

Background: Wnt5a is involved in inflammation.
Results: Wnt5a promotes an inflammatory response by up-regulating chemokines and cytokines via the NF-κB and MAPK pathways in HDPCs, leading to macrophage migration.
Conclusion: Wnt5a, as an inflammatory mediator, drives the integration of cytokines and chemokines to control dental pulp inflammation.
Significance: Wnt5a-mediated inflammation occurs downstream of TNF-α.
Keywords: Chemokine, Cytokine, Inflammation, Stromal Cell, Wnt Signaling, Wnt5a, Dental Pulp Cells
Abstract
Wnt5a has been found recently to be involved in inflammation regulation through a mechanism that remains unclear. Immunohistochemical staining of infected human dental pulp and tissue from experimental dental pulpitis in rats showed that Wnt5a levels were increased. In vitro, Wnt5a was increased 8-fold in human dental pulp cells (HDPCs) after TNF-α stimulation compared with control cells. We then investigated the role of Wnt5a in HDPCs. In the presence of TNF-α, Wnt5a further increased the production of cytokines/chemokines, whereas Wnt5a knockdown markedly reduced cytokine/chemokine production induced by TNF-α. In addition, in HDPCs, Wnt5a efficiently induced cytokine/chemokine expression and, in particular, expression of IL-8 (14.5-fold) and CCL2 (25.5-fold), as assessed by a Luminex assay. The cytokine subsets regulated by Wnt5a overlap partially with those induced by TNF-α. However, no TNF-α and IL-1β was detected after Wnt5a treatment. We then found that Wnt5a alone and the supernatants of Wnt5a-treated HDPCs significantly increased macrophage migration, which supports a role for Wnt5a in macrophage recruitment and as an inflammatory mediator in human dental pulp inflammation. Finally, Wnt5a participates in dental pulp inflammation in a MAPK-dependent (p38-, JNK-, and ERK-dependent) and NF-κB-dependent manner. Our data suggest that Wnt5a, as an inflammatory mediator that drives the integration of cytokines and chemokines, acts downstream of TNF-α.
Introduction
Wnt signaling is involved in myriad biological processes, including embryonic development, tissue regeneration, and human disease (1–4). The signaling pathway is initiated by binding of Wnts to complex Frizzled receptors (5, 6). Wnt signaling pathways can be classified into β-catenin-dependent (canonical) or β-catenin-independent (non-canonical) (1, 7, 8). The β-catenin-dependent pathway regulates the stability of β-catenin as well as specific Lef/Tcf family transcription factors and, ultimately, induces the transcription of many target genes (8–10). The β-catenin-independent pathway involves the calcium/nuclear factor of activated T cells, PKC, and JNK for signal transduction (2, 11, 12).
Wnt5a participates in a β-catenin-independent pathway and has been found recently to be involved in inflammation (13). Wnt5a has been shown to be expressed in several inflammatory diseases, such as atherosclerosis, rheumatoid arthritis, and periodontitis (14–17). In synoviocytes from rheumatoid arthritis patients, the expression of Wnt5a and Frizzled5 (Fzd5) was enhanced significantly (15), and its blockade inhibited synoviocyte activation (18). More recently, Wnt5a was found to be highly expressed in synovial tissues in a mouse model of rheumatoid arthritis where inhibition of Wnt5a-Ror2 signaling suppressed bone loss (19). These results indicate that Wnt5a may be a therapeutic target for the treatment of rheumatoid arthritis and osteoporosis.
Wnt5a expression can be induced in activated macrophages, antigen-presenting cells, endothelial cells, and bone marrow mesenchymal stem cells (BMSCs)2 after inflammatory stimulation (20–24). Besides its induction during inflammation, Wnt5a itself has been found to have regulatory functions in infection (53). Several studies indicate a pathobiological role for Wnt5a in inflammatory diseases, where it promotes the production of proinflammatory cytokines (IL-1β, IL-6, IL-12, and IL-15) that are dependent on activation of the NF-κB pathway and, in turn, increased macrophage migration. Paradoxically, a number of articles have recently claimed that Wnt5a induces anti-inflammatory cytokines, such as IL-10, in macrophages and dendritic cells (21, 25, 26). Bergenfelz et al. (25) demonstrated that, under proinflammatory conditions, Wnt5a induces immunosuppressive macrophages. The suppressive phenotype induced by Wnt5a is associated with the induction of IL-10 and inhibition of the classical TLR4-NF-κB signaling pathway. In addition, they found that Wnt5a-induced inhibition is active both in LPS-stimulated macrophages and in patients with sepsis (25). Meanwhile, Oderup et al. (26) found that Wnt5a directly stimulated IL-10 expression and inhibited IL-6 expression in murine dendritic cells in response to viral mimics. The complex effects and mechanisms of Wnt5a in regulating inflammation remain to be clarified.
Dental pulp inflammation is mainly caused by dental caries that results from bacterial infection. Such inflammation or infection nearly always causes pain and suffering and can result in serious fatal systemic infections if left untreated (28). Dental pulp inflammation may initiate repair of dentinogenesis to eliminate the insult and block the route of infection (29, 30). However, if the source of infection (i.e. caries) is not eliminated, regardless of how minute, the continued infection can promote pulpitis and result in total pulp necrosis that can lead to total loss of the entire dental pulp. Despite the reported clinical success of root canal treatments, endodontically treated teeth often become devitalized and brittle and, thus, susceptible to postoperative fracture and other complications, including reinfection because of coronal leakage or microleakage (31, 32).
In pulpitis tissue, HDPCs, the main dental pulp component, can interact with immune cells to secrete significant amounts of inflammatory cytokines and chemokines locally. These cytokines and chemokines may promote pulpitis progression, tissue destruction, or regulate inflammatory responses to eliminate pathogens (33, 34). These finely tuned cytokine networks can preserve the balance between pro- and anti-inflammation to create a favorable environment for tissue repair (35, 36, 54).
Several studies report that Wnt5a plays a critical role in inflammatory processes associated with skeletal tissue (19, 23, 37). Osteoblast cells express Wnt5a, whereas osteoclast precursors express Ror2. Wnt5a-Ror2 signaling, together, contributes to bone resorption in both physiological and pathological states (19). Dental pulp cells have similar properties as bone cells (38), with HDPCs participating in dental pulp inflammation (34) in a way that is similar to osteoblastic cells (40, 41). However, little is known about the role of Wnt5a in dental pulp inflammation. We hypothesize that inflammatory stimuli could induce Wnt5a expression in dental pulp and that the resulting increased Wnt5a levels could play important roles in dental pulp inflammation.
In this study we show that Wnt5a levels were increased in tissues from dental pulpitis patients and also in an experimental model of dental pulpitis in rats. Similarly, TNF-α highly induced Wnt5a expression in HDPCs in vitro. By applying rhWNT5A, we found that Wnt5a increased the expression of cytokines (IL-6 and IL-8) and chemokines (CCL2 and CCL5) through distinct signaling pathways. Notably, TNF-α and IL-1β were not detected by a Luminex assay after human/mouse recombinant WNT5A (rhWNT5A) treatment. Wnt5a promoted basal TNF-α-induced cytokine/chemokine production, whereas Wnt5a knockdown markedly reduced cytokine/chemokine levels. Lastly, Wnt5a alone and supernatants of Wnt5a-treated HDPCs promoted macrophage migration. Together, these results indicate that Wnt5a-driven integration of cytokine and chemokine production occurs downstream of TNF-α action and in a MAPK-dependent (p38-, JNK-, and ERK-dependent) and NF-κB-dependent manner. Thus, this study reveals a critical role for Wnt5a in regulating dental inflammatory processes and points to its underlying molecular mechanism.
EXPERIMENTAL PROCEDURES
Collection of Human Dental Pulp Tissue
The entire study was approved by the human research committee of the West China School of Stomatology, Sichuan University, and performed after written informed consent from patients was obtained. Dental pulp samples were collected from five patients with dental pulpitis. The diagnosis of irreversible pulpitis was determined by endodontic specialists on the basis of clinical assessment, including a history of spontaneous pain and intense, lingering pain in response to cold stimulus, and by subsequent H&E staining. Pulp tissue was also collected from healthy control subjects undergoing orthodontic tooth extraction.
Immunohistochemical Staining
Immunostaining was performed on formalin-fixed, paraffin-embedded tissue. For immunohistochemistry, paraffin sections were dewaxed in xylene, rehydrated with distilled water, and then subjected to antigen retrieval for 30 min at 95 °C. The slides were subsequently incubated overnight at 4 °C with the following antibodies: Wnt5a (1:13, R&D Systems, Minneapolis, MN), Wnt5a (1:25, Abcam, Cambridge, MA), TNF-α (1:800, Abcam), CD68 (1:100, Dako Cytomation, Glostrup, Denmark), Vimentin (1:100, Abcam), and isotype IgG antibody (1:5000, Sigma-Aldrich). Slides were then treated with an anti-rabbit secondary antibody (ZSGB-Biology, Beijing, China) and developed using avidin-conjugated HRP with diaminobenzidine as a substrate (ZSGB-Biology), followed by hematoxylin counterstaining.
Construction of a Dental Pulp Inflammatory Model in Rats
A well characterized rat experimental pulp infection model was established (42). Fifteen Wistar rats were used and divided into five groups with three rats each. Rats were classified according to the sacrifice time after operation: 1 day for group 1, 3 days for group 2, 5 days for group 3, 7 days for group 4, and group 5 was not operated on as the control. The animals were submitted to general intraperitoneal anesthesia. For each animal, a cavity was made on the occlusal face of the mandibular first molars with a spherical burr size of ¼ at high speed without cooling (43). Rats were sacrificed by decapitation, and the jaws were fixed promptly in 4% formaldehyde. The local ethical committee at Sichuan University approved all laboratory animal procedures.
Cultivation and Treatment of Human Dental Pulp Cells
Primary HDPCs were collected from healthy donors (aged 18–25 years, mixed gender) and cultured according to a modified version of a method reported previously (44). Cells were maintained in DMEM (Corning, Manassas, VA) with 10% fetal calf serum (Invitrogen), and passages 3–5 were used. Cells were treated with human/mouse recombinant WNT5A (rhWNT5A, 500 ng/ml, R&D Systems) or recombinant human TNF-α (rhTNF-α, 10 ng/ml, Invivogen, San Diego, CA) for the given times. In the specified experiments, cells were pretreated with one of the following specific pathway inhibitors: pyrrolidinedithiocarbamic acid (an NF-κB inhibitor, 10 mm, Beyotime Institute of Biotechnology, Shanghai, China), BAY11-7082 (an IκB phosphorylation inhibitor, 1 μm, Selleckchem, Houston, TX), SP600125 (a JNK inhibitor, 10 μm, Merck), U0126 (an ERK inhibitor, 10 μm, Merck), and SB203580 (a p38 MAPK inhibitor, 20 μm, Cell Signaling Technology, Danvers, MA).
Isolation of Human Monocyte-derived Macrophages
Peripheral blood mononuclear cells from healthy volunteers at Sichuan University were isolated by Ficoll density gradient centrifugation and purified with human CD14+ MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany) following the instructions of the manufacturer. Monocytes were cultured in RPMI 1640 medium supplemented with 10% fetal calf serum for 7–10 days until differentiation into macrophages.
Wnt5a RNA Interference
Scrambled (Scr) shRNA and Wnt5a-shRNA HDPCs were generated and maintained as described previously (45). An oligoribonucleotide with the following sequence was used: 5′-CCGGCCCTGTTCAGATGTCAGAATTCAAGAGATTCTGACATCTGAACAGGGTTTTTTG-3′. The entire sequences were derived from the sequence of human Wnt5a mRNA (NM_003392). Wnt5a knockdown lentivirus was purchased from Sunbio Medical Biotechnology (Shanghai, China). The GFP-tagged lentiviral vector pLVT553-LV for Wnt5a-RNAi was constructed by inserting the annealing nucleotides into the AgeI and EcoRI site of the plasmid pMAGic 7.1 (Sunbio). HDPCs were plated at 2 × 105 cells/well of a 24-well cell culture plate and infected with lentivirus at a multiplicity of infection of 100. Cells infected with pLVT553-LV and CMV-GFP-pLVT7-LV (blank lentiviral vector pMAGic 7.1) are referred to as Wnt5a-shRNA and Scr-shRNA, respectively. Stable cell lines were selected by culturing cells in 3 μg/ml puromycin (Sigma-Aldrich) for 1 week. Real-time PCR and Western blot analysis were used to determine the effects of knockdown.
RNA Isolation and Reverse Transcription and Real-time PCR
Total RNA from cell cultures were isolated using TRIzolTM reagent (Invitrogen) according to the protocol of the manufacturer. 1 μg of mRNA was reverse-transcribed using the RT regent kit (Takara Biotechnology, Dalian, Liaoning, China) and subsequently used in SsoAdvancedTM SYBR® Green Super Mix real-time PCR reactions (Bio-Rad) using a standard protocol. Primers were designed to generate products of less than 200 bp for efficient analysis and were as follows: Wnt5a, 5′-CAGTTCAAGACCGTGCAGAC-3′ (forward) and 5′-GCACCCACTACTTGCACACA-3′ (reverse);CCL2, 5′-CTGCTCATAGCAGCCACCTT-3′ (forward) and 5′-CAGGTGACTGGGGCATTGAT-3′ (reverse); CCL5, 5′-CACAGCCTCTCTCCCACAGGTA-3′ (forward) and 5′-GAGCACTTGCCACTGGTGTA-3′ (reverse); CXCL1, 5′-ACTGGTGGCTGTTCCTGAAG-3′ (forward) and 5′-CTTCTCCTAAGCGATGCTCAA-3′ (reverse); IL-1β, 5′-AAGGCGGCCAGGATATAACT-3′ (forward) and 5′-TACGGCCTAAGGCAGGCAGTTG-3′ (reverse); IL-6, 5′-ACCTTCCAAAGATGGCTGAA-3′ (forward) and 5′-GCTCTGGCTTGTTCCTCACT-3′ (reverse); IL-8, 5′-CACTCCATAAGGCACAAACTTTC-3′ (forward) and 5′-GCCAGCTTGGAAGTCATGTT-3′ (reverse); and GAPDH, 5′-TCAACAGCGACACCCACTC-3′ (forward) and 5′-GCTGTAGCCAAATTCGTTGTC-3′ (reverse). PCR conditions were as follows: 95 °C for 30 s; followed by 40 cycles of 95 °C for 5 s, 60 °C for 30 s, 95 °C for 15 s, 60 °C for 15 s, and 95 °C for 15 s. The melting curve was assessed in the following program: 60 °C for 1 min and 95 °C continuous. The results were calculated applying the ΔΔCT method and presented as fold increases relative to GAPDH.
Western Blot Analysis
Cells were washed twice with PBS and scraped in lysis buffer with proteinase inhibitor mixture (Roche R&D Center China, Shanghai, China). The total protein concentration in the supernatant was measured by BCA protein assay kit (Thermo Scientific, Rockford, IL). 20–40 μg were loaded onto a 10% polyacrylamide gel and separated by electrophoresis. After blocking for 1 h with 5% nonfat dry milk in Tris-buffered saline with 1% Tween 20 (TBST), PVDF membranes were incubated with an anti-human Wnt5a antibody (1:1000, Santa Cruz Biotechnology, Dallas, TX) or an anti-human GAPDH antibody (1:1000, Cell Signaling Technology) at 4 °C overnight and washed three times with TBST, followed by 1-h incubation with appropriate HRP-conjugated IgG antibodies (1:2000, Cell Signaling Technology). PVDF membranes were washed with TBST, and proteins were visualized with Super Signal-enhanced (Thermo Scientific) chemiluminescence. For evaluation of signaling pathway activation, antibodies from Cell Signaling Technology were used and applied according to the provided recommendations: phospho-ERK (catalog no. 4370), ERK (catalog no. 4695), phospho-p65 (catalog no. 3033), p65 (catalog no. 8242), phospho-p38 (catalog no. 4511), p38 (catalog no. 9212), phospho-JNK (catalog no. 4668), and JNK (catalog no. 9258).
Cytokine and Chemokine Analyses
IL-1β, IL-6, IL-8, CCL2, CCL5, and TNF-α were measured in the supernatants of Wnt5a-treated and untreated HDPC cultures using Luminex xMAP technology (Millipore). Luminex xMAP assays for a panel of human cytokines and chemokines (Milliplex MAP human cytokine/chemokine panel, Millipore, Billerica, MA) were performed (46).
Cell Migration Assay
The migration capacity of macrophages was measured in Transwell chambers (3-μm pore, Corning). HDPC monolayers were incubated with serum-free DMEM with or without rhWTN5A treatment for 48 h in a 5% CO2 incubator at 37 °C before collection of supernatants. After overnight culture in serum-free RPMI1640 medium, 200 μl of MACS-purified cells were resuspended (2 × 106/ml) and added to the upper chamber in serum-free RPMI 1640 medium, and 600 μl of the supernatant of untreated, WNT5A-treated HDPCs was placed into the bottom wells. Various concentrations of rhWnt5A were also applied to the bottom well. After 4-h incubation, non-migrating cells on the upper surface of the membrane were removed, and the cells that migrated to the underside of the polycarbonate membrane were fixed with ethanol and stained with 1% crystal violet for 30 min. The number of migrating cells was then determined from five independent microscopic fields. The mean of triplicate assays for each experimental condition was used for analysis.
Statistical Analysis
Results are presented as mean ± S.E. All experiments were repeated at least three times. Significance was determined using analysis of variance. p < 0.05 was considered significant. Error bars show the mean ± S.D. or S.E. across duplicate or triplicate experiments.
RESULTS
Wnt5a Is Increased in Human and Rat Dental Pulpitis Specimens
We first investigated whether Wnt5a levels were increased during dental pulp inflammation. We examined Wnt5a expression levels in tissues from healthy control subjects undergoing orthodontic tooth extraction and dental pulpitis patients by immunohistochemistry staining with an anti-Wnt5a antibody. Wnt5a was found to be only expressed in the odontoblast layer of normal dental pulp. In contrast, in inflamed dental pulp tissue sections, Wnt5a expression was present at the local inflammation site (Fig. 1, E and F). In addition, Wnt5a mRNA levels were increased 1.2-fold over that of normal pulp tissue (data not shown). We also investigated the expression of TNF-α and CD68 in normal and dental pulpitis tissues and found that both were expressed in the local inflammatory region (Fig. 1, G–L). Together, these data indicate that Wnt5a is increased in human dental pulp inflammation.
FIGURE 1.

Wnt5a is increased in human dental pulpitis tissue. A–C, H&E-stained sections of normal and inflamed human dental pulp. In contrast to normal human dental pulp (D), increased Wnt5a levels are clearly visible in human inflamed dental pulp (E and F) by immunohistochemical staining. Anti-TNF-α (G–I), anti-CD68 (J–L), anti-vimentin (M–O), and isotype antibody (P–R) are shown by immunohistochemistry. Cell nuclei are visualized with hematoxylin.
We next performed immunohistochemistry with an anti-Wnt5a antibody to examine the expression pattern of Wnt5a during different pathological stages in the rat dental pulpitis model. Pulp tissue samples were stained with H&E, and infection levels were classified into different pathological stages: normal, mild, moderate, and severe (Fig. 2). Similar to human tissues, Wnt5a in the rat model was only expressed in the odontoblast layer of normal pulp tissues and was increased in the local inflammatory tissues of the infected dental pulp. A similar TNF-α profile was also observed. One representative image for each group is shown in Fig. 2. Our findings indicate that Wnt5a is increased during dental pulp inflammation both in dental pulpitis patients and in a rat model.
FIGURE 2.
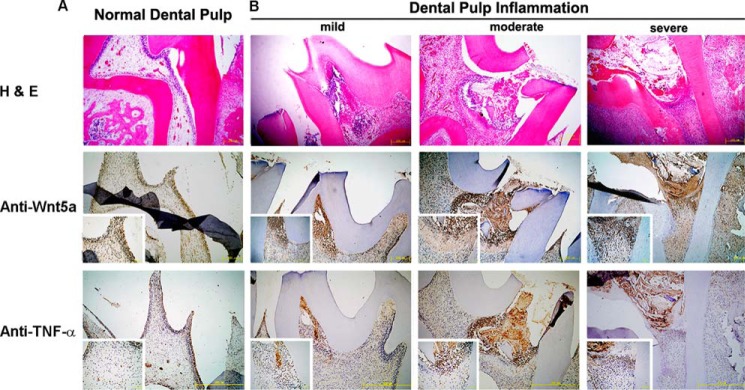
Wnt5a levels are increased in dental pulpitis tissue from rats. A and B, Wnt5a levels are increased in infected rat dental pulp sections, and a similar TNF-α effect is observed. A, H&E, anti-Wnt5a, and anti-TNF-α were stained in normal rat dental pulp. Contrasting the normal with inflamed dental pulp, the number of Wnt5a-postitive cells is increased, as is the enclosed inflammation area through the inflammatory process of dental pulp. B, localization of Wnt5a-positive cells at various inflammation stages after dental pulp infection. Sections of dental pulp inflammation are divided into mild, moderate, and severe stages at days 2, 4, and 6 post-injury for Wnt5a staining by immunohistochemistry. The top row shows representative H&E-stained sections, the center row shows staining with anti-TNF-α, and the bottom row shows immunohistochemistry staining of anti-Wnt5a in normal and dental pulpitis tissue of rats. The insets show higher magnification images (scale bars = 50 μm). Cell nuclei were visualized with hematoxylin.
TNF-α Induced Wnt5a Expression in Dental Pulp Cells
We next assessed how Wnt5a was involved in dental pulp inflammation. Wnt5a expression was low by real-time PCR in HDPCs under the culture conditions. HDPCs were then treated using 10 ng/ml rhTNF-α for up to 6 h and, in real-time PCR analysis, showed an 8-fold increase inWnt5a mRNA levels (Fig. 3A). We also confirmed the results at the protein level by Western blot analysis (Fig. 3B). Expression of cytokines associated with inflammation, including IL-1β, IL-6, IL-8 (CXCL8), and NF-κB were increased after stimulation with TNF-α, which activated the ERK, JNK, and p38 MAPK pathways, as well as the NF-κB pathway, within 30–60 min (Fig. 3C). The TNF-α-mediated induction of Wnt5a expression decreased significantly by treatment with an NF-κB inhibitor (BAY11-7082) or specific JNK (SP600125), p38 (SB203580), and ERK (U0126) inhibitors (Fig. 3, D and E). Incubation of HDPCs with these inhibitors alone did not significantly alter gene expression levels (data not shown). Thus, these findings suggest that expression of Wnt5a was induced in HDPCs upon TNF-α stimulation in an NF-κB and MAPK-dependent (ERK-, p38-, and JNK-dependent) manner.
FIGURE 3.

Wnt5a expression is induced by TNF-α in a MAPK- and NF-κB-dependent manner. Shown are real-time PCR (A) and Western blot analysis (B) of Wnt5a expression in HDPCs treated with TNF-α (n = 3–4). The gene expression level was normalized to GAPDH (n = 3–6). *, p < 0.05 versus control (CO). C, HDPCs were incubated for the indicated time points with TNF-α. Then, NF-κB, ERK, JNK, and p38 activation were assessed by Western blot analysis of total and phosphorylated signaling proteins. GAPDH was used as a control. pp65, phospho-p65; pp38, phospho-p38; pJNK, phospho-JNK; pERK, phospho-ERK. D, HDPCs were preincubated with pyrrolidinedithiocarbamic acid (PDTC, an NF-κB inhibitor), BAY11-7082 (an NF-κB inhibitor), SP600125 (a JNK inhibitor), SB203580 (a p38 inhibitor), or U0126 (an ERK inhibitor) before treatment with TNF-α. Real-time PCR (D) and Western blot analysis (E) of Wnt5a expression are also shown. **, p < 0.01 versus control; #, p < 0.05 versus TNF-α.
Wnt5a-treated HDPCs Enhanced Macrophage Migration
After confirming the induction of Wnt5a expression by inflammation in vitro and in vivo, we next questioned the role of Wnt5a in the inflammatory response. We investigated the effect of Wnt5a on macrophage migration, which is a hallmark of the immune response for disposal of invasions. rhWNT5A at 125, 250, and 500 ng/ml enhanced the migration of human macrophages in a concentration-dependent manner (Fig. 4A). We also examined the chemotactic migration of human macrophages in response to supernatants of HDPCs that were either left untreated or treated with Wnt5a by a Transwell migration assay. The migration of macrophages toward the supernatant of Wnt5a-treated HDPCs was enhanced by 54.2% compared with Wnt5a-untreated HDPCs supernatants (Fig. 4B). The chemotactic activity of the supernatant of Wnt5a-treated HDPCs was increased 58.4% when compared with Wnt5a (500 ng/ml) (Fig. 4). Because sFPR5 binds to Wnt5a and blocks the action of Wnt5a to its specific receptors, we examined the effect of sFRP5 on Wnt5a-induced human macrophage chemotaxis (47). As shown in Fig. 4, pretreatment of sFRP5 did not inhibit the chemotaxis of macrophages that was induced by Wnt5a. These data indicate that Wnt5a itself is a chemoattractant, whereas the chemotactic activity of macrophages exposed to supernatants of Wnt5a-treated HDPCs was even better.
FIGURE 4.

Chemotactic activity of Wnt5a and Wnt5a-treated HDPCs on human macrophages. A, Transwell assay showing that rhWNT5A at different concentrations (125, 250, 500 ng/ml) promotes human macrophage migration. CO, control (without rhWnt5A). **, p < 0.01. B, Transwell assay measuring macrophage migration in response to treatment with supernatants of Wnt5a-treated or untreated HDPCs. Human macrophages were incubated with or without 1 μg/ml sFRP5 for 1 h. Cells were used in a chemotaxis assay using 500 ng/ml Wnt5a. Cell migration was suppressed by treatment with sFPR5 recombinant protein (1 μg/ml). The number of migrating cells was counted with Image-Pro Plus 6.0 (n = 4). *, p < 0.05; **, p < 0.01 versus control (Wnt5a-untreated HDPC supernatant). Error bars represent the mean ± S.D.
Wnt5a-regulated Cytokines/Chemokines Mostly Act Downstream of TNF-α
Our data suggest that Wnt5a-regulated cytokines/chemokines are almost downstream of TNF-α. To investigate these regulatory patterns further, we compared the Wnt5a and TNF-α regulation of several cytokines using real-time PCR and Luminex. We examined the Wnt5a-induced expression profile of inflammatory cytokines and chemokines in HDPCs by real-time PCR and Luminex X-map. Upon treatment of HDPCs with 500 ng/ml rhWNT5A for 1, 4, and 6 h and subsequent real-time PCR,IL-6 (110-fold), IL-8 (7-fold), CCL2 (13.5-fold), CXCL1 (67.9-fold), and CXCL2 (6-fold) were all up-regulated compared with the untreated control (Fig. 5) after 1 h of treatment. A Luminex X-map assay showed that CCL5 (99-fold), CCL2 (25.5-fold), IL-8 (14.5-fold), and IL-6(6.7-fold) expression in HDPCs was also enhanced after 4 h of rhWnt5A treatment (Fig. 6).
FIGURE 5.

Wnt5a regulates cytokine/chemokine production induced by TNF-α. A, Wnt5a-shRNA and Scr-shRNA HDPCs were treated with TNF-α or left untreated. Real-time PCR using RNA isolated from Scr-shRNA HDPCs and Wnt5a-shRNA HDPCs showed that Wnt5a mRNA levels were reduced by at least 70% in Wnt5a-shRNA HDPCs, which was confirmed by Western blot analysis (B). C–F, Wnt5a promotes TNF-α-induced production of cytokines and chemokines. HDPCs were stimulated with Wnt5a or Wnt5a/TNF-α. IL-6 (C), IL-8 (D), CXCL1 (E), and CCL2 (F) gene expression levels were determined by real-time PCR and normalized to GAPDH (n = 3). G–J, Wnt5a knockdown decreased TNF-α-induced production of cytokines and chemokines. Wnt5a-shRNA and Scr-shRNA HDPCs were treated with TNF-α or left untreated. IL-6 (G), IL-8 (H), CXCL1 (I), and CCL2 (J) gene expression levels were determined by real-time PCR and normalized to GAPDH (n = 3). *, p < 0.05, **, p < 0.01 versus control.
FIGURE 6.

Wnt5a positively regulates cytokine and chemokine expression in HDPCs. IL-6 (A), IL-8 (B), CCL2 (C), and CCL5 (D) expression levels were determined using a Luminex assay (n = 3). **, p < 0.01. CO, control.
Compared with TNF-α stimulation, rhTNF-α (10 ng/ml) also induced the expression of cytokines/chemokines such as IL-6, IL-8, CCL2, CCL5, CXCL1, CXCL2, and CXCL5 in HDPCs (data not shown). Although TNF-α and Wnt5a had similar cytokine/chemokine regulation profiles, Wnt5a induced expression of some chemokines (IL-8, CCL2, CXCL1, and CXCL2) to reach a maximum in 1 h, whereas maximum values after TNF-α stimulation were not achieved until 4 h, as assessed by real-time PCR. This difference suggests a major role for Wnt5a in CC and CXC chemokine secretion in HDPCs after TNF stimulation. IL-8 (CXCL8), CXCL1, and CXCL2 are implicated in neutrophil chemotaxis for acute inflammatory reactions (48–49). CC chemokines attract mononuclear cells to sites of chronic inflammation, and the most thoroughly characterized CC chemokine is CCL2 (49). To quantitatively measure the chemokines that are induced by Wnt5a, we used a Luminex assay, which showed that CCL2 expression was enhanced robustly to more than 20 ng/sample (Fig. 6C). CCL5 was also up-regulated rapidly (Fig. 6D). Notably, although TNF-α and IL-1β mRNA levels were up-regulated mildly after Wnt5a treatment, at the protein level, they were not detected in Wnt5a-treated supernatants. In contrast, IL-8 and IL-6 were both up-regulated efficiently by Wnt5a treatment. Therefore, our data suggest that HDPCs might have different capabilities to induce and/or promote local inflammation.
In the presence of TNF-α, Wnt5a treatment further increased the production of cytokines and chemokines, including IL-6, IL-8, CXCL1, CCL2, and CCL5 (Figs. 5 and 6). We also used Wnt5a knockdown to verify the regulation of these cytokines and chemokines by TNF-α. Wnt5a knockdown reduced Wnt5a mRNA levels by 73% in HDPCs and 76% in TNF-α-treated HDPCs (Fig. 5A). The protein level of Wnt5a was also reduced after Wnt5a knockdown, as assessed by Western blot analysis (Fig. 5B). Compared with Scr-shRNA cells, Wnt5a knockdown significantly decreased the expression levels of IL-8, CXCL1, CCL2, and CCL5 after stimulation with TNF-α in HDPCs(Figs. 5 and 6). However, we found that, even with Wnt5a knockdown, levels of those cytokines/chemokines induced by TNF-α were still significantly higher than in untreated cells, which suggests that a Wnt5a-independent pathway is involved in the induction of these cytokines/chemokines by TNF-α.
Wnt5a-induced Cytokine and Chemokine Expression Is Dependent on the NF-κB and MAPK Pathways
To gain further insight into the mechanism underlying Wnt5a-induced cytokine/chemokine expression in HDPCs, we examined the NF-κB and MAPK pathways, which have been suggested by previous reports to be involved in modulating cytokine/chemokine expression (20, 23). Wnt5a treatment led to the activation of the NF-κB, ERK, JNK, and p38 MAPK pathways, as demonstrated by phosphorylation of those signaling molecules after 15–60 min of stimulation (Fig. 7A). HDPCs were then preincubated with specific inhibitors of the NF-κB (pyrrolidinedithiocarbamic acid, BAY11-7082), ERK (U0126), p38 MAPK (SB203580), and JNK pathways (SP600125) before stimulation with rhWNT5A. Our results show that the induction of IL-6 by Wnt5a is dependent on the MAPK and NF-κB pathways. Wnt5a induction of IL-8 is dependent on MAPK, whereas the induction of CCL2 and CCL5 is dependent on IκBα activity and MAPK (Fig. 7, B–D). Incubation of HDPCs with the inhibitors alone did not significantly alter gene expression levels (data not shown). Thus, these data suggest that Wnt5a increases cytokine/chemokine expression by different signaling pathways in HDPCs.
FIGURE 7.

Wnt5a-induced cytokine/chemokine expression is mediated by the NF-κB and MAPK pathways. A, HDPCs were incubated for the indicated time points with rhWNT5A, and NF-KB, ERK, JNK, and p38 activation was assessed by Western blot analysis of total and phosphorylated signaling proteins. GAPDH was used as a control. A representative Western blot analysis is shown. HDPCs were preincubated with pyrrolidinedithiocarbamic acid (PDTC, an NF-κB inhibitor), BAY11-7082 (an IκBα inhibitor), SB203580 (a p38 inhibitor), SP600125 (a JNK inhibitor) or U0126 (an ERK inhibitor) before treatment with Wnt5a. pNF-κB, phospho-NF-κB; pp38, phospho-p38; pJNK, phospho-JNK; pERK, phospho-ERK. IL-8 (B), CCL2 (C), and CCL5 (D) expression was determined by real-time PCR and Luminex technology (n = 3). *, p < 0.05, **, p < 0.01.
DISCUSSION
The role of Wnt5a in dental pulp inflammation remains undetermined, and no study has yet been done that concerns infected dental pulp tissue. Several recent studies have highlighted Wnt5a as a regulator of cytokine production in cells of monocytic lineage (e.g. monocytes and macrophages) (20, 50), neutrophils (51), and endothelial cells (22). These reports show that Wnt5a has an essential role in modulating inflammation that originates in immune cells. In human pulpitis sections, we found that the area of Wnt5a-positive expression is similar to, but larger than, CD68, a monocyte and macrophage marker (Fig. 1, F and L), which indicates that Wnt5a is secreted at the local inflammation area and from both immune cells (antigen-presenting cells) and pulp fibroblasts in dental pulpitis. Therefore, immune cells may communicate with adjacent mesenchymal cells in a paracrine manner to induce Wnt5a express in pulpitis. A previous study found that, in primary human gingival fibroblasts, Wnt5a mRNA expression remained constant after stimulation with Porphyromonas gingivalis LPS (16). However, in our study, Wnt5a gene and protein expression in primary HDPCs were both up-regulated after TNF-α stimulation, although differences in the functional setting may have given rise to this discrepancy. Because HDPCs are more similar to osteoblastic cells than they are to gingival fibroblasts, the different immunophenotype patterns may reflect a functional difference between these two cells (40, 41). In addition, the activity of Wnt signaling is likely highly dependent on the cellular context.
TNF-α is a well known inflammatory cytokine. Both trauma or bacteria can induce TNF-α secretion and cause dental pulp inflammation (52). Here we demonstrated that TNF-α induced mRNA and protein secretion of Wnt5a in primary cultures of HDPCs. TNF-α has been reported previously to enhance the expression of both Wnt5a and its receptor in BMSCs. BMSC-derived Wnt5a is considered to act during a critical step in the regulation of inflammatory processes (23, 37). In inflamed dental pulp tissue, high levels of TNF-α and IL-1β have been detected (27). We also demonstrated that Wnt5a was increased in HDPCs after TNF-α stimulation. Therefore, it is conceivable that TNF-α-induced Wnt5a expression and secretion in HDPCs could regulate dental pulp inflammation processes. Our data showed that TNF-α induced HDPCs to secrete Wnt5a and proinflammatory cytokines. In addition, samples from both humans and an experimental animal model of dental pulpitis confirmed that Wnt5a was expressed consistently in the local dental pulp of the inflammatory area and that increases in Wnt5a expression in HDPCs following TNF-α stimulation occurred through a MAPK- and NF-κB-dependent pathway (Fig. 3). We also demonstrated that increased Wnt5a and Wnt5a-induced chemokines could promote macrophage migration. These findings are supported by previous reports showing that enhanced Wnt5a expression in immune cells exposed to bacterial products orchestrated inflammatory reactions and identified NF-κB and MAPK as being associated with Wnt5a induction (23). Together, these results confirm the role of Wnt5a in the regulation of dental pulp inflammation.
The subtle relationship between TNF-α and Wnt5a in inflammation remains to be clarified. Kim et al. (22) showed previously that Wnt5a and TNF-α regulated subsets of downstream genes with functions that partially overlap in endothelial inflammation and that Wnt5a-mediated regulation was independent of TNF-α. Endothelial inflammation has also been suggested to be regulated by a dual system consisting of β-catenin-independent Wnt signaling and TNF-α-mediated signaling (22). Rauner et al. (23) showed that TNF-α increased the expression of Wnt5a in BMSCs and that Wnt5a regulated the basal and LPS-induced production of cytokines and chemokines. They also showed that TNF-α was not influenced by Wnt5a in their whole-genome studies of murine primary osteoblasts, which suggested that Wnt5a may not influence TNF-α function, although this was not verified by further experimentation (23). However, they did demonstrate that TNF-α influences Wnt5a secretion in rheumatoid arthritis but lacks the effects of Wnt5a on TNF-α.
In this study, we showed that Wnt5a, as an inflammatory mediator, drives the integration of cytokines and chemokines that function downstream of TNF-α. First, we found that TNF-α- and Wnt5a-induced downstream cytokine/chemokine expression patterns are similar and that their induced downstream profiles almost overlapped. Notably, TNF-α and IL-1β gene expression was not detected in the supernatants of Wnt5a-treated HDPCs. Second, we found that Wnt5a alone not only increased the basal expression of cytokines and chemokines but also further increased the expression level of cytokines/chemokines induced by TNF-α (Figs. 5 and 6). Third, Wnt5a knockdown significantly reduced the expression level of cytokines and chemokines induced by TNF-α (Fig. 5). Together, these results suggest that Wnt5a is influenced by TNF-α and that Wnt5a functions downstream of TNF-α. However, we should note that Wnt5a knockdown did not inhibit the expression of all of the cytokines induced by TNF-α. The remaining cytokines were still expressed to levels higher than the control, which might suggest that Wnt5a is not critical for the function of this TNF-α-dependent subset of cytokines, indicating the complexity involved in inflammation. We also found that Wnt5a knockdown in HDPCs treated with TNF-α increased IL-6 mRNA expression compared with the control. This inconsistent change in IL-6 level was through an unknown mechanism.
In our study, the Wnt5a-induced inflammation response in human dental pulp appears to represent a collaboration of the Wnt, NF-κB, and MAPK pathways. Wnt5a stimulated the phosphorylation of three MAPKs (p38, JNK, and ERK) and NF-κB, whereas inhibition of MAPK or NF-κB by specific inhibitors induced a dramatic reduction in Wnt5a-induced cytokine/chemokine production, suggesting that they all have some roles in Wnt5a-mediated inflammation (Fig. 3). These results are in accordance with previous studies showing that Wnt5a activated THP-1 cells via JNK-dependent NF-κB activation (20). In BMSCs, Wnt5a could regulate the production of proinflammatory cytokines and chemokines via distinct pathways (e.g. NF-κB, MAPK, Akt, and calcium/nuclear factor of activated T cells) (23). More recently, a report showed that Wnt5a stimulated the phosphorylation of three MAPKs (ERK, p38 MAPK, and JNK) and contributed to neutrophil recruitment (51).
The important task for immunoregulation in inflammatory sites is the ingestion and disposal of invading pathogens. Although phagocytosis is indeed a vital host defense mechanism (39), here we investigated whether Wnt5a could increase macrophage migration. Our data showed that different concentrations of rhWnt5a affected macrophage motility compared with untreated control cells and that supernatants from Wnt5a-treated HDPCs significantly enhanced macrophage migration (Fig. 4). This result suggests that Wnt5a itself acts as a chemoattractant for macrophage migration and that Wnt5a-induced CC and CXC chemokines provoke immune cell attraction (Fig. 4). The notion that Wnt5a strengthens the chemokine-mediated migration of immune cells is supported by several studies (23, 39). Recently, Wnt5a has been shown to contribute to neutrophil recruitment that promotes production of CXCL8 and CCL2 (51). Moreover, a recent report demonstrated that Wnt5a enhances macrophages to internalize bacteria but does not induce bacterial killing (48). These results may suggest that Wnt5a promotes innate immunity by enhancing cytokine and chemokine production, which, in turn, recruits neutrophils and macrophages to internalize bacteria while not facilitating bacteria killing. Further study will be required to support this line of thinking.
In summary, the results of this study show that Wnt5a plays an important role in dental pulp inflammation and significantly extends the previous conclusion that Wnt5a acts downstream of TNF-α. To our knowledge, this is the first study to describe the role of Wnt5a in dental pulp inflammation. Furthermore, chemokines that are induced by Wnt5a may mainly contribute to its positive regulatory role in inflammation. Further experiments will be necessary to examine the potential of Wnt5a as a therapeutic target for dental pulpitis and other mesenchymal inflammation.
Acknowledgments
We thank Xian-Ming Mo, Wen-Tong Meng, and the State Key Laboratory of Biotherapy and Stem Cell Biology for technical assistance.
This work was supported by National Nature Science Foundation of China Grants 81070801 and 813220170, by Science and Technology Planning Project of Sichuan Province Grant 2012SZ0034, by Innovative Research Team of Education Department of Sichuan Province Grant 13TD0038, and by Program of International S&T Cooperation Grant 2014DFA31990.
- BMSC
- bone marrow mesenchymal stem cell
- HDPC
- human dental pulp cell
- rhWNT5A
- human/mouse recombinant WNT5A
- rhTNF-α
- recombinant human TNF-α
- Scr
- scrambled.
REFERENCES
- 1. Freese J. L., Pino D., Pleasure S. J. (2010) Wnt signaling in development and disease. Neurobiol. Dis. 38, 148–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bouldin C. M., Kimelman D. (2012) Taking a bite out of Wnts. Cell Res. 22, 1621–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Petersen C. P., Reddien P. W. (2009) Wnt signaling and the polarity of the primary body axis. Cell 139, 1056–1068 [DOI] [PubMed] [Google Scholar]
- 4. Whyte J. L., Smith A. A., Helms J. A. (2012) Wnt signaling and injury repair. Cold Spring Harb. Perspect. Biol. 4, a008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. van Amerongen R. (2012) Alternative Wnt pathways and receptors. Cold Spring Harb. Perspect. Biol. 4, a007914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sato A., Yamamoto H., Sakane H., Koyama H., Kikuchi A. (2010) Wnt5a regulates distinct signalling pathways by binding to Frizzled2. EMBO J. 29, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lai S. L., Chien A. J., Moon R. T. (2009) Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 19, 532–545 [DOI] [PubMed] [Google Scholar]
- 8. Nusse R. (2012) Wnt signaling. Cold Spring Harb. Perspect. Biol. 4, a011163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Huang H., He X. (2008) Wnt/β-catenin signaling: new (and old) players and new insights. Curr. Opin. Cell Biol. 20, 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gordon M. D., Nusse R. (2006) Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J. Biol. Chem. 281, 22429–22433 [DOI] [PubMed] [Google Scholar]
- 11. Slusarski D. C., Pelegri F. (2007) Calcium signaling in vertebrate embryonic patterning and morphogenesis. Dev. Biol. 307, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kilander M. B., Dijksterhuis J. P., Ganji R. S., Bryja V., Schulte G. (2011) WNT-5A stimulates the GDP/GTP exchange at pertussis toxin-sensitive heterotrimeric G proteins. Cell Signal. 23, 550–554 [DOI] [PubMed] [Google Scholar]
- 13. Pereira C. P., Bachli E. B., Schoedon G. (2009) The Wnt pathway: a macrophage effector molecule that triggers inflammation. Curr. Atheroscler. Rep. 11, 236–242 [DOI] [PubMed] [Google Scholar]
- 14. Christman M. A., 2nd, Goetz D. J., Dickerson E., McCall K. D., Lewis C. J., Benencia F., Silver M. J., Kohn L. D., Malgor R. (2008) Wnt5a is expressed in murine and human atherosclerotic lesions. Am. J. Physiol. Heart Circ. Physiol. 294, H2864–H2870 [DOI] [PubMed] [Google Scholar]
- 15. Sen M., Lauterbach K., El-Gabalawy H., Firestein G. S., Corr M., Carson D. A. (2000) Expression and function of wingless and frizzled homologs in rheumatoid arthritis. Proc. Natl. Acad. Sci. U.S.A. 97, 2791–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nanbara H., Wara-aswapati N., Nagasawa T., Yoshida Y., Yashiro R., Bando Y., Kobayashi H., Khongcharoensuk J., Hormdee D., Pitiphat W., Boch J. A., Izumi Y. (2012) Modulation of Wnt5a expression by periodontopathic bacteria. PloS ONE 7, e34434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li B., Shi Y., Shu J., Gao J., Wu P., Tang S. J. (2013) Wingless-type mammary tumor virus integration site family, member 5A (Wnt5a) regulates human immunodeficiency virus type 1 (HIV-1) envelope glycoprotein 120 (gp120)-induced expression of pro-inflammatory cytokines via the Ca2+/calmodulin-dependent protein kinase II (CaMKII) and c-Jun N-terminal kinase (JNK) signaling pathways. J. Biol. Chem. 288, 13610–13619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sen M., Chamorro M., Reifert J., Corr M., Carson D. A. (2001) Blockade of Wnt-5A/frizzled 5 signaling inhibits rheumatoid synoviocyte activation. Arthritis Rheum. 44, 772–781 [DOI] [PubMed] [Google Scholar]
- 19. Maeda K., Kobayashi Y., Udagawa N., Uehara S., Ishihara A., Mizoguchi T., Kikuchi Y., Takada I., Kato S., Kani S., Nishita M., Marumo K., Martin T. J., Minami Y., Takahashi N. (2012) WNT5A-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat Med. 18, 405–412 [DOI] [PubMed] [Google Scholar]
- 20. Kim J., Chang W., Jung Y., Song K., Lee I. (2012) Wnt5a activates THP-1 monocytic cells via a β-catenin-independent pathway involving JNK and NF-κB activation. Cytokine 60, 242–248 [DOI] [PubMed] [Google Scholar]
- 21. Pereira C., Schaer D. J., Bachli E. B., Kurrer M. O., Schoedon G. (2008) Wnt5A/CaMKII signaling contributes to the inflammatory response of macrophages and is a target for the anti inflammatory action of activated protein C and interleukin-10. Arterioscler. Thromb. Vasc. Biol. 28, 504–510 [DOI] [PubMed] [Google Scholar]
- 22. Kim J., Kim J., Kim D. W., Ha Y., Ihm M. H., Kim H., Song K., Lee I. (2010) Wnt5a induces endothelial inflammation via β-catenin independent signaling. J. Immunol. 185, 1274–1282 [DOI] [PubMed] [Google Scholar]
- 23. Rauner M., Stein N., Winzer M., Goettsch C., Zwerina J., Schett G., Distler J. H., Albers J., Schulze J., Schinke T., Bornhäuser M., Platzbecker U., Hofbauer L. C. (2012) WNT5A is induced by inflammatory mediators in bone marrow stromal cells and regulates cytokine and chemokine production. J. Bone Miner. Res. 27, 575–585 [DOI] [PubMed] [Google Scholar]
- 24. Pukrop T., Klemm F., Hagemann T., Gradl D., Schulz M., Siemes S., Trümper L., Binder C. (2006) Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc. Natl. Acad. Sci. U.S.A. 103, 5454–5459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bergenfelz C., Medrek C., Ekström E., Jirström K., Janols H., Wullt M., Bredberg A., Leandersson K. (2012) Wnt5a induces a tolerogenic phenotype of macrophages in sepsis and breast cancer patients. J. Immunol. 188, 5448–5458 [DOI] [PubMed] [Google Scholar]
- 26. Oderup C., LaJevic M., Butcher E. C. (2013) Canonical and noncanonical Wnt proteins program dendritic cell responses for tolerance. J. Immunol. 190, 6126–6134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Briolay A., Lencel P., Bessueille L., Caverzasio J., Buchet R., Magne D. (2007) Irreversible but not reversible pulpitis is associated with up-regulation of tumour necrosis factor-α gene expression in human pulp. Int. Endod. J. 40, 198–203 [DOI] [PubMed] [Google Scholar]
- 28. Chen L., Wen Y. M. (2011) The role of bacterial biofilm in persistent infections and control strategies. Int. J. Oral Sci. 3, 66–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cooper P. R., McLachlan J. L., Simon S., Graham L. W., Smith A. J. (2011) Mediators of inflammation and regeneration. Adv. Dent. Res. 23, 290–295 [DOI] [PubMed] [Google Scholar]
- 30. Schmalz G., Galler K. M. (2011) Tissue injury and pulp regeneration. J. Dent. Res. 90, 828–829 [DOI] [PubMed] [Google Scholar]
- 31. Dammaschke T., Steven D., Kaup M., Ott K. H. (2003) Long-term survival of root-canal-treated teeth: a retrospective study over 10 years. J. Endod. 29, 638–643 [DOI] [PubMed] [Google Scholar]
- 32. Kim J. Y., Xin X., Moioli E. K., Chung J., Lee C. H., Chen M., Fu S. Y., Koch P. D., Mao J. J. (2010) Regeneration of dental-pulp-like tissue by chemotaxis-induced cell homing. Tissue Eng. Part A 16, 3023–3031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hahn C. L., Liewehr F. R. (2007) Update on the adaptive immune responses of the dental pulp. J. Endod. 33, 773–781 [DOI] [PubMed] [Google Scholar]
- 34. Staquet M. J., Durand S. H., Colomb E., Roméas A., Vincent C., Bleicher F., Lebecque S., Farges J. C. (2008) Different roles of odontoblasts and fibroblasts in immunity. J. Dent. Res. 87, 256–261 [DOI] [PubMed] [Google Scholar]
- 35. O'Shea J. J., Ma A., Lipsky P. (2002) Cytokines and autoimmunity. Nat. Rev. Immunol. 2, 37–45 [DOI] [PubMed] [Google Scholar]
- 36. Horst O. V., Horst J. A., Samudrala R., Dale B. A. (2011) Caries induced cytokine network in the odontoblast layer of human teeth. BMC Immunol. 10.1186/1471-2172-12-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Briolay A., Lencel P., Bessueille L., Caverzasio J., Buchet R., Magne D. (2013) Autocrine stimulation of osteoblast activity by Wnt5a in response to TNF-α in human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 430, 1072–1077 [DOI] [PubMed] [Google Scholar]
- 38. Komada Y., Yamane T., Kadota D., Isono K., Takakura N., Hayashi S., Yamazaki H. (2012) Origins and properties of dental, thymic, and bone marrow mesenchymal cells and their stem cells. PLoS ONE 7, e46436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ryan G. B., Majno G. (1977) Acute inflammation. Am. J. Pathol. 86, 183–276 [PMC free article] [PubMed] [Google Scholar]
- 40. Martinez E. F., Araújo V. C. (2004) In vitro immunoexpression of extracellular matrix proteins in dental pulpal and gingival human fibroblasts. Int. Endod. J. 37, 749–755 [DOI] [PubMed] [Google Scholar]
- 41. Martinez E. F., Donato T. A., Arana-Chavez V. E. (2012) In vitro effects of ascorbic acid and β-glycerophosphate on human gingival fibroblast cells. Tissue Cell. 44, 325–331 [DOI] [PubMed] [Google Scholar]
- 42. Ohshima H., Sato O., Kawahara I., Maeda T., Takano Y. (1995) Responses of immunocompetent cells to cavity preparation in rat molars: an immunohistochemical study using OX6-monoclonal antibody. Connect Tissue Res. 32, 303–311 [DOI] [PubMed] [Google Scholar]
- 43. Tagger M., Massler M., (1975) Periapical tissue reactions after pulp exposure in rat molars. Oral Surg. Oral Med. Oral Pathol. 39, 304–317 [DOI] [PubMed] [Google Scholar]
- 44. Ye L., Peng L., Tan H., Zhou X. (2006) HGF enhanced proliferation and differentiation of dental pulp cells. J. Endod. 32, 736–741 [DOI] [PubMed] [Google Scholar]
- 45. Zhou W., Wang L., Gou S. M., Wang T. L., Zhang M., Liu T., Wang C. Y. (2012) ShRNA silencing glycogen synthase kinase-3 β inhibits tumor growth and angiogenesis in pancreatic cancer. Cancer Lett. 316, 178–186 [DOI] [PubMed] [Google Scholar]
- 46. Zhang B., Ho Y. W., Huang Q., Maeda T., Lin A., Lee S. U., Hair A., Holyoake T. L., Huettner C., Bhatia R. (2012) Altered microenvironmental regulation of leukemic and normal stem cells in chronic myelogenous leukemia. Cancer Cell. 21, 577–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhao C., Bu X., Wang W., Ma T, Ma H., (2014) GEC-derived SFRP5 inhibits Wnt5a-induced macrophage chemotaxis and activation. PLoS ONE 9, e85058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maiti G., Naskar D., Sen M. (2012) The Wingless homolog Wnt5a stimulates phagocytosis but not bacterial killing. Proc. Natl. Acad. Sci. U.S.A. 109, 16600–16605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Charo I. F., Ransohoff R. M. (2006) The many roles of chemokines and chemokine receptors in inflammation. N. Engl. J. Med. 354, 610–621 [DOI] [PubMed] [Google Scholar]
- 50. Blumenthal A., Ehlers S., Lauber J., Buer J., Lange C., Goldmann T., Heine H., Brandt E., Reiling N. (2006) The Wingless homolog WNT5A and its receptor Frizzled-5 regulate inflammatory responses of human mononuclear cells induced by microbial stimulation. Blood 108, 965–973 [DOI] [PubMed] [Google Scholar]
- 51. Jung Y. S., Lee H. Y., Kim S. D., Park J. S., Kim J. K., Suh P. G., Bae Y. S. (2013) Wnt5a stimulates chemotactic migration and chemokine production in human neutrophils. Exp. Mol. Med. 14, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Pezelj-Ribaric S., Anic I., Brekalo I., Miletic I., Hasan M., Simunovic-Soskic M. (2002) Detection of tumor necrosis factor α in normal and inflamed human dental pulps. Arch. Med. Res. 33, 482–484 [DOI] [PubMed] [Google Scholar]
- 53. Ghosh M. C., Collins G. D., Vandanmagsar B., Patel K., Brill M., Carter A., Lustig A., Becker K. G., Wood W. W., 3rd., Emeche C. D., French A. D., O'Connell M. P., Xu M., Weeraratna A. T., Taub D. D. (2009) Activation of Wnt5A signaling is required for CXC chemokine ligand 12-mediated T-cell migration. Blood 114, 1366–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kim J. M., Kang S. W., Shin S. M., Su Kim D., Choi K. K., Kim E. C., Kim S. Y. (2013) Inhibition of matrix metalloproteinases expression in human dental pulp cells by all-trans retinoic acid. Int. J. Oral Sci. 10.1038/ijos.2013.63 [DOI] [PMC free article] [PubMed] [Google Scholar]