

Background: Eukaryotic elongation factor 1A (eEF1A) interacts with the actin cytoskeleton.
Results: Mutations in eEF1A that cause actin disorganization affect translation at both the initiation and elongation steps.
Conclusion: Reduced aminoacyl-tRNA binding by eEF1A and increased eukaryotic initiation factor 2α (eIF2α) phosphorylation are connected through Gcn2p.
Significance: Alternative eEF1A functions can affect the interplay between translation steps and other cellular processes.
Keywords: Actin, Protein Synthesis, Transfer RNA (tRNA), Translation Elongation Factor, Translation Initiation Factor, Gcn2 Kinase
Abstract
Apart from its canonical function in translation elongation, eukaryotic translation elongation factor 1A (eEF1A) has been shown to interact with the actin cytoskeleton. Amino acid substitutions in eEF1A that reduce its ability to bind and bundle actin in vitro cause improper actin organization in vivo and reduce total translation. Initial in vivo analysis indicated the reduced translation was through initiation. The mutant strains exhibit increased levels of phosphorylated initiation factor 2α (eIF2α) dependent on the presence of the general control nonderepressible 2 (Gcn2p) protein kinase. Gcn2p causes down-regulation of total protein synthesis at initiation in response to increases in deacylated tRNA levels in the cell. Increased levels of eIF2α phosphorylation are not due to a general reduction in translation elongation as eEF2 and eEF3 mutants do not exhibit this effect. Deletion of GCN2 from the eEF1A actin bundling mutant strains revealed a second defect in translation. The eEF1A actin-bundling proteins exhibit changes in their elongation activity at the level of aminoacyl-tRNA binding in vitro. These findings implicate eEF1A in a feedback mechanism for regulating translation at initiation.
Introduction
Yeast cells use the hierarchy of monomer-based structures formed by the actin cytoskeleton to facilitate transport of numerous cellular components. Organization of filamentous actin (F-actin) produces cortical spots (patches) and long fibers (cables), the two most recognizable actin-based structures (1). Together these structures provide the structural network required for maintenance of cell morphology, polarization, and growth. Proteins that can bundle, cross-link, and/or stabilize actin filaments regulate the dynamics of actin cytoskeletal reorganization (2). Changes in the organization of the actin cytoskeleton in response to environmental stresses such as glucose deprivation (3, 4) and osmotic stress (5) have been documented. Regulation of protein synthesis also occurs under similar stresses (6, 7), indicating that a functional link likely exists between the actin cytoskeleton and translation. The link is strengthened by reports of components of the translational machinery such as aminoacyl-tRNA synthetases (8), initiation and elongation factors (9, 10) being associated with the actin cytoskeleton.
During the elongation phase of translation, aminoacyl-tRNAs (aa-tRNAs)2 are bound and delivered to the A-site of the ribosome by eEF1A in its GTP-bound form. When the correct match between the anticodon stem of the cognate-tRNA and the mRNA codon is formed, a conformational change in the ribosome occurs that induces GTP hydrolysis and release of eEF1A·GDP (11). Reactivation of eEF1A·GDP is stimulated by the guanine nucleotide exchange factor complex eEF1Bαβγ (eEF1Bαγ in yeast). Aside from its canonical function in translation elongation, eEF1A has been shown to play a role in actin binding and bundling. Since the discovery of eEF1A as an actin-binding protein (12), an extensive number of in vitro studies have been done to characterize the interaction. Competitive binding experiments on eEF1A with F-actin and aa-tRNA showed that binding is mutually exclusive and pH-dependent (13). eEF1A has two pH-sensitive actin-binding domains that could be important for modulating the cellular response to external stimuli by allowing the reorganization of the actin cytoskeleton and the associated translational machinery (13). These two actin-binding sites were further defined to be the first 49 residues of the N terminus and residues 403–456 at the C terminus of Dictyostelium discoideum eEF1A, with domain III containing higher actin binding activity (14). Because eEF1A interacts with Bni1p/She5p, a downstream target of Rho1p that can regulate actin reorganization (15), it is possible that eEF1A acts as a bridge between the cytoskeleton and actin modulators.
Overexpression of eEF1A affects the organization of the actin cytoskeleton (16). Mutational analysis of eEF1A revealed two classes of mutants that suppressed the overexpression phenotype. The first class, characterized by the eEF1A-Ura3p N305S and N329S mutations, displayed severe defects in the organization of the actin cytoskeleton yet demonstrated normal total translation (17). However, the second class of mutants demonstrated cytoskeletal defects and reduced total translation. Surprisingly, polyribosome profile analysis of eEF1A-Ura3p F308L and S405P mutant strains revealed an accumulation of ribosomes in the 80 S peak, indicating reduced translation was through a block at the initiation step (18). However, given the effect on growth seen in the wild-type eEF1A-Ura3p fusion strain (17), it remains unclear if translation elongation activity of the fusion protein is also affected.
One of the major regulatory mechanisms for translational control is phosphorylation of the α subunit of initiation factor 2 (eIF2α) at serine 51 (19). GTP-bound eIF2 is responsible for delivering the initiator methionyl-tRNA (Met-tRNAi) as part of the 43 S subunit complex, allowing for initiation codon recognition and subsequent assembly of the fully active 80 S ribosome at the start codon (20). Four types of protein kinases have been identified that phosphorylate eIF2α depending on the stress condition. Heme deficiency, endoplasmic reticulum (ER) stress, virus infection, and nutrient starvation activate the eIF2α kinases heme-regulated inhibitor of translation, protein kinase RNA-like endoplasmic reticulum kinase, dsRNA-activated protein kinase, and Gcn2p (general control nonderepressible 2), respectively (21). Of these, Gcn2p is the only known eIF2α kinase identified in yeast. Although different stresses have been shown to activate Gcn2p, the predominant signal for full Gcn2p activation is accumulation of uncharged tRNAs as a result of starvation (22, 23).
Utilizing the eEF1A-Ura3p F308L and S405P actin bundling mutant strains, we investigated the mechanism behind the eEF1A-directed translation initiation defect. Our results indicate that eEF1A-Ura3p F308L and S405P mutant strains have increased levels of eIF2α phosphorylation. The specificity of eIF2α phosphorylation, the effects of deleting GCN2 in the eEF1A actin bundling mutant strains, and analysis of the elongation activities independent of initiation demonstrate that eEF1A mutants that alter the actin cytoskeleton affect initiation by eIF2α phosphorylation but also have underlying elongation defects, indicating the link between these two functions of eEF1A.
EXPERIMENTAL PROCEDURES
Yeast Techniques and Mutant Preparation
Saccharomyces cerevisiae strains used in this study are listed in Table 1. The disruption of GCN2 (YDR283C) in strains TKY1585, TKY1586, TKY1587, and TKY1588 was obtained by PCR of genomic DNA from the Open Biosystem GCN2 gene deletion collection strain (Open Biosystem, AL) using primers 400 nucleotides 5′ and 3′ of the open reading frame and transformation of the PCR fragment using the Frozen-EZ Yeast Transformation II kit (Zymo Research, CA). Cells in which in vivo recombination had occurred were selected on medium containing 200 μg/ml of G418 sulfate. Plasmid pTKB731 (TEF1 TRP1 2μ) served as a template to generate putative eEF1A tRNA binding mutants using the QuikChange Site-directed Mutagenesis kit (Stratagene) as per the manufacturer's recommendations. To obtain strains TKY1717, TKY1723, TKY1724, TKY1727, TKY1735, and TKY1740, strain TKY895 was initially transformed with pTKB907 (TEF1 URA3) using the lithium acetate method (24) and selected on media containing 5-fluoroanthranilic acid for loss of TEF1 TRP1 to obtain TKY1712. TKY1712 was transformed with pTKB929 (wild-type eEF1A), pTKB1207 (S18A), pTKB1208 (S18D), pTKB1220 (T430C), pTKB1221 (L77H), and pTKB1227 (E291A) and loss of TEF1 URA3 was monitored by growth on 5-fluoroorotic acid. Yeast cells were grown in either yeast extract-peptone-dextrose (YEPD; 1% Bacto-yeast extract, 2% Bacto-tryptone, 2% dextrose) or defined synthetic complete medium (C) supplemented with 2% dextrose as a carbon source. Growth assays were performed by streaking cells onto YEPD plates and incubating at 30 °C for 2–3 days.
TABLE 1.
S. cerevisiae strains used in this study
| Strain | Genotype | Ref. |
|---|---|---|
| MC214 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 met2–1 his4–713 tef1::LEU2 tef2Δ pTEF2 TRP1 | 48 |
| TKY225 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 met2–1 his4–713 tef1::LEU2 tef2Δ ptef2–17 TRP1 (D156N) | 49 |
| TKY226 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 met2–1 his4–713 tef1::LEU2 tef2Δ ptef2–19 TRP1 (N153T) | 49 |
| TKY675 | MATa ade2 leu2 ura3 his3 leu2 trp1 eft1::HIS3 eft2::TRP1 pEFT2-His6 LEU2 CEN | 50 |
| TKY825 | MATa ade2 leu2 ura3 his3 leu2 trp1 eft1::HIS3 eft2::TRP1 pEFT2-His6 D696A LEU2 CEN | 29 |
| TKY895 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 | 17 |
| TKY896 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1-URA3 | 17 |
| TKY901 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1-URA3 TRP1 (F308L) | 18 |
| TKY902 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1-URA3 TRP1 (N305S) | 17 |
| TKY903 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1-URA3 TRP1 (S405P) | 18 |
| TKY904 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1-URA3 TRP1 (N329S) | 17 |
| TKY1585 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ gcn2Δ::kanMX6 pTEF1 TRP1 | This study |
| TKY1586 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ gcn2Δ::kanMX6 pTEF1 TRP1-URA3 | This study |
| TKY1587 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ gcn2Δ::kanMX6 pTEF1-URA3 TRP1 (F308L) | This study |
| TKY1588 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ gcn2Δ::kanMX6 pTEF1-URA3 TRP1 (S405P) | This study |
| TKY1653 | MATα leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 yef3::HIS3 His6 pYEF3 TRP1 | 30 |
| TKY1655 | MATα leu2–3,112 trp1–1 can1–100 ura3–1 ade2–1 his3–11,15 yef3::HIS3 His6 pYEF3 TRP1 (R803A F806A) | 30 |
| TKY1717 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 CEN | This study |
| TKY1723 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 (L77H) | This study |
| TKY1724 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 (E291A) | This study |
| TKY1727 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 (T430C) | This study |
| TKY1735 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 CEN (S18A) | This study |
| TKY1740 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 CEN (S18D) | This study |
| TKY1744 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 (F308L) | This study |
| TKY1746 | MATα ura3–52 leu2–3,112 trp1-Δ1 lys2–20 MET2 his4–713 tef1::LEU2 tef2Δ pTEF1 TRP1 (N305S) | This study |
Global Translation Assays
Total translation was monitored by in vivo [35S]methionine incorporation as previously described (17). Briefly, liquid cultures (100 ml) were grown in C-Met at 30 °C to an A600 of 0.5–0.7, followed by addition of 50 mm unlabeled methionine and [35S]methionine to a final concentration of 1 mCi/ml. Samples were taken in triplicate at 15-min intervals for optical density (A600) determination and trichloroacetic acid (TCA) precipitation to monitor methionine incorporation.
Actin Phalloidin Staining
Yeast cultures were grown in YEPD to an A600 of 0.5–0.7 and fixed by adding formaldehyde and Triton X-100 to a final concentration of 4.4% (v/v) and 0.04% (v/v), respectively, and incubated for 1 h at room temperature. Cells were centrifuged, washed once with wash buffer (1× PBS, 1 mg/ml of bovine serum albumin, and 0.1% Triton X-100), and resuspended in wash buffer containing Alexa Fluor® 594 Phalloidin (Invitrogen) at a final concentration of 0.6 μm. Samples were vortexed at room temperature for 45 min in the dark. Samples were centrifuged, washed two times with wash buffer lacking Triton X-100, and resuspended in 1× mounting medium (90% (v/v) glycerol, 0.1% (v/v) PBS, 92 mm p-phenylmediane). Images were captured with an IX70 inverted fluorescence microscope (Olympus) equipped with a HiQ fluorescein filter set (excitation wavelength 450–492 nm), a Planapochromatic 100× oil immersion objective lens, and a 100-W mercury lamp. Images were collected and analyzed with a Princeton Instruments 5-MHz MicroMax cooled CCD camera, a shutter and controller unit, and IPLab software (version 3.5, Scanalytics). Cell size populations were quantified for an average of 500 cells per sample.
Aminoacylation of Phenylalanyl-tRNA
Approximately 120 ng of tRNAPhe (Sigma) were aminoacylated in a 100-μl reaction containing 0.12 A280 units of unfractionated yeast tRNA synthetases, 1.8 μCi of [14C]phenylalanine in aminoacylation buffer (200 mm Tris-HCl, pH 8.0, 10 mm ATP, 100 mm MgOAc, 5 mm EDTA, 6 mm β-mercaptoethanol) for 20 min at 37 °C. The reaction mixture was sequentially extracted with phenol (pH 4) and chloroform:isoamyl alcohol (24:1), then ethanol precipitated. The Phe-tRNAPhe was resuspended in 5 mm NaOAc, pH 4.8.
Purification of Wild-type and Mutant eEF1A Proteins
Wild-type and mutant eEF1A strains were grown to an A600 of 1.0 in YEPD. Cells were centrifuged, resuspended in 10 mm Tris-HCl, pH 7.5, and frozen by dripping into liquid nitrogen. Cells were lysed by grinding using a 6870 Freezer/Mill® (SPEX SamplePrep) and dissolved by stirring on ice for 1 h in a volume of lysis buffer (50 mm Tris-HCl, pH 7.5, 50 mm NH4Cl, 5 mm MgCl2, 0.1 mm EDTA, pH 8.0, 10% glycerol, 1 mm dithiothreitol, and 0.2 mm phenylmethylsulfonyl fluoride) equal to half the wet weight of the cell pellet. The lysate was centrifuged for 30 min at 10,000 × g and 90 min at 50,000 × g. The supernatant was passed through 0.80- and 0.22-μm pore size filters, and glycerol was added to a final concentration of 25%. The cleared lysate was passed through Q- and SP-Sepharose Fast Flow columns (Amersham Biosciences) pre-equilibrated with buffer A (20 mm Tris, pH 7.5, 50 mm KCl, 0.1 mm EDTA, pH 8.0, 25% glycerol, 1 mm dithiothreitol, and 0.2 mm phenylmethylsulfonyl fluoride). eEF1A proteins were eluted from the SP-Sepharose column using a linear gradient of buffer B (20 mm Tris, pH 7.5, 500 mm KCl, 0.1 mm EDTA, pH 8.0, 25% glycerol, 1 mm dithiothreitol, and 0.2 mm phenylmethylsulfonyl fluoride). Fractions containing eEF1A were pooled, concentrated using Amicon Ultracel®-3K centrifugal filter units (EMD Millipore) by centrifuging at 4,800 × g until samples reached a volume of 2–4 ml. Proteins were further purified by gel filtration using a HiLoad® 16/60 Superdex® 200 PG column (GE Healthcare) in buffer C (20 mm Tris, pH 7.5, 200 mm KCl, 0.1 mm EDTA, pH 8.0, 25% glycerol, 1 mm dithiothreitol). The appropriate fractions were pooled, concentrated as above, and the protein concentration was determined using a NanoDrop 2000c Spectrophotometer (Thermo Scientific); concentrations were adjusted according to the estimated extinction coefficient of each protein.
Western Blot Analysis
Yeast cells were cultured in YEPD and grown to an A600 of 0.5–0.7. Cells were collected by centrifugation, washed with ice-cold water, and resuspended in lysis buffer containing 20% glycerol, 100 mm Tris-HCl, pH 8.0, 1 mm dithiothreitol, and 1 mm phenylmethylsulfonyl fluoride. Cells were lysed with glass beads and vortexing for 2 min. Samples were centrifuged to remove glass beads and cell debris. Equal amounts of each protein sample were separated by electrophoresis in a 12% SDS-polyacrylamide gel and transferred to nitrocellulose filters. Immunoblot analyses were carried out using a polyclonal antibody that specifically recognizes eIF2α phosphorylated at Ser-51 (Cell Signaling). Total eIF2α levels were measured using a rabbit polyclonal antibody against recombinant yeast eIF2α (25).
Poly(U)-directed Poly(Phe) Synthesis
Polyphenylalanine synthesis reactions (50 μl) contained 100 mm KCl, 20 mm Tris-HCl, pH 7.5, 10 mm MgCl2, 2 mm GTP, 2.1 mm creatine phosphate, 80 μg/ml of creatine kinase, 1 mm dithiothreitol, 0.125–0.2 A260 units of poly(U) RNA, 0.3 A260 units of 80 S ribosomes, 60 pmol of [14C]Phe-tRNAPhe, 10 pmol of eEF1A, 7.5 pmol of eEF2, and 6.5 pmol of eEF3. Reactions were incubated at 37 °C for 20 min followed by trichloroacetic acid (TCA) precipitation. Samples were pipetted onto nitrocellulose filters (Whatman GF/C 25-mm) that were soaked in 5% TCA. Samples were washed two times with 3 ml of 5% TCA and once with 3 ml of 95% ethanol on a Millipore vacuum manifold. Membrane filters were air-dried for 15–30 min and subjected to liquid scintillation counting.
Phe-tRNAPhe Binding Assay
Phe-tRNAPhe binding experiments were performed as previously described (26) with some modifications. Binding reactions (50 μl) containing 60 pmol of wild-type or mutant eEF1A and 30 pmol of [14C]Phe-tRNAPhe in Buffer TC (20 mm Tris-HCl, pH 7.5, 50 mm KCl, 100 μm GTP, 1 mm dithiothreitol) were incubated on ice for 10 min followed by 10 min at 37 °C. After incubation, samples were pipetted onto nitrocellulose filters (EMD Millipore, 0.45 μm HA) that were soaked in Buffer TC. Samples were washed three times with 2 ml of Wash Buffer (20 mm Tris-HCl, pH 7.5, 100 mm KCl) on a Millipore vacuum manifold. Membrane filters were air-dried and subjected to liquid scintillation counting.
Mant-GTP Binding Assay
The binding affinity for mant-GTP to wild-type or mutant forms of eEF1A was performed using a FluoroMax-3 spectrofluorimeter (Horiba Jobin Yvon Inc.) as previously described (27). Briefly, wild-type or mutant forms of eEF1A (1 μm) in 2.5 ml of binding buffer (10% glycerol, 50 mm Tris-HCl, pH 8.0, 50 mm KCl, and 5 mm MgCl2) were placed in a 10 × 10 × 40-mm quartz cuvette with a magnetic stirring bar. All assays were performed at 25 °C. Increasing concentrations of mant-GTP (Molecular Probes) were added with continuous stirring. Fluorescence changes of the mant-nucleotides (Fobs) were monitored upon indirect excitation via fluorescence resonance energy transfer from excited tryptophans or tyrosines of eEF1A at a wavelength of 280 nm, and emission from the mant moiety at a wavelength of 444 nm.
RESULTS
eEF1A-Ura3p F308L and S405P Strains Exhibit Increased Phosphorylation of eIF2α
The slow growth and accumulation of cells in the G1 phase of cell division as a result of eEF1A overexpression in S. cerevisiae are proposed to occur due to increased interaction between eEF1A and the actin cytoskeleton (17). We designed a screen to identify mutations in eEF1A that would impair the actin binding function of the protein and inhibit the overexpression phenotype. Given that domain III of eEF1A contains the major actin-binding site, we fused the nutritional selection marker URA3 to the C terminus of the protein. This prevented isolation of false-positive suppressors that would arise from reduced eEF1A levels resulting from a truncated form of the protein. The screen gave rise to a class of eEF1A mutants (eEF1A-Ura3p F308L and S405P) that exhibited changes in global translation. Surprisingly, initial analysis of the eEF1A-Ura3p F308L and S405P mutants suggested that the defect in global translation was at the initiation step, which seemed counterintuitive given that eEF1A is an elongation factor. We sought to determine how mutations in an elongation factor could feed back to affect translation at initiation. One of the major regulatory mechanisms for translational control at initiation is the phosphorylation of eIF2α by the protein kinase Gcn2p. When cells undergo nutritional stress, there is an accumulation of deacylated tRNAs. Gcn2p interacts with the deacylated tRNAs via its His-tRNA synthetase homology domain, causing its full activation and subsequent phosphorylation of eIF2α. In addition, Gcn2p is the only eIF2α kinase in yeast, therefore, eIF2α phosphorylation was monitored in the eEF1A actin bundling mutant strains. Western blot analysis showed that eEF1A-Ura3p F308L and S405P mutant strains exhibit increased levels of eIF2α phosphorylation (Fig. 1A). Deletion of Gcn2p prevents phosphorylation confirming that phosphorylation is due to this kinase.
FIGURE 1.

eEF1A-Ura3p F308L and S405P mutants have increased eIF2α phosphorylation. A, wild-type and eEF1A-Ura3p mutant strains (TKY895, TKY896, TKY901, TKY903) with and without GCN2 (TKY1585, TKY1586, TKY1587, TKY1588) were grown to mid-log phase in YEPD at 30 °C. Total protein was extracted and equal protein amounts as determined by Bradford reagent were separated by SDS-PAGE. Phosphorylated and total eIF2α were detected by Western blot analysis using polyclonal antibodies. B, protein extracts from strains MC214 (eEF1A), TKY225 (eEF1A D156N), TKY226 (eEF1A N153T), TKY675 (eEF2), TKY825 (eEF2 D696A), TKY1653 (eEF3), and TKY1655 (eEF3 R803A/F806A) were made and eIF2α phosphorylation analyzed as in A. Bar graphs represent the fold-change compared with the respective wild-type strain in phosphorylated versus total eIF2α. Significant differences in eIF2α phosphorylation relative to the wild-type strain are indicated by an asterisk (p < 0.05; Student's t test).
To determine whether the increased eIF2α phosphorylation was specific to this class of eEF1A mutants, we assessed phosphorylation levels in other untagged eEF1A mutant strains not linked to actin related effects (28). We did not observe appreciable changes in the level of eIF2α phosphorylation in the eEF1A N153T and D156N mutant strains (Fig. 1B). To determine whether the phosphorylation effect was due to a general inhibition of translation, we analyzed the level of eIF2α phosphorylation in eEF2 and eEF3 mutants with established translation elongation defects (29, 30). Neither of the eEF2 and eEF3 mutants gave rise to increased eIF2α phosphorylation (Fig. 1B). These results suggest that the increase in eIF2α phosphorylation is specific to the F308L-Ura3p and S405P-Ura3p mutants and that a general inhibition of translation elongation is not sufficient to cause this effect. It is important to note that the eEF1A-Ura3p strain exhibited a slight increase in phosphorylation, which correlates with the effect the Ura3p fusion has on growth and modest effects on cell morphology. This also supports that the C terminus of eEF1A plays a role in the phenotypes observed in the eEF1A-Ura3p F308L and S405P mutant strains.
eEF1A-Ura3p F308L and S405P Mutant Strains Have an Elongation Defect
As Gcn2p is responsible for phosphorylating eIF2α, we deleted GCN2 to determine whether removal of the kinase would be enough to rescue the translational defect in the eEF1A-Ura3p F308L and S405P strains. Deletion of GCN2 causes an increase in growth rate in the wild-type as well as the eEF1A-Ura3p and F308L-Ura3p strains (Fig. 2A). We did not observe a detectable difference in growth rate for the S405P-Ura3p strain in the presence or absence of Gcn2p. Staining of the actin cytoskeleton with rhodamine phalloidin showed that deleting GCN2 partially rescues the large cell phenotype of the F308L- and S405P-Ura3p mutant strains (Fig. 2B). However, the wild-type and eEF1A-Ura3p strains also exhibited a reduction in the large cell population, indicating that the effect on growth caused by removal of GCN2 may not be specific to these mutants. Analysis of global translation by [35S]methionine incorporation revealed that whereas removal of Gcn2p increased the translation rate in the wild-type strain, the eEF1A-Ura3p or F308L- and S405P-Ura3p strains showed no change relative to the wild-type GCN2 background (Fig. 3). This result indicates that removal of Gcn2p unmasks a second defect in translation that is present in these strains.
FIGURE 2.

Deleting GCN2 in eEF1A-Ura3p mutant strains partially rescues growth and actin disorganization. A, wild-type and eEF1A-Ura3p mutant strains (TKY895, TKY896, TKY901, TKY903) with and without GCN2 (TKY1585, TKY1586, TKY1587, TKY1588) were struck on a YEPD plate and incubated at 30 °C for 2–3 days. The strains were also cultured in liquid YEPD media and their growth rates quantified as shown in the table. B, wild-type and eEF1A-Ura3p mutant strains with and without GCN2 as in A were fixed and stained with rhodamine phalloidin. Images were captured with an IX70 Olympus inverted fluorescence microscope.
FIGURE 3.

Deleting GCN2 does not rescue the translation defect in eEF1A-Ura3p mutants. Total translation was monitored in exponentially growing wild-type (A) and eEF1A-Ura3p mutant strains (B, C, and D) with and without GCN2 as described in the legend to Fig. 2 by measuring [35S]methionine incorporation for the indicated times. [35S]Methionine incorporation is expressed as cpm/A600 unit of cells.
To determine whether the elongation function of eEF1A was compromised in the eEF1A-Ura3p strains, we assessed activity of the proteins in vitro using the initiation-independent polyphenylalanine synthesis assay with purified translation components from yeast (Fig. 4A). The eEF1A-Ura3p mutant protein exhibited decreased elongation activity compared with the wild-type protein, whereas the F308L-Ura3p and S405P-Ura3p mutant proteins showed a further reduction in activity. Interestingly, two actin bundling mutants initially isolated in the same screen, N305S-Ura3p and N329S-Ura3p, also showed decreased elongation activity in vitro despite not having a detectable defect in global translation in vivo (17). As the canonical role of eEF1A in translation is to deliver aminoacylated-tRNAs to the ribosome, we sought to determine which property of eEF1A was affected in the eEF1A-Ura3p mutants. Analysis of nucleotide binding using mant-GTP showed no significant changes in affinity among the wild-type and mutant proteins (Fig. 4B). Dissociation constants for all proteins were ∼0.2 μm. When aa-tRNA binding was analyzed using radiolabeled Phe-tRNAPhe, a marked reduction in binding was seen with the F308L- and S405P-Ura3p mutant proteins (Fig. 4C). The eEF1A-Ura3p mutant did not exhibit reduced aa-tRNA binding, although it has decreased elongation activity. This could also suggest that the Ura3p fusion might also be causing reduced elongation activity by altering the interaction between eEF1A and the ribosome.
FIGURE 4.
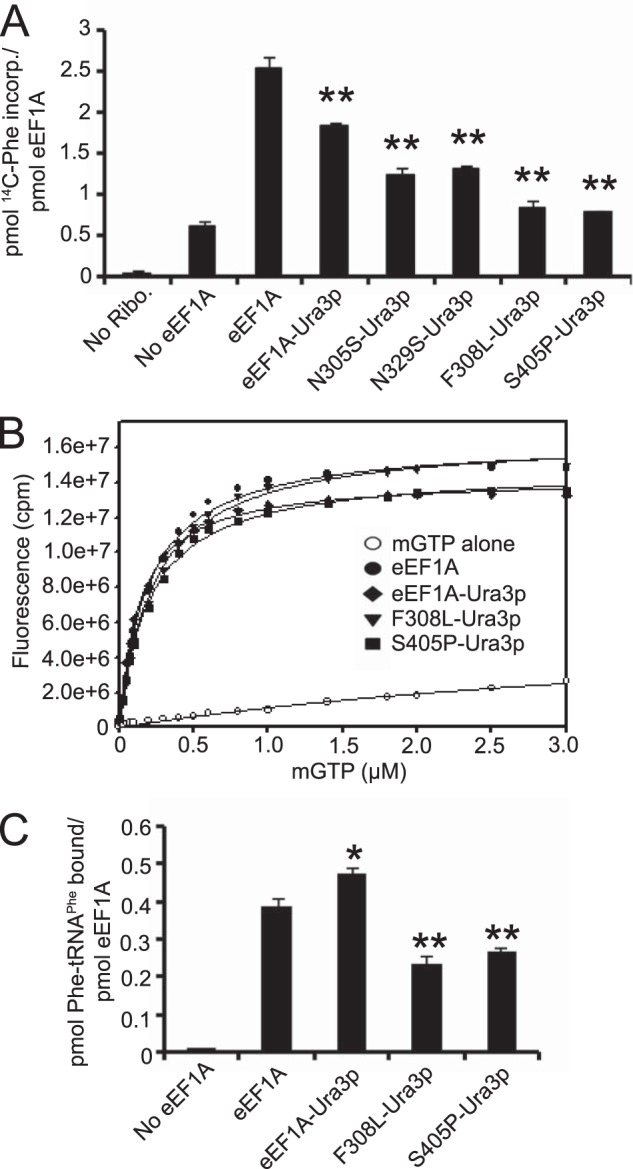
eEF1A-Ura3p mutant proteins have a defect in translation elongation. A, wild-type eEF1A, eEF1A-Ura3p, N305S-Ura3p, N329S-Ura3p, F308L-Ura3p, and S405P-Ura3p fusion proteins were purified and subjected to the poly(U)-dependent poly(Phe) synthesis assay at 37 °C for 20 min. Samples were collected on Whatman GF/C 25-mm nitrocellulose filters and the amount of radioactive phenylalanine incorporated was measured by scintillation counting. B, increasing concentrations of mant-GTP were added to 1 μm wild-type eEF1A, eEF1A-Ura3p, F308L-Ura3p, or S405P-Ura3p in binding buffer and mixed. Fluorescence was measured by FRET via excitation at 280 nm and emission at 444 nm for the mant moiety. C, purified proteins were incubated with [14C]Phe-tRNAPhe, collected on nitrocellulose filters (EMD Millipore, 0.45 μm HA) and the amount of radioactive tRNA bound was measured by scintillation counting. Significant differences relative to the wild-type protein are indicated by either one (p < 0.05) or two asterisks (p < 0.01; Student's t test).
Because the translation elongation defect in eEF1A-Ura3p mutants appears to require the presence of the Ura3p fusion, we wanted to determine what the relative contributions of the amino acid substitutions and the Ura3p fusion were toward eliciting the phenotype. The Ura3p fusion was removed from one eEF1A mutant that showed the translation defect in vivo (F308L) and one that did not (N305S). Removal of the Ura3p fusion allows the mutant strains to grow like the wild-type strain (Fig. 5A). The purified untagged N305S and F308L mutant proteins did not exhibit statistically significant differences in elongation activity in the polyphenylalanine synthesis assay compared with the wild-type, although a trend toward reduced activity was observed with the Ura3p mutant proteins that correlates with the translation effects in vivo (Fig. 5B). Thus, the effects on actin bundling and cell morphology are the result of synthetic interactions between the tag and the amino acid substitution.
FIGURE 5.
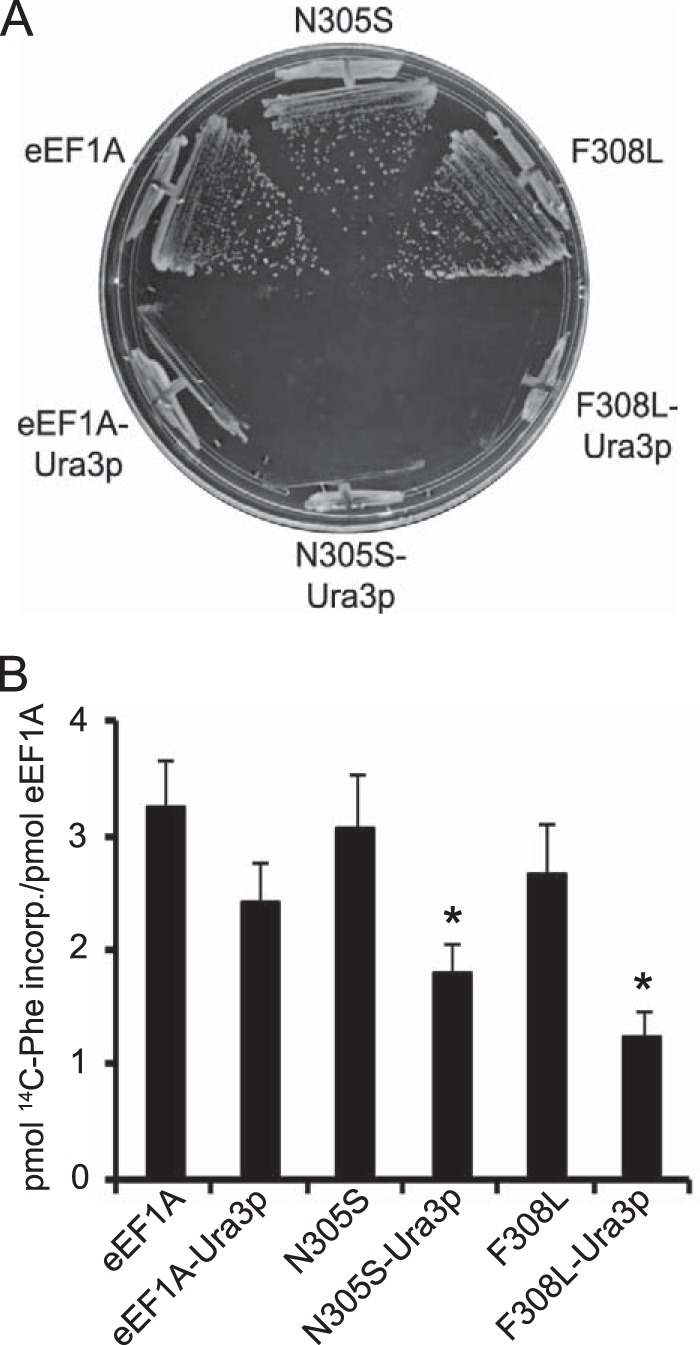
Removal of Ura3p fusion restores growth and elongation activity of the eEF1A-Ura3p strains. A, wild-type, N305S- and F308L-eEF1A mutant strains with (TKY896, TKY902, TKY901) and without the Ura3p fusion (TKY895, TKY1746, TKY1744) were struck on a YEPD plate and incubated at 30 °C for 2–3 days. B, purified Ura3p fusion and non-fusion wild-type eEF1A N305S and F308L proteins were subjected to the poly(U)-dependent poly(Phe) synthesis assay at 37 °C for 20 min. Samples were collected on Whatman GF/C 25-mm nitrocellulose filters and the amount of radioactive phenylalanine incorporated was measured by scintillation counting. Significant differences relative to the eEF1A-Ura3p protein are indicated by an asterisk (p < 0.05; Student's t test).
Putative eEF1A tRNA Binding Mutants Do Not Exhibit Elongation Defects
The eEF1A-Ura3p F308L and S405P mutants exhibited a significant increase in eIF2α phosphorylation and both initiation and elongation effects. Because the reduced elongation activity is linked to defective aa-tRNA binding by eEF1A, we sought to determine whether significant alterations in aa-tRNA binding without affecting actin bundling would be enough to induce eIF2α phosphorylation in the absence of the Ura3p fusion. Altered forms of untagged eEF1A were made by mutating one non-conserved (Leu-77) and two conserved (Glu-291 and Thr-430) residues predicted to be involved in aa-tRNA binding in bacterial EF-Tu (31–33), as aa-tRNA binding mutants are not recorded for eEF1A. Strains harboring these mutants as the only form of eEF1A exhibited a slow growth phenotype compared with the wild-type (Fig. 6A). However, none of the mutant proteins exhibited reduced elongation (Fig. 6B) or Phe-tRNAPhe binding activity (Fig. 6C). As eEF1A makes substantive contacts with the tRNA body, it is possible that mutations at individual amino acids would not be sufficient to give rise to a aa-tRNA binding defect.
FIGURE 6.
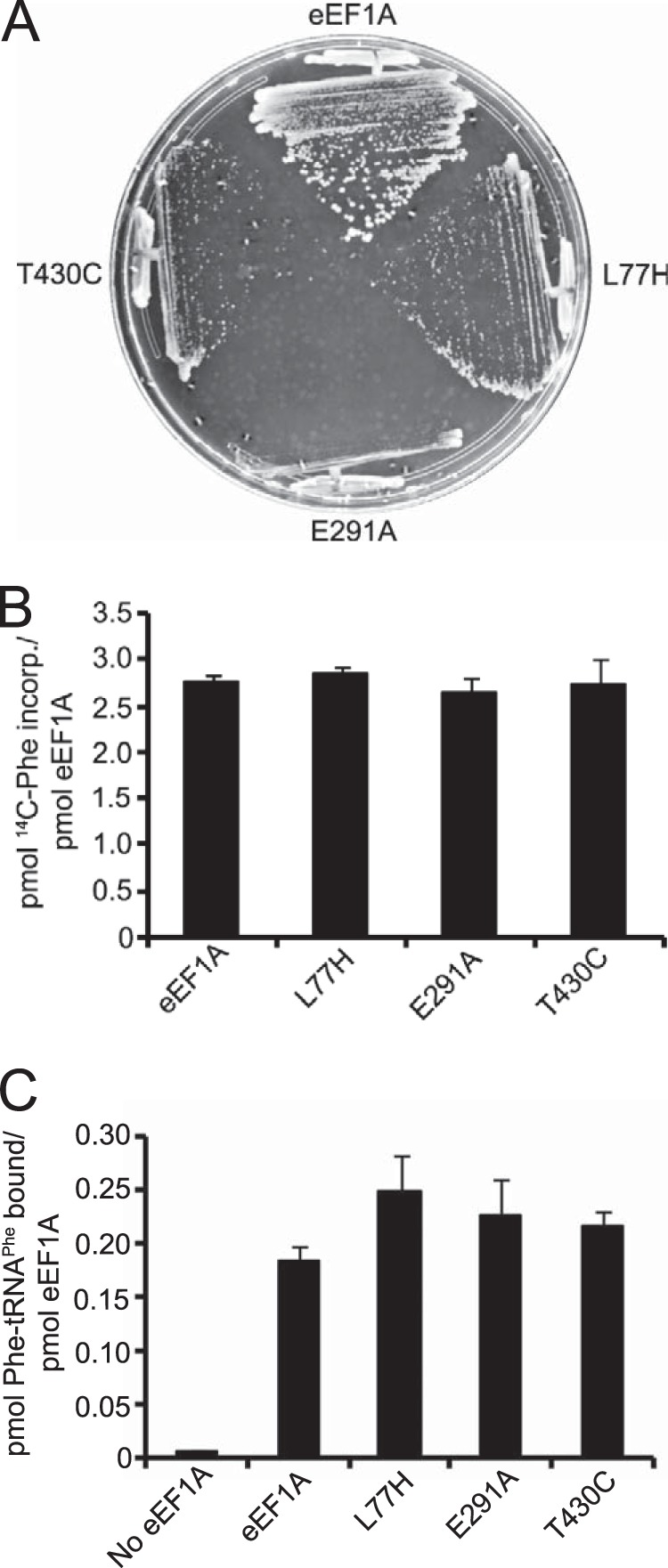
Putative tRNA binding residue mutations in eEF1A causes a slow growth phenotype but do not give rise to translation defects in vitro. A, wild-type (TKY895), L77H (TKY1723), E291A (TKY1724), and T430C (TKY1727) eEF1A mutant strains were struck on a YEPD plate and incubated at 30 °C for 2 days. Purified eEF1A, L77H, E291A, and T430C proteins were subjected to the poly(U)-dependent poly(Phe) synthesis assay (B) and Phe-tRNAPhe filter binding assay (C).
A Putative Phosphomimetic eEF1A Mutant Exhibits Increased eIF2α Phosphorylation
It remains unclear if reduced eEF1A activity triggers eIF2α phosphorylation under physiological conditions. Previous studies have shown that eEF1A is phosphorylated in response to different growth conditions (34, 35). The negative impact on eEF1A function as a result of phosphorylation may be through defective aa-tRNA binding (36). Phosphoproteomic studies in S. cerevisiae have identified several conserved residues that are phosphorylated (37–39). Based on these studies, we created an eEF1A mutant with either alanine or aspartic acid substitutions at Ser-18 to determine the effects of this putative phosphorylation on eEF1A activity. The eEF1A S18D mutant exhibited a strong growth defect (Fig. 7A). Therefore, we sought to determine whether this phosphomimetic eEF1A mutant could trigger eIF2α phosphorylation. Western blot analysis revealed an increase in eIF2α phosphorylation in the eEF1A S18D mutant strain (Fig. 7B). The mutant protein also exhibited decreased Phe-tRNAPhe binding in vitro (Fig. 7C). These defects are not associated with reduced expression of eEF1A as the wild-type and S18D mutant are expressed at similar levels (data not shown). Taken together, these findings suggest that conditions that significantly alter the aa-tRNA binding activity of eEF1A lead to increased eIF2α phosphorylation.
FIGURE 7.
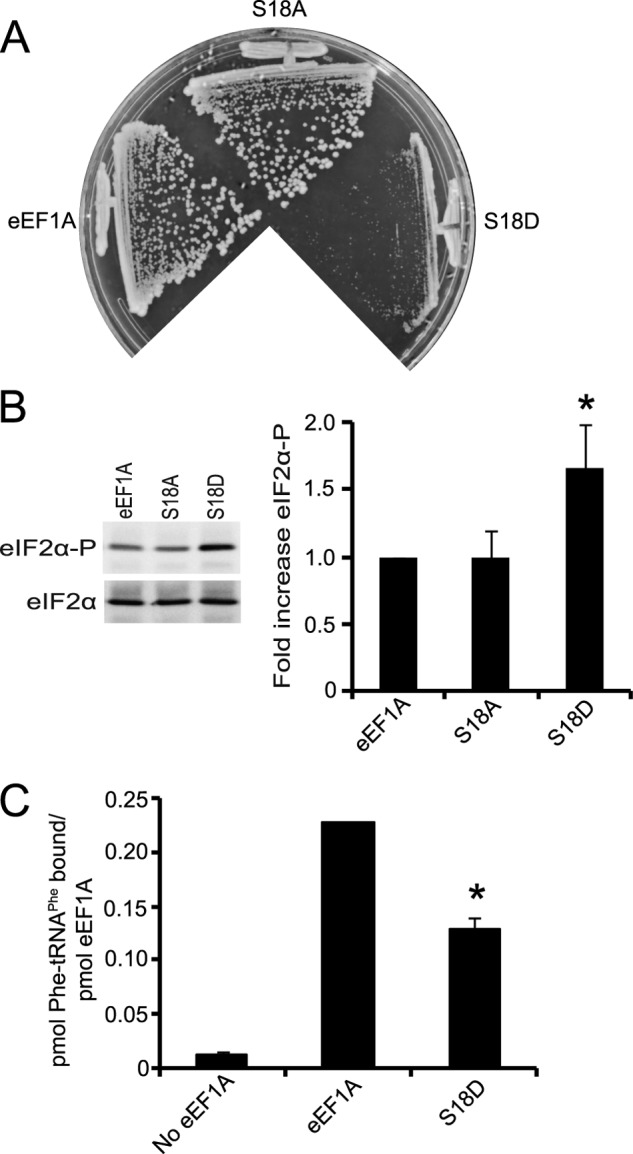
A putative phosphomimetic eEF1A mutant exhibits increased eIF2α phosphorylation. A, wild-type (TKY1717), eEF1A S18A (TKY1735), and eEF1A S18D (TKY1740) were struck on a YEPD plate and incubated at 30 °C for 2–3 days. B, wild-type and mutant eEF1A strains were grown to mid-log phase in YEPD at 30 °C. Total protein was extracted and equal amounts were separated by SDS-PAGE. Phosphorylated and total eIF2α were detected by Western blot analysis using polyclonal antibodies. C, purified proteins were subjected to the Phe-tRNAPhe filter binding assay. Significant differences in B and C relative to the wild-type are indicated by an asterisk (p < 0.05; Student's t test).
DISCUSSION
The identification of eEF1A as an actin-binding protein (12) provided a direct link between the actin cytoskeleton and the translational machinery. Although the eEF1A-actin interaction has been well characterized in vitro (13, 40), the consequences of disrupting the interaction in vivo are not fully understood. Overexpression of eEF1A in S. cerevisiae causes a slow growth phenotype and concurrent large cell phenotype arising from disorganization of the actin cytoskeleton (16). We developed a genetic screen to identify mutations in eEF1A that suppress the overexpression phenotype and reduce actin disorganization. To prevent false-positive suppressors, the TEF1 gene encoding eEF1A was fused to the nutritional selection marker URA3. The screen generated two classes of eEF1A mutants. The class one mutants (N305S- and N329S-Ura3p) exhibited disorganization of the actin cytoskeleton whereas maintaining normal levels of global translation (17). The class two mutants (F308L- and S405P-Ura3p) exhibited both cytoskeletal and translational defects. Surprisingly, the translational defect in the F308L- and S405P-Ura3p mutants appeared to be at initiation rather than elongation (18). We wanted to characterize the eEF1A-Ura3p F308L and S405P mutants to gain understanding of how mutations in an elongation factor can feed back and affect translation initiation.
One of the major regulatory mechanisms for controlling gene expression through inhibition of translation is the phosphorylation of eIF2α. Although higher eukaryotes possess four different eIF2α kinases (21), only the Gcn2p kinase is present in S. cerevisiae. The F308L- and S405P-Ura3p mutants exhibited increased levels of eIF2α phosphorylation (Fig. 1A). The idea that defects at one phase of translation can have a feedback effect on a different phase has been discussed (41). Therefore, we wanted to determine whether a general defect in translation would be enough to cause an increase in eIF2α phosphorylation levels. Analysis of other eEF1A as well as eEF2 and eEF3 mutants known to affect translation elongation (29, 30) showed that increased eIF2α phosphorylation does not result from a general reduction in translation elongation (Fig. 1B). We also analyzed strains harboring mutations in actin (act1–2, act1–122) or lacking actin-associated proteins (abp1Δ, mdm20Δ, sac6Δ, tpm1Δ) but did not observe a correlation between the severity of actin defects and eIF2α phosphorylation levels (data not shown). These findings suggest that increased eIF2α phosphorylation is specific to the F308L- and S405P-Ura3p mutants and may be directly related to the canonical function of eEF1A in translation.
Because the actin cytoskeletal and translational defects in the F308L- and S405P-Ura3p mutants appeared to be linked, we removed Gcn2p in an attempt to rescue the defects in vivo. Deletion of GCN2 inhibits eIF2α phosphorylation, confirming that Gcn2p is the only eIF2α kinase in yeast (Fig. 1A). One of the hallmarks of disrupting the cytoskeleton in yeast is an increase in cell size. We found that although deletion of GCN2 partially suppresses the slow growth phenotype (Fig. 2A) and reduces the population of large cells in F308L- and S405P-Ura3p cultures, the same effect is seen in wild-type and eEF1A-Ura3p cultures (Fig. 2B). This suggests that the in vivo effects of deleting GCN2 in all of the strains is a result of removing the translational block at initiation rather than the direct rescue of the cytoskeletal defects. Surprisingly, the removal of GCN2 unmasked a second translation defect (Fig. 3). In vitro analysis confirmed that the unmasked defect was at the elongation phase (Fig. 4A). As the mutations isolated from the screen cluster to domain II of eEF1A, it is not surprising that the elongation defect is linked to aa-tRNA binding (Fig. 4C). Although the N305S- and N329S-Ura3p mutants also exhibit reduced elongation activity in vitro, they appear to function better than the F308L- and S405P-Ura3p mutant proteins in a defined system. This suggests that the level of elongation activity exhibited by the N305S- and N329S-Ura3p mutants is enough to be above the threshold required to see defects in global translation rates (normalized for growth rate) by [35S]methionine incorporation (17).
To understand the contributions of the mutations isolated from the screen and the relationship of the Ura3p fusion to the phenotypes, we created untagged N305S and F308L eEF1A mutant strains. Removal of the Ura3p fusion essentially suppresses the slow growth phenotype and elongation defect of the mutants (Fig. 5). However, it is clear that there is a combinatorial effect of having the Ura3p fusion and the mutations together. These findings show that the Ura3p fusion is required to exacerbate the translation elongation defects seen in these eEF1A mutants. Despite having a reduction in elongation activity compared with the wild-type protein, the eEF1A-Ura3p fusion protein did not display defective aa-tRNA binding indicating that the fusion may be causing a secondary effect. Given the size of the Ura3p fusion (∼29 kDa), it is possible that it could be interfering with the ability of eEF1A to efficiently interact with the ribosome thereby reducing the rate of elongation.
In an attempt to understand the relationship between reduced aa-tRNA binding by eEF1A and the eIF2α phosphorylation seen in the F308L- and S405P-Ura3p mutants, we generated untagged putative aa-tRNA binding mutants. Due to the lack of structural information regarding the interaction between eEF1A and aa-tRNAs as well as limited genetic information, we chose residues to mutate based on the structure of EF-Tu with Phe-tRNAPhe. The conserved Glu-291 residue was chosen for its location within the binding pocket formed by domain II for the aminoacyl end of the tRNA (32). This residue is also close to aa-tRNA cross-links to rabbit eEF1A (31). The Thr-430 residue was chosen because mutation of the corresponding T. thermophilus EF-Tu residue (Thr-394) was shown to inhibit aa-tRNA binding (33). A non-conserved residue (Leu-77) was also chosen because the corresponding Escherichia coli EF-Tu residue (His-66) has been cross-linked to ϵ-bromo-Lys-tRNA (42). Although mutating these residues induces a slow growth phenotype (Fig. 6A), we did not observe changes in elongation activity or aa-tRNA binding (Fig. 6, B and C). Given the substantial contacts EF-Tu makes with Phe-tRNAPhe (32), it is possible that mutating individual amino acids is not enough to significantly reduce the ability of eEF1A to bind aa-tRNAs or that more significant differences than anticipated exist between the yeast and bacterial elongation factor.
As there is some data supporting sites where eEF1A is regulated via phosphorylation in vivo (43), we assessed the effect of mimicking this modification on its function. We found that phosphorylation at Ser-18 may be important in regulating eEF1A function as the S18D mutant exhibited a strong growth defect, decreased aa-tRNA binding, and increased eIF2α phosphorylation (Fig. 7). Taken together, the findings of this study indicate that a significant reduction in aa-tRNA binding by eEF1A is associated with increased eIF2α phosphorylation. However, it is important to note that not all eEF1A mutants exhibit increased levels of eIF2α phosphorylation, making it difficult to determine the physiological conditions that trigger this feedback regulation. Aside from phosphorylation, virus infection may also trigger eIF2α phosphorylation through exploitation of the aa-tRNA binding property of eEF1A. eEF1A interacts with the 3′-end of several positive strand RNA viruses, which have predicted secondary structures that closely resemble that of tRNA (44–46). By sequestering eEF1A away from the translational machinery, these viruses would effectively alter the amount of aa-tRNA bound by eEF1A. This in turn could trigger activation of Gcn2p in parallel to the activation of the dsRNA-activated protein kinase as a result of the virus infection.
Although further support is needed to determine whether drastic alterations in aa-tRNA binding by eEF1A are enough to induce eIF2α phosphorylation, the increased levels seen in the F308L-Ura3p, S405P-Ura3p, and S18D eEF1A mutants is a result of Gcn2p activation. There are two scenarios that may help explain this phenomenon. In the first scenario, reduced aa-tRNA binding by eEF1A would increase the population of unbound aa-tRNAs and possibly lead to deacylation of these tRNAs over time in vivo. The increase in deacylated tRNAs would trigger activation of Gcn2p and subsequent eIF2α phosphorylation. To that extent, we assessed aminoacylation of His-tRNA by Northern blot analysis but did not observe significant changes between the wild-type and eEF1A-Ura3p mutants (data not shown). It is possible that there may be other aa-tRNAs that are being affected or that only a small fraction of possibly many tRNAs are being deacylated. Therefore, more sensitive methods like the tRNA microarray (23) may have to be applied to determine the aminoacylation levels of all tRNAs. The second scenario stems from the recent finding that eEF1A and Gcn2p directly interact (47). eEF1A binding to Gcn2p inhibits its activation by deacylated tRNAs. If the eEF1A-Ura3p mutants have lost this ability, then Gcn2p would become activated by the basal level of deacylated tRNAs in the cell.
Although the eEF1A-Ura3p mutants were isolated as actin bundling mutants, we now know that these mutants also exhibit translation elongation defects. The fact that both eEF1Bα and aa-tRNAs interact with eEF1A via domain II and the clustering of the mutations from the screen to this region of the protein highlights the importance of this domain for eEF1A functions. The interactions between eEF1A and its binding partners being mutually exclusive (13) also suggests the presence of overlapping binding sites, making it more difficult to determine which residues are involved in actin bundling. These findings also indicate that the overlapping sites may play a role in balancing the functions of eEF1A and their outcomes in the cell. As previous studies have already determined that domain III of eEF1A contains the dominant actin binding site (14), future work should focus on mutational analysis of domain III whereas steering clear of residues predicted to be important for other functions of eEF1A.
Acknowledgments
We thank Paul R. Copeland and members of the Kinzy lab for helpful suggestions and discussions especially Maria Mateyak for supporting the preliminary work on the S18D mutant. We also thank Thomas Dever (National Institutes of Health, NICHD) for the eIF2α antibody.
This work was supported, in whole or in part, by National Institutes of Health Grants GM57483 (to T. G. K.) and T32AI007403 and R25 GM055145 (Initiative to Maximize Student Diversity) (to W. B. P).
- aa-tRNA
- aminoacyl-tRNAs
- Gcn2p
- general control nonderepressible 2
- eEF1A
- eukaryotic translation elongation factor 1A.
REFERENCES
- 1. Moseley J. B., Goode B. L. (2006) The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol. Mol. Biol. Rev. 70, 605–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bretscher A. (2003) Polarized growth and organelle segregation in yeast: the tracks, motors, and receptors. J. Cell Biol. 160, 811–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashe M. P., De Long S. K., Sachs A. B. (2000) Glucose depletion rapidly inhibits translation initiation in yeast. Mol. Biol. Cell 11, 833–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Uesono Y., Ashe M. P., Toh-E A. (2004) Simultaneous yet independent regulation of actin cytoskeletal organization and translation initiation by glucose in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 1544–1556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chowdhury S., Smith K. W., Gustin M. C. (1992) Osmotic stress and the yeast cytoskeleton: phenotype-specific suppression of an actin mutation. J. Cell Biol. 118, 561–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Muaddi H., Majumder M., Peidis P., Papadakis A. I., Holcik M., Scheuner D., Kaufman R. J., Hatzoglou M., Koromilas A. E. (2010) Phosphorylation of eIF2alpha at serine 51 is an important determinant of cell survival and adaptation to glucose deficiency. Mol. Biol. Cell 21, 3220–3231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uesono Y., Toh-E A. (2002) Transient inhibition of translation initiation by osmotic stress. J. Biol. Chem. 277, 13848–13855 [DOI] [PubMed] [Google Scholar]
- 8. Dang C. V., Yang D. C., Pollard T. D. (1983) Association of methionyl-tRNA synthetase with detergent-insoluble components of the rough endoplasmic reticulum. J. Cell Biol. 96, 1138–1147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bekta[uncomma]s M., Nurten R., Gürel Z., Sayers Z., Bermek E. (1994) Interactions of eukaryotic elongation factor 2 with actin: a possible link between protein synthetic machinery and cytoskeleton. FEBS Lett. 356, 89–93 [DOI] [PubMed] [Google Scholar]
- 10. Howe J. G., Hershey J. W. (1984) Translational initiation factor and ribosome association with the cytoskeletal framework fraction from HeLa cells. Cell 37, 85–93 [DOI] [PubMed] [Google Scholar]
- 11. Ogle J. M., Carter A. P., Ramakrishnan V. (2003) Insights into the decoding mechanism from recent ribosome structures. Trends Biochem. Sci. 28, 259–266 [DOI] [PubMed] [Google Scholar]
- 12. Yang F., Demma M., Warren V., Dharmawardhane S., Condeelis J. (1990) Identification of an actin-binding protein from Dictyostelium as elongation factor 1a. Nature 347, 494–496 [DOI] [PubMed] [Google Scholar]
- 13. Liu G., Tang J., Edmonds B. T., Murray J., Levin S., Condeelis J. (1996) F-actin sequesters elongation factor 1a from interaction with aminoacyl-tRNA in a pH-dependent reaction. J. Cell Biol. 135, 953–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu G., Grant W. M., Persky D., Latham V. M., Jr., Singer R. H., Condeelis J. (2002) Interactions of elongation factor 1alpha with F-actin and β-actin mRNA: implications for anchoring mRNA in cell protrusions. Mol. Biol. Cell 13, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Umikawa M., Tanaka K., Kamei T., Shimizu K., Imamura H., Sasaki T., Takai Y. (1998) Interaction of Pho1p target Bni1p with F-actin-binding elongation factor 1a: implications in Rho1p-regulated reorganization of the actin cytoskeleton in Saccharomyces cerevisiae. Oncogene 16, 2011–2016 [DOI] [PubMed] [Google Scholar]
- 16. Munshi R., Kandl K. A., Carr-Schmid A., Whitacre J. L., Adams A. E., Kinzy T. G. (2001) Overexpression of translation elongation factor 1α affects the organization and function of the actin cytoskeleton in yeast. Genetics 157, 1425–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gross S. R., Kinzy T. G. (2005) Translation elongation factor 1A is essential for regulation of the actin cytoskeleton and cell morphology. Nat. Struct. Mol. Biol. 12, 772–778 [DOI] [PubMed] [Google Scholar]
- 18. Gross S. R., Kinzy T. G. (2007) Improper organization of the actin cytoskeleton affects protein synthesis at initiation. Mol. Cell. Biol. 27, 1974–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baird T. D., Wek R. C. (2012) Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 3, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jackson R. J., Hellen C. U., Pestova T. V. (2010) The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 11, 113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hinnebusch A. G. (2005) Translational regulation of GCN4 and the general amino acid control of yeast. Annu. Rev. Microbiol. 59, 407–450 [DOI] [PubMed] [Google Scholar]
- 22. Dong J., Qiu H., Garcia-Barrio M., Anderson J., Hinnebusch A. G. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279 [DOI] [PubMed] [Google Scholar]
- 23. Zaborske J. M., Narasimhan J., Jiang L., Wek S. A., Dittmar K. A., Freimoser F., Pan T., Wek R. C. (2009) Genome-wide analysis of tRNA charging and activation of the eIF2 kinase Gcn2p. J. Biol. Chem. 284, 25254–25267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ito H., Fukuda Y., Murata K., Kimura A. (1983) Transformation of intact yeast cells treated with alkali cations. J. Bacteriol. 153, 163–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krishnamoorthy T., Pavitt G. D., Zhang F., Dever T. E., Hinnebusch A. G. (2001) Tight binding of the phosphorylated α subunit of initiation factor 2 (eIF2α) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol. Cell. Biol. 21, 5018–5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Safer B., Adams S. L., Anderson W. F., Merrick W. C. (1975) Binding of MET-TRNAf and GTP to homogeneous initiation factor MP. J. Biol. Chem. 250, 9076–9082 [PubMed] [Google Scholar]
- 27. Ozturk S. B., Kinzy T. G. (2008) Guanine nucleotide exchange factor independence of the G-protein eEF1A through novel mutant forms and biochemical properties. J. Biol. Chem. 283, 23244–23253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Carr-Schmid A., Durko N., Cavallius J., Merrick W. C., Kinzy T. G. (1999) Mutations in a GTP-binding motif of eEF1A reduce both translational fidelity and the requirement for nucleotide exchange. J. Biol. Chem. 274, 30297–30302 [DOI] [PubMed] [Google Scholar]
- 29. Ortiz P. A., Ulloque R., Kihara G. K., Zheng H., Kinzy T. G. (2006) Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J. Biol. Chem. 281, 32639–32648 [DOI] [PubMed] [Google Scholar]
- 30. Sasikumar A. N., Kinzy T. G. (2014) Mutations in the chromodomain-like insertion of translation elongation factor 3 compromise protein synthesis through reduced ATPase activity. J. Biol. Chem. 289, 4853–4860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kinzy T. G., Freeman J. P., Johnson A. E., Merrick W. C. (1992) A model of the aminoacyl-tRNA binding site of eukaryotic elongation factor 1a. J. Biol. Chem. 267, 1623–1632 [PubMed] [Google Scholar]
- 32. Nissen P., Kjeldgaard M., Thirup S., Polekhina G., Reshetnikova L., Clark B. F., Nyborg J. (1995) Crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu, and a GTP analog. Science 270, 1464–1472 [DOI] [PubMed] [Google Scholar]
- 33. Shin B. S., Kim J. R., Walker S. E., Dong J., Lorsch J. R., Dever T. E. (2011) Initiation factor eIF2γ promotes eIF2-GTP-Met-tRNAi(Met) ternary complex binding to the 40S ribosome. Nat. Struct. Mol. Biol. 18, 1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang Y. W., Traugh J. A. (1998) Insulin stimulation of phosphorylation of elongation factor 1 (eEF-1) enhances elongation activity. Eur. J. Biochem. 251, 201–207 [DOI] [PubMed] [Google Scholar]
- 35. Peters H. I., Chang Y.-W., Traugh J. A. (1995) Phosphorylation of elongation factor 1 (EF-1) by protein kinase C stimulates GDP/GTP-exchange activity. Eur. J. Biochem. 234, 550–556 [DOI] [PubMed] [Google Scholar]
- 36. Alexander C., Bilgin N., Lindschau C., Mesters J. R., Kraal B., Hilgenfeld R., Erdmann V. A., Lippmann C. (1995) Phosphorylation of elongation factor Tu prevents ternary complex formation. J. Biol. Chem. 270, 14541–14547 [DOI] [PubMed] [Google Scholar]
- 37. Albuquerque C. P., Smolka M. B., Payne S. H., Bafna V., Eng J., Zhou H. (2008) A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol. Cell. Proteomics 7, 1389–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chi A., Huttenhower C., Geer L. Y., Coon J. J., Syka J. E., Bai D. L., Shabanowitz J., Burke D. J., Troyanskaya O. G., Hunt D. F. (2007) Analysis of phosphorylation sites on proteins from Saccharomyces cerevisiae by electron transfer dissociation (ETD) mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2193–2198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Holt L. J., Tuch B. B., Villén J., Johnson A. D., Gygi S. P., Morgan D. O. (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325, 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Edmonds B. T., Bell A., Wyckoff J., Condeelis J., Leyh T. S. (1998) The effect of F-actin on the binding and hydrolysis of guanine nucleotide by Dictyostelium elongation factor 1A. J. Biol. Chem. 273, 10288–10295 [DOI] [PubMed] [Google Scholar]
- 41. Beznosková P., Cuchalová L., Wagner S., Shoemaker C. J., Gunis̆ová S., von der Haar T., Valás̆ek L. S. (2013) Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS Genet. 9, e1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Duffy L. K., Gerber L., Johnson A. E., Miller D. L. (1981) Identification of a histidine residue near the aminoacyl transfer ribonucleic acid binding site of elongation factor Tu. Biochemistry 20, 4663–4666 [DOI] [PubMed] [Google Scholar]
- 43. Lin K. W., Yakymovych I., Jia M., Yakymovych M., Souchelnytskyi S. (2010) Phosphorylation of eEF1A1 at Ser300 by TβR-I results in inhibition of mRNA translation. Curr. Biol. 20, 1615–1625 [DOI] [PubMed] [Google Scholar]
- 44. Blackwell J. L., Brinton M. A. (1997) Translation elongation factor-1 alpha interacts with the 3′ stem-loop region of West Nile virus genomic RNA. J. Virol. 71, 6433–6444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li Z., Pogany J., Tupman S., Esposito A. M., Kinzy T. G., Nagy P. D. (2010) Translation elongation factor 1A facilitates the assembly of the tombusvirus replicase and stimulates minus-strand synthesis. PLoS Pathog. 6, e1001175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matsuda D., Yoshinari S., Dreher T. W. (2004) eEF1A binding to aminoacylated viral RNA represses minus strand synthesis by TYMV RNA-dependent RNA polymerase. Virology 321, 47–56 [DOI] [PubMed] [Google Scholar]
- 47. Visweswaraiah J., Lageix S., Castilho B. A., Izotova L., Kinzy T. G., Hinnebusch A. G., Sattlegger E. (2011) Evidence that eukaryotic translation elongation factor 1A (eEF1A) binds the Gcn2 protein C terminus and inhibits Gcn2 activity. J. Biol. Chem. 286, 36568–36579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sandbaken M. G., Culbertson M. R. (1988) Mutations in elongation factor EF-1a affect the frequency of frameshifting and amino acid misincorporation in Saccharomyces cerevisiae. Genetics 120, 923–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cavallius J., Merrick W. C. (1998) Site-directed mutagenesis of yeast eEF1A: viable mutants with altered nucleotide specificity. J. Biol. Chem. 273, 28752–28758 [DOI] [PubMed] [Google Scholar]
- 50. Jørgensen R., Carr-Schmid A., Ortiz P. A., Kinzy T. G., Andersen G. R. (2002) Purification and crystallization of the yeast elongation factor eEF2. Acta Crystallogr. D Biol. Crystallogr. 58, 712–715 [DOI] [PubMed] [Google Scholar]