

Background: Non-visual arrestins regulate the signaling of hundreds of GPCRs.
Results: Receptor binding-induced conformational changes in non-visual arrestins partially overlap with those in visual arrestin-1.
Conclusion: Some receptor binding-induced conformational changes are conserved between arrestin-1, -2, and -3.
Significance: Characterization of receptor-induced conformational changes will help identify how the non-visual arrestins interact with hundreds of receptors.
Keywords: Arrestin, Electron Paramagnetic Resonance (EPR), G Protein-coupled Receptor (GPCR), Rhodopsin, Spectroscopy
Abstract
The non-visual arrestins, arrestin-2 and arrestin-3, belong to a small family of multifunctional cytosolic proteins. Non-visual arrestins interact with hundreds of G protein-coupled receptors (GPCRs) and regulate GPCR desensitization by binding active phosphorylated GPCRs and uncoupling them from heterotrimeric G proteins. Recently, non-visual arrestins have been shown to mediate G protein-independent signaling by serving as adaptors and scaffolds that assemble multiprotein complexes. By recruiting various partners, including trafficking and signaling proteins, directly to GPCRs, non-visual arrestins connect activated receptors to diverse signaling pathways. To investigate arrestin-mediated signaling, a structural understanding of arrestin activation and interaction with GPCRs is essential. Here we identified global and local conformational changes in the non-visual arrestins upon binding to the model GPCR rhodopsin. To detect conformational changes, pairs of spin labels were introduced into arrestin-2 and arrestin-3, and the interspin distances in the absence and presence of the receptor were measured by double electron electron resonance spectroscopy. Our data indicate that both non-visual arrestins undergo several conformational changes similar to arrestin-1, including the finger loop moving toward the predicted location of the receptor in the complex as well as the C-tail release upon receptor binding. The arrestin-2 results also suggest that there is no clam shell-like closure of the N- and C-domains and that the loop containing residue 136 (homolog of 139 in arrestin-1) has high flexibility in both free and receptor-bound states.
Introduction
G protein-coupled receptors (GPCRs)2 represent the largest, most versatile, and ubiquitous class of membrane receptors, with more than 800 members in the human genome (1). Upon activation, they regulate a variety of intracellular signaling pathways to produce appropriate cellular responses, such as cell growth, differentiation, and metabolism, and mediate smell, vision, and taste (2). Arrestins are a small family of proteins that preferentially bind active phosphorylated GPCRs, block G protein-mediated signaling, and facilitate receptor trafficking (3–6). There are four arrestin subtypes expressed in mammals, and they clearly fall into two categories. One is the visual arrestins, which include arrestin-1 and arrestin-4 and are exclusively expressed at high levels in photoreceptors (7–9). Arrestin-1 demonstrates high specificity for its cognate receptor rhodopsin (10–13). The other category is the non-visual arrestins or β-arrestins, which include arrestin-2 (β-arrestin-1) and arrestin-3 (β-arrestin-2).3 Non-visual arrestins are expressed ubiquitously in all cells and tissues, with the highest expression levels in mature neurons (14–17). The broad distribution and receptor specificity of non-visual arrestins suggest that these two subtypes recognize and regulate the vast majority of GPCRs (18–20).
The four mammalian arrestin subtypes have greater than 50% amino acid conservation and a similar elongated two-domain structure in the basal state (4, 6, 14, 21–24). The two conserved domains, the N-domain and C-domain, are connected through a hinge region (Fig. 1). In the basal state, the C terminus folds back toward the N-domain and forms a highly conserved tripartite interaction with the N-domain, involving β-strand I and α-helix I (4, 21, 25, 26). Upon binding to its cognate GPCR rhodopsin, several conformational changes in arrestin-1 have been proposed and confirmed by studies using NMR, fluorescence quenching, and site-directed spin labeling electron paramagnetic resonance (EPR) spectroscopy techniques (25, 27–33). An EPR study using double electron electron resonance (DEER) combined with RosettaEPR modeling provided a global picture of the phosphorylated activated rhodopsin (PR*)-induced conformational changes in arrestin-1, which involve the release of the C-tail, the movement of the finger loop, the movement of a loop containing residue 139, and smaller changes in distal loops containing residues 157 and 344 at the tips of the N- and C-domains (31). Moreover, the DEER study found no interdomain movement of arrestin-1 induced by PR* binding, which rejects the closing “clam shell” model (31, 34). However, a recent crystal structure of preactivated p44, a naturally occurring variant of arrestin-1 in which the final 35 amino acid residues of the C terminus are replaced by a single alanine residue, revealed an unexpected 21° twist between the N- and C-domains (35). A model of the receptor-bound state of non-visual arrestins is necessary to elucidate the structural basis of arrestin-mediated signaling. In addition, truncated arrestin-2 was recently co-crystallized with the multiphosphorylated C terminus of the vasopressin V2 receptor (36). Although this structure was not obtained by co-crystallization of arrestin-2 with a receptor, the crystal structure of arrestin-2 in complex with an antibody fragment (Fab30) and the phosphorylated peptide (V2Rpp) provided the first glimpse of the possible active form of arrestin-2. Compared with the basal state of arrestin-2, this complex captured a conformation of arrestin-2 that involves a 20° twist between the N- and C-domains and conformational changes in the “finger loop” (residues 63–75), the loop containing residue 136 (residues 129–140), and the “lariat loop” (residues 274–300) (36). The observed consistency of interdomain rotation between the two crystal structures suggests that the activation mechanism may be conserved among all arrestins. Interestingly, a similar domain rotation was proposed in 2006 by modeling (37) on the basis of earlier findings that hinge deletions in arrestins impede receptor binding (38, 39).
FIGURE 1.

Crystal structure of arrestin-2 in the basal conformation (PDB entry 1G4M) with the residues studied by DEER spectroscopy shown as red CαCPK models. The backbone structure of the N domain is shown in gray, and the backbone structure of the C domain is shown in black. The major structural features of arrestin-2 are indicated.
Although crystal structures yield atomic details, there are still questions remaining: whether truncated arrestin-2 in complex with V2Rpp exhibits the same conformational changes as WT arrestin-2 in complex with full length receptor and to what extent the crystal-packing and the extra antibody fragment (Fab30) contribute to the observed conformational changes (40). Thus, to investigate the proteins under more physiological conditions, we utilized DEER spectroscopy, which provided distance measurements between two attached spin labels in arrestin-2 and arrestin-3 in the presence and absence of bound full-length phosphorylated, light-activated rhodopsin (PR*). DEER spectroscopy is a pulse EPR technique that detects the spin-spin interaction between two spin-labeled sites in the range of 20–80 Å. Because of the ability to measure long distances, DEER spectroscopy is advantageous to the study of protein conformational transitions and biomolecular associations. In this study, the intramolecular distance data give a global picture of the receptor-induced conformational changes in the non-visual arrestins. Multiple elements of the non-visual arrestins were specifically investigated, and the data revealed essential receptor binding-induced movements.
EXPERIMENTAL PROCEDURES
Site-directed Mutagenesis
Single cysteine substitutions were introduced into cysteineless arrestin-2 and arrestin-3 using QuikChange PCR (Stratagene). Double cysteine substitutions were created by introducing one cysteine mutation first and then using the single cysteine vector as a base to introduce the second mutation. All PCR primers were designed by us and purchased from IDT DNA. Mutations were verified by Retrogen Sequencing. Cysteineless arrestin-2 and arrestin-3 base mutants are fully functional in terms of receptor binding (39).
Protein Expression and Purification
Arrestin-2 and arrestin-3 were expressed and purified as described (22, 25, 41). Escherichia coli BL21 (DE3) cells were transformed with plasmids encoding each mutant and grown in Luria broth/ampicillin (100 mg/liter) at 30 °C and induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside. After lysis and ammonium sulfate precipitation, the protein was dissolved in buffer, and arrestin was purified by sequential chromatography on heparin-Sepharose, Q-Sepharose, and SP-Sepharose (GE Healthcare). Protein purity was verified by 15% SDS-PAGE, and concentrations were determined using a BCA assay (Pierce) with bovine serum albumin (BSA) as a standard. Proteins were concentrated to the final desired concentration with YM-10 centrifugal concentrators (Amicon). Rhodopsin was isolated from bovine rod outer segments, as described (42). Phosphorylation of rhodopsin by endogenous GRK1 in purified rod outer segments yielded 2.6 phosphates/rhodopsin (44).
Arrestin-Rhodopsin Binding Assay
Rhodopsin binding of arrestins was tested in the buffer used for the EPR experiments (50 mm MOPS, 100 mm NaCl, pH 7.0) as described (41). Purified spin-labeled arrestin mutants (4.5 μm) were incubated with 9 μm phosphorylated rhodopsin. After a 5-min incubation in room light at 37 °C, the arrestin/rhodopsin mixture was loaded onto a 100-μl 0.2 m sucrose cushion to separate bound and free arrestin by centrifugation for 20 min at 350,000 × g in an ultracentrifuge (Sorvall). Supernatants were carefully removed, and the rhodopsin pellets were resuspended in binding buffer (50 mm MOPS, 100 mm NaCl, pH 7.0). Equal amounts of arrestin/PR* input, pellet, and supernatant were subjected to 10% SDS-PAGE for direct comparison.
Spin Labeling
Purified double arrestin mutants were spin-labeled with a 40-fold molar excess of 4-maleimido-TEMPO (MAL-6) spin label and agitated for 12 h at 4 °C. Excess spin label was removed by extensive dialysis into buffer consisting of 50 mm MOPS, 100 mm NaCl, pH 7.0.
DEER Spectroscopy
DEER data were collected on a Q-band Bruker ELEXSYS E580 equipped with an EN5107D2 resonator with overcoupling at 80 K. Samples contained 20% deuterated glycerol as a cryoprotectant and were flash-frozen in a dry ice and acetone mixture. Acquired raw data after phase correction were background-corrected, plotted, and analyzed using DeerAnalysis2011 software freely available at the ETH Zürich Web site (43) and the LongDistances software program (44) provided by C. Altenbach (UCLA, Los Angeles, CA). Distance distributions were determined by fitting the corrected dipolar evolution data using model-free Tikhonov regularization (43) or the algorithms included in the LongDistances program. For distance distributions that are broad or complex, the mean distance values were used in the table to analyze conformational changes. To obtain the mean distances, the distance distributions were integrated and then normalized to the maximum amplitude. The mean distance was estimated as the value at 0.5 of the integrated intensity, and the values were reported in Tables 1 and 2. The upper reliable distance limit for each sample was determined based on the maximum data collection time (t) used in each of the DEER experiments according to the equation, d ≈ 5(t/2)⅓ (45) and is reflected in the x axis of each distance distribution plot. The expected distance between arrestin-2 pairs was predicted via the PRONOX program (46) by measuring the distances between rotameric configurations of the two spin labels attached to the highest resolution arrestin-2 crystal structure available (24) based on an experimentally derived library of allowed rotameric configurations. For comparison, free and receptor-bound state distances were determined for the free state crystal structure of arrestin-2 (PDB entry 1G4M) and the crystal structure of arrestin-2 in the presence of V2Rpp (PDB entry 4JQI), respectively.
TABLE 1.
Receptor binding-induced changes for 17 interspin distances in arrestin-2
Summary of receptor binding-induced changes for 17 interspin distances in arrestin-2. The expected distances between spin-labeled side chains were calculated using PRONOX, and the experimental DEER distances were determined from the best fit to the dipolar evolution curves (see Figs. 3–5). Arr2-free, basal state of arrestin-2; Arr2-V2Rpp, crystal structure of truncated arrestin-2 bound to the multiphosphorylated C-terminal peptide of V2 vasopressin receptor (36); Arr2+PR*, arrestin-2 bound to phosphorylated, light-activated rhodopsin. ΔDistance, distance change between the two residues upon PR* binding. ND, not determined.
| Arrestin-2 doubles | PRONOX Arr2-free | PRONOX Arr2 + V2Rpp | ΔDistance | DEER Arr2-free | DEER Arr2 + PR* | ΔDistance |
|---|---|---|---|---|---|---|
| Å | Å | Å | Å | Å | Å | |
| C-tail | ||||||
| 12/392 | 15 | ND | ND | 21 | 55 | 34 |
| 192/392 | 63 | ND | ND | 65 | 63 | −2 |
| Interdomain | ||||||
| 47/257 | 67 | 67 | 0 | 65 | 65 | 0 |
| 81/339 | 65 | 56 | −9 | 58 | 62 | 4 |
| 167/340 | 48 | 55 | 7 | 44 | 44 | 0 |
| 136 loop | ||||||
| 33/136 | 42 | 46 | 4 | 49 | 49 | 0 |
| 47/136 | 39 | 34 | −5 | 45 | 44 | −1 |
| 136/167 | 15 | 18 | 3 | 24 | 23 | −1 |
| 136/238 | 31 | 39 | 8 | 34 | 38 | 4 |
| 136/246 | 31 | 38 | 7 | 22, 35 | 23, 36 | 1, 1 |
| Finger loop | ||||||
| 68/167 | 14 | 26 | 12 | 21 | 25 | 4 |
| N-domain | ||||||
| 47/167 | 29 | 24 | −5 | 29 | 34 | 5 |
| 158/167 | 17 | 17 | 0 | 24 | 24 | 0 |
| C-domain | ||||||
| 192/238 | 32 | 39 | 7 | 32 | 40 | 8 |
| 192/339 | 17 | 22 | 5 | 25 | 29 | 4 |
| 234/340 | 13 | 19 | 6 | 27 | 25 | −2 |
| 238/339 | 27 | 30 | 3 | 30 | 33 | 3 |
TABLE 2.
Receptor binding-induced changes for 2 interspin distances in arrestin-3
Distance changes for two double-labeled arrestin-3 mutants. Arr3-free, inactive state of arrestin-3; Arr3 + PR*, arrestin-3 upon binding to light-activated and phosphorylated rhodopsin. ΔDistance, distance change between the two residues upon PR* binding.
| Arrestin-3 doubles | PRONOX, Arr3-free | DEER, Arr3-free | DEER, Arr3 + PR* | ΔDistance | |
|---|---|---|---|---|---|
| Å | Å | Å | Å | ||
| C-tail | 13/392 | 15 | 22 | 31, 43, 54 | 4, 16, 27 |
| Finger loop | 68/168 | 7 | 22, 43 | 24, 43 | 2, 0 |
RESULTS
Conformational Changes in Arrestin-2 upon Binding to PR*
To identify the conformational changes in arrestin-2 upon receptor binding, 17 pairs of residues were selected for the introduction of spin labels to measure intramolecular distances. Each double cysteine mutant was purified and spin-labeled. The 17 double mutants contain pairs of spin labels that fall into six groups and sample a variety of potential structural changes: three interdomain pairs, one pair to report on the finger loop, two to report on the C-tail movement, five pairs to report on the 136 loop, two within the N-domain, and four within the C-domain (Fig. 1 and Table 1). Because rhodopsin is a prototypical GPCR available as a purified protein in native membranes, PR* was used as the model receptor in these studies. Both non-visual arrestins specifically bind PR* (47). First, the functional consequences of the cysteine mutations and spin labeling of arrestin-2 were assessed by testing the ability of the purified spin-labeled arrestin-2 double mutants to bind to PR*. We found that all spin-labeled arrestin-2 mutants retain the ability to bind PR* at 80–100% of the wild type (WT) level except for 68/167, which binds PR* at ∼67% of WT level, based on densitometry analysis (Fig. 2). In addition, all arrestin proteins tested remained in the supernatant in the absence of PR* (data not shown). Thus, each of the selected double arrestin-2 mutants was able to functionally bind PR* at the molar ratios and concentrations used for the DEER experiments.
FIGURE 2.
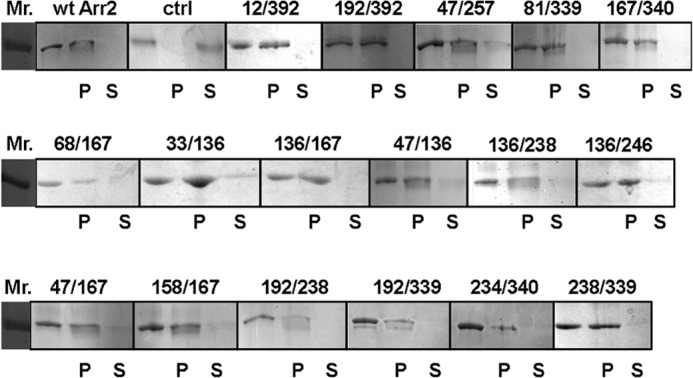
PR* binding of all spin-labeled arrestin-2 cysteine mutants used in this study. The ability of each arrestin-2 mutant to bind to PR* was tested. Wild-type arrestin-2 in the presence (wt Arr2) and absence (ctrl) of PR* are shown as positive and negative controls, respectively. Added arrestin-2 (blank), arrestin-2 in the pellet fraction (P), and arrestin-2 in the supernatant (S) fraction are shown for each protein. Arrestin binding to PR* is indicated by the presence of this protein in the pellet, whereas unbound arrestin is found in the supernatant fraction.
DEER data were collected for each double-cysteine arrestin-2 mutant for both the free and PR*-bound states. The distance distributions yield the range of distances between the two labels, and, the width and shape of the distribution contain information on the structural heterogeneity of the protein or the contribution of rotameric equilibria of the spin labeled side chains. For each mutant, the mean distance of the distribution was determined and shown as experimental distances in Table 1. The positive ΔDistance values indicate a distance increase between the two spin-labeled sites of arrestin-2 upon PR* binding, whereas a negative number indicates a decrease in the distance between sites. The results from each of the six groups of mutants are described in detail below.
The Release of the C-tail
The most dramatic distance change was observed for the C-tail of arrestin-2, which was probed by the spin-labeled double mutant A12C/A392C (Fig. 3A). In arrestin-1, the C-tail was shown to be released from the N-domain upon rhodopsin binding (25, 29, 31); therefore, the 12/392 mutant was chosen to monitor the C-tail movement in arrestin-2. Residue 392 is on the arrestin-2 C-tail, whereas residue 12 is located in β-strand I in the N-domain (Fig. 3F). The distance between positions 12 and 392 in the basal state of free arrestin-2 is short because the C-tail is anchored to the N-domain (24). Upon the addition of PR*, there is a 34-Å increase in the distance between 12 and 392, indicating that the C-tail is released. Interestingly, the width of the PR*-bound distance distribution is narrow, suggesting that the released C-tail may dock to a specific location elsewhere on the structure. This is in contrast to a very wide distance distribution upon PR* binding between homologous sites in arrestin-1, which suggested that the released arrestin-1 C-tail does not have a fixed position in the receptor-bound form (25, 29). To test whether the C-tail of arrestin-2 interacts with the C-domain upon receptor binding, another pair (M192C/A392C) was designed to identify the position of the released C-tail. Residue 192 is located on the edge of the C-domain, 63 Å away from 392 in the free state. If the C-tail folds back to the C-domain after its release from the N-domain, the distance between 192 and 392 was expected to become significantly shorter upon PR* binding. The free state DEER distance of 192/392 has a very long component at about 68 Å and a short population at 43 Å (Fig. 3B and Table 1). Upon PR* binding, we found no dramatic interspin distance change between 192 and 392; the DEER distances of the receptor-bound state still have similar distributions around 43 Å and a very long distance population at 63 Å. The absence of a dramatic distance decrease between 192 and 392 suggests that the released C-tail does not fold back to the C-domain.
FIGURE 3.

DEER analysis of the movement of the C-tail and arrestin-2 domains. Fits to the free (black) and PR*-bound (red) background-corrected dipolar evolution data (gray dots) are plotted on the left for the C-tail (A and B) and interdomain (C–E) mutants to illustrate the data quality and support the distance distribution data. The corresponding distance distributions are shown on the right as overlays for the free (black) and PR*-bound (red) states. F, the free state crystal structure (Protein Data Bank entry 1G4M) of arrestin-2 is labeled with each pair of double mutants (red spheres). Measured distances are shown as dotted lines.
Interdomain Distances
Global conformational changes in arrestin-2 were evaluated by monitoring interdomain distances between Y47C/E257C, V81C/A339C, and V167C/S340C. Spin labels at sites 47, 81, and 167 served as reference points on the N-domain, whereas sites 257, 339, and 340 were chosen as reference points on the C-domain. The DEER data did not reveal any obvious interspin distance change for the 47/257 and 167/340 pairs, suggesting that PR* binding does not induce a dramatic movement of the two domains relative to each other (Fig. 3, C and E). Moreover, we found slightly increased 81/339 interspin distances (Fig. 3D). Thus, the DEER data suggest that the N-domain and the C-domain do not undergo a “clam shell-like” closing movement.
Finger Loop Extension
As shown in Fig. 4A, a small distance change was observed between L68C and V167C, which is expected to reflect the finger loop movement induced by PR* binding. Site 68 is at the tip of the finger loop, and 167 is a rigid reference point on the N-domain. The free state distance is short because the finger loop is partially folded back onto the N-domain (Fig. 4A). Upon PR* binding, the distance between 68 and 167 was increased by 4 Å, suggesting that the finger loop extends. Despite dislocation of the finger loop, the narrow width of the distance distribution indicates that this loop has a relatively restricted motion upon binding to PR*.
FIGURE 4.

DEER analysis of the movement of the finger loop and the loop containing residue 136. Fits to the free (black) and PR*-bound (red) background-corrected dipolar evolution data (gray dots) are plotted on the left for the finger loop (A) and the loop containing residue 136 mutants (B–F) to illustrate the data quality and support the distance distribution data. The corresponding distance distributions are shown on the right as overlays for the free (black) and PR*-bound (red) states. G, the positions of the spin labels are shown as red CPK models on the crystal structure of the free state (PDB entry 1G4M) of arrestin-2. Measured distances are shown as dotted lines.
Movement of Loop Containing 136
To identify the movement of the loop containing residue 136, several double mutants were analyzed as shown in Fig. 4: L33C/T136C, Y47C/T136C, T136C/V167C, T136C/Y238C, and T136C/T246C. Residue 136 is located at the tip of a central loop, adjacent to the finger loop (Fig. 4G), and is predicted to be in a flexible region, especially upon receptor binding. According to the DEER results, the distance distribution between positions 33 and 136 on the N-domain did not change upon PR* binding (Fig. 4B and Table 1), but the distance distributions between positions 47 and 136 and 136 and 167 both demonstrated a small decrease upon PR* binding (Fig. 4, C and D). There is also a significant broadening of the distance distribution for 136/167 or a second peak at 32 Å. Residue 167 is in the middle of β-strand X in the N-domain, which is expected to be rigid, whereas 47 is located on a flexible loop at the tip of the N-domain (Fig. 4G). Decreased distances for the 47/136 and 136/167 pairs suggested that the 136 loop slightly moves toward the N-domain, and the broad distributions suggest that this loop remains highly flexible upon receptor binding. Spin labels at sites 238 and 246, which are both located in the C-domain, were chosen to investigate the distance changes between the central loop and the C-domain (Fig. 4, E and F). Broad distance distributions were observed for these two pairs. Specifically, two major distances were observed between 136 and 246 both in the absence and presence of PR*, and both distance populations were found to be increased by 1 Å upon PR* binding. Because residue 246 is located in the loop in the C-domain and adjacent to the 136 loop, the movement between 136 and 246 indicated a slight translation of the 136 loop along with a slight narrowing of the two distance distributions. It was also found that a distance increase of 4 Å occurred between 136 and 238. Taken together, these data suggest a limited movement of the 136 loop toward the N-domain and away from the C-domain upon PR* binding.
Intradomain Distances
Possible conformational changes induced by PR* binding within each domain were investigated using distances between two spin labels located either in the N-domain or in the C-domain. Intra-N-domain conformational changes induced by PR* binding were examined by monitoring the distances between the N-domain loops (Y47C and I158C) and the center of the N-domain (V167C) (Fig. 5G). We detected a 5-Å distance increase between sites 47 and 167 and no change in the distance between sites 158 and 167 (Fig. 5, A and B). As shown in Fig. 5G, M192C/Y238C, Y238C/A339C, and S234C/S340C were selected to measure distances from the tips of the C-domain loops to the center of the C-domain. With reference point 238 at the center of the C-domain, both 192/238 and 238/339 showed longer interspin distances upon the addition of PR*, with a clear 8- and 3-Å increase, respectively, indicating that the C-domain loops move away from 238 upon PR* binding (Fig. 5, C and D). However, the interspin distance between positions 234 and 340 showed a slight decrease upon PR* binding (Fig. 5E). Moreover, the movement between loops of the C-domain was detected by measuring the distance change between 192 and 339, which are located on the tips of adjacent loops (Fig. 5F). The distance distributions for 192/339 in the absence and presence of PR* were found to be broad with multiple populations. Because these populations are not well separated, the mean distances are determined for 192/339 in the absence and presence of PR* (Table 1) in order to include all distances and structural information. As a result, the mean distances for 192/339 revealed a 4-Å increase upon the addition of PR*. Interestingly, the distance distributions associated with these C-domain loops (192/238, 238/339, and 234/340) became narrower upon PR* binding, suggesting that receptor binding immobilized these C-domain loop residues. Overall, PR* binding results in small conformational changes in some of the loops within the C-domain and within the N-domain.
FIGURE 5.

DEER analysis of the conformational changes in the N- and C-domains of arrestin-2. Fits to the free (black) and PR*-bound (red) background-corrected dipolar evolution data (gray dots) are plotted on the left for the N-domain (A and B) and the C-domain mutants (C–F) to illustrate the data quality and support the distance distribution data. The corresponding distance distributions are shown on the right as overlays for the free (black) and PR*-bound (red) states. G, the positions of the spin labels are shown as red CPK models on the crystal structure of the free state (PDB entry 1G4M) of arrestin-2. Measured distances are shown as dotted lines.
Conformational Changes in Arrestin-3 upon Binding to PR*
For comparison, we also tested the other non-visual subtype, arrestin-3, using DEER to detect two possible conformational changes induced by receptor binding. Although arrestin-3 has a largely similar basal structure (22) and has some overlapping functions with arrestin-2 (6), receptor-induced structural changes in the two subtypes were reported to be distinct (47). To identify two key conformational changes upon receptor binding, spin-labeled S13C/A392C and D68C/V168C arrestin-3 mutants were used to monitor the movement of the C-tail and finger loop, respectively (Fig. 6B). Because the C-tail release and opening of the finger loop have been found in both arrestin-1 (31) and arrestin-2 (reported here), it is of great interest to determine whether these conformational changes are conserved. As shown in Fig. 6C, both spin-labeled arrestin-3 proteins retained the ability to bind PR*, which was again used as the model GPCR. The DEER data reveal that the interspin distance between positions 13 and 392 became longer when arrestin-3 binds to PR*, suggesting that the C-tail is released (Fig. 6A). Interestingly, three distinct distance populations coexist between spin labels at 13 and 392 in the receptor-bound conformation, which suggests that the C-tail of arrestin-3 probably adopts several specific conformations, in contrast to arrestin-2 with a single distance (Fig. 3) and to the completely flexible and free C-tail in receptor-bound arrestin-1 (29, 31, 33). Conformational changes induced by PR* binding in the finger loop were tested using 68/168 arrestin-3. Similar to arrestin-2, a small distance increase of 2 Å between positions 68 and 168 was observed, supporting the model in which the finger loop is slightly dislocated upon PR* binding. The relatively broad distance distribution of 68/168 in both free and PR*-bound state suggests that the finger loop in arrestin-3 is not fully extended and maintains a flexible range of positions even when it is bound to the receptor. This is similar to the movement of this loop in arrestin-2 (Fig. 4) and arrestin-1 (31).
FIGURE 6.
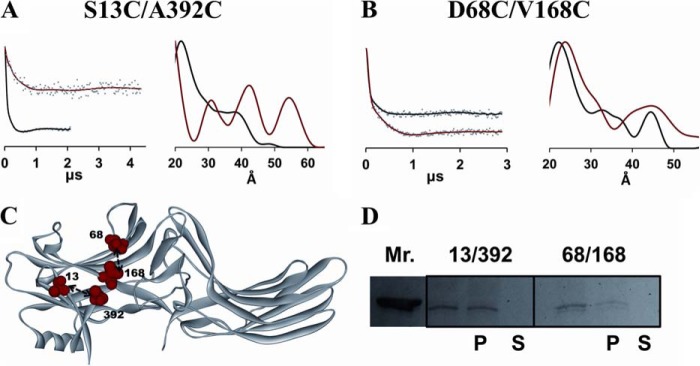
A and B, DEER analysis for two arrestin-3 spin-labeled double mutants. Fits to the free (black) and PR*-bound (red) background-corrected dipolar evolution data (gray dots) are plotted on the left to illustrate the data quality and support the distance distribution data. The corresponding distance distributions are shown on the right as overlays for the free (black) and PR*-bound (red) states. C, the free state crystal structure of arrestin-3 (PDB entry 3P2D) is labeled with the double mutants (red spheres) used for the DEER study. D, pull-down assay results for the spin-labeled arrestin-3 protein pairs.
DISCUSSION
Conformational changes in signaling proteins regulate their interactions with their binding partners. G protein-mediated signaling by the majority of GPCRs is terminated by a conserved two-step mechanism: selective phosphorylation of active receptors by G protein-coupled receptor kinases (48), followed by arrestin binding to the phosphorylated activated receptor (15, 34). In recent years, many studies have revealed a novel role for non-visual arrestins as multifunctional signaling scaffolds (49–51). By recruiting various non-receptor binding partners, such as trafficking and signaling proteins, to the receptor, non-visual arrestins connect activated GPCRs to diverse signaling pathways, which leads to the phosphorylation of numerous intracellular targets (52, 53). Interestingly, some non-receptor signaling proteins, such as ERK2, preferentially bind to GPCR-associated rather than to free arrestins (54, 55). Thus, understanding receptor binding-induced structural changes in non-visual arrestins is necessary to elucidate how receptor-bound non-visual arrestins mediate the second wave of signaling. Previous studies suggested that key mechanisms of arrestin activation by GPCRs are conserved in all subtypes and that it is highly possible that receptor binding induces similar small domain movements in non-visual arrestins (56–58). Here we used DEER distance measurements to identify the conformations of receptor-bound arrestin-2 and -3. Along with previous studies of receptor-induced changes in arrestin-1 (31, 33), the data provide an important comparison of the activation mechanism among these three arrestin subtypes. We found that both non-visual arrestins undergo major conformational changes similar to arrestin-1 upon PR* binding, which include the release of the C-tail and the opening up of the finger loop. However, the data suggest that arrestin-2 undergoes somewhat different structural changes upon PR* binding compared with arrestin-1, which might contribute to its lower receptor specificity (59), less pronounced selectivity for PR* (47, 56, 57), and ability to bind numerous non-receptor partners. The changes induced by PR* binding in arrestin-3 are also distinct, particularly in the position of the C-tail upon its release.
As shown in Table 1, in free arrestin-2, the DEER distances for most mutants are in reasonable agreement with those predicted from the crystal structure (24). However, DEER distances in the PR*-bound arrestin-2 are distinct from those in the crystal structure of truncated arrestin-2 bound to the multiphosphorylated vasopressin receptor peptide (V2Rpp) (36), with a few sites rendering up to 10-Å differences. The reasons for these differences probably include the absence of the full-length receptor in the structure of the arrestin-2·V2Rpp complex; the presence of the Fab fragment of the antibody, which turned out to be necessary to crystallize it (36); the non-physiological conditions required for crystallization; crystal packing forces; or all of the above. In addition, here we used full-length arrestin-2, in contrast to the crystal structure, where a truncated arrestin-2-(1–393), which was previously shown to be preactivated (56), was used for crystallization (36).
Previous EPR studies demonstrated that the C-tail of arrestin-1 becomes dynamically disordered upon receptor binding (25, 29, 31). Here we show that in both arrestin-2 and arrestin-3, the C-tail is released upon PR* binding. The consistency of this conformational change induced by receptor binding strongly suggests that all subtypes of arrestins share the same activation mechanism because the C-tail is involved in the activation process of arrestins. The arrestin C-tail, which is anchored to the N-domain via the three-element interaction in the free state, is important for constraining the inactive conformation of arrestins (29, 61). Here we show directly for the first time that the C termini of both non-visual arrestins are released upon receptor binding. This makes the release of the C-tail a hallmark of arrestin activation. Although EPR (25, 29, 31) and NMR studies (33, 62) of arrestin-1 suggest that the arrestin-1 C-tail becomes dynamically disordered upon PR* binding (25, 29, 31), the DEER results presented here for arrestin-2 and arrestin-3 suggest that the C-tail may adopt specific conformations after release. Thus, whereas the receptor induced C-tail release is conserved in arrestin-1, -2, and -3, the resulting position of this element in different subtypes is probably distinct. In the case of arrestin-2, upon release, the C-tail appears to assume a specific conformation, being located at a fixed distance from β-strand I, which is unlikely to be near the C-domain (Fig. 3) and may instead be located on rhodopsin. In contrast, in arrestin-3, there appear to be at least three distinct positions of the released C-tail (Fig. 6). Regardless of the location of the C-tail after its release, it is likely that this receptor-induced conformational change exposes binding motifs of the C-tail that interact with clathrin (63) and clathrin adaptor AP2 (64) and possibly some signaling proteins. Fixed position(s) of the C-tail in both non-visual subtypes can promote interactions with clathrin and other partners that arrestin-1 does not bind. Interestingly, even the C-tail of arrestin-1 can interact with AP2 upon its release by receptor binding (65).
The N- and C-domains in arrestin are connected by a hinge, the length of which is essential for receptor binding (38, 39). Because progressive deletions in the interdomain hinge reduced the ability of all arrestins to bind receptors, the “clam shell” model of arrestin activation, where the two domains move relative to each other closing in on the receptor, was proposed (34, 38). However, no evidence of such domain movement in arrestin-1 was found by DEER distance measurements (31). Here we also found no evidence of the clam shell-like motion of the N- and C-domains in arrestin-2 when it binds to PR*. Our data only reveal subtle changes in the interdomain distances, with only 81/339 showing small distance increases and both 47/257 and 167/340 revealing no interdomain distances changes. Notably, 47/257 and 167/340 double mutants bind PR* as well as the wild type arrestin-2 (Fig. 2). Thus, the absence of interdomain distance changes is not due to a binding deficiency. Domain rotation was predicted earlier based on hinge deletion data by molecular modeling (37). A long hinge provides the flexibility for an interdomain rotation upon arrestin activation, which can expose additional surfaces for interactions with non-receptor partners (66). Recent crystal structures of a preactivated short splice variant of arrestin-1 (35) and arrestin-2 in complex with multiphosphorylated vasopressin receptor peptide (36) support this new possibility of the interdomain rotation. In the overlay of the free (24) and bound to phosphorylated V2Rpp arrestin-2 crystal structures (36) (Fig. 7), the twisting and translation of the C-domain relative to the N-domain is evident. However, the interdomain distance changes predicted by these two crystal structures are not supported by the DEER results because the two spin-labeled pairs (81/339 and 167/340) specifically designed to detect the interdomain twisting revealed different distance changes compared with the crystal structure prediction (Table 1). Arrestins are highly flexible (67); therefore, it is possible that the crystal structure only captured one possible conformation. It is also possible that because bound full-length receptor was absent in both crystal structures of “active” arrestins, they do not represent the physiologically relevant arrestin·receptor complex.
FIGURE 7.
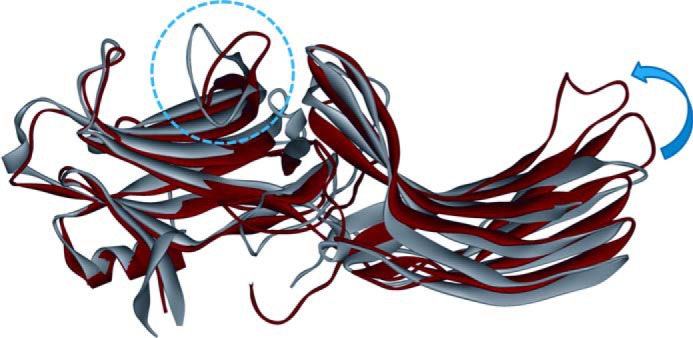
The crystal structures of inactive arrestin-2 (PDB entry 1G4M) and V2Rpp-bound arrestin-2 (PDB entry 4JQI) overlaid by alignment of the N domains. The inactive structure is shown in gray, and the phosphorylated receptor peptide-bound structure is shown in red. The conformational change of the finger loop is highlighted by the blue dotted circle. The interdomain rotation observed in the crystal structure is indicated by the blue arrow.
Another conformational change that was detected in both arrestin-2 and arrestin-3 upon PR* binding is the opening up of the finger loop (Tables 1 and 2). DEER results for 68/167 of arrestin-2 and 68/168 of arrestin-3 both show relatively small increases in distance upon PR* binding, indicating that the finger loop where 68 is located is extending. Because the finger loop dislocation has also been observed in arrestin-1 upon PR* binding (25, 31), this consistency among arrestin subtypes indicates that this conformational change is conserved. However, the conformation of the finger loop induced by receptor binding might be distinct in different arrestin subtypes. In the basal state, the finger loops of arrestin-1, -2, and -3 all form a folded conformation, which is confirmed by the shorter distances observed by DEER in the free state (27) (Figs. 4 and 6). Specifically, the DEER-derived distances for homologous spin-labeled pairs in arrestin-1 (72/173) (27), arrestin-2 (68/167), and arrestin-3 (68/168) all manifested similar results in the free state: 19, 21, and 22 Å, respectively. However, in the crystal structures of free arrestin-1, -2, and -3, the finger loops adopt a fully bent conformation (at least in some protomers in the arrestin-1 crystal tetramer), and the corresponding crystal structure-derived distances are predicted to be 12, 14, and 7 Å, respectively. Thus, the DEER measurements suggest that the finger loop is not fully bent in the free state, and this is true for arrestin-1, -2, and -3. Upon binding to PR*, the arrestin-1 finger loop was observed to move slightly toward the expected location of the receptor, and it is proposed to form a helical conformation and bind in or near the central cavity of the activated receptor (25, 30–32). As for arrestin-2, previous continuous wave EPR studies (12) identified that the finger loop is part of the arrestin-2 receptor binding surface by having more side chain flexibility in the free than in the receptor-bound state (12). This is also confirmed by our DEER data showing a narrower distance distribution in the PR*-bound state. Our DEER data also showed that the arrestin-2 finger loop extends slightly upon PR* binding (Fig. 4), which resembles the slight movement of the arrestin-1 finger loop (31). A similar movement was also detected in the arrestin-3 finger loop (Fig. 6). This is the first direct detection of the conformational change of the finger loop in arrestin-2 and -3. Small distance changes between the finger loop and the receptor-binding surface of the N-domain revealed by DEER measurements are virtually the same in arrestin-1 (3 Å), arrestin-2 (4 Å), and arrestin-3 (2 Å). Thus, upon PR* binding, the finger loop of arrestin-1, -2, and -3 undergoes a transition from a partially folded to more extended conformation. This conformational change can be visualized in the alignment of the inactive arrestin-2 and V2Rpp-bound arrestin-2 crystal structures (Fig. 7), wherein the finger loop is shown to open up slightly upon V2Rpp binding. Notably, the distance distributions involving finger loops in homologous spin-labeled pairs of arrestin-1, -2, and -3 revealed the plasticity of the finger loop. Unlike arrestin-1 and -2, the finger loop of arrestin-3 is found to be more flexible in both the free and receptor-bound states. Therefore, it is likely that the finger loop of arrestin-1 and -2 is more essential in their interaction with incoming receptors by contacting the receptors directly, whereas the finger loop of arrestin-3 is only slightly affected by the receptor binding and may play a less important role.
The other striking conformational change in arrestins that has drawn a lot of attention is the movement of the central loop containing residue 139 or 136, in arrestin-1 and -2, respectively. In arrestin-1, this loop has been designated as the 139-loop and well investigated by several studies (25, 31, 68). The 139-loop of arrestin-1 is located in the center of the receptor binding surface, next to several elements directly engaged by PR*. It was proposed that the 139-loop probably undergoes a large conformational change in order to facilitate receptor binding by making the finger loop and adjacent elements more accessible (25, 31). Notably, there is a remarkable structural conservation of this loop within the arrestin family (4, 22–24), so it is likely that the central loop in arrestin-2 and -3 also enhances their stability and selectivity. Here we focused on the central loop (136-loop) of arrestin-2 by measuring distance changes between it and different reference sites on both the N- and C-domains in the absence and presence of PR*. Our DEER data for the arrestin-2 136-loop do not show structural changes upon PR* binding as dramatic as those observed in arrestin-1 (31). We found that the 136-loop in arrestin-2 moved slightly toward the N-domain and away from the C-domain upon PR* binding. Moreover, broad distance distributions between 136 and the reference sites in the absence and presence of PR* strongly suggest that this loop has high flexibility in both the basal and active states. The predicted crystal structure distances (36) (Table 1) reveal larger changes at the central loop than the distance changes reflected by the DEER data measured for arrestin-2 in solution. Experimental conditions used to collect DEER data more closely resemble physiological conditions and reveal the dynamic flexibility of the central loop in both the basal state and the receptor bound state. This kind of flexibility of the central loop may help arrestin-2 to interact with hundreds of GPCRs by fine-tuning its structure (Fig. 4 and Table 1).
Finally, the intradomain conformational changes in arrestin-2 induced by PR* binding were investigated using distances between pairs of spin-labeled residues located in the N- or the C-domain. Residues 47, 158, 192, 339, and 340 are all located in plastic loops of arrestin-2 (Fig. 5) that are likely to undergo large conformational changes during arrestin-receptor interaction. Previous continuous wave EPR studies mapped the receptor-binding surface of arrestin-2, which is quite similar to that of arrestin-1, covering the concave sides of both domains (12, 13, 18, 25, 60). The distal loops are on the periphery of the receptor-binding surface of both arrestin-2 domains, which suggests that they may not be in direct contact with the receptor. Interestingly, our DEER data revealed dramatic structural changes at the distal tips of both the N-domain (47/167) and C-domain (192/238, 238/339, 192/339, and 234/340) (Table 1). Compared with the V2Rpp-bound crystal structure of arrestin-2, the relatively large structural rearrangement of C-domain loops based on the DEER data is also observed in the overlay of the crystal structures (Fig. 7). It is tempting to speculate that these plastic loops in the C-domain might be involved in interactions with other protein partners after the dramatic conformational change upon receptor binding. The movement of the N-domain loops indicated by the DEER data is also observed in the crystal structure overlay (Fig. 7), suggesting that the entire N-domain of arrestin-2 does not need to undergo large structural rearrangement to either facilitate receptor binding or contribute to further interaction with other proteins.
Here we used site-directed spin labeling and DEER spectroscopy to measure intramolecular distances in arrestin-2 and arrestin-3 in the basal state and bound to the model GPCR rhodopsin. We provided evidence that arrestin binding to phosphorylated activated rhodopsin induces similar conformational changes in arrestin-2 and arrestin-3, including the C-tail release and partial extension of the finger loop. We did not detect “clam shell” interdomain movement or domain twisting in arrestin-2 upon PR* binding. We found that the 136-loop of arrestin-2 undergoes only a small movement toward the N-domain and maintains its structural flexibility upon PR* binding. This may help to fine-tune the arrestin-2 structure to facilitate its binding to a wide variety of GPCRs, in contrast to strict specificity of arrestin-1 for rhodopsin (12, 13, 47, 59). Last, our data reveal that the distal loops on the C- and the N-domain, which are unlikely to contact the receptor directly, undergo more significant conformational changes upon PR* binding than those detected in homologous regions of arrestin-1 (31). These changes might prepare the non-visual arrestins for binding numerous non-receptor partners. In summary, DEER data providing dynamic structural information, complemented by the crystal structures, give a comprehensive overview of receptor-induced conformational changes in arrestin-2 and provide a comparison of the receptor binding-induced changes in different members of the arrestin family. Comparisons of conformational changes induced in arrestin-2 or arrestin-3 by their binding to different GPCRs will be necessary to identify how these two non-visual subtypes interact with hundreds of different receptors and dozens of non-receptor partners.
This work was supported, in whole or in part, by National Institutes of Health (NIH) Grants GM077561, EY011500, and GM081756 (to V. V. G.). The DEER instrumentation was supported by NIH Grants S10RR022422 and S10OD011937, and data were collected at the National Biomedical EPR Center (supported by NIH Grant P41EB001980).
We use the systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48-kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database).
- GPCR
- G protein-coupled receptor
- DEER
- double electron electron resonance
- PR*
- phosphorylated activated rhodopsin
- PDB
- Protein Data Bank.
REFERENCES
- 1. Lagerström M. C., Schiöth H. B. (2008) Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat. Rev. Drug Discov. 7, 339–357 [DOI] [PubMed] [Google Scholar]
- 2. Pierce K. L., Premont R. T., Lefkowitz R. J. (2002) Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 3, 639–650 [DOI] [PubMed] [Google Scholar]
- 3. Lefkowitz R. J., Shenoy S. K. (2005) Transduction of receptor signals by β-arrestins. Science 308, 512–517 [DOI] [PubMed] [Google Scholar]
- 4. Hirsch J. A., Schubert C., Gurevich V. V., Sigler P. B. (1999) A model for arrestin's regulation: the 2.8 Å crystal structure of visual arrestin. Cell 97, 257–269 [DOI] [PubMed] [Google Scholar]
- 5. DeWire S. M., Ahn S., Lefkowitz R. J., Shenoy S. K. (2007) β-Arrestins and Cell Signaling. Annu. Rev. Physiol. 69, 483–510 [DOI] [PubMed] [Google Scholar]
- 6. Gurevich E. V., Gurevich V. V. (2006) Arrestins are ubiquitous regulators of cellular signaling pathways. Genome Biol. 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hanson S. M., Gurevich E. V., Vishnivetskiy S. A., Ahmed M. R., Song X., Gurevich V. V. (2007) Each rhodopsin molecule binds its own arrestin. Proc. Natl. Acad. Sci. U.S.A. 104, 3125–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Strissel K. J., Sokolov M., Trieu L. H., Arshavsky V. Y. (2006) Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J. Neurosci. 26, 1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Song X., Vishnivetskiy S. A., Seo J., Chen J., Gurevich E. V., Gurevich V. V. (2011) Arrestin-1 expression in rods: balancing functional performance and photoreceptor health. Neuroscience 174, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Maeda T., Imanishi Y., Palczewski K. (2003) Rhodopsin phosphorylation: 30 years later. Prog. Retin. Eye Res. 22, 417–434 [DOI] [PubMed] [Google Scholar]
- 11. Gurevich V. V., Hanson S. M., Song X., Vishnivetskiy S. A., Gurevich E. V. (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog. Retin. Eye Res. 30, 405–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vishnivetskiy S. A., Gimenez L. E., Francis D. J., Hanson S. M., Hubbell W. L., Klug C. S., Gurevich V. V. (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J. Biol. Chem. 286, 24288–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vishnivetskiy S. A., Hosey M. M., Benovic J. L., Gurevich V. V. (2004) Mapping the arrestin-receptor interface. J. Biol. Chem. 279, 1262–1268 [DOI] [PubMed] [Google Scholar]
- 14. Attramadal H., Arriza J. L., Aoki C., Dawson T. M., Codina J., Kwatra M. M., Snyder S. H., Caron M. G., Lefkowitz R. J. (1992) β-Arrestin2, a novel member of the arrestin/β-arrestin gene family. J. Biol. Chem. 267, 17882–17890 [PubMed] [Google Scholar]
- 15. Lohse M. J., Benovic J. L., Codina J., Caron M. G., Lefkowitz R. J. (1990) β-Arrestin: a protein that regulates β-adrenergic receptor function. Science 248, 1547–1550 [DOI] [PubMed] [Google Scholar]
- 16. Gurevich E. V., Benovic J. L., Gurevich V. V. (2002) Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience 109, 421–436 [DOI] [PubMed] [Google Scholar]
- 17. Gurevich E. V., Benovic J. L., Gurevich V. V. (2004) Arrestin2 expression selectively increases during neural differentiation. J. Neurochem. 91, 1404–1416 [DOI] [PubMed] [Google Scholar]
- 18. Gurevich V. V., Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G protein-coupled receptors. Pharmacol. Ther. 110, 465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krupnick J. G., Goodman O. B., Jr., Keen J. H., Benovic J. L. (1997) Arrestin/clathrin interaction: localization of the clathrin binding domain of nonvisual arrestins to the carboxy terminus. J. Biol. Chem. 272, 15011–15016 [DOI] [PubMed] [Google Scholar]
- 20. Pan L., Gurevich E. V., Gurevich V. V. (2003) The nature of the arrestin·receptor complex determines the ultimate fate of the internalized receptor. J. Biol. Chem. 278, 11623–11632 [DOI] [PubMed] [Google Scholar]
- 21. Milano S. K., Pace H. C., Kim Y.-M., Brenner C., Benovic J. L. (2002) Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry 41, 3321–3328 [DOI] [PubMed] [Google Scholar]
- 22. Zhan X., Gimenez L. E., Gurevich V. V., Spiller B. W. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., Gurevich V. V. (2005) Crystal structure of cone arrestin at 2.3 Å: evolution of receptor specificity. J. Mol. Biol. 354, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 24. Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., Schubert C. (2001) Crystal structure of β-arrestin at 1.9 Å: possible mechanism of receptor binding and membrane translocation. Structure 9, 869–880 [DOI] [PubMed] [Google Scholar]
- 25. Hanson S. M., Francis D. J., Vishnivetskiy S. A., Kolobova E. A., Hubbell W. L., Klug C. S., Gurevich V. V. (2006) Differential interaction of spin-labeled arrestin with inactive and active phosphorhodopsin. Proc. Natl. Acad. Sci. U.S.A. 103, 4900–4905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vishnivetskiy S. A., Paz C. L., Schubert C., Hirsch J. A., Sigler P. B., Gurevich V. V. (1999) How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 274, 11451–11454 [DOI] [PubMed] [Google Scholar]
- 27. Palczewski K., Riazance-Lawrence J. H., Johnson W. C., Jr. (1992) Structural properties of arrestin studied by chemical modification and circular dichroism. Biochemistry 31, 3902–3906 [DOI] [PubMed] [Google Scholar]
- 28. Ohguro H., Palczewski K., Walsh K. A., Johnson R. S. (1994) Topographic study of arrestin using differential chemical modifications and hydrogen/deuterium exchange. Protein Sci. 3, 2428–24234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vishnivetskiy S. A., Francis D., Van Eps N., Kim M., Hanson S. M., Klug C. S., Hubbell W. L., Gurevich V. V. (2010) The role of arrestin α-helix I in receptor binding. J. Mol. Biol. 395, 42–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sommer M. E., Farrens D. L., McDowell J. H., Weber L. A., Smith W. C. (2007) Dynamics of arrestin-rhodopsin interactions: loop movement is involved in arrestin activation and receptor binding. J. Biol. Chem. 282, 25560–25568 [DOI] [PubMed] [Google Scholar]
- 31. Kim M., Vishnivetskiy S. A., Van Eps N., Alexander N. S., Cleghorn W. M., Zhan X., Hanson S. M., Morizumi T., Ernst O. P., Meiler J., Gurevich V. V., Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Feuerstein S. E., Pulvermüller A., Hartmann R., Granzin J., Stoldt M., Henklein P., Ernst O. P., Heck M., Willbold D., Koenig B. W. (2009) Helix formation in arrestin accompanies recognition of photoactivated rhodopsin. Biochemistry 48, 10733–10742 [DOI] [PubMed] [Google Scholar]
- 33. Zhuang T., Chen Q., Cho M.-K., Vishnivetskiy S. A., Iverson T. M., Gurevich V. V., Sanders C. R. (2013) Involvement of distinct arrestin-1 elements in binding to different functional forms of rhodopsin. Proc. Natl. Acad. Sci. U.S.A. 110, 942–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gurevich V. V., Gurevich E. V. (2004) The molecular acrobatics of arrestin activation. Trends Pharmacol. Sci. 25, 105–111 [DOI] [PubMed] [Google Scholar]
- 35. Kim Y. J., Hofmann K. P., Ernst O. P., Scheerer P., Choe H. W., Sommer M. E. (2013) Crystal structure of pre-activated arrestin p44. Nature 497, 142–146 [DOI] [PubMed] [Google Scholar]
- 36. Shukla A. K., Manglik A., Kruse A. C., Xiao K., Reis R. I., Tseng W. C., Staus D. P., Hilger D., Uysal S., Huang L. Y., Paduch M., Tripathi-Shukla P., Koide A., Koide S., Weis W. I., Kossiakoff A. A., Kobilka B. K., Lefkowitz R. J. (2013) Structure of active β-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature 497, 137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Modzelewska A., Filipek S., Palczewski K., Park P. S. (2006) Arrestin interaction with rhodopsin: conceptual models. Cell Biochem. Biophys. 46, 1–15 [DOI] [PubMed] [Google Scholar]
- 38. Vishnivetskiy S. A., Hirsch J. A., Velez M.-G., Gurevich Y. V., Gurevich V. V. (2002) Transition of arrestin into the active receptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 277, 43961–43967 [DOI] [PubMed] [Google Scholar]
- 39. Hanson S. M., Cleghorn W. M., Francis D. J., Vishnivetskiy S. A., Raman D., Song X., Nair K. S., Slepak V. Z., Klug C. S., Gurevich V. V. (2007) Arrestin mobilizes signaling proteins to the cytoskeleton and redirects their activity. J. Mol. Biol. 368, 375–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rapp C. S., Pollack R. M. (2005) Crystal packing effects on protein loops. Proteins 60, 103–109 [DOI] [PubMed] [Google Scholar]
- 41. Gurevich V. V., Benovic J. L. (2000) Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 315, 422–437 [DOI] [PubMed] [Google Scholar]
- 42. Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., Gurevich V. V. (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J. Biol. Chem. 282, 32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jeschke G., Chechik V., Ionita P., Godt A., Zimmermann H., Banham J., Timmel C., Hilger D., Jung H. (2006) DeerAnalysis2006: a comprehensive software package for analyzing pulsed ELDOR data. Appl. Magn. Reson. 30, 473–498 [Google Scholar]
- 44. Toledo Warshaviak D., Khramtsov V. V., Cascio D., Altenbach C., Hubbell W. L. (2013) Structure and dynamics of an imidazoline nitroxide side chain with strongly hindered internal motion in proteins. J. Magn. Reson. 232, 53–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jeschke G. (2012) DEER Distance Measurements on Proteins. Annu. Rev. Phys. Chem. 63, 419–446 [DOI] [PubMed] [Google Scholar]
- 46. Hatmal M. M., Li Y., Hegde B. G., Hegde P. B., Jao C. C., Langen R., Haworth I. S. (2012) Computer modeling of nitroxide spin labels on proteins. Biopolymers 97, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Gurevich V. V., Dion S. B., Onorato J. J., Ptasienski J., Kim C. M., Sterne-Marr R., Hosey M. M., Benovic J. L. (1995) Arrestin interactions with G protein-coupled receptors: direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 270, 720–731 [DOI] [PubMed] [Google Scholar]
- 48. Gurevich E. V., Tesmer J. J., Mushegian A., Gurevich V. V. (2012) G protein-coupled receptor kinases: more than just kinases and not only for GPCRs. Pharmacol. Ther. 133, 40–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wei H., Ahn S., Shenoy S. K., Karnik S. S., Hunyady L., Luttrell L. M., Lefkowitz R. J. (2003) Independent β-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. U.S.A. 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. DeFea K. A. (2007) Stop that cell! β-Arrestin-dependent chemotaxis: a tale of localized actin assembly and receptor desensitization. Annu. Rev. Physiol. 69, 535–560 [DOI] [PubMed] [Google Scholar]
- 51. Shenoy S. K., McDonald P. H., Kohout T. A., Lefkowitz R. J. (2001) Regulation of receptor fate by ubiquitination of activated β2-adrenergic receptor and β-arrestin. Science 294, 1307–1313 [DOI] [PubMed] [Google Scholar]
- 52. Xiao K., McClatchy D. B., Shukla A. K., Zhao Y., Chen M., Shenoy S. K., Yates J. R., 3rd, Lefkowitz R. J. (2007) Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U.S.A. 104, 12011–12016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Xiao K., Sun J., Kim J., Rajagopal S., Zhai B., Villén J., Haas W., Kovacs J. J., Shukla A. K., Hara M. R., Hernandez M., Lachmann A., Zhao S., Lin Y., Cheng Y., Mizuno K., Ma'ayan A., Gygi S. P., Lefkowitz R. J. (2010) Global phosphorylation analysis of β-arrestin-mediated signaling downstream of a seven transmembrane receptor (7TMR). Proc. Natl. Acad. Sci. U.S.A. 107, 15299–15304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Coffa S., Breitman M., Hanson S. M., Callaway K., Kook S., Dalby K. N., Gurevich V. V. (2011) The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One 6, e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Luttrell L. M., Roudabush F. L., Choy E. W., Miller W. E., Field M. E., Pierce K. L., Lefkowitz R. J. (2001) Activation and targeting of extracellular signal-regulated kinases by β-arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A. 98, 2449–2454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kovoor A., Celver J., Abdryashitov R. I., Chavkin C., Gurevich V. V. (1999) Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 274, 6831–6834 [DOI] [PubMed] [Google Scholar]
- 57. Celver J., Vishnivetskiy S. A., Chavkin C., Gurevich V. V. (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043–9048 [DOI] [PubMed] [Google Scholar]
- 58. Breitman M., Kook S., Gimenez L. E., Lizama B. N., Palazzo M. C., Gurevich E. V., Gurevich V. V. (2012) Silent scaffolds: inhibition of JNK3 activity in the cell by a dominant-negative arrestin-3 mutant. J. Biol. Chem. 287, 19653–19664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gimenez L. E., Vishnivetskiy S. A., Baameur F., Gurevich V. V. (2012) Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J. Biol. Chem. 287, 29495–29505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Skegro D., Pulvermüller A., Krafft B., Granzin J., Hofmann K. P., Büldt G., Schlesinger R. (2007) N-terminal and C-terminal domains of arrestin both contribute in binding to rhodopsin. Photochem. Photobiol. 83, 385–392 [DOI] [PubMed] [Google Scholar]
- 61. Vishnivetskiy S. A., Schubert C., Climaco G. C., Gurevich Y. V., Velez M. G., Gurevich V. V. (2000) An additional phosphate-binding element in arrestin molecule: implications for the mechanism of arrestin activation. J. Biol. Chem. 275, 41049–41057 [DOI] [PubMed] [Google Scholar]
- 62. Zhuang T., Vishnivetskiy S. A., Gurevich V. V., Sanders C. R. (2010) Elucidation of IP6 and heparin interaction sites and conformational changes in arrestin-1 by solution NMR. Biochemistry 49, 10473–10485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Goodman O. B., Jr., Krupnick J. G., Santini F., Gurevich V. V., Penn R. B., Gagnon A. W., Keen J. H., Benovic J. L. (1996) β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383, 447–450 [DOI] [PubMed] [Google Scholar]
- 64. Laporte S. A., Oakley R. H., Zhang J., Holt J. A., Ferguson S. S., Caron M. G., Barak L. S. (1999) The β2-adrenergic receptor/βarrestin complex recruits the clathrin adaptor AP-2 during endocytosis. Proc. Natl. Acad. Sci. U.S.A. 96, 3712–3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moaven H., Koike Y., Jao C. C., Gurevich V. V., Langen R., Chen J. (2013) Visual arrestin interaction with clathrin adaptor AP-2 regulates photoreceptor survival in the vertebrate retina. Proc. Natl. Acad. Sci. U.S.A. 110, 9463–9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gurevich V. V., Gurevich E. V. (2003) The new face of active receptor bound arrestin attracts new partners. Structure 11, 1037–1042 [DOI] [PubMed] [Google Scholar]
- 67. Carter J. M., Gurevich V. V., Prossnitz E. R., Engen J. R. (2005) Conformational differences between arrestin2 and pre-activated mutants as revealed by hydrogen exchange mass spectrometry. J. Mol. Biol. 351, 865–878 [DOI] [PubMed] [Google Scholar]
- 68. Vishnivetskiy S. A., Baameur F., Findley K. R., Gurevich V. V. (2013) Critical role of the central 139-loop in stability and binding selectivity of arrestin-1. J. Biol. Chem. 288, 11741–11750 [DOI] [PMC free article] [PubMed] [Google Scholar]