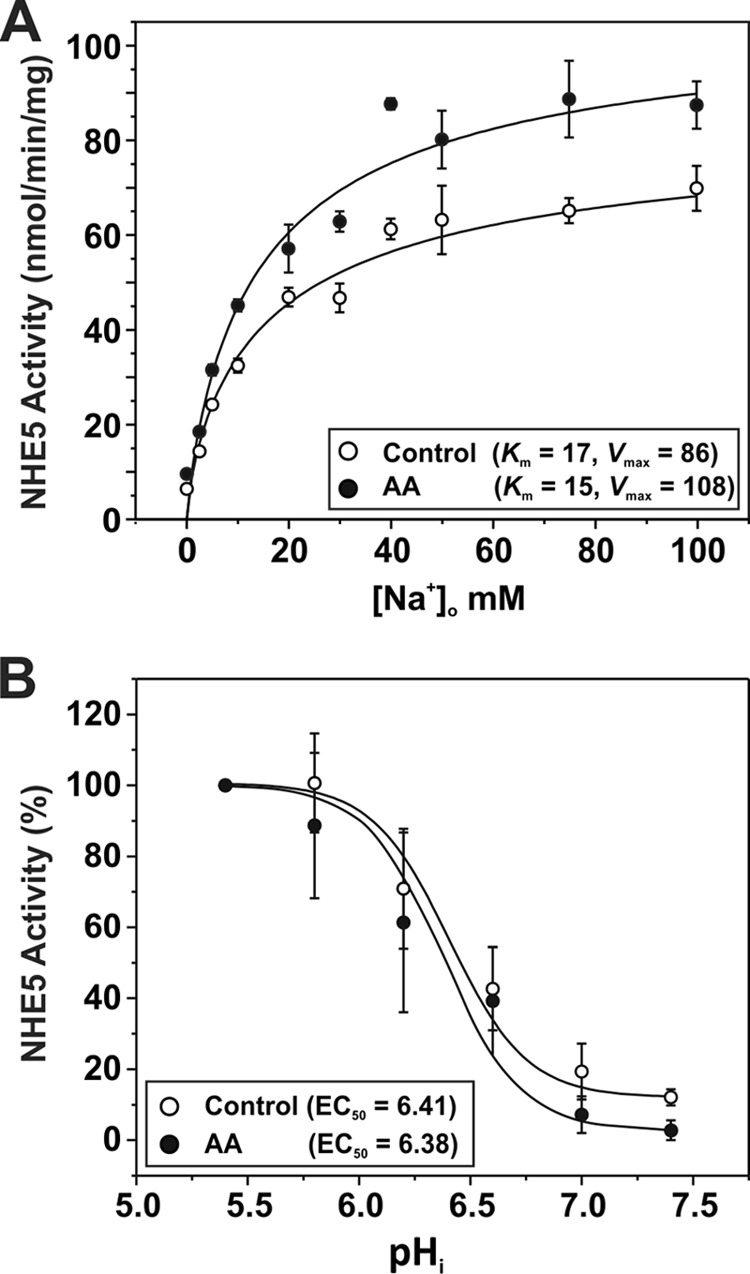
Effect of AA-induced metabolic stress on the kinetic properties of NHE5. Stably expressing AP1/NHE5HA3 cells were untreated or exposed to AA (1 μg/ml) for a 60-min period. A, to measure the Na+ affinity (Km) and maximal transport velocity (Vmax) of NHE5, cells were acidified to ∼pH 6 by prepulsing the cells with 50 mm NH4Cl, and then the rate of EIPA-inhibitable 22Na+ influx (nmol/min/mg of protein) was measured at different concentrations of Na+ (adjusted with non-radioactive Na+) during the linear phase (1 min uptake) of pHi recovery. The data were fitted using a Hill function (y = Vmax × xn/kn + xn). B, to measure H+ affinity, AP1/NHE5HA3 cells were clamped at various pHi values using the K+-nigericin technique, and the rates of EIPA-inhibitable 22Na+ uptake (in the presence of nominal unlabeled Na+) were measured. The data were fit with a dose-response function (y = A1 + (A2-A1)/1 + 10(Logx0-x)p), where Logx0 represents the EC50). In this particular experiment, the data were normalized to a percentage of the maximal rate of transport at pHi 5.4. The data are presented as the mean ± S.E. of three independent experiments, with each data point done in triplicate.
FIGURE 12.

Antimycin A-induced metabolic stress increases the abundance of NHE5 at the cell surface that is dependent on activation of AMPK. Stably expressing AP-1/NHE5HA3 cells were treated with (A and B) AA (1 μg/ml) alone or (C and D) in combination with the AMPK inhibitor CC (20 μm) over a 60-min period. Surface proteins were labeled with sulfo-NHS-SS-biotin (0.5 mg/ml) at 15-min intervals followed by quenching with glycine to remove unbound sulfo-NHS-SS-biotin. Cells were lysed and biotin-labeled proteins were extracted by binding to neutravidin-agarose beads. Total cell lysates and biotin-conjugated proteins were resolved by SDS-PAGE and immunoblotted with a polyclonal anti-HA antibody to detect NHE5HA3 and an anti-GAPDH antibody to control for protein loading. B and D, quantification of surface NHE5 was determined by densitometry (n = 7). Surface NHE5HA3 in panel B is significantly different at 45 and 60 min of AA treatment when compared with 0 min (asterisks indicate p < 0.05, one-way ANOVA).
FIGURE 13.

Metabolic stress-induced activation of NHE5 activity is impaired in the AMPK phosphorylation-defective NHE5(S575A/S576A/S577A) mutant. A, AP-1 cells stably expressing NHE5HA3 mutants S575A, S577A, S575/S576A/S577A (S575–7A), or S851A/S853A/S855A/S856A (S851–6A) were untreated or incubated with AA (1 μg/ml) for 60 min and assayed for NHE5 activity as measured by EIPA-inhibitable 22Na+ influx (nmol/min/mg of protein) and normalized as a percentage of the basal transport rate for each condition. Data represent the mean ± S.E. (n = 4, each performed in quadruplicate). Asterisks indicate statistical significance from control values (p < 0.05, Student's t test). B, AP-1/NHE5HA3(S575–7A) cells were treated with AA (1 μg/ml) over a total of 60 min. Surface proteins were biotinylated as described in the legend to Fig. 10. Total cell lysates and biotin-conjugated proteins were resolved by SDS-PAGE and immunoblotted with a polyclonal anti-HA antibody to detect NHE5HA3 and an anti-GAPDH antibody to control for protein loading. C, quantification of surface NHE5HA3 was determined by densitometry (n = 7). D, AP-1 cells were transiently transfected with NHE5GFP wild-type (upper panels) and mutant S575A/S576A/S577A (lower panels) constructs. After 48 h, the cells were then transferred and maintained at 37 °C in a Live Cell Instruments environment chamber fitted to a Zeiss LSM710 confocal microscope. The cells were treated with AA (1 μg/ml) and monitored every 15 min over a 60-min period. Data are representative of at least three independent experiments. Scale bar represents 20 μm.
AMPK Regulation of NHE5 in Hippocampal Neurons
To validate this regulatory behavior under more native conditions, we selected newborn (0–2-day-old) mouse hippocampal neurons for further study in vitro as they express high levels of AMPK, especially AMPKα2 (30, 50), and NHE5 (24). Importantly, hippocampal tissue at this developmental stage contains comparatively few glial cells and the neurons are capable of undergoing significant growth and differentiation under defined culture conditions to form extensive, synaptically connected networks that, whereas less complex than neural tissue, make them an ideal preparation for investigating the function and trafficking of neuronal proteins (37). However, as NHE1 activity has also been detected in freshly dissociated hippocampal neurons, albeit from more mature postnatal mice (21–30 days old) (51), it was necessary to appraise the NHE composition of our preparation of neonatal mouse hippocampal neurons. We chose pharmacological and single-cell imaging approaches to distinguish the NHE composition of individual neurons instead of indiscriminate RT-PCR of hippocampal culture lysates because of concerns of residual “contaminating” glia cells that are known to express NHE1 (52). To this end, we took advantage of the marked differential potency of HOE694 to block the various NHE isoforms that, as demonstrated herein (see Fig. 7) and elsewhere (22), is more highly selective for NHE1 relative to NHE5. For comparison with AP-1 cells, the neurons were also initially examined in HEPES-buffered nominally HCO3−/CO2-free ACSF media to minimize potential regulation by bicarbonate transporters. As shown in Fig. 14A, a low concentration of HOE694 (10 μm) sufficient to completely block NHE1 activity was ineffective, whereas a much higher concentration of HOE694 (1000 μm) was required to fully suppress Na+-dependent pHi recovery, consistent with NHE5 activity. Moreover, in resting cells, 1000 μm HOE694, but not 10 μm HOE694, induced a significant and sustained reduction of steady-state pHi (7.2 → 6.7) (Fig. 14B). Hence, these results combined with previous findings using analogous rat preparations (21, 25, 53, 54) indicate that NHE5 is the principal Na+/H+ exchanger responsible for cytoplasmic pH homeostasis, at least in fetal or neonatal hippocampal neurons.
FIGURE 14.
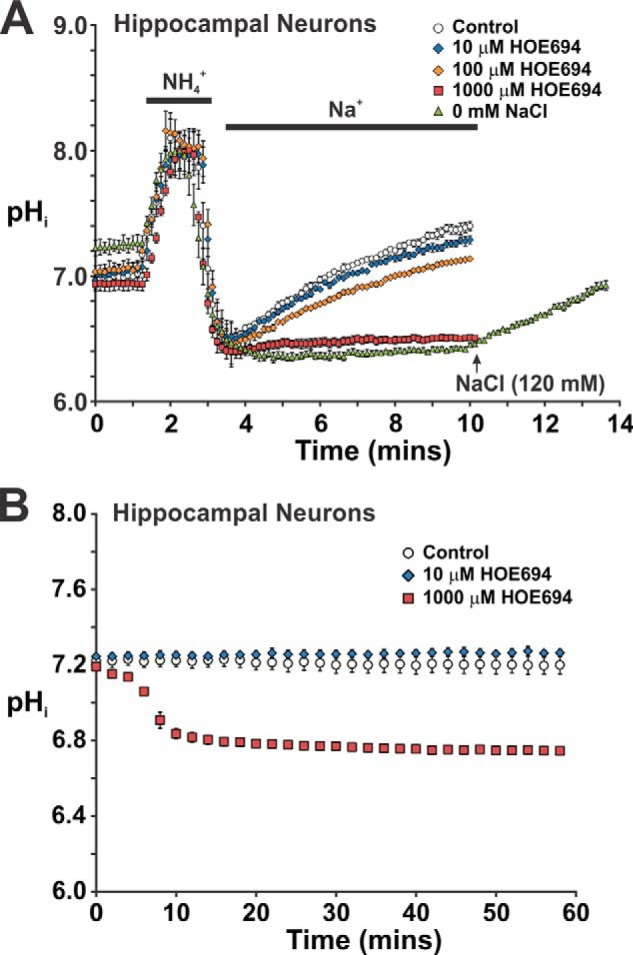
Effect of the NHE antagonist HOE694 on pH recovery from an intracellular acid load in mouse primary hippocampal neurons. Primary cultures of mouse hippocampal neurons (14–21 days in vitro) were grown on 12-mm coverslips and loaded for 15 min with the pH-sensitive fluorescent dye BCECF-AM (50 μm). Coverslips were placed into a Live Cell Instruments magnetic chamber and incubated with HEPES-buffered ACSF (contains 126 mm NaCl). The cells were examined at 37 °C using a Zeiss LSM710 confocal microscope. A, ACSF was perfused onto the cells at ∼2 ml/min for 1 min prior to acid loading with NH4Cl. Following NH4Cl treatment, the cells were allowed to recover in Na+-rich ACSF medium containing increasing concentrations (10–1000 μm) of the NHE-specific antagonist HOE694 as indicated, or in some cases ACSF medium where the NaCl was substituted with molar equivalents of choline chloride (i.e. 0 mm NaCl). The rate of pHi recovery following acidification was monitored as a function of time. B, steady-state pHi of cultured hippocampal neurons was monitored as a function of time following addition of diluent (control) or increasing concentrations of HOE694. The data represent the mean ± S.E. of three independent experiments, with ∼10 cells per experiment.
To test whether NHE5 is regulated by AMPK in hippocampal neurons, the cells were treated with AICAR (1 mm) or AA (1 μg/ml) over a 60-min period while monitoring the subcellular distribution of NHE5 and changes in steady-state pHi. In untreated neurons, NHE5 exhibited a punctate distribution throughout the soma and dendrites (Fig. 15A). By contrast, acute exposure to AICAR caused a noticeable redistribution of endogenous vesicular NHE5 immediately adjacent to or at the plasma membrane (Fig. 15B), concomitant with a gradual alkalinization of steady-state pHi (pH ∼ 7.55) that was prevented by CC (Fig. 15D). The AICAR-induced alkalinization was also blocked by 1000 μm HOE694, which elicited a further decrease in steady-state pHi to pHi ∼ 6.6–6.7. Similarly, AA-treated neurons showed a comparable recruitment of NHE5 to the cell surface (Fig. 15C). However, unlike AICAR-treated neurons, AA-treated neurons showed an initial decrease in pHi over the first 15 min to pH ∼ 6.75, followed by a gradual recovery of pHi (pH ∼ 7.5) that exceeded steady-state pHi of control neurons (pH ∼ 7.2) (Fig. 15E). Significantly, the pHi recovery was effectively blocked by CC and HOE694 (1000 μm). These data mirror the results obtained in heterologous AP-1/NHE5 cells.
FIGURE 15.

Activation of AMPK stimulates the plasma membrane accumulation of native NHE5 and regulation of intracellular pH in cultured hippocampal neurons. Primary cultures of mouse hippocampal neurons (14–21 days in vitro) were grown on 12-mm coverslips and untreated (A) or treated with (B) AICAR (1 mm) or (C) AA (1 μg/ml) for 1 h prior to fixation and immunolabeling for endogenous NHE5 using a rabbit polyclonal isoform-specific NHE5 antibody. Images were obtained by confocal microscopy. The scale bar represents 10 μm. D–G, primary cultures of mouse hippocampal neurons were loaded for 15 min with the pH-sensitive fluorescent dye BCECF-AM (50 μm) in either 10 mm HEPES-buffered ACSF (D and E) or 22 mm sodium bicarbonate-buffered ACSF (F and G) perfused with 5% CO2 balanced with air. Intracellular pH was measured as described under ”Experimental Procedures.“ Intracellular pH in the somatodendritic region of cultured hippocampal neurons was monitored as a function of time following addition of (D and F) diluent, AICAR (1 mm), AICAR plus CC (20 μm) or AICAR plus HOE694 (1000 μm) and (E and G) diluent, AA (1 μg/ml), AA plus CC (20 μm), or AA plus HOE694 (1000 μm). The data represent the mean ± S.E. of 3–4 independent experiments, with at least 10 different cells per experiment.
To further evaluate the relationship between NHE5 and AMPK in hippocampal neurons under more physiological buffer conditions, we repeated the above measurements in neurons maintained in media that contains bicarbonate (22 mm NaHCO3 buffered ACSF equilibrated with 5% CO2, 95% air). As shown in Fig. 15, F and G, activation of AMPK by AICAR and AA, respectively, modulated pHi in HCO3−/CO2-buffered media in a manner qualitatively similar to that observed in HEPES-buffered media, with some quantitative differences. First, in HCO3−/CO2-buffered media, the resting pHi was noticeably higher (∼7.36 ± 0.07 versus 7.20 ± 0.08; p < 0.05, Student's paired t test), as might be expected. Second, detectable increases in pHi induced by AICAR in neurons maintained in HCO3−/CO2-buffered media were slightly delayed and the extent of the ΔpH was not as great compared with cells in HEPES-buffered media, although the final pHi attained after 60 min treatment was comparable (HCO3−/CO2: 7.60 ± 0.04 versus HEPES: 7.55 ± 0.08). Third, AA-induced acidification as well as the pHi recovery in neurons maintained in HCO3−/CO2-buffered media were also slightly delayed and not as robust as compared with cells in HEPES-buffered media within the time frame of the experiment. Notwithstanding, these effects were blocked by the AMPK antagonist CC and NHE5 antagonist HOE694 in HCO3−/CO2-buffered media. Collectively, these findings support a physiologically relevant relationship between native NHE5 and AMPK in regulating pHi in hippocampal neurons during acute cellular energy stress.
DISCUSSION
The milieu of the central and peripheral nervous systems undergoes significant fluctuations in pH in response to depolarizing stimuli or metabolic stress and must be effectively controlled to maintain neuronal excitability, function, and survival (1, 55, 56). This regulation is largely mediated by Na+/H+ exchangers alongside bicarbonate transporters and carbonic anhydrases (12). Na+/H+ exchangers constitute a diverse family of at least 11 members (13, 57), many of which are expressed in brain based on in situ mRNA analysis (58). However, with the exception of NHE1, their individual cellular/subcellular distributions and functional properties have not been extensively documented.
In this report, we focused on the NHE5 isoform because it is highly expressed in the central and peripheral nervous systems relative to other tissues (19, 20, 24). It is especially prevalent in hippocampal neurons that seemingly lack measureable NHE1 activity during early development (i.e. fetal and neonatal stages) (21, 53, 54), although NHE1 can be detected in hippocampal neurons isolated from more mature animals (51, 59). Unlike NHE1, which resides exclusively at the cell surface, the majority of NHE5 accumulates in a latent pool of intracellular vesicles with only a minor fraction active at the plasma membrane (23, 25). Here, we demonstrate that NHE5 forms a complex with the heterotrimeric AMPKα1- and α2-containing isozymes that serve as vital sensors and regulators of biochemical energy (i.e. ATP) levels at the cellular and organismal levels (27–29). Furthermore, purified liver AMPK enzyme robustly phosphorylates the cytoplasmic C terminus of NHE5 in vitro at two main putative AMPK phospho-recognition motifs, one of which (i.e. the proximal site -571LLREpSGpSGACL581-) conferred sensitivity to regulation by AMPK in intact cells. The significance, if any, of phosphorylation of the more distal site located between amino acids 849 and 859 is presently unknown.
In NHE5-transfected Chinese hamster ovary AP-1 cells, direct activation of endogenous AMPK by AICAR gradually increased NHE5 activity in a linear manner as a function of time and enhanced the rate of pHi recovery from an imposed intracellular acidification relative to untreated control cells. We also investigated the effects of indirect activation of AMPK by partially depleting cellular ATP levels (∼50%) with AA, which blocks mitochondrial oxidative phosphorylation and places a greater reliance on glycolysis, resulting in cytoplasmic acidification due to increased production of metabolic acids such as lactate. Under these conditions, AMPK also increased NHE5 activity, but in a more pronounced biphasic manner. The early phase coincided with AA-elicited acidification and is best accounted for by an increase in the transmembrane H+ gradient that drives the activity of existing plasmalemmal NHE5 transporters. The later phase of accelerated NHE5 activity correlated with elevated cell surface abundance of NHE5 and gradual restoration of pHi to near resting levels. Significantly, these effects were blocked by the AMPK antagonist CC. Moreover, mutations that block phosphorylation of the proximal AMPK recognition site prevented the AA-induced increase in NHE5 surface expression and activity. These findings complement earlier observations that AMPK can regulate membrane trafficking and surface levels of other solute carriers linked to energy and electrolyte homeostasis, including glucose transporters GLUT3 (60) and GLUT4 (61) and the Na+/K+-ATPase pump (62). Moreover, this interaction was relatively specific to the NHE5 isoform in so far as AMPKα1 or AMPKα2 oligomers did not form a complex with NHE1 and acute AICAR activation of AMPK did not regulate NHE1 activity in heterologous cell systems. An earlier study indicated that sustained AICAR stimulation of AMPK (i.e. 24 h) significantly enhanced NHE1 activity in HEK293 cells (44). However, the increased NHE1 activity appeared to be an indirect consequence of prolonged AMPK activation that correlated with increased NHE1 gene expression and protein abundance coupled with enhanced glucose uptake and excessive lactic acidosis.
In addition to heterologous systems, we demonstrated that activation of AMPK by AICAR or metabolic stress on NHE5 activity and membrane abundance were largely mimicked in primary cultures of neonatal mouse hippocampal neurons maintained in HEPES- or more physiological HCO3−/CO2-buffered media. The only notable differences between the two media conditions were higher resting pHi values and less robust changes in pHi in response to AICAR or following AA-induced acidification in HCO3−/CO2- versus HEPES-buffered media. This may be due in part to the higher buffering capacity of neurons maintained in HCO3−/CO2-buffered media, but may also reflect less recruitment of NHE5 to the cell surface upon activation of AMPK when the initial resting pHi is more alkaline. Although this remains to be tested, it is noteworthy that the catalytic activity of AMPK is sensitive to pH within the physiological range (i.e. pH 6.6–7.3), with lesser activity at more alkaline levels (63). Hence, pHi could be a determinant of AMPK activity and its ability to activate downstream targets such as NHE5. These data also suggest that Na+-dependent HCO3− transporters do not make major contributions to pHi regulation of primary cultures of neonatal mouse hippocampal neurons maintained under our experimental conditions, as their presence would have been anticipated to accelerate pHi recovery following acidification. These findings are consistent with earlier studies of hippocampal neurons that showed that pHi recovery from an imposed acid load is accomplished to a significant extent by an amiloride-insensitive NHE, although other NHEs (e.g. NHE1) and Na+-dependent bicarbonate transporters can make important contributions depending on the age of the animals, their preparation (i.e. freshly dissociated versus cultured), and temperature (ambient versus 37 °C) (21, 53, 54, 64, 65). For example, Baxter and Church (53) reported that acid extrusion in cultured fetal rat hippocampal neurons maintained at 37 °C involves primarily amiloride- or HOE694-insensitive Na+/H+ exchange, whereas at room temperature both Na+/H+ exchange and Na+-dependent HCO3−/Cl− exchange participate in the maintenance of steady-state pHi at room temperature. The basis for this temperature-dependent effect remains unclear. In contrast, in acutely dissociated CA1 hippocampal neurons from postnatal rats (4–14-days), Na+-dependent HCO3−/Cl− exchange appears to play a more prominent role in the maintenance of resting pHi and acid extrusion, whereas amiloride-insensitive Na+/H+ exchange makes a lesser contribution (66). However, the relative involvement of Na+-dependent HCO3−/Cl− exchange in pHi regulation during metabolic stress is unknown.
One potential caveat to the use of pharmacological agents such as AICAR and CC to manipulate AMPK activity is that they have been reported to have off-target effects that could potentially affect interpretation of the results (67–70). AICAR is an adenosine analogue that is carried into cells by the equilibrative nucleoside transporter and then phosphorylated to ZMP, an AMP analogue. In the CA1 region of rat hippocampus, AICAR has been found to activate not only AMPK but also to compete with extracellular adenosine for the nucleoside transporter (67). This results in accumulation of extracellular adenosine that activates adenosine A1 receptors, although AICAR per se does not, which in turn depresses excitatory synaptic transmission (67). Therefore, in principle, an AICAR-mediated reduction in synaptic activity might lead to elevated pHi not only through activation of an AMPK-NHE5 pathway but also by reducing energy consumption and the generation of metabolic acid. Likewise, the AMPK antagonist CC, which acts as a competitive inhibitor of ATP binding to AMPK, also inhibits the adenosine transporter (71) as well as other unrelated protein kinases, including ERK8, MNK1, PHK, MELK, DYRK, HIPK2, Src, and Lck (70). Hence, the ability of CC to block the effects of AICAR on the AMPK-NHE5 pathway in neurons might be due to reduced uptake of AICAR in addition to inhibiting AMPK activity, although the net effect would be the same. CC inhibition of other kinases might also impact this signaling pathway, although at present there is no evidence to implicate their involvement. We have attempted to minimize some of these concerns by activating AMPK by an alternate means independent of nucleoside uptake, such as by inducing metabolic stress with the mitochondrial inhibitor AA. This also activates NHE5, an effect that was blocked by CC. However, perhaps the most compelling evidence in support of an AMPK-NHE5 pathway are the mutations that abolished phosphorylation of the proximal AMPK recognition site in NHE5 and prevented AA-induced increase in NHE5 surface expression and activity in AP-1 cells. Future experiments that manipulate AMPK expression in neurons by gene knock-out or siRNA knockdown approaches should provide more definitive proof and are ongoing.
Our findings implicate a role for AMPK and NHE5 in protecting hippocampal neurons from acidosis during episodes of metabolic stress. Cellular insults such as hypoxia-ischemia decrease not only cellular ATP levels but also induce anaerobic glycolysis that produces a pronounced lactic acidosis followed by H+ extrusion and external acidification. In turn, this leads to excess accumulation of Nai+ (via the Na+/H+ exchanger) and Ca2+i (via the Na+/Ca2+ exchanger acting in reverse mode) (72–74), which if sufficiently severe, can lead to neuronal and glial cell death (9, 75–78). Hippocampal pyramidal neurons are especially susceptible to damage from hypoxic-ischemic episodes (79–82). However, mild acidosis during less severe energy stress or seizure has been proposed to have protective effects by inhibiting the conductances of various pH-sensitive neurotransmitter- and voltage-gated ion channels (2–4, 83–85), thereby limiting Ca2+ overload and damping membrane excitability associated with excitotoxicity, while at the same time permitting the gradual restoration of intra- and extracellular pH to resting values. With regard to the latter, Diarra et al. (86) demonstrated that exposure of cultured postnatal rat hippocampal neurons to acute chemical anoxia induced a triphasic change in pHi consisting of an initial fall, subsequent rise, and further increase upon the return to normoxia, and implicated NHE activity as a contributing factor in the post-anoxic restoration of pHi. Our measurements of pHi in metabolically stressed cultured neonatal mouse hippocampal neurons complement and extend these earlier findings by showing that the gradual pHi recovery following acidification is likely dependent, at least in part, on AMPK and the recruitment/retention of NHE5 at the cell surface.
Various depolarizing stimuli are also known to cause an influx in Ca2+ that is also accompanied by a significant decrease in pHi in hippocampal neurons as well as other nerve cells, followed by active extrusion of excess H+ over several minutes to restore pHi (87–93). More recently, Diering and colleagues (25) extended these observations by showing that NMDA receptor-induced neural activity initiates a modest acidification of dendritic spines (i.e. the postsynaptic compartment) of cultured rat fetal hippocampal neurons over a 5–10-min period, followed by a time-dependent alkalinization of spine pHi that reached a new steady-state level (i.e. pHi ∼ 7.6) significantly above resting values (i.e. pHi ∼ 7.2). Notably, this increase was dependent on the recruitment of NHE5 to the membrane surface of dendritic spines and was also found to suppress activity-dependent growth of spine density and size. It was proposed that NHE5-mediated alkalinization of the dendritic spine produces a concurrent acidification of the synaptic cleft, which may serve as an autocrine feedback mechanism to regulate pH-sensitive proteins on the pre- and post-synaptic membranes, such as voltage-gated Ca2+ channels and NMDA receptors, respectively. How activation of NMDA receptors stimulated this translocation process was not examined. However, it is tempting to speculate that this may involve activity-dependent activation of AMPK, as has recently been described in hippocampal neurons (94). Activity-driven increases in intracellular Ca2+ can activate AMPK indirectly by stimulating its upstream regulator CaMKKβ, which is highly abundant in brain (95). Moreover, a CaMKKβ/CaM-kinase I/βPIX signaling pathway has been implicated in the promotion of spine growth and synaptogenesis in hippocampal neurons (96). Hence, CaMKKβ may serve as a common regulatory hub that counterbalances the NMDA-induced spine growth promoting effects of the CaMKKβ/CaM-kinase I/βPIX signaling cascade versus the potential suppressive effects of a CaMKKβ/AMPK/NHE5 pathway.
In conclusion, we have demonstrated that AMPK directly binds and phosphorylates NHE5, which promotes its accumulation at the cell surface to regulate cytoplasmic pH. Overall, our data reveal a new signaling pathway for integrating pHi homeostasis with energy metabolism in hippocampal neurons and potentially other nerve cells in response to metabolic stressors such as hypoxia-ischemia, and may also play a role in regulating pHi during prolonged neural activity.
Acknowledgments
We thank Annie Boucher, Claire M. Brown, Emily C. Deane, Micaela Das Gupta, Khalid Hassan, Alina Ilie, Viktoria Lukashova, and Hans Zaun for technical assistance. Images were collected and/or image processing and analysis was performed in the McGill University Life Sciences Complex Advanced BioImaging Facility (ABIF). Purchase of equipment in the facility was made possible with funding from the Canadian Foundation for Innovation (CFI) and the Ministère du développement économique, innovation et exportation Québec (MDEIE). We also acknowledge the services provided by Genome Quebec.
This work was supported by Canadian Institutes of Health Research Grant MOP-11221 (to J. O.).
- NHE
- Na+/H+ exchanger
- AMPK
- AMP-activated protein kinase
- AICAR
- 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside
- EIPA
- ethylisopropylamiloride
- ChFP
- cherry fluorescent protein
- MEM
- minimal essential medium
- ACSF
- artificial cerebrospinal fluid
- BCECF-AM
- 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethylester
- ANOVA
- analysis of variance
- AA
- antimycin A
- CC
- compound C
- HOE694
- (3-Methanesulfonyl-4-piperidinobenzoyl)guanidine hydrochloride.
REFERENCES
- 1. Chesler M., Kaila K. (1992) Modulation of pH by neuronal activity. Trends Neurosci. 15, 396–402 [DOI] [PubMed] [Google Scholar]
- 2. Traynelis S. F., Cull-Candy S. G. (1991) Pharmacological properties and H+ sensitivity of excitatory amino acid receptor channels in rat cerebellar granule neurones. J. Physiol. 433, 727–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Tombaugh G. C., Somjen G. G. (1996) Effects of extracellular pH on voltage-gated Na+, K+ and Ca2+ currents in isolated rat CA1 neurons. J. Physiol. 493, 719–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Church J., Baxter K. A., McLarnon J. G. (1998) pH modulation of Ca2+ responses and a Ca2+-dependent K+ channel in cultured rat hippocampal neurones. J. Physiol. 511, 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Waldmann R., Lazdunski M. (1998) H+-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr. Opin. Neurobiol. 8, 418–424 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi K. I., Copenhagen D. R. (1996) Modulation of neuronal function by intracellular pH. Neurosci. Res. 24, 109–116 [DOI] [PubMed] [Google Scholar]
- 7. Velísek L. (1998) Extracellular acidosis and high levels of carbon dioxide suppress synaptic transmission and prevent the induction of long-term potentiation in the CA1 region of rat hippocampal slices. Hippocampus 8, 24–32 [DOI] [PubMed] [Google Scholar]
- 8. DeVries S. H. (2001) Exocytosed protons feedback to suppress the Ca2+ current in mammalian cone photoreceptors. Neuron 32, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 9. Lipton P. (1999) Ischemic cell death in brain neurons. Physiol. Rev. 79, 1431–1568 [DOI] [PubMed] [Google Scholar]
- 10. Siesjö B. K., von Hanwehr R., Nergelius G., Nevander G., Ingvar M. (1985) Extra- and intracellular pH in the brain during seizures and in the recovery period following the arrest of seizure activity. J. Cereb. Blood Flow Metab. 5, 47–57 [DOI] [PubMed] [Google Scholar]
- 11. Xiong Z. Q., Saggau P., Stringer J. L. (2000) Activity-dependent intracellular acidification correlates with the duration of seizure activity. J. Neurosci. 20, 1290–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chesler M. (2003) Regulation and modulation of pH in the brain. Physiol. Rev. 83, 1183–1221 [DOI] [PubMed] [Google Scholar]
- 13. Casey J. R., Grinstein S., Orlowski J. (2010) Sensors and regulators of intracellular pH. Nat. Rev. Mol. Cell Biol. 11, 50–61 [DOI] [PubMed] [Google Scholar]
- 14. Jang I. S., Brodwick M. S., Wang Z. M., Jeong H. J., Choi B. J., Akaike N. (2006) The Na+/H+ exchanger is a major pH regulator in GABAergic presynaptic nerve terminals synapsing onto rat CA3 pyramidal neurons. J. Neurochem. 99, 1224–1236 [DOI] [PubMed] [Google Scholar]
- 15. Dietrich C. J., Morad M. (2010) Synaptic acidification enhances GABAA signaling. J. Neurosci. 30, 16044–16052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cox G. A., Lutz C. M., Yang C. L., Biemesderfer D., Bronson R. T., Fu A., Aronson P. S., Noebels J. L., Frankel W. N. (1997) Sodium/hydrogen exchanger gene defect in slow-wave epilepsy mutant mice. Cell 91, 139–148 [DOI] [PubMed] [Google Scholar]
- 17. Bell S. M., Schreiner C. M., Schultheis P. J., Miller M. L., Evans R. L., Vorhees C. V., Shull G. E., Scott W. J. (1999) Targeted disruption of the murine Nhe1 locus induces ataxia, growth retardation, and seizures. Am. J. Physiol. 276, C788–C795 [DOI] [PubMed] [Google Scholar]
- 18. Liu Y., Zaun H. C., Orlowski J., Ackerman S. L. (2013) CHP1-mediated NHE1 biosynthetic maturation is required for Purkinje cell axon homeostasis. J. Neurosci. 33, 12656–12669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baird N. R., Orlowski J., Szabó E. Z., Zaun H. C., Schultheis P. J., Menon A. G., Shull G. E. (1999) Molecular cloning, genomic organization, and functional expression of Na+/H+ exchanger isoform 5 (NHE5) from human brain. J. Biol. Chem. 274, 4377–4382 [DOI] [PubMed] [Google Scholar]
- 20. Attaphitaya S., Park K., Melvin J. E. (1999) Molecular cloning and functional expression of a rat Na+/H+ exchanger (NHE5) highly expressed in brain. J. Biol. Chem. 274, 4383–4388 [DOI] [PubMed] [Google Scholar]
- 21. Raley-Susman K. M., Cragoe E. J., Jr., Sapolsky R. M., Kopito R. R. (1991) Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. J. Biol. Chem. 266, 2739–2745 [PubMed] [Google Scholar]
- 22. Szabó E. Z., Numata M., Shull G. E., Orlowski J. (2000) Kinetic and pharmacological properties of human brain Na+/H+ exchanger isoform 5 stably expressed in Chinese hamster ovary cells. J. Biol. Chem. 275, 6302–6307 [DOI] [PubMed] [Google Scholar]
- 23. Szaszi K., Paulsen A., Szabo E. Z., Numata M., Grinstein S., Orlowski J. (2002) Clathrin-mediated endocytosis and recycling of the neuron-specific Na+/H+ exchanger NHE5 isoform: regulation by phosphatidylinositol 3′-kinase and the actin cytoskeleton. J. Biol. Chem. 277, 42623–42632 [DOI] [PubMed] [Google Scholar]
- 24. Lukashova V., Jinadasa T., Ilie A., Verbich D., Cooper E., Orlowski J. (2013) The Na+/H+ exchanger NHE5 is sorted to discrete intracellular vesicles in the central and peripheral nervous systems. Adv. Exp. Med. Biol. 961, 397–410 [DOI] [PubMed] [Google Scholar]
- 25. Diering G. H., Mills F., Bamji S. X., Numata M. (2011) Regulation of dendritic spine growth through activity-dependent recruitment of brain-enriched Na+/H+ exchanger NHE5. Mol. Biol. Cell 22, 2246–2257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Spasi M. R., Callaerts P., Norga K. K. (2009) AMP-activated protein kinase (AMPK) molecular crossroad for metabolic control and survival of neurons. Neuroscientist. 15, 309–316 [DOI] [PubMed] [Google Scholar]
- 27. Carling D., Mayer F. V., Sanders M. J., Gamblin S. J. (2011) AMP-activated protein kinase: nature's energy sensor. Nat. Chem. Biol. 7, 512–518 [DOI] [PubMed] [Google Scholar]
- 28. Hardie D. G., Ross F. A., Hawley S. A. (2012) AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, 251–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Steinberg G. R., Kemp B. E. (2009) AMPK in health and disease. Physiol. Rev. 89, 1025–1078 [DOI] [PubMed] [Google Scholar]
- 30. Turnley A. M., Stapleton D., Mann R. J., Witters L. A., Kemp B. E., Bartlett P. F. (1999) Cellular distribution and developmental expression of AMP-activated protein kinase isoforms in mouse central nervous system. J. Neurochem. 72, 1707–1716 [DOI] [PubMed] [Google Scholar]
- 31. Orlowski J. (1993) Heterologous expression and functional properties of the amiloride high affinity (NHE-1) and low affinity (NHE-3) isoforms of the rat Na/H exchanger. J. Biol. Chem. 268, 16369–16377 [PubMed] [Google Scholar]
- 32. Rotin D., Grinstein S. (1989) Impaired cell volume regulation in Na+-H+ exchange-deficient mutants. Am. J. Physiol. 257, C1158–C1165 [DOI] [PubMed] [Google Scholar]
- 33. Brewer G. J., Torricelli J. R. (2007) Isolation and culture of adult neurons and neurospheres. Nat. Protoc. 2, 1490–1498 [DOI] [PubMed] [Google Scholar]
- 34. Broussard J. A., Rappaz B., Webb D. J., Brown C. M. (2013) Fluorescence resonance energy transfer microscopy as demonstrated by measuring the activation of the serine/threonine kinase Akt. Nat. Protoc. 8, 265–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thomas J. A., Buchsbaum R. N., Zimniak A., Racker E. (1979) Intracellular pH measurements in Ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 18, 2210–2218 [DOI] [PubMed] [Google Scholar]
- 36. Aharonovitz O., Zaun H. C., Balla T., York J. D., Orlowski J., Grinstein S. (2000) Intracellular pH regulation by Na+/H+ exchange requires phosphatidylinositol 4,5-bisphosphate. J. Cell Biol. 150, 213–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deane E. C., Ilie A. E., Sizdahkhani S., Das Gupta M., Orlowski J., McKinney R. A. (2013) Enhanced recruitment of endosomal Na+/H+ exchanger NHE6 into dendritic spines of hippocampal pyramidal neurons during NMDA receptor-dependent long-term potentiation. J. Neurosci. 33, 595–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Le Bivic A., Real F. X., Rodriguez-Boulan E. (1989) Vectorial targeting of apical and basolateral plasma membrane proteins in a human adenocarcinoma epithelial cell line. Proc. Natl. Acad. Sci. U.S.A. 86, 9313–9317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Szabó E. Z., Numata M., Lukashova V., Iannuzzi P., Orlowski J. (2005) β-Arrestins bind and decrease cell-surface abundance of the Na+/H+ exchanger NHE5 isoform. Proc. Natl. Acad. Sci. U.S.A. 102, 2790–2795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chien C.-T., Bartel P. L., Sternglanz R., Fields S. (1991) The two-hybrid system: a method to identify and clone genes for proteins that interact with a protein of interest. Proc. Natl. Acad. Sci. U.S.A. 88, 9578–9582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hardie D. G. (2011) AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 25, 1895–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salt I., Celler J. W., Hawley S. A., Prescott A., Woods A., Carling D., Hardie D. G. (1998) AMP-activated protein kinase: greater AMP dependence, and preferential nuclear localization, of complexes containing the α2 isoform. Biochem. J. 334, 177–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Day R. N., Davidson M. W. (2012) Fluorescent proteins for FRET microscopy: monitoring protein interactions in living cells. Bioessays 34, 341–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rotte A., Pasham V., Eichenmüller M., Bhandaru M., Föller M., Lang F. (2010) Upregulation of Na+/H+ exchanger by the AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 398, 677–682 [DOI] [PubMed] [Google Scholar]
- 45. Boron W. F., De Weer P. (1976) Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. J. Gen. Physiol. 67, 91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Witters L. A., Nordlund A. C., Marshall L. (1991) Regulation of intracellular acetyl-CoA carboxylase by ATP depletors mimics the action of the 5′-AMP-activated protein kinase. Biochem. Biophys. Res. Commun. 181, 1486–1492 [DOI] [PubMed] [Google Scholar]
- 47. Wick A. N., Drury D. R., Nakada H. I., Wolfe J. B. (1957) Localization of the primary metabolic block produced by 2-deoxyglucose. J. Biol. Chem. 224, 963–969 [PubMed] [Google Scholar]
- 48. Kim M. S., Lee K. U. (2005) Role of hypothalamic 5′-AMP-activated protein kinase in the regulation of food intake and energy homeostasis. J. Mol. Med. 83, 514–520 [DOI] [PubMed] [Google Scholar]
- 49. Handa N., Takagi T., Saijo S., Kishishita S., Takaya D., Toyama M., Terada T., Shirouzu M., Suzuki A., Lee S., Yamauchi T., Okada-Iwabu M., Iwabu M., Kadowaki T., Minokoshi Y., Yokoyama S. (2011) Structural basis for compound C inhibition of the human AMP-activated protein kinase alpha2 subunit kinase domain. Acta Crystallogr. D Biol. Crystallogr. 67, 480–487 [DOI] [PubMed] [Google Scholar]
- 50. Potter W. B., O'Riordan K. J., Barnett D., Osting S. M., Wagoner M., Burger C., Roopra A. (2010) Metabolic regulation of neuronal plasticity by the energy sensor AMPK. PLoS ONE 5, e8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yao H., Ma E., Gu X. Q., Haddad G. G. (1999) Intracellular pH regulation of CA1 neurons in Na+/H+ isoform 1 mutant mice. J. Clin. Invest. 104, 637–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pizzonia J. H., Ransom B. R., Pappas C. A. (1996) Characterization of Na+/H+ exchange activity in cultured rat hippocampal astrocytes. J. Neurosci. Res. 44, 191–198 [DOI] [PubMed] [Google Scholar]
- 53. Baxter K. A., Church J. (1996) Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. J. Physiol. 493, 457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Bevensee M. O., Cummins T. R., Haddad G. G., Boron W. F., Boyarsky G. (1996) pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. J. Physiol. 494, 315–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Putnam R. W., Filosa J. A., Ritucci N. A. (2004) Cellular mechanisms involved in CO2 and acid signaling in chemosensitive neurons. Am. J. Physiol. Cell Physiol. 287, C1493–C1526 [DOI] [PubMed] [Google Scholar]
- 56. Rossi D. J., Brady J. D., Mohr C. (2007) Astrocyte metabolism and signaling during brain ischemia. Nat. Neurosci. 10, 1377–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Brett C. L., Donowitz M., Rao R. (2005) Evolutionary origins of eukaryotic sodium/proton exchangers. Am. J. Physiol. Cell Physiol. 288, C223–C239 [DOI] [PubMed] [Google Scholar]
- 58. Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T. M., Chin M. C., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H. W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K. R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R. (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 59. Ma E., Haddad G. G. (1997) Expression and localization of Na+/H+ exchangers in rat central nervous system. Neuroscience 79, 591–603 [DOI] [PubMed] [Google Scholar]
- 60. Weisová P., Concannon C. G., Devocelle M., Prehn J. H., Ward M. W. (2009) Regulation of glucose transporter 3 surface expression by the AMP-activated protein kinase mediates tolerance to glutamate excitation in neurons. J. Neurosci. 29, 2997–3008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kurth-Kraczek E. J., Hirshman M. F., Goodyear L. J., Winder W. W. (1999) 5′ AMP-activated protein kinase activation causes GLUT4 translocation in skeletal muscle. Diabetes 48, 1667–1671 [DOI] [PubMed] [Google Scholar]
- 62. Benziane B., Björnholm M., Pirkmajer S., Austin R. L., Kotova O., Viollet B., Zierath J. R., Chibalin A. V. (2012) Activation of AMP-activated protein kinase stimulates Na+,K+-ATPase activity in skeletal muscle cells. J. Biol. Chem. 287, 23451–23463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ponticos M., Lu Q. L., Morgan J. E., Hardie D. G., Partridge T. A., Carling D. (1998) Dual regulation of the AMP-activated protein kinase provides a novel mechanism for the control of creatine kinase in skeletal muscle. EMBO J. 17, 1688–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Raley-Susman K. M., Sapolsky R. M., Kopito R. R. (1993) Cl−/HCO3− exchange function differs in adult and fetal rat hippocampal neurons. Brain Res. 614, 308–314 [DOI] [PubMed] [Google Scholar]
- 65. Chen L. M., Kelly M. L., Parker M. D., Bouyer P., Gill H. S., Felie J. M., Davis B. A., Boron W. F. (2008) Expression and localization of Na-driven Cl-HCO3− exchanger (SLC4A8) in rodent CNS. Neuroscience 153, 162–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schwiening C. J., Boron W. F. (1994) Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl−-HCO3− exchange. J. Physiol. 475, 59–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gadalla A. E., Pearson T., Currie A. J., Dale N., Hawley S. A., Sheehan M., Hirst W., Michel A. D., Randall A., Hardie D. G., Frenguelli B. G. (2004) AICA riboside both activates AMP-activated protein kinase and competes with adenosine for the nucleoside transporter in the CA1 region of the rat hippocampus. J. Neurochem. 88, 1272–1282 [DOI] [PubMed] [Google Scholar]
- 68. Fryer L. G., Parbu-Patel A., Carling D. (2002) Protein kinase inhibitors block the stimulation of the AMP-activated protein kinase by 5-amino-4-imidazolecarboxamide riboside. FEBS Lett. 531, 189–192 [DOI] [PubMed] [Google Scholar]
- 69. Emerling B. M., Viollet B., Tormos K. V., Chandel N. S. (2007) Compound C inhibits hypoxic activation of HIF-1 independent of AMPK. FEBS Lett. 581, 5727–5731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bain J., Plater L., Elliott M., Shpiro N., Hastie C. J., McLauchlan H., Klevernic I., Arthur J. S., Alessi D. R., Cohen P. (2007) The selectivity of protein kinase inhibitors: a further update. Biochem. J. 408, 297–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fryer L. G., Parbu-Patel A., Carling D. (2002) The anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J. Biol. Chem. 277, 25226–25232 [DOI] [PubMed] [Google Scholar]
- 72. Stys P. K., Waxman S. G., Ransom B. R. (1991) Reverse operation of the Na+-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian CNS white matter. Ann. N.Y. Acad. Sci. 639, 328–332 [DOI] [PubMed] [Google Scholar]
- 73. Matsuda T., Takuma K., Nishiguchi E., Hashimoto H., Azuma J., Baba A. (1996) Involvement of Na+-Ca2+ exchanger in reperfusion-induced delayed cell death of cultured rat astrocytes. Eur. J. Neurosci. 8, 951–958 [DOI] [PubMed] [Google Scholar]
- 74. Kintner D. B., Chen X., Currie J., Chanana V., Ferrazzano P., Baba A., Matsuda T., Cohen M., Orlowski J., Chiu S. Y., Taunton J., Sun D. (2010) Excessive Na+/H+ exchange in disruption of dendritic Na+ and Ca2+ homeostasis and mitochondrial dysfunction following in vitro ischemia. J. Biol. Chem. 285, 35155–35168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nedergaard M., Goldman S. A., Desai S., Pulsinelli W. A. (1991) Acid-induced death in neurons and glia. J. Neurosci. 11, 2489–2497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Nedergaard M., Kraig R. P., Tanabe J., Pulsinelli W. A. (1991) Dynamics of interstitial and intracellular pH in evolving brain infarct. Am. J. Physiol. 260, R581–R588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Silver I. A., Deas J., Erecińska M. (1997) Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 78, 589–601 [DOI] [PubMed] [Google Scholar]
- 78. Ying W., Han S. K., Miller J. W., Swanson R. A. (1999) Acidosis potentiates oxidative neuronal death by multiple mechanisms. J. Neurochem. 73, 1549–1556 [DOI] [PubMed] [Google Scholar]
- 79. Simon R. P., Griffiths T., Evans M. C., Swan J. H., Meldrum B. S. (1984) Calcium overload in selectively vulnerable neurons of the hippocampus during and after ischemia: an electron microscopy study in the rat. J. Cereb. Blood Flow Metab. 4, 350–361 [DOI] [PubMed] [Google Scholar]
- 80. Xiang Z., Bergold P. J. (2000) Synaptic depression and neuronal loss in transiently acidic hippocampal slice cultures. Brain Res. 881, 77–87 [DOI] [PubMed] [Google Scholar]
- 81. Ding D., Moskowitz S. I., Li R., Lee S. B., Esteban M., Tomaselli K., Chan J., Bergold P. J. (2000) Acidosis induces necrosis and apoptosis of cultured hippocampal neurons. Exp. Neurol. 162, 1–12 [DOI] [PubMed] [Google Scholar]
- 82. Liu C. L., Siesjö B. K., Hu B. R. (2004) Pathogenesis of hippocampal neuronal death after hypoxia-ischemia changes during brain development. Neuroscience 127, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Tombaugh G. C., Sapolsky R. M. (1990) Mild acidosis protects hippocampal neurons from injury induced by oxygen and glucose deprivation. Brain Res. 506, 343–345 [DOI] [PubMed] [Google Scholar]
- 84. Saybasili H. (1998) The protective role of mild acidic pH shifts on synaptic NMDA current in hippocampal slices. Brain Res. 786, 128–132 [DOI] [PubMed] [Google Scholar]
- 85. Tang C. M., Dichter M., Morad M. (1990) Modulation of the N-methyl-d-aspartate channel by extracellular H+. Proc. Natl. Acad. Sci. U.S.A. 87, 6445–6449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Diarra A., Sheldon C., Brett C. L., Baimbridge K. G., Church J. (1999) Anoxia-evoked intracellular pH and Ca2+ concentration changes in cultured postnatal rat hippocampal neurons. Neuroscience 93, 1003–1016 [DOI] [PubMed] [Google Scholar]
- 87. Amato A., Ballerini L., Attwell D. (1994) Intracellular pH changes produced by glutamate uptake in rat hippocampal slices. J. Neurophysiol. 72, 1686–1696 [DOI] [PubMed] [Google Scholar]
- 88. Irwin R. P., Lin S. Z., Long R. T., Paul S. M. (1994) N-Methyl-d-aspartate induces a rapid, reversible, and calcium-dependent intracellular acidosis in cultured fetal rat hippocampal neurons. J. Neurosci. 14, 1352–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zhan R. Z., Fujiwara N., Tanaka E., Shimoji K. (1998) Intracellular acidification induced by membrane depolarization in rat hippocampal slices: roles of intracellular Ca2+ and glycolysis. Brain Res. 780, 86–94 [DOI] [PubMed] [Google Scholar]
- 90. Endres W., Ballanyi K., Serve G., Grafe P. (1986) Excitatory amino acids and intracellular pH in motoneurons of the isolated frog spinal cord. Neurosci. Lett. 72, 54–58 [DOI] [PubMed] [Google Scholar]
- 91. Trapp S., Lückermann M., Brooks P. A., Ballanyi K. (1996) Acidosis of rat dorsal vagal neurons in situ during spontaneous and evoked activity. J. Physiol. 496, 695–710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Lückermann M., Trapp S., Ballanyi K. (1997) GABA- and glycine-mediated fall of intracellular pH in rat medullary neurons in situ. J. Neurophysiol. 77, 1844–1852 [DOI] [PubMed] [Google Scholar]
- 93. Amos B. J., Mathie A., Richards C. D. (1998) Activation of group I metabotropic glutamate receptors elicits pH changes in cultured rat cortical glia and neurons. Neuroscience 86, 1109–1120 [DOI] [PubMed] [Google Scholar]
- 94. Terunuma M., Vargas K. J., Wilkins M. E., Ramírez O. A., Jaureguiberry-Bravo M., Pangalos M. N., Smart T. G., Moss S. J., Couve A. (2010) Prolonged activation of NMDA receptors promotes dephosphorylation and alters postendocytic sorting of GABAB receptors. Proc. Natl. Acad. Sci. U.S.A. 107, 13918–13923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Woods A., Dickerson K., Heath R., Hong S. P., Momcilovic M., Johnstone S. R., Carlson M., Carling D. (2005) Ca2+/calmodulin-dependent protein kinase kinase-β acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2, 21–33 [DOI] [PubMed] [Google Scholar]
- 96. Saneyoshi T., Wayman G., Fortin D., Davare M., Hoshi N., Nozaki N., Natsume T., Soderling T. R. (2008) Activity-dependent synaptogenesis: regulation by a CaM-kinase kinase/CaM-kinase I/βPIX signaling complex. Neuron 57, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Gietz R. D., Schiestl R. H., Willems A. R., Woods R. A. (1995) Studies on the transformation of intact yeast cells by the LiAc/SS-DNA/PEG procedure. Yeast 11, 355–360 [DOI] [PubMed] [Google Scholar]