

Background: Myosin polymerizes into filaments that move on actin.
Results: ATPase and moving velocities of filaments may be limited by the weak to strong transition.
Conclusion: Filaments moving on top of actin may have fewer drag heads than actin filaments moving on myosin monomers.
Significance: Understanding kinetics of intact myosin filaments with actin is important to understand muscle mechanics.
Keywords: ATPase, Myosin, Pre-steady-state Kinetics, Protein Cross-linking, Smooth Muscle, Filaments, Acto-myosin Mechanochemistry, Fluorescence Microscopy, In Vitro Motility, Nonlinear Elasticity
Abstract
Actin-myosin interactions are well studied using soluble myosin fragments, but little is known about effects of myosin filament structure on mechanochemistry. We stabilized unphosphorylated smooth muscle myosin (SMM) and phosphorylated smooth muscle myosin (pSMM) filaments against ATP-induced depolymerization using a cross-linker and attached fluorescent rhodamine (XL-Rh-SMM). Electron micrographs showed that these side polar filaments are very similar to unmodified filaments. They are ∼0.63 μm long and contain ∼176 molecules. Rate constants for ATP-induced dissociation and ADP release from acto-myosin for filaments and S1 heads were similar. Actin-activated ATPases of SMM and XL-Rh-SMM were similarly regulated. XL-Rh-pSMM filaments moved processively on F-actin that was bound to a PEG brush surface. ATP dependence of filament velocities was similar to that for solution ATPases at high [actin], suggesting that both processes are limited by the same kinetic step (weak to strong transition) and therefore are attachment-limited. This differs from actin sliding over myosin monomers, which is primarily detachment-limited. Fitting filament data to an attachment-limited model showed that approximately half of the heads are available to move the filament, consistent with a side polar structure. We suggest the low stiffness subfragment 2 (S2) domain remains unhindered during filament motion in our assay. Actin-bound negatively displaced heads will impart minimal drag force because of S2 buckling. Given the ADP release rate, the velocity, and the length of S2, these heads will detach from actin before slack is taken up into a backwardly displaced high stiffness position. This mechanism explains the lack of detachment-limited kinetics at physiological [ATP]. These findings address how nonlinear elasticity in assemblies of motors leads to efficient collective force generation.
Introduction
Muscles contract when myosin filaments slide past actin filaments. Determining the kinetics underlying this sliding furthers understanding of the basic mechanisms of muscle contraction and regulation of contraction. Actin-myosin mechanochemistry is well studied in assays where actin filaments interact with soluble myosin fragments; however, little is known about how this mechanochemistry is altered when myosin molecules are incorporated into their native filamentous form. Here, we prepare stable smooth muscle myosin filaments to determine how the mechanochemistry of actin filament-myosin filament interaction differs from that of actin filament-myosin single-molecule interactions.
Myosin II is a class of molecular motors found in skeletal, cardiac, and smooth muscles (1, 2) in addition to nonmuscle cells. Myosin II is a double-headed molecule containing two heavy chains that each form a globular motor domain that binds actin and ATP, an α-helical lever arm, and an α-helical rod or tail domain. An essential light chain and an RLC2 bind to each lever arm domain, whereas the two rod regions homodimerize in a parallel manner through ionic interactions to form the coiled-coil tail.
In smooth muscle, phosphorylation of Ser-19 of the RLC of SMM by MLCK (3) activates the actin-activated ATPase activity, resulting in ATP-dependent cross-bridge cycling between pSMM and actin (4) and smooth muscle contraction. A similar phosphorylation-dependent activation occurs with nonmuscle myosins in nonmuscle cells (5).
The kinetic cycle of SMM, like other myosin IIs, is limited by phosphate release associated with the weak to strong actin-binding transition. This means that SMM spends most (∼95%) of its time in weak actin-binding states, making SMM a low duty ratio nonprocessive motor. Many SMM molecules are needed to generate persistent motion or physiological levels of force. Thus, the form of SMM that produces force in the cell is the filament, which consists of hundreds of myosin monomers that interact through their coiled-coil tails through ionic bonds. Unlike skeletal, cardiac, and nonmuscle myosins, which all form bipolar filaments, SMM forms side polar filaments (6–11). Side polar filaments have opposite polarity on each side of the filament, which may be suited to the loose organization of contractile machinery and plasticity seen in some smooth muscle cells (12, 13), because they can depolymerize and repolymerize from both ends (14).
For the past 40 years, virtually all kinetic studies of nucleotide-dependent acto-myosin interactions have been carried out with the soluble fragments of myosin, S1 or HMM, neither of which can form filaments. S1 contains a single head domain, whereas HMM contains both heads and approximately one-third of the tail. We are interested in understanding the more physiological nucleotide-dependent actin filament-myosin filament interactions. Comparing the sliding velocities and kinetics with those of actin filament-myosin single molecule interactions will provide insights into the effects of the myosin filaments on actin-myosin mechanochemistry (15). Unfortunately, SMM filaments are not stable structures under physiological conditions in the presence of ATP, and for this reason, little is known about the interaction of these filaments with actin and nucleotides. They are in dynamic equilibrium with the intramolecularly folded monomer called 10S, which has unique kinetic properties. The position of the equilibrium is affected by many factors including phosphorylation of the RLC (16, 17), ionic strength, pH, concentration of SMM (17, 18), Mg2+ ion concentration (19), and the presence of actin (20). These factors are the same ones that would be interesting and necessary to vary to understand the mechanochemical interactions of filaments with actin and nucleotides using solution kinetics and in vitro motility assays.
To address this problem, we have developed a simple method to stabilize SMM filaments against depolymerization while retaining the known structural and functional properties of SMM. We show that these filaments, prepared from purified chicken gizzard myosin, are stable under the very conditions that strongly promote depolymerization of unmodified SMM, such as in the presence of ATP, in the unphosphorylated state, at low protein concentrations, and under near physiological ionic strength (148 mm). These stabilized SMM filaments: 1) are similar in length and shape to unmodified filaments, 2) are largely stable to ATP-induced depolymerization, 3) dissociate from actin in an ATP-dependent manner and interact with ADP similarly to S1, 4) retain their regulation by phosphorylation, and 5) show similar steady-state ATPase kinetics to unmodified SMM.
By fluorescently labeling both SMM and actin filaments, we directly observed SMM filaments binding to actin and sliding using TIRF microscopy. Our results show that the kinetic steps limiting the unloaded velocity of motion powered by myosin filaments differs fundamentally from that powered by myosin cross-bridges unincorporated into filaments. We suggest that the low stiffness of the relatively flexible S2 region in filaments influences the kinetics of myosin cross-bridge interactions during motion on actin, making the process limited by attachment rather than detachment kinetics.
EXPERIMENTAL PROCEDURES
Buffers
The following buffers were used: conjugation buffer (20 mm HEPES, pH 7.2, 0.5 m NaCl, 0.1 mm EGTA, 5 mm DTT, 30 nm NaN3); filament buffer (10 mm sodium phosphate, pH 7.0, 5 mm MgCl2, 125 mm NaCl, 0.1 mm EGTA, 1 mm DTT, 30 nm NaN3); imaging filament buffer, filament buffer plus 0.5% methylcellulose and an oxygen scavenger system (0.1 mg ml−1 glucose oxidase, 0.018 mg ml−1 catalase, 2.3 mg ml−1 glucose); phosphorylation buffer (10 mm MOPS, pH 7.0, 50 mm NaCl, 2 mm MgCl2, 3 mm CaCl2, 0.2 mm EGTA, 10 μm ATP, 30 nm NaN3); myosin buffer (25 mm imidazole, pH 7.4, 300 mm KCl, 4 mm MgCl2, 1 mm EGTA, 5 mm DTT, 30 nm NaN3); and actin buffer (50 mm imidazole, pH 7.0, 50 mm KCl, 2 mm EGTA, 8 mm MgCl2, and 10 mm DTT).
Rhodamine Labeling and EDC Cross-linking of SMM Filaments
SMM was purified from frozen chicken gizzards (Pel-Freez Biologicals, Rogers, AR) (21) except that the last polymerization-depolymerization step was omitted. After dialysis into conjugation buffer overnight, NHS-rhodamine (Thermo Scientific, Rockford, IL) was added at a 10:1 molar ratio of dye to SMM (8 mg ml−1) and reacted for 2 h at 4 °C with slow rotation. After a 20-min 164,000 × g spin at 4 °C to pellet any aggregated SMM, the monomeric rhodamine-SMM (Rh-SMM) in the supernatant was dialyzed into conjugation buffer overnight to remove unconjugated dye, followed by DTT-free filament buffer. The SMM concentration and degree of labeling were measured in a high ionic strength buffer using the manufacturer's protocol and the Ε280 nm (0.1%) of 0.56 for SMM. Typical labeling stoichiometry was 0.24 mol rhodamine/mol SMM monomer.
Rh-SMM or SMM (8 mg/ml) was cross-linked with EDC (Thermo Scientific) at a final concentration of 5 mm in DTT-free filament buffer for 30 min at room temperature with slow rotation or as indicated in the figure legends. The reaction was quenched with 5 mm DTT (Soltec Ventures, Beverly, MA), and the filaments were pelleted for 20 min at 164,000 × g at 4 °C. The supernatant was removed, and its SMM concentration was measured at 280 and 555 nm and subtracted from the total SMM to calculate the protein concentration of the filament pellet. The pellet, typically containing more than 90% of the total protein, was then resuspended and dialyzed into filament buffer (with 1 mm DTT). The pSMM samples were phosphorylated prior to EDC treatment.
An Improved Protocol for Phosphorylation of SMM with ATP and CaM-MLCK
SMM at 0.5 mg ml−1 or less in 10 mm MOPS, pH 7.2, 50 mm NaCl, 0.2 mm EGTA, 1 mm DTT was prepared for phosphorylation by adding these reagents to the following final concentrations: 1 mm ATP (Sigma; A3377; this ATP has low levels of ADP), 10 μg ml−1 CaM (Sigma; P1431), 4 μg ml−1 MLCK (22), 3 mm CaCl2. Phosphorylation was initiated by dialysis (3500 molecular weight cutoff cassette; Pierce) against 1 liter of 10 mm MOPS, pH 7.2, 50 mm NaCl, 0.2 mm EGTA, 1 mm DTT, 1 mm ATP (see above), 3 mm CaCl2, 2 mm MgCl2 overnight. After dialysis, 100 pm microcystin LR (Sigma; M2912) was added to inhibit endogenous myosin phosphatase activity. Samples remained fully phosphorylated for at least 2 weeks.
We have found that this method always gives 100% phosphorylation, whereas our previous method (23) gave intermittent results, often with ∼50% phosphorylation. The new method uses a higher purity ATP (low ADP) and also allows any ADP generated during the phosphorylation reaction to be diluted by dialysis. The above protocol will give less than full phosphory lation if a different source of ATP is used (e.g. AMRESCO ultrapure grade #0220). ADP is a common contaminant in ATP and is difficult to completely remove. It has been shown that the MLCK reaction can be driven in reverse to dephosphorylate SMM with ADP to make ATP (24). Other critical aspects are to phosphorylate at no higher than 0.5 mg ml−1 SMM and to expose the reaction mixture to MgCl2 last.
Phosphorylation was assessed using 10% or 4–20% Tris-glycine gels (10 cm × 10 cm, 12 lanes; Invitrogen) with standard Tris-glycine running buffer. This type of gel gave superior results to urea and/or urea-glycerol gels (30, 31). The samples were precipitated with 3 volumes of cold acetone prior to the addition of sample buffer (8 m urea ultrapure; Research Organics), 33 mm Tris-glycine, pH 8.6, 0.17 mm EDTA, 10 mm DTT (added immediately before use), and bromphenol blue), so that the final concentration of SMM was 6–7 mg ml−1, and 40 μg of SMM was applied to the gel. Concentrated samples gave better resolution than more dilute ones. The samples were not heated. Gels were stained with Coomassie Blue using an automated gel stainer (E Stain 2.0; Genscript). The samples were fully phosphorylated as evidenced by the lack of an unphosphorylated RLC band.
Removal of ATP from Protein Solutions
To remove ATP from pSMM and actin for assays requiring ATP-free protein, samples were dialyzed (10,000 molecular weight cutoff dialysis cassette; Pierce) with rapid stirring against 2 liters of the respective protein buffer containing ∼1 g of Dowex resin (1 × 8 Cl− form, 200–400 mesh; Sigma, 44340) and 100 pm microcystin LR (for pSMM) for 8 h and then transferred to a fresh 2 liters of buffer containing Dowex and microcystin LR for overnight dialysis.
Actin
Rabbit skeletal muscle actin (25) was used for all experiments. For biotination, F-actin (24 μm) was dialyzed into 10 mm MOPS, pH 7.6, 100 mm KCL, 0.5 mm ATP, 0.1 mm CaCl2, 1 mm MgCl2, 30 nm NaN3, and labeled using a 4-fold molar excess of EZ-Link maleimide PEG-2 Biotin (Thermo Scientific; 21901), suspended in dimethyl formamide (Sigma-Aldrich; 319937). After mixing, actin was incubated at room temperature for 1.5 h and put on ice for no less than 8 h, pelleted at 275,000 × g for 90 min, and pellets were homogenized into G buffer (2 mm MOPS, pH 7.6, 0.1 mm CaCl2, 0.2 mm ATP, 1 mm DTT, 30 nm NaN3). Actin was depolymerized by dialysis into 2 × 4 liters of G buffer for no less than 24 h total, and the supernatants were collected after centrifugation at 275,000 × g for 90 min. Actin was then mixed with unlabeled G-actin to a final ratio of 5% biotinylation (mol/mol) and polymerized by addition of 100 mm KCl and 2 mm MgCl2. To stabilize against depolymerization at low concentrations, actin was incubated with equimolar phalloidin, Alexa 488-Phalloidin, or TRITC-Phalloidin (Alexis Corp., San Diego, CA) as indicated.
Electron Microscopy of SMM Filaments
Electron microscopy was performed at the Oregon Health Sciences University Multi-scale Microscopy Core using a Field Emission, Inc. Tecnai Spirit transmission electron microscope at 120 Kv. The images were collected as 2048 × 2048 pixel, 16-bit gray scale files using the transmission electron microscope imaging and analysis (Field Emission, Inc.) interface on an Eagle TM 2K CCD multiscan camera. Grids were ultrathin carbon film on holey carbon support film (400 mesh copper; Ted Pella, 01824). SMM specimens at 83 μg ml−1 in filament buffer were applied in a drop for 30 s, after which the buffer was wicked off the grid with filter paper. Grids were washed with five drops of buffer and wicked off again. The specimens were stained and fixed with 1% freshly filtered uranyl acetate. Air-dried grids were promptly visualized.
Steady-state Actin-activated ATPase Assays
An NADH-coupled assay (26) was used to measure steady-state actin-activated ATPase activities (see Fig. 7). The reaction was performed in ATPase buffer (10 mm MOPS, pH 7.0, 125 mm NaCl, 0.2 mm EGTA, 2 mm MgCl2, 1 mm DTT, 1 mm ATP, 1 mm NADH, 100 units ml−1 lactate dehydrogenase, 500 units ml−1 pyruvate kinase, 2.5 mm phospho(enol)-pyruvate) at 25 °C. All ATPase reagents were from Sigma-Aldrich. Separate stock solutions of ATP-free actin and ATP-free myosin filaments were prepared by dialysis to 10 mm MOPS, pH 7.0, 125 mm NaCl, 0.2 mm EGTA, 2 mm MgCl2, 1 mm DTT. For the assay, the order of addition of reagents is important to reduce light scattering from the two filamentous proteins. All mixing was done with a positive pressure pipette (Gilson Microman). The procedure for preparation of the actin is particularly important. Best results are obtained if stock actin is added to a glass test tube first and mixed with volume of 5× reaction mix containing the NADH-coupled enzymes and substrates. Next, buffer (see above) is added to the actin in small increments with extensive mixing each time so that a homogeneous actin solution is obtained. Large single dilutions of actin should be avoided. Homogeneity of the actin can be checked by holding the test tube up to a light source to look for Schleiren lines, which should be minimal if mixing is sufficient. If bubbles form, a low speed spin may be required for higher actin concentrations. More rather than less mixing is usually required. If needed, actin solutions can be incubated for ∼10 min to promote homogeneity and or to remove bubbles. After adding the required amount of ATP to the actin and adjusting the MgCl2 concentration (if necessary), the reaction is started by addition with further mixing of SMM or pSMM to 0.6 or 0.1 mg/ml, respectively. These myosin concentrations gave minimal light scattering while still giving ΔAU > 0.1. Myosin must be added last to avoid the rigor state, which will give high light scattering. The solution was transferred to a cuvette, the absorbance at 340 nm was continuously monitored in a UV-visible spectrophotometer for 10 min, and the data were fit to a line to obtain the slope for conversion to ATPase activity. Calculated slopes were typically ±10% after fitting. Reactions lacking SMM were used for background subtraction.
FIGURE 7.

Steady-state actin-activated ATPase rates. A, determination of KATPase and vmax. XL-Rh-pSMM (squares, dashed line) and pSMM (circles, solid line) data were fit to the Michaelis-Menten equation (see Table 1 for kinetic parameters). XL-Rh-SMM (crosses) and SMM (diamonds) data were not fit. Most points are from a single determination, but experiments at 5, 30, and 75 μm actin were repeated using another protein preparation, with error bars showing the range of the two points. B, determination of KATP. Actin-activated ATPase activity at 75 μm actin at varying [ATP]. The data were fit to the Michaelis-Menten equation (see Table 1 for kinetic parameters. pSMM (circles, solid line) and XL-Rh-pSMM (squares, dashed line)). Points with error bars show the averages and S.D. for triplicate assays using three different myosin preparations. For XL-Rh-SMM filaments, three points without error bars are from a single determination. The inset shows expanded plot of points at low [ATP]. Assays were performed at room temperature in ATPase buffer (see Table 1 for data summary).
Standard Geometry in Vitro Motility Assay
Standard in vitro motility assays were performed at room temperature using a Nikon TE2000 epifluorescence microscope (Technical Instruments, Burlingame, CA) and a Roper Cascade 512B camera (Princeton Instruments, Trenton, NJ) with wide field excitation at 532 nm with a Hg-Xe lamp, a Nikon 565 nm (TRITC) G-2E/C band pass emission filter, and a 100× objective. Each field of view was 54 μm2 or 512 pixels2, at 106 nm pixel−1. The following treatments were applied in sequence to a nitrocellulose-coated coverslip: pSMM monomers (in myosin buffer) or XL-Rh-pSMM filaments (in filament buffer) (100 μl of 100 μg/ml) for 1 min, wash with 2 × 80 μl of respective buffer, block for 1 min with 80 μl of 5 mg ml−1 BSA (Fraction V; Sigma; A-3059) in actin buffer, 80 μl of 20 nm TRITC-phalloidin-labeled actin in actin buffer for 1 min, and wash with 2 × 80 μl of actin buffer. Imaging filament buffer was added before image sequences (0.1–0.2 s of exposure) were captured in 1–4 fields (54 × 54 μm) for 1 min each, constituting one data set. Actin filament trajectories moving over monomeric and filamentous myosin were analyzed using the Spot Tracker 2D (27) and the JFilament (28) plug-ins for ImageJ (National Institutes of Health), respectively.
Preparation of Biotin-PEG Coverslips
Glass coverslips (18 × 18 mm #1.5; Fisher Scientific) were cleaned as follows: sonicated 1 h in ddH2O with 1% Versaclean detergent (Fisher Scientific); extensively rinsed with ddH2O; sonicated for 15 min each in 1 m HCl, 1 m NaOH, ethanol for 1 h; extensively rinsed in 60 °C ddH2O; and dried using a stream of N2. Cleaned coverslips could be stored for up to 1 month with desiccant to remove moisture. The day prior to the experiment, cleaned coverslips were rinsed with ddH2O and dried with a stream of N2 before silanizing with 100 μl of a mixture of 2 mg ml−1 mPEG-silane MW 2,000 and 2 μg ml−1 biotin-PEG-silane MW 3,400 (both (Laysan Bio, Inc. Arab, AL)) in 80% ethanol, pH 2.0, and reacted by baking at 70 °C for 16 h. Coverslips were then extensively rinsed with ddH2O and dried with N2. Flow cells were constructed using two strips of double-sided tape (3M, St. Paul, MN) between the coverslip and an ethanol-rinsed microscope slide (3 inch × 1 inch - 1 mm; Fisher Scientific) with a final flow cell volume of ∼80 μl.
Inverted Geometry in Vitro Motility Assay
Prior to each experiment, biotin-PEG flow cells were incubated with 10 mg ml−1 BSA (Sigma; A3059) in PBS (13 mm sodium phosphate, pH 7.4, 150 mm NaCl, 30 nm NaN3) for 2 min, and then 4 μg ml−1 streptavidin (Invitrogen; 434302) diluted in actin buffer for 15 s, followed by washing with 80 μl of PBS and 10 mg ml−1 BSA. Alexa 488-phalloidin-labeled 5% biotinylated actin (80 μl of 100 nm) in actin buffer was added and incubated for 2 min. The flow cell was washed three times with 1 ml of filament buffer followed by XL-Rh-pSMM (80 μl of 0.01 mg ml−1) diluted in phosphorylation buffer immediately followed by 80 μl of imaging filament buffer with indicated ATP. Image sequences (0.1 s of exposure) were immediately collected with wide field excitation to image the moving XL-Rh-pSMM filaments for 3 min followed by a still image of the actin filaments using TIRF (488-nm excitation laser), which was overlaid with the image sequence of the SMM filaments using ImageJ. Data obtained from three fields (20–30 trajectories) at each ATP concentration constituted one experiment. Filament motion was analyzed using the Spot Tracker 2D plug-in for ImageJ (27).
Visualization of XL-Rh-SMM Filaments Bound to Surface-attached Actin Using TIRF Microscopy
Biotin-PEG flow cells were prepared and treated identically to the inverted geometry motility assay protocol up until the three 1-ml washes of filament buffer. After the washes, 80 μl of 0.02 mg ml−1 XL-Rh-SMM with or without 1 mm ATP in filament buffer were added to the flow cell and incubated for 5 min. Flow cells were then washed three times with 80 μl of filament buffer to remove unbound XL-Rh-SMM filaments. Eighty μl of imaging filament buffer was added, and the data were collected and processed as above.
Transient Kinetics
All transient kinetic experiments were performed at 25 °C in filament buffer using a High-Tech SF61MX stopped flow spectrofluorometer with a mercury/xenon excitation lamp. The reported concentrations are those after mixing, and the SMM concentrations are that of myosin heads (two heads per SMM). S1 was prepared from chicken gizzard myosin by digestion with Staphylococcus aureus protease (29). F-actin was labeled with N-(1-pyrene) iodoacetamide (Invitrogen; P-29) using the procedure for biotination of F-actin (see above) without mixing with unlabeled actin. The pyrene-actin was mixed with equimolar phalloidin (Invitrogen; P-3457; from a 100 μm stock solution dissolved in methanol) at least 1 h before use. Phalloidin-stabilized actin and pyrene-actin were extensively dialyzed to remove any ATP (see above).
Monitoring the ATP-induced Transition from Strongly to Weakly Bound Acto-SMM and ATP-induced Dissociation of Acto-SMM
ATP interactions with acto-SMM were monitored at 25 °C either by light scattering, with excitation and emission at 406 nm and no emission filter, or by pyrene-actin fluorescence with excitation at 365 nm and a KV-399 cutoff filter (Schott). Unlabeled acto-SMM gave no signal change upon mixing with ATP using the instrument settings for the pyrene experiments, showing that changes in light scattering did not contribute to the pyrene signal changes. Acto-myosin (molar ratio of 1 actin to 1 head) was prepared by adding actin in small aliquots to XL-SMM filaments with extensive mixing by hand to give 0.5 μm heads. Acto-SMM was mixed in the stopped flow with and equal volume of ATP, giving a final myosin head concentration of 0.25 μm. Rates at each ATP concentration were taken from an average of at least four traces. Pyrene fluorescence data were fit to a single exponential plus a line. The slope of the line reflected the small change in fluorescence after mixing actin alone with ATP. Light scattering data were fit to a double-exponential model. The fast phase was dependent upon the ATP concentration and was therefore taken to reflect the dissociation of acto-SMM. A slow phase ranging from 0.30 to 0.94 s−1 was not correlated to the ATP concentration and was not further analyzed.
Kinetics of ADP Interactions with Acto-SMM and Acto-S1
KADP and k−ADP (see Scheme 2) were measured as in Ref. 30. XL-Rh-SMM filaments or S1 were first mixed with ADP and then equimolar (to SMM heads) phalloidin-stabilized pyrene-actin. This acto-SMM-ADP complex was incubated for 30 min and then mixed in the stopped flow with ATP. Final concentrations of ATP were 2 mm for XL-Rh-SMM filaments and 200 μm for S1. Transients at each ADP concentration were an average of at least four traces and were fit to a double exponential model. The rate of the fast phase (∼200 s−1 for filaments and 90 s−1 for S1) was determined from a mix of acto-SMM with no ADP against ATP. This value was fixed for the fits to transients containing ADP. The ADP release rate (k−ADP; see Scheme 2) is given by the slow phase of the transient and was calculated from an average of all the rates that could be reliably measured (where the amplitude of the slow phase was at least 10% of the entire transient). The amplitudes of the slow and fast phases, represented as relative percentages of the total amplitude, were plotted against the ADP concentration and fit to a Michaelis-Menten equation to determine the KADP for both XL-Rh-SMM filaments and S1.
SCHEME 2.
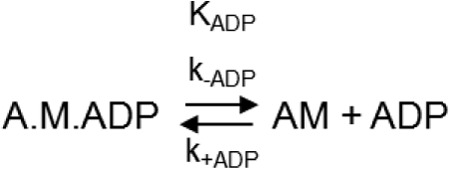
Mechanism for ADP interactions with acto-myosin.
RESULTS
Preparation and Fluorescence Labeling of SMM Filaments
SMM filaments were prepared by dialysis from a high ionic strength buffer, in which the protein is monomeric, into filament buffer to allow filaments to form relatively slowly. This buffer and protocol were chosen because 1) the final ionic strength (148 mm) and pH is close to physiological, 2) most of the SMM formed filaments, 3) the filaments were relatively uniform in length, and 4) the samples lacked large aggregates (see below). In cases where we needed to visualize and measure the velocity of single filaments moving along actin using TIRF microscopy, exposed amines on monomeric SMM were substoichiometrically labeled with NHS-rhodamine prior to filament formation (Rh-SMM).
EDC Cross-links the Heavy Chains of SMM Filaments
To stabilize filaments against depolymerization, SMM or Rh-SMM filaments were treated with the zero-length cross-linker, EDC, which cross-links the carboxyl groups on glutamate and aspartate side chains to the amine groups on lysine side chains. These amino acids are abundant and in close proximity in the SMM tail region, which is known to form a stable α-helical coiled-coil for much of the LMM region. Fig. 1A shows an image of a polyacrylamide gel designed to separate high molecular mass proteins, comparing an uncross-linked (no XL) to a 30-min EDC-treated (XL) Rh-SMM sample. The lanes were overloaded to visualize the three prominent bands resulting from the cross-linking (asterisks). Fig. 1C shows a plot of the molecular mass standards (circles) versus distance migrated along with a linear fit (dotted line). The three crosses indicate the calculated masses of one, two, or three HC. The squares show the measured positions of the HC and the higher bands indicated in Fig. 1A (asterisks) and the calculated masses from the standard curve. The HC migrates at ∼ 231 kDa. The two bands (* and **) and the density between them (bracketed) span the calculated mass for 2 HC and therefore may represent different conformations of two heavy chains cross-linked together (see below). The band labeled *** is very close to the calculated mass of 3HC. Fig. 1B shows a split image of a single gel (designed to separate all subunits) loaded with Rh-SMM filament samples generated from an EDC cross-linking time course. The upper image shows the disappearance of the heavy chain and the appearance of cross-linked heavy chains with time. The lower image shows that there was little change in the intensity of either light chain band up to 30 min of cross-linking. Fig. 1D shows a quantitative analysis of the combined results from eight time course experiments as in Fig. 1B. The density of the heavy chain band (percentage of the uncross-linked control; circles) decreases in a nearly linear manner with cross-linking time, reaching a loss of 38% after 30 min and ∼ 70% after 60 min. In contrast, there was little change in the RLC (squares) or essential light chain (diamonds) bands after 30 min and a ∼10% loss after 60 min. To avoid light chain cross-linking, which may adversely affect the phosphorylation-dependent regulation of the ATPase activity, we further characterized the properties of the preparation after 30 min of cross-linking.
FIGURE 1.

EDC cross-linking of Rh-SMM filaments. A, image of Coomassie-stained 3–8% Tris-acetate gel (Invitrogen) showing HiMark prestained high molecular weight protein standards (Stds; Invitrogen), 5 μg of Rh-SMM (No XL), and 5 μg of XL-Rh-SMM (XL; 30-min EDC reaction). Less than 5% of the protein in this sample remained in the supernatant after pelleting the filaments. Un-cross-linked HC, cross-linked heavy chain bands are labeled with asterisks. Light chains are not seen on this gel. B, split image of one 4–20% Tris-glycine gel (Invitrogen) showing time course of EDC cross-linking of Rh-SMM filaments quenched with 5 mm DTT at specified times. Upper panel, bands labeled HC XL correspond to those from A, although they are not well resolved; lower panel, light chain region. C, plot of molecular mass standards (circles) fit to a line (dotted). Crosses are the calculated masses for one, two, or three HC as indicated. Squares show measured band positions corresponding to gel in A, using the standard curve. D, plot of pixel density of bands averaged from eight different EDC cross-linking time courses from three independent Rh-SMM preparations. HC (circles), RLC (squares), and essential light chain (ELC, diamonds). Error bars are ± S.D. E, cartoon showing mode of SMM molecule packing in a single side polar filament sheet. The tails are shown as gray rods with the assembly domains in black. The S2 region is connected to the light meromyosin (LMM) region of the rod at a known bending region (32). Filament stability comes from intermolecular interactions between assembly domains. F, cartoon of possible types of inter- and intramolecular cross-links induced by EDC treatment. The positions of the cross-links (dotted vertical lines) are shown in the LMM region of the heavy chains for illustration only. Actual cross-link locations are unknown except that they only involve the heavy chains and that some cross-links must be forming between the assembly domains of adjacent molecules to explain filament stability.
Fig. 1E shows a cartoon of a single sheet of a SMM side polar filament depicting the packing model proposed by Cross and Engel (9). A single molecule overlaps two parallel partners with a stagger of ± 14.3 nm and overlaps one antiparallel partner by 14.3 nm. The stability of the filament is attributable to the antiparallel intermolecular interactions, through the contacts within the assembly competent domain (black). This domain contains 28 residues near the C-terminal end of the predicted α-helical coiled-coil but does not include the terminal nonhelical tailpiece (31) (not shown). Although other interactions are possible, without this domain, filaments do not form (31). Fig. 1F depicts the types of cross-links (not the specific locations) that could reasonably explain our gel data. Two types of cross-links are possible to explain the two HC band; intramolecular cross-links (top panel) would not likely lead to filament stability, but intermolecular cross-links between the assembly competent domains of adjacent antiparallel molecules (second panel from top) would stabilize the filament against depolymerization (see below). These two different types of dimers (and their possible variations) may have different conformations that could lead to the bracketed bands in Fig. 1A. Example cross-link locations are shown to explain the trimer band (*** in Fig. 1A). Cross-link(s) are most likely within the LMM region of the coiled-coiled tail domain, because it is more stably dimerized than the S2 region (31, 33–35). However, our data do not directly show the locations of any cross-link, except that they primarily involve the heavy chains.
Imaging of SMM Filament Ultrastructure by Electron Microscopy
Fig. 2 shows electron micrographs of representative fields of negatively stained SMM, Rh-SMM, XL-SMM, and XL-Rh-SMM filaments in panels A–D, respectively. Inspection of a large number of fields showed no significant difference in general morphology caused by either rhodamine labeling, EDC cross-linking, or the combination of the two modifications. The average length of XL-Rh-SMM filaments was 0.63 ± 0.02 μm (S.E.) (n = 167); the median was 0.57 μm. From this population, only one filament was clearly bipolar, whereas the rest were apparently side polar. The side polar structure is particularly evident for the two cross-linked samples (Fig. 2, C and D) because the amount of monomeric myosin (black arrow) in the background is much lower (as expected) than the uncross-linked samples (Fig. 2, A and B). A halo of what appear to be the heads (S1; white arrows) and the S2 region of the rods (white arrowheads) can be seen projecting along both sides of the main filament backbone in all four images.
FIGURE 2.

Electron micrographs of negatively stained SMM filaments in the absence of ATP. A, SMM. B, Rh-SMM. C, XL-SMM. D, XL-Rh-SMM. Note side polar structures in all images. Scale bars, 100 nm. Black arrow in D, monomeric myosin; white arrowheads in B and C, S2 projecting from filament backbone; three white arrows in C, halo of S1/S2 around filament backbone.
Stability of XL-SMM Filaments against ATP-induced Depolymerization
Rh-SMM (not cross-linked) was attached to a glass coverslip and imaged by fluorescence microscopy (Fig. 3A). The fields contain oblong structures with dimensions expected of individual filaments (see arrow), along with a small number of filament aggregates. In Fig. 3C, surface-attached Rh-SMM filaments were treated with ATP. Except for a few remaining filament aggregates, most myosin no longer appears as discrete filaments. Fig. 3B shows XL-Rh-SMM filaments in the absence of ATP, which appear to be similar to Rh-SMM in Fig. 3A, but with less background fluorescence. Unlike Rh-SMM, XL-Rh-SMM remains filamentous in the presence of ATP (Fig. 3D) even after 30 min. Although actual filaments lengths cannot be obtained from these images because of the diffraction limit, we found no significant difference between the apparent lengths of Rh-SMM (0.85 ± 0.24 μm) and XL-Rh-SMM (0.94 ± 0.26 μm), respectively, without ATP (n = 200). In summary, we conclude that the XL-Rh-SMM filaments appear indistinguishable from Rh-SMM filaments with regard to length, shape, and levels of aggregation, except that they are stable enough to be imaged in the presence of ATP.
FIGURE 3.

Assessment of SMM filament stability. A–D, imaging of SMM filaments by TIRF microscopy. Rh-SMM (A) or XL-Rh-SMM (B) at 100 μg ml−1 in filament buffer was added to a bare glass flow cell followed by imaging filament buffer. White arrows indicate single filaments. Rh-SMM (C) and XL-Rh-SMM (D) filaments are as in A and B except that 1 mm ATP was added to the flow cell after filament binding to the surface. Notice that the XL-Rh-SMM filaments retain their structure in the presence of ATP (D), whereas the Rh-SMM filaments (C) largely depolymerize. The smallest structure resolvable is 106 nm long (1 pixel). E and F, dilution-induced SMM filament depolymerization. Light scattering was monitored with excitation and emission at 406 nm with no emission filter. All proteins were assayed at 1 μm heads (final concentration). ATP was removed from phosphorylated samples (see “Experimental Procedures”). Light scattering transients after rapidly mixing 2 μm SMM heads with an equal volume of filament buffer with or without 100 μm ATP (final [ATP] = 50 μm). E, XL-SMM (top two traces) and SMM (middle two traces). Traces at bottom labeled high ionic strength are shots against a 50 mm MOPS, pH 7.0, 500 mm KCl without ATP (final concentration, 0.25 m KCl) The process was essentially too fast to measure within the dead time of the instrument. F, XL-pSMM (top two traces) and pSMM (bottom two traces). Experiments were performed in filament buffer at 25 °C. The data in E and F reflect the raw data and were not normalized in any way.
We also measured the relative stability of SMM versus XL-Rh-SMM (Fig. 3E) or pSMM versus XL-Rh-pSMM filaments in solution using light scattering after perturbing the equilibrium between filaments and monomers by rapid dilution in the stopped flow spectrophotometer. For both phosphorylation states, 1) the initial signal of XL-SMM was higher than SMM, showing that cross-linking stabilized filaments against the depolymerization prior to the stopped flow shots; 2) after rapid dilution without ATP, cross-linked filaments depolymerized to a much smaller extent but at a similar rate to unmodified SMM filaments, showing that molecules are able to leave the cross-linked filaments at the normal rate as might be expected because not all the molecules were intermolecularly cross-linked; and 3) the transients with ATP show a rapid early phase (within 5 s) with small amplitude corresponding to the effect of ATP to promote depolymerization as expected. Phosphorylated filaments (Fig. 3F) were more stable than unphosphorylated filaments (Fig. 3E) as expected (36). A low level of scattering was observed at high ionic strength showing that filaments, even if cross-linked, depolymerized under these conditions. This is consistent with the fact that, for XL-SMM, at most 35% of the heavy chains were intermolecularly cross-linked (Fig. 1).
Visualization of Actin-bound XL-Rh-SMM Filaments by Fluorescence Microscopy
We used fluorescence microscopy to visualize specific interactions of individual XL-Rh-SMM filaments with F-actin. To obtain interpretable results, conditions had to be such that SMM filaments could not bundle actin into large aggregates. We treated cleaned glass coverslips with a mixture of PEG-silane and biotin-PEG-silane to create a covalently attached PEG brush surface. Exposed biotin groups were saturated with streptavidin to provide binding sites for 5% biotinylated TRITC-F-actin. This method allowed us to effectively wash away any unbound actin that would otherwise be available for bundling. The remaining actin filaments were firmly attached to the surface as evidenced by lack of Brownian motion (data not shown). XL-Rh-SMM filaments were added and allowed to bind to the surface-attached actin in rigor (Fig. 4A). Single XL-Rh-SMM filaments (green) can be seen bound to actin filaments (red), with little nonspecific binding to the PEG surface. Two modes of binding can be seen. Most of the filaments bind with one end or side projecting perpendicularly from the actin (arrows), but some appear to be bound lengthwise or parallel to actin (arrowheads). Single actin filaments are often decorated with multiple SMM filaments. The perpendicular mode of binding (Fig. 4A, right panels) is similar to that observed with nonmuscle myosin IIb, although those filaments are bipolar (37, 38). In the presence of 1 mm ATP (Fig. 4B), fewer filaments were bound per unit length of actin, although some SMM filaments remained bound after several minutes of incubation with ATP. Averaged data from three different images (54 × 54 μm) showed that adding ATP reduced the number of bound SMM filaments per μm length of actin from 1.5 to 0.5 (data not shown). Most but not all of these remaining filaments appeared to bind lengthwise (Fig. 4B, right panels). The structural basis of the modes of binding is not clear at this time and will require further study. These data show that XL-Rh-filaments are stable enough to be clearly seen as individual filaments bound to actin, and their binding to actin is diminished in the presence of ATP as expected.
FIGURE 4.
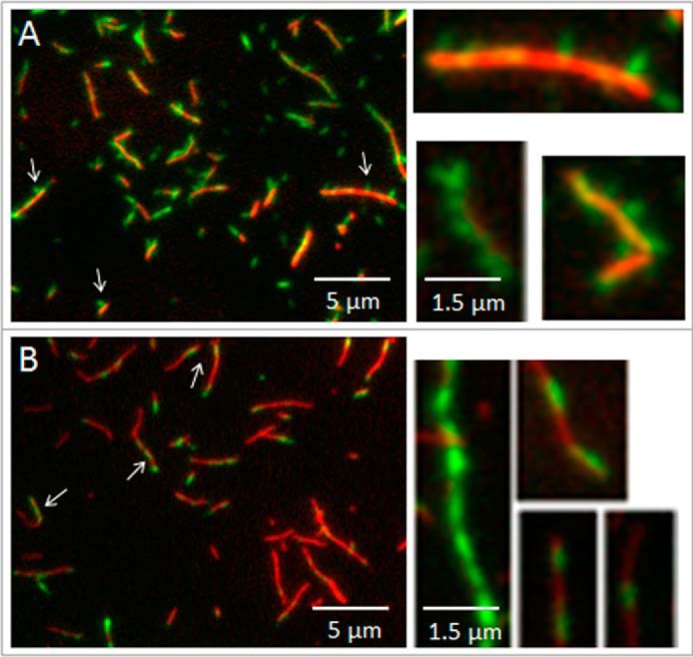
Visualization of single XL-Rh-SMM filaments binding to F-actin using fluorescence microscopy. A, dual channel overlaid image. F-actin filaments (red) attached to a PEG brush surface (see “Experimental Procedures”) interacting with XL-Rh-SMM filaments (green) in the absence of ATP. Arrows and arrowheads point to single SMM filaments that appear to be attached to actin by only one end (perpendicular) or along the length of the filament (parallel), respectively. Magnified selected images at right show the perpendicular mode of filament attachment. B, same as A except that SMM was added to the actin on the surface in the presence of 1 mm ATP. Note fewer SMM filaments are bound to actin compared with A. Arrows and arrowheads as in A. Magnified selected images at right show the parallel mode of filament attachment.
Kinetics of Interaction of ATP with Acto-myosin
Stopped flow spectrometry was used to characterize the kinetics of ATP interactions with the XL-SMM-actin complex in solution. Experiments with S1 were done in parallel. The fluorescence of pyrene-labeled actin is quenched by strong myosin binding (39). Fig. 5A shows representative transients of pyrene fluorescence enhancement upon mixing ATP with a stoichiometric mixture of pyrene-actin with either S1 or XL-SMM filaments. Both transients followed single exponentials, but the amplitude of the S1 was greater. This is probably due to the fact that not all heads in a given filament can bind to actin because of steric constraints. These constraints would be minimal with the head fragment S1. For both filaments and S1, the observed rate constants (kobs) depended linearly on the [ATP] over the range tested (Fig. 5B, circles and triangles, respectively). The mechanism was modeled as a two-step process described in Scheme 1 (40). The low fluorescence strong actin-binding actin myosin (AM) complex is in rapid equilibrium (K1 = k+1/k−1) with low fluorescence strong actin-binding A.M.ATP collision complex which isomerizes (k+2) to the high fluorescence (indicated by A*) weak actin-binding A*-M.ATP state that rapidly dissociates to actin and myosin-ATP. Under these conditions (kdiss ≫ k+2 + k−2), Equation 1 applies (41, 42).
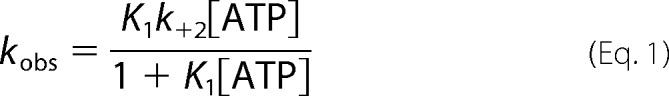 |
At low [ATP], the second order rate constant (K1k+2) is approximated by the initial slope of the plot in Fig. 5B giving K1k+2 = 0.27 μm−1 s−1 for filaments and 0.44 μm−1 s−1 for S1. The S1 value is essentially the same as previously measured under similar buffer conditions (0.47 μm−1 s−1) (30). For both, 1/K1 can be estimated to be >1.5 mm. All kinetic data are summarized in Table 1.
FIGURE 5.

Kinetics of interaction of ATP with acto-myosin. A, representative averaged transients of pyrene fluorescence after mixing pyrene-acto-myosin with 25 μm ATP (final concentration) for S1 and XL-SMM filaments as indicated. The initial values were normalized to 1.0. The data were fit to a single-exponential plus a line. The kobs from the single-exponential fits (lines) are 13 and 11 s−1, respectively, and the slopes of the lines are 0.006 and 0.089 for S1 and filaments, respectively. B, the observed rates (kobs) for light scattering and pyrene-actin fluorescence changes after mixing ATP with acto-myosin. ATP concentrations after mixing are plotted. K1k+2 (Scheme 1) was obtained from the slope of the linear fits. Pyrene-actin fluorescence (triangles) and light scattering (crosses) for smooth muscle myosin S1. Pyrene-actin fluorescence (circles) and light scattering (squares) for XL-SMM filaments. All experiments were performed in filament buffer at 25 °C (see Table 1 for a summary of data).
SCHEME 1.
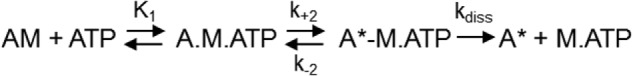
Mechanism for ATP interactions with acto-myosin.
TABLE 1.
Summary of kinetic data
K1k+2 is defined in Scheme I, KADP and k−ADP in Scheme II. KATPase is the [actin] at half-maximal v, where v is the actin-activated ATPase rate, and vmax is the maximal v from a fit to the Michaelis-Menten equation. KATP is the [ATP] at half maximal actin-activated ATPase measured at 75 μm actin. KATP mot is the [ATP] at half maximal V, where V is the rate of relative motion of actin and myosin, and Vmax is the maximal V from a fit to a rectangular hyperbola.
Because the kinetics of XL-Rh-SMM filament interactions with actin have not been previously characterized, we determined whether or not the conversion to the weak actin-binding state (A*-M.ATP) was sufficient to dissociate filaments from actin and to confirm that Equation 1 applies (kdiss ≫ k+2 + k−2) under these conditions. We repeated the experiment with unlabeled actin and monitored the change in light scattering. In this case, scattering signal comes from both actin and SMM filaments, but the acto-SMM complex scatters more intensely. Therefore, the decrease in the light scattering signal (kobs) after mixing ATP should proceed at the rate that limits the dissociation of actin and myosin (k+2). The decrease in light scattering signal after mixing ATP fit well to a double-exponential with the exception of an early phase (<0.1 s) showing a small rise in signal. A fast phase depended upon [ATP] in a nearly linear manner as shown in Fig. 5B (squares), and a slower phase ranged from 0.30 to 0.94 s−1 but was not correlated to the ATP concentration (not plotted). The slope of a linear fit to the scattering data gave K1k+2 = 0.10 μm−1 s−1 or a factor of 2.7 slower than K1k+2 measured by pyrene-actin fluorescence. This slower rate may be related to myosin filaments detaching from more than one actin filament, because it was not evident in experiments with S1. For S1, the scattering transients (Fig. 5B, crosses) were similarly fit as above, except that the initial small rise was not observed, suggesting that it is unique to the filament results. K1k+2 for the scattering signal was 0.50 μm−1 s−1, which is essentially the same as K1k+2 for the pyrene-actin signal (Fig. 5B, triangles). The similarity between the slopes for the pyrene-actin fluorescence and the light scattering shows that the rate of isomerization to the weak actin-binding state A*-M.ATP partially limits the observed rate of dissociation of the filaments from actin.
Kinetics of ADP Interactions with Acto-myosin
Fig. 6 (A and B) shows representative fluorescence transients for XL-SMM filaments and S1, respectively, after ATP was rapidly mixed in the stopped flow spectrophotometer with pyrene acto-myosin containing ADP at the indicated concentrations. The data were fit (lines) to a double-exponential model, with the fast phase giving the rate of ATP binding to acto-myosin and the slow phase giving the ADP release rate, k−ADP (Scheme 2) (30). Fig. 6 (C and D) shows a plot of the amplitudes of the fast and slow phases versus [ADP] for XL-filaments and S1 (respectively). The relative amplitudes of the two phases varied in a reciprocal manner, reflecting the fractional binding of ADP to myosin. A fit to the plots using the Michaelis-Menten equation gives KADP = k−ADP/k+ADP (Scheme 2). The data for S1 gave k−ADP = 28 ± 4 s−1 and KADP = 27.7 ± 5.1 μm, which are similar to previously measured values (22 s−1 and 5 μm (30) under slightly different conditions). For XL-SMM filaments, the data were very similar to those for S1 with k−ADP = 21 ± 3 s−1 and KADP = 38.5 ± 7.6 μm. These data suggest that ADP interactions of myosin heads incorporated into acto-SMM filaments are little affected by the environment of the filament under these conditions.
FIGURE 6.

Kinetics of ADP release from pyrene-acto-myosin. A and C, XL-Rh-SMM filaments. B and D, S1. A and B, representative averaged pyrene fluorescence transients at indicated [ADP] with fits to a double-exponential model (lines). The initial values were normalized to 1.0. For filaments, the fast phase was fixed to 200 s−1, and the slow phase was 30, 31, and 23 s−1, for 10, 50, and 200 μm ADP, respectively. For S1, the fast phase was fixed to 70 s−1, and the slow phase was 16 and 24 s−1 for 10 and 100 μm ADP, respectively. C and D, relative amplitudes of the two phases are plotted as a percentage of the total amplitude of the transients. The fast phase (squares) reports the rate of binding of ATP to acto-myosin, whereas the slow phase reports the ADP release rate (k−ADP). The data were fit to the Michaelis-Menten equation to obtain KADP (lines). All experiments were performed in filament buffer at 25 °C (see Scheme 2 for definitions of rate constants and Table 1 for values).
Steady-state Actin-activated MgATPase Measurements
Fig. 7A shows the ability of actin to stimulate the steady-state MgATPase activity of pSMM (circles, solid line) and XL-Rh-pSMM filaments (squares, dashed line). Lines show the fit to the Michaelis-Menten equation. For pSMM, the KATPase = 10.5 ± 1.0 μm, and vmax = 0.48 ± 0.01 s−1 head−1. For XL-Rh-pSMM filaments the KATPase = 12.9 ± 0.8 μm, and vmax = 0.49 ± 0.01 s−1 head−1. Variables are defined in and summarized in Table 1. These data show that neither cross-linking nor rhodamine labeling had a measureable effect on the actin-activated ATPase rates in the phosphorylated states. As expected for fully regulated SMM (43), the unphosphorylated samples, regardless of modification, had a very low ATPase activity that was not activated appreciably by actin. This suggests that the XL-Rh-SMM shows normal regulation of the actin-activated MgATPase activity by phosphorylation of the RLC.
Fig. 7B shows the [ATP] dependence of actin-activated steady-state MgATPase activity of pSMM (circles, solid line) and XL-Rh-pSMM filaments (squares, dashed line) at 75 μm actin. A fit to the Michaelis-Menten equation gave KATP = 4.8 ± 1.7 and 9.2 ± 3.0 μm, and vmax = 0.48 ± 0.01 and 0.49 ± 0.01 s−1 head−1, for pSMM and XL-Rh-pSMM, respectively. These data suggest that there is no significant difference between pSMM and the XL-Rh-pSMM filaments with regard to the ATP dependence of the steady-state actin-activated ATPase.
ATP Dependence of the Rates of Motion of Myosin Moving Actin or Moving on Actin in Vitro
We compared the ability of XL-Rh-pSMM filaments and monomeric pSMM to move actin by vitro motility assays under the same buffer conditions used in the ATPase assays. First, we used a standard geometry motility assay to determine the velocities of actin moving over surface-attached (nitrocellulose) monomeric pSMM versus [ATP] (Fig. 8A, circles). Monomeric pSMM was obtained by applying the protein to a nitrocellulose-covered coverslip in high ionic strength buffer, which dissolves all filaments. Actin was added, and the motility was measured in imaging filament buffer. A fit to a Michaelis-Menten equation (solid line) using Equation 2 gave Vmax mot = 0.48 ± 0.03 μm s−1 and KATP mot = 16.7 ± 2.6 μm.
 |
These data are similar to those of Harris and Warshaw (44), who found Vmax mot = 0.77 μm s−1 and KATP mot = 46 μm at lower ionic strength (25 mm KCl).
FIGURE 8.

In vitro motility assays measuring relative motion of actin and myosin. A, ATP dependence of velocities of XL-Rh-pSMM filaments moving on surface-attached actin (inverted geometry; squares, solid line fit), actin filaments moving over surface-attached XL-Rh-pSMM filaments (Xs; dashed line fit), or surface-attached monomeric pSMM (circles, solid line fit). The lines show fits to Equation 2. Error bars are S.D. (see Table 1 for kinetic parameters, Vmax mot and KATP mot). The inset shows low ATP region of plot. Schematics compare the geometries of the standard motility assay measuring sliding velocities of fluorescent actin (green/yellow) on top of monomeric pSMM (green; only showing a few molecules for clarity) bound to a coverslip (blue) and the geometry of the inverted motility assay measuring sliding velocities of XL-Rh-pSMM filaments (green) on actin filaments bound to a PEG brush (blue) on a coverslip. Cartoons are not meant to indicate the details of the heads during motion. Arrows indicate whether actin or myosin is moving (the direction of the arrow is arbitrary). B, dual channel overlaid image time course showing the movement of a single XL-Rh-pSMM filament (green, solid arrow) moving along a single actin filament (red). After 300 s, the SMM filament still remains bound (parked) at the end of the actin and does not dissociate. The dotted arrow points to a collection of previously parked SMM filaments at the end of the actin filament. The [ATP] was 1 mm (see supplemental Movie S1 for actual image sequence). Scale bar, 2 μm. C, double reciprocal plot of the data in A, showing filament velocities measured in inverted assay (squares) and actin velocities driven by monomeric myosin (circles and circles containing dots). The line is a linear weighted fit to the monomer data from 5 to 50 μm ATP (circles). The fit gives k−ADP = 32 s−1 and kATP = 6 μm−1 s−1 with r = 0.84. The remaining points at [ATP] > 50 μm (circles with dots) indicate the hypermotile region that is not solely detachment-limited (46–48). D, plot of filament velocities measured in the inverted assay (V) from A versus ATPase activity (v; units converted to s−1 per filament) at high actin from Fig. 7B at various [ATP]. The line is a linear fit to the data giving the equation V = 0.036 + 0.0051x; r = 0.96), where 0.0051 = Nfilament·d. Using d = 0.01 μm gives Nfilament = 0.51.
A similar experiment was performed to determine the velocity of actin powered by surface-attached XL-Rh-pSMM filaments (Fig. 8A, crosses). The surface density of the filaments was adjusted so that most actin filaments were moved by a single SMM filament. Actin filaments moved over myosin filaments without detaching (see supplemental Movie S4 for a representative image sequence). No directed actin motion was observed in areas that did not have an attached myosin filament, suggesting that any small amount of surface-attached monomeric myosin was not sufficient to generate motion. The Vmax (0.45 ± 0.03 μm s−1) of the observed moving actin was similar to the velocity measured for myosin monomers, but the KATP mot was much lower at 4.1 ± 1.3 μm.
We used an inverted geometry motility assay to measure the velocity of XL-Rh-pSMM filaments moving over surface-attached F-actin. The ATP dependence of the motion (Fig. 8A, squares) gave a Vmax mot = 0.78 ± 0.02 μm s−1 and KATP mot = 8.5 ± 1.4 μm. Fig. 8B shows an example of a typical XL-Rh-SMM filament (solid arrow) moving along fluorescent actin that was firmly attached to a PEG brush surface (as described above) (see supplemental Movies S1–S3 for the corresponding image sequence and other examples of such motion, respectively). We noted several interesting features of the motion that were common to essentially all moving filaments: 1) Filaments moved along actin without detaching. Run lengths were such that we were unable to capture the detachment of moving XL-Rh-pSMM filaments from actin (from ∼100 trajectories). 2) When filaments reached the end of actin, they remained attached (parked). This is evident in Fig. 8B, after 300 s the filament is still attached to the end of the actin as were several other SMM filaments having already moved and parked at the end of the actin filament (dashed arrow). 3) The addition of phosphatase and wortmannin (kinase inhibitor) in the presence of ATP stopped motility (as expected upon dephosphorylation) but did not lead to detectably fewer SMM filaments attached to the actin (data not shown), even if only a single filament was parked at a given actin end.
Interestingly, parking was also observed in the standard geometry experiments with actin moving over surface-attached XL-Rh-pSMM filaments (see above). Typically, actin parked when it reached its end, after which it remained attached to the XL-Rh-pSMM filaments. These parked actin filaments were usually dynamic except for the point of attachment at the myosin filament, suggesting that interactions with the surface were not the reason for parking. Observations from both the inverted and standard geometry assays together suggest that filament parking is caused by an attribute of the XL-Rh-pSMM filaments. Interestingly, this parking behavior was not observed with skeletal myosin filaments (data not shown). Further work is needed to determine the mechanism of this parking behavior.
We were unable to determine whether or not the parking behavior was due to the presence of the cross-links in the filaments because for both the inverted and standard geometry assays, cross-linking was required to be able to observe motion. Without cross-linking, even if filaments were phosphorylated before adding them to the motility assay, filaments disassembled because of the low protein concentration and the presence of ATP.
DISCUSSION
To measure the mechanochemistry of actin filament-myosin filament sliding, we first developed a method to prepare fluorescent cross-linked SMM filaments that are largely stable to depolymerization by ATP in a near physiological buffer. The stability of the filaments is due to EDC-induced zero-length cross-linking of the heavy chains not the light chains (Fig. 1). Only ∼35% of the heavy chains were cross-linked to other heavy chains. Although cross-link locations were not determined, intermolecular cross-linking between heavy chains of antiparallel adjacent molecules within the filament assembly domains of the coiled-coil tails (Fig. 1D) is consistent with our gel data (Fig. 1, A–C) and the resulting filament stability (Fig. 3). Other intrasubunit cross-links may be present that we cannot detect using SDS gel electrophoresis.
Electron micrographs of negatively stained samples showed that Rh-SMM, XL-SMM, and XL-Rh-SMM filaments are side polar and highly similar to unmodified filaments (Fig. 2). Side polar SMM filaments are known to have four molecules per 14.3-nm repeat (9, 10). This corresponds to two side polar sheets stacked upon one another (a single sheet is depicted in Fig. 1E). From the micrographs, our side polar filaments are on average ∼0.63 μm in length and therefore on average have ∼176 molecules or ∼352 heads.
We used two approaches to assess the stability of XL-SMM filaments. First, we showed that individual XL-Rh-SMM filaments were fluorescent and stable enough to be easily imaged under the highly dilute conditions of fluorescence microscopy. These experiments were in the presence of ATP and in the unphosphorylated state (Fig. 3), conditions that most strongly promote myosin depolymerization. Rh-XL-SMM filaments were strikingly more stable in ATP than SMM filaments (Fig. 3). Second, by rapidly diluting filaments with ATP using stopped flow mixing (Fig. 3, E and F) and monitoring the transition to a new equilibrium position with light scattering, we showed that XL-Rh-SMM filaments depolymerized to a much smaller extent but at a similar rate to unmodified SMM filaments.
The stability of the XL-SMM filaments allowed us to examine ATP interactions with acto-myosin. For the unphosphorylated filaments, we used TIRF microscopy to show that the number of filaments bound to actin decreases with added ATP, as expected for a native actin-myosin interaction (Fig. 4). Also, we used stopped flow spectrophotometry to characterize the kinetics of ATP interactions using standard methods that have historically been limited to soluble fragments of myosin. The interaction of ATP with the rigor complex of pyrene-actin with XL-SMM filaments (Scheme 1 and Fig. 5) gave two interesting findings. First, the second order rate constant for ATP binding, K1k+2, for filaments and S1 was very similar, showing that the myosin heads in a XL-SMM filament act much like single myosin heads with regard to the transition from the strong to weak actin-bound state. Second, the transition was accompanied by filament detachment from actin as evidenced light scattering kinetics. This showed that the combined interaction of hundreds of weakly bound heads with actin is not sufficient to keep the filament bound to actin under these conditions.
We also used stopped flow methods to determine KADP and k−ADP (Scheme 2) for unphosphorylated filaments and compared these values to those for S1. The myosin heads in a filament act much like single myosin heads with regard to the kinetics of ADP interactions. Importantly, the k−ADP was ∼20–30 s−1 for both S1 and filaments.
The XL-Rh-SMM and unmodified myosin had very similar steady-state MgATPase kinetics (Fig. 7A. Specifically, the XL-Rh-SMM filaments behave similarly to unphosphorylated HMM (29, 43, 45), which is inhibited and not activated by actin. Also, XL-Rh-pSMM filaments, as for pHMM, become activated in the presence of actin (Fig. 7A. These data agree with Trybus (16), who showed that SMM filaments stabilized with an antibody were regulated by phosphorylation, meaning that for full-length myosin, regulation can occur independently of filament assembly. Our data show that the chemical modifications present in XL-Rh-pSMM filaments do not significantly alter actin interactions or the conformational changes required for inhibition or activation of the ATPase. This allows us to be reasonably confident that mechanochemical behaviors described below are representative of the “native” SMM filament.
To determine the biochemistry underlying the enhanced velocities of filament-filament sliding, we measured the ATP dependence of the velocity of phosphorylated filaments interacting with actin using motility assays for both the standard and inverted geometries. We fit these data to Equation 2, which has a Michaelis-Menten form that is derived from the kinetic mechanism in Scheme 3.
 |
Equation 3 applies only if we assume that velocity is limited by the rate that actin and myosin can detach from each other (46–48). Drag heads that are at the end of the ATPase cycle in the ADP state still bound to actin restrict or limit the stroke size of the driving heads and thus limit the velocity. The detachment-limited model predicts that V = d·kdet, or V = d·τon−1, where d is the myosin step size (10 nm) (49, 50) and kdet is the effective actin-myosin detachment rate, which is limited by the rate of ADP release from acto-myosin at high ATP (15). The meaning of KATP mot is clear in this context. KATP mot·kATP = 1/τon, where τon is the attached time at high ATP and kATP is the second order rate constant for ATP binding. This is the point where the attached time is doubled (2τon), and the velocity is therefore cut in half. Both assays using SMM filaments gave very similar KATP mot, which were significantly lower than that for the standard assay using myosin monomers (4–8 μm versus 16.7 μm; Table 1). Therefore, the comparatively low KATP mot appears to be a unique property of the filaments that is not shared by the monomers. More importantly, the KATP of the steady-state ATPase (Fig. 7B; 9.2 μm) is strikingly similar to the KATP mot of the inverted and standard geometry filament sliding (Fig. 8A; 8.5 and 4.1 μm, respectively).
SCHEME 3.
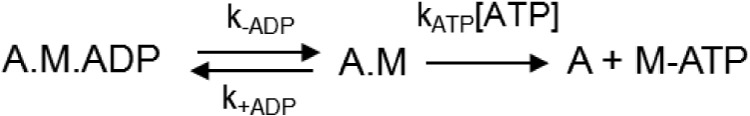
Mechanism for detachment-limited kinetics controlling actin-sliding velocity.
Below we make three arguments that strongly suggest that the rate of unloaded SMM filament movement on actin is not primarily limited by detachment kinetics but rather are primarily limited by attachment kinetics (weak to strong binding kinetics). The arguments have to do with 1) the duty ratio, 2) the shape of the velocity versus ATP curves, and 3) the relationship between ATPase activities and velocities. Finally we propose a structural reason for the attachment-limited kinetics observed in filaments. In this section, we will restrict the discussion to the velocity data obtained in the inverted geometry, because we feel it best represents how the SMM filament would behave during unloaded shortening of muscle. However, the same fundamental mechanisms would also apply to filament behaviors in the standard geometry.
Duty Ratio Considerations
It has been known for many years that the ATP concentration needed for half-maximal ATPase (KATP) is much lower than the ATP concentration needed for half-maximal actin sliding velocity in the motility assay (KATP mot) using myosin II monomers attached to a surface. For example, for skeletal muscle myosin, the respective values are 10 and 200 μm (51). Using the classic interpretation that the speed of motion is completely limited by detachment kinetics and that the ATPase rate is controlled by weak to strong binding kinetics (52, 53) (attachment-limited), the fraction of time a myosin spends strongly bound to actin, or duty ratio, r, can be calculated from Equation 4 (51). According to this equation, skeletal muscle myosin has a relatively low duty ratio of ∼0.05, which is consistent with other measurements. The duty ratios determined for SMM monomers are similar (54).
 |
Unlike myosin monomer-based motility, here we have shown that KATP mot obtained using myosin filament-based motility is similar to KATP. Applying the detachment-limited model (Equation 3) to these data gives a duty ratio of 0.9 that cannot be reconciled with previous estimates for SMM of ∼0.05 (15). This duty ratio analysis strongly suggests that the data for SMM filaments do not fit a detachment-limited model. A closer look at our filament velocity data also supports this conclusion.
Velocity versus ATP curve
Fig. 8C shows a double-reciprocal plot of the data in Fig. 8A, comparing the SMM filament velocities measured in the inverted assay (squares) to the actin velocities from monomeric SMM (circles). As has been previously shown for both skeletal myosin and SMM monomers (46–48), the velocity is not detachment-limited over the entire range of [ATP] values. The line is a linear fit to the monomer data from 5 to 50 μm ATP (circles) and represents the region of the curve that can be explained by detachment limited kinetics (Michaelis-Menten plot is linear in double-reciprocal format). The fit gives k−ADP = 32 s−1 and kATP = 6 μm−1 s−1 with r = 0.84, consistent with prior work (48). The remaining points at [ATP] > 50 μm (circles with dots) fall below the curve, indicating a hypermotile region that is not solely detachment-limited (46–48). Note the strikingly different and peculiar shape of the data for the SMM filament velocities (squares). At high ATP, velocities are completely independent of ATP, showing that they do not fit a detachment-limited model. Only at low ATP, below 20 μm, does V begin to depend upon ATP, thus revealing detachment-limited conditions where enough heads are in rigor to slow the velocities.
The Relationship between ATPase Activities and Velocities
Our observation that KATP mot and KATP for filaments are similar implies that V and v are limited by the same kinetic steps. Specifically, V, like v, is limited by the rate of weak to strong binding (≈ v) rather than the detachment rate (kdet) (55). In a purely attachment-limited model, for every ATP molecule hydrolyzed, a head in a given thick filament moves that filament a distance, d, along an actin filament and the predicted V = Nheads·d·v (where v is expressed in s−1 head−1). Here d is the step size, and Nheads is the number of heads in the filament system available to cycle with actin. The measured filament ATPase activity implicitly accounts for geometrical considerations such as the helical structure of the actin filament and the orientation of the myosin heads in the thick filament with respect to actin. There is no requirement that the Nheads simultaneously interact at any given instant in time. Alternatively, V = Nfilament vfilament·d, where vfilament is simply v converted to the units of s−1 filament−1, where Nfilament is simply the number of filaments working to generate velocity. If Nfilament = 1, then all the heads in the filament are available to work. In our case, this conversion is possible because we know the average number of heads per filament (352 ± 11 heads). Fig. 8D shows a plot of V for the filaments in the inverted assay versus vfilament from Fig. 7B at matching [ATP]. The slope of this plot V/vfilament should = Nfilament d, giving Nfilament = 0.51 ± 0.02. This means that on average in the inverted assay V is generated by half of a filament or half of the heads in the filament. Again this does not require that all these heads are simultaneously interacting with actin, rather that they are available for actin interaction at some time during filament movement. Using this model, the slower maximal velocity of the filaments in the standard geometry compared with the inverted geometry (Fig. 8A) suggests that N has been decreased. This might be due to surface interactions of some of the heads that inhibits their normal actin interactions.
The above calculations using the attachment-limited model say that when a filament is moving over actin in the inverted assay, it is using approximately half of the total heads in the filament. This makes good geometrical sense, given the side polar structure of SMM filaments, where half of the heads are of opposite polarity to the other half. In contrast, in the ATPase assay (where [actin] ≫ [myosin]) at any ATP a given myosin filament will most likely interact with more than one actin filament, one on each side of the side polar structure. Therefore, it makes sense that vfilament ∼2V. Of course other effects could contribute to this result. Some strongly bound myosin heads in the SMM filament could also contributing to slowing actin sliding velocities; in other words, there could be a detachment-limited kinetic component to the velocities, especially at the low [ATP]. In any case, the observation that Vmax < vfilament·d but exceeds d·τon−1 and that the KATP for V and vfilament are similar is consistent with a model in which vfilament significantly influences V.
Structural Reason for the Attachment-limited Kinetics in SMM Filaments
We suggest that elements of the myosin filament structure are the reason for the attachment-influenced kinetics observed here. In filaments, heads are attached to the flexible S2 region, which has a relatively low stiffness of ∼0.01–0.02 pN nm−1 (56, 57). Importantly, in our experiments these S2 regions are not interacting with a surface because in the inverted geometry motility assay, the SMM filaments are moving unhindered on top of actin filaments. When heads in a filament are moved into a drag position by filament sliding, they will contribute very little drag force because of the “buckling” of their S2 regions (56, 57). This is the basis of the nonlinear elasticity of myosins incorporated into a co-filament over a wide range of positive and negative strains (56, 57). Our data show that these drag heads detach from actin too quickly to be pulled into a high negative stiffness state that would decrease the effective working stroke distance of the driving heads, ultimately slowing velocity. Vmax mot = 0.78 ± 0.02 μm s−1 for filaments in the inverted assay, which is 3.5-fold higher than expected for the detachment-limited model V = d·k−ADP = 0.01 μm·22 s−1 = 0.22 μm s−1, where k−ADP is the ADP release rate from acto-myosin measured here for SMM filaments in solution. For this model to work, the k−ADP would have to be 3.5-fold faster, or 77 s−1, which is outside the variability of our measurements. According to Veigel and coworkers (58), a negative force of ∼2 pN is necessary to increase the rate of ADP release 3.5-fold. In a co-filament, Kaya and Higuchi (56) estimate that a backward displacement of ∼40 nm (caused by the S2 region) would be required to generate a negative force of 2 pN, which far exceeds the average distance heads are dragged before detaching (V·τon = 780 nm s−1·0.013 s = 10 nm). This analysis suggests that on average heads will not remain attached to actin long enough in the ADP state to experience sufficient negative force to accelerate their ADP release rate sufficiently for a detachment-limited model to explain our filament velocities.
In contrast to myosin in a filament, the stiffness of myosin heads (S1) bound directly to a solid support is very high at ∼ 2 pN nm−1 (56, 59, 60). These heads will contribute a significant drag force that slows velocity, the magnitude of which is influenced by the rate of detachment from actin. This is also partially true for full-length SMM monomers attached to nitrocellulose, where interaction of the S2 region with the surface may hinder the normal S2 conformational changes expected in the filament moving on top of an actin filament as in our inverted assay.
If our suggestion about the role of the S2 in filament motion is correct, then the motion of other types of filaments on actin may also not be limited by detachment kinetics. This appears to be the case for motion of nonmuscle myosin IIb filaments on top of surface-attached actin filaments, where the rate of movement is much faster than predicted from a detachment-limited model (37).
It is important to consider and compare the general models or mechanisms by which an S2 domain might enhance shortening speeds. If movement of this domain enhances the driving force of motility through a biased diffusion mechanism similar to that proposed by Huxley in 1957 (61), then we would expect a force-distance relationship for a myosin head to follow the plot illustrated in Fig. 9A. The corresponding energy landscape is shown in Fig. 9B. Here, the myosin S2 domain moves from a high force state to a low force state over a distance x that is larger than the known myosin step size because of the lever arm alone (an enhanced apparent step size of 30 nm because of the length of S2). To our knowledge there is no direct experimental support for the concept that the S2 domain enhances the myosin step size. In contrast, if S2 decreases forces that resist shortening through a mechanism similar to that proposed by Kaya and Higuchi (56) as discussed above, then we would expect a force-distance relationship for a myosin head to follow that illustrated in Fig. 9C with the corresponding energy landscape shown in Fig. 9D. Here, driving force is exerted over a distance equal to the 10-nm myosin step size, and movement of the myosin head beyond this point results in minimal resistive (negative) forces. In fact, this force-distance relationship has been experimentally observed by Kaya and Higuchi in myosin filaments.
FIGURE 9.
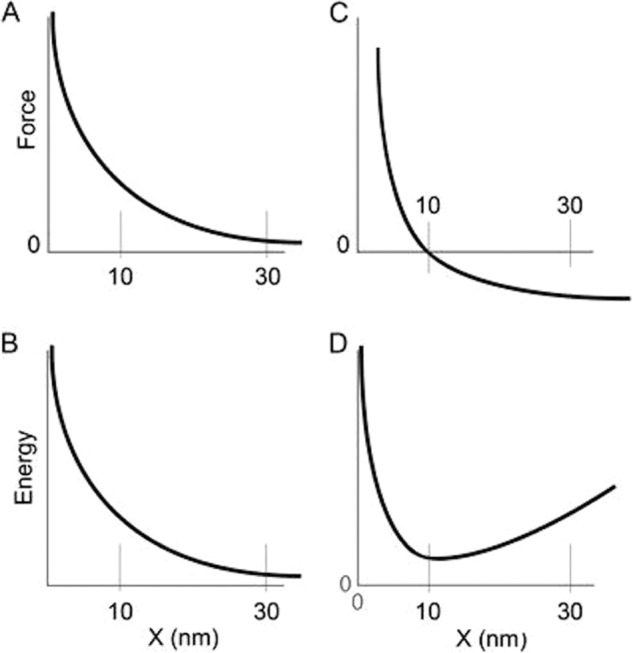
Comparison of two models or mechanisms by which an S2 domain might enhance shortening speeds. A and B, force-distance plot (A) and energy-distance plot (B) if the S2 domain moves from a high force to a low force state over a distance x. Note that the apparent step size is larger than that because of the heads alone (10 nm) because of the involvement of the S2 (30 nm). C and D are the corresponding plots if S2 decreases forces that resist shortening through a mechanism similar to that proposed by Kaya and Higuchi (56). The driving force is exerted over a distance equal to the 10-nm myosin step size, and movement beyond this point results in minimal resistive (negative) forces.
In summary, according to the sliding filament theory of muscle contraction, muscles contract when myosin thick filaments slide past actin thin filaments. The kinetics underlying this sliding has been inferred from mechanochemical studies of actin filaments interacting with monomeric myosin or myosin subfragments that are not able to form filaments. Here we present the first direct measurements of the mechanochemistry of actin-myosin filament interactions. Our results show that the kinetic steps limiting the unloaded velocity of motion powered by myosin filaments differs fundamentally from that powered by myosin cross-bridges unincorporated into filaments. We suggest that the low stiffness of the relatively flexible S2 region in filaments influences the kinetics of myosin cross-bridge interactions during motion on actin, making the process limited by attachment rather than detachment kinetics. These findings address the mechanism of how assemblies of motors generate force collectively and minimize the interference between motor molecules.
Acknowledgments
We thank Dr. Masumi Eto (Department of Molecular Physiology and Biophysics and Kimmel Cancer Center, Thomas Jefferson University, Philadelphia, PA) for the kind gift of myosin phosphatase, Dr. Michael Walsh (Departments of Biochemistry and Molecular Biology and Physiology and Pharmacology, Smooth Muscle Research Group, University of Calgary, Calgary, Canada) for gizzard MLCK, and Diego Alcala for technical assistance.
This work was supported, in whole or in part, by National Institutes of Health Grant 5R01HL110214 (to C. R. C., K. C. F., and J. E. B.).

This article contains supplemental Movies S1–S4.
- RLC
- regulatory light chain
- SMM and pSMM
- unphosphorylated and phosphorylated smooth muscle myosin with no modifications
- Rh
- rhodamine
- Rh-SMM and Rh-pSMM
- rhodamine-labeled myosins
- EDC
- 1-ethyl-3-(-3-dimethylaminopropyl) carbodiimide hydrochloride
- XL-SMM or XL-pSMM
- EDC cross-linked myosin in filaments (30 min of treatment unless indicated otherwise)
- XL-Rh-SMM and XL-Rh-pSMM
- EDC cross-linked and rhodamine labeled myosins in filaments
- MLCK
- smooth muscle myosin light chain kinase
- TRITC
- tetramethylrhodamine isothiocyanate
- LMM
- light meromyosin region of the myosin tail
- S2
- S2 region of the myosin tail
- S1
- myosin head with light chains
- HMM
- heavy meromyosin region of myosin
- HC
- myosin heavy chain
- ddH2O
- double distilled H2O.
REFERENCES
- 1. Cremo C. R., Hartshorne D. J. (2008) Smooth muscle myosin II. In Myosins: A Superfamily of Molecular Motors (Coluccio L. M., ed) pp. 171–222, Springer, Dordrecht, The Netherlands [Google Scholar]
- 2. Eddinger T. J., Meer D. P. (2007) Myosin II isoforms in smooth muscle: heterogeneity and function. Am. J. Physiol. Cell Physiol. 293, C493–C508 [DOI] [PubMed] [Google Scholar]
- 3. Hong F., Haldeman B. D., Jackson D., Carter M., Baker J. E., Cremo C. R. (2011) Biochemistry of smooth muscle myosin light chain kinase. Arch. Biochem. Biophys. 510, 135–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chacko S., Conti M. A., Adelstein R. S. (1977) Effect of phosphorylation of smooth muscle myosin on actin activation and Ca2+ regulation. Proc. Natl. Acad. Sci. U.S.A. 74, 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Heissler S. M., Manstein D. J. (2013) Nonmuscle myosin-2: mix and match. Cell Mol. Life Sci. 70, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Craig R., Megerman J. (1977) Assembly of smooth muscle myosin into side-polar filaments. J. Cell Biol. 75, 990–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cooke P. H., Fay F. S., Craig R. (1989) Myosin filaments isolated from skinned amphibian smooth cells are side-polar. J. Muscle. Res. Cell Motil. 10, 206–220 [DOI] [PubMed] [Google Scholar]
- 8. Xu J.-Q., Harder B. A., Uman P., Craig R. (1996) Myosin filament structure in vertebrate smooth muscle. J. Cell Biol. 134, 53–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cross R. A., Engel A. (1991) Scanning transmission electron microscopic mass determination of in vitro self-assembled smooth muscle myosin filaments. J. Mol. Biol. 222, 455–458 [DOI] [PubMed] [Google Scholar]
- 10. Tonino P., Simon M., Craig R. (2002) Mass determination of native smooth muscle myosin filaments by scanning transmission electron microscopy. J. Mol. Biol. 318, 999–1007 [DOI] [PubMed] [Google Scholar]
- 11. Trybus K. M. (1991) Assembly of cytoplasmic and smooth muscle myosins. Curr. Opin. Cell Biol. 3, 105–111 [DOI] [PubMed] [Google Scholar]
- 12. Seow C. Y. (2005) Myosin filament assembly in an ever-changing myofilament lattice of smooth muscle. Am. J. Physiol. Cell Physiol. 289, C1363–C1368 [DOI] [PubMed] [Google Scholar]
- 13. Milton D. L., Schneck A. N., Ziech D. A., Ba M., Facemyer K. C., Halayko A. J., Baker J. E., Gerthoffer W. T., Cremo C. R. (2011) Direct evidence for functional smooth muscle myosin II in the 10S self-inhibited monomeric conformation in airway smooth muscle cells. Proc. Natl. Acad. Sci. U.S.A. 108, 1421–1426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cross R. A., Geeves M. A., Kendrick-Jones J. (1991) A nucleation-elongation mechanism for the self-assembly of side polar sheets of smooth muscle myosin. EMBO J. 10, 747–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tyska M. J., Warshaw D. M. (2002) The myosin power stroke. Cell Motil. Cytoskeleton 51, 1–15 [DOI] [PubMed] [Google Scholar]
- 16. Trybus K. M. (1989) Filamentous smooth muscle myosin is regulated by phosphorylation. J. Cell Biol. 109, 2887–2894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kendrick-Jones J., Smith R. C., Craig R., Citi S. (1987) Polymerization of vertebrate non-muscle and smooth muscle myosins. J. Mol. Biol. 198, 241–252 [DOI] [PubMed] [Google Scholar]
- 18. Craig R., Padrón R., Kendrick-Jones J. (1987) Structural changes accompanying phosphorylation of tarantula muscle myosin filaments. J. Cell Biol. 105, 1319–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ikebe M., Barsotti R. J., Hinkins S., Hartshorne D. J. (1984) Effects of magnesium chloride on smooth muscle actomyosin adenosine-5′-triphosphatase activity, myosin conformation, and tension development in glycerinated smooth muscle fibers. Biochemistry 23, 5062–5068 [DOI] [PubMed] [Google Scholar]
- 20. Applegate D., Pardee J. D. (1992) Actin-facilitated assembly of smooth muscle myosin induces formation of actomyosin fibrils. J. Cell Biol. 117, 1223–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ikebe M., Hartshorne D. J. (1985) Effects of Ca2+ on the conformation and enzymatic activity of smooth muscle myosin. J. Biol. Chem. 260, 13146–13153 [PubMed] [Google Scholar]
- 22. Walsh M. P., Hinkins S, Dabrowska R., Hartshorne D. J. (1983) Smooth muscle myosin light chain kinase. Methods Enzymol. 99, 279–288 [DOI] [PubMed] [Google Scholar]
- 23. Ellison P. A., DePew Z. S., Cremo C. R. (2003) Both heads of tissue-derived smooth muscle heavy meromyosin bind to actin in the presence of ADP. J. Biol. Chem. 278, 4410–4415 [DOI] [PubMed] [Google Scholar]
- 24. Ikebe M., Hartshorne D. J. (1986) Reverse reaction of smooth muscle myosin light chain kinase. Formation of ATP from phosphorylated light chain plus ADP. J. Biol. Chem. 261, 8249–8253 [PubMed] [Google Scholar]
- 25. Spudich J. A., Watt S. (1971) The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J. Biol. Chem. 246, 4866–4871 [PubMed] [Google Scholar]
- 26. De La Cruz E. M., Sweeney H. L., Ostap E. M. (2000) ADP inhibition of myosin V ATPase activity. Biophys. J. 79, 1524–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sage D., Neumann F. R., Hediger F., Gasser S. M., Unser M. (2005) Automatic tracking of individual fluorescence particles: application to the study of chromosome dynamics. IEEE Trans. Image Process 14, 1372–1383 [DOI] [PubMed] [Google Scholar]
- 28. Smith M. B., Li H., Shen T., Huang X., Yusuf E., Vavylonis D. (2010) Segmentation and tracking of cytoskeletal filaments using open active contours. Cytoskeleton 67, 693–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ikebe M., Hartshorne D. J. (1985) Proteolysis of smooth muscle myosin by Staphylococcus aureus protease: preparation of heavy meromyosin and subfragment 1 with intact 20,000-Dalton light chains. Biochemistry 24, 2380–2387 [DOI] [PubMed] [Google Scholar]
- 30. Cremo C. R., Geeves M. A. (1998) Interaction of actin and ADP with the head domain of smooth muscle myosin: implications for strain-dependent ADP release in smooth muscle. Biochemistry 37, 1969–1978 [DOI] [PubMed] [Google Scholar]
- 31. Ikebe M., Komatsu S., Woodhead J. L., Mabuchi K., Ikebe R., Saito J., Craig R., Higashihara M. (2001) The tip of the coiled-coil rod determines the filament formation of smooth muscle and nonmuscle myosin. J. Biol. Chem. 276, 30293–30300 [DOI] [PubMed] [Google Scholar]
- 32. Cross R. A. (1984) Bending of smooth muscle myosin rod. FEBS Lett. 176, 197–201 [DOI] [PubMed] [Google Scholar]
- 33. Trybus K. M., Freyzon Y., Faust L. Z., Sweeney H. L. (1997) Spare the rod, spoil the regulation: necessity for a myosin rod. Proc. Natl. Acad. Sci. U.S.A. 94, 48–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ikebe M., Yamada M., Mabuchi K., Kambara T., Ikebe R. (1999) Registration of the rod is not critical for the phosphorylation-dependent regulation of smooth muscle myosin. Biochemistry 38, 10768–10774 [DOI] [PubMed] [Google Scholar]
- 35. Kumon A., Kuba M., Murakami N., Yasuda S., Takashima T., Matsumura S., Suezaki Y. (1986) Regions necessary for pH-dependent assembly of gizzard myosin rod. Eur. J. Biochem. 160, 499–506 [DOI] [PubMed] [Google Scholar]
- 36. Ikebe M., Koretz J., Hartshorne D. J. (1988) Effects of phosphorylation of light chain residues threonine 18 and serine 19 on the properties and conformation of smooth muscle myosin. J. Biol. Chem. 263, 6432–6437 [PubMed] [Google Scholar]
- 37. Nagy A., Takagi Y., Billington N., Sun S. A., Hong D. K., Homsher E., Wang A., Sellers J. R. (2013) Kinetic characterization of nonmuscle myosin IIb at the single molecule level. J. Biol. Chem. 288, 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Billington N., Wang A., Mao J., Adelstein R. S., Sellers J. R. (2013) Characterization of three full-length human nonmuscle myosin II paralogs. J. Biol. Chem. 288, 33398–33410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Criddle A. H., Geeves M. A., Jeffries T. (1985) The use of actin labelled with N-(1-pyrenyl)iodoacetamide to study the interaction of actin with myosin subfragments and troponin/tropomyosin. Biochem. J. 232, 343–349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coates J. H., Criddle A. H., Geeves M. A. (1985) Pressure-relaxation studies of pyrene-labelled actin and myosin subfragment 1 from rabbit skeletal muscle: evidence for two states of acto-subfragment 1. Biochem. J. 232, 351–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robblee J. P., Olivares A. O., de la Cruz E. M. (2004) Mechanism of nucleotide binding to actomyosin VI: evidence for allosteric head-head communication. J. Biol. Chem. 279, 38608–38617 [DOI] [PubMed] [Google Scholar]
- 42. Robblee J. P., Cao W., Henn A., Hannemann D. E., De La Cruz E. M. (2005) Thermodynamics of nucleotide binding to actomyosin V and VI: a positive heat capacity change accompanies strong ADP binding. Biochemistry 44, 10238–10249 [DOI] [PubMed] [Google Scholar]
- 43. Ellison P. A., Sellers J. R., Cremo C. R. (2000) Kinetics of smooth muscle heavy meromyosin with one thiophosphorylated head. J. Biol. Chem. 275, 15142–15151 [DOI] [PubMed] [Google Scholar]
- 44. Harris D. E., Warshaw D. M. (1993) Smooth and skeletal muscle actin are mechanically indistinguishable in the in vitro motility assay. Circ. Res. 72, 219–224 [DOI] [PubMed] [Google Scholar]
- 45. Sellers J. R. (1985) Mechanism of the phosphorylation-dependent regulation of smooth muscle heavy meromyosin. J. Biol. Chem. 260, 15815–15819 [PubMed] [Google Scholar]
- 46. Baker J. E., Brosseau C., Joel P. B., Warshaw D. M. (2002) The biochemical kinetics underlying actin movement generated by one and many skeletal muscle myosin molecules. Biophys. J. 82, 2134–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hooft A. M., Maki E. J., Cox K. K., Baker J. E. (2007) An accelerated state of myosin-based actin motility. Biochemistry 46, 3513–3520 [DOI] [PubMed] [Google Scholar]
- 48. Baker J. E., Brosseau C., Fagnant P., Warshaw D. M. (2003) The unique properties of tonic smooth muscle emerge from intrinsic as well as intermolecular behaviors of myosin molecules. J. Biol. Chem. 278, 28533–28539 [DOI] [PubMed] [Google Scholar]
- 49. Tyska M. J., Dupuis D. E., Guilford W. H., Patlak J. B., Waller G. S., Trybus K. M., Warshaw D. M., Lowey S. (1999) Two heads of myosin are better than one for generating force and motion. Proc. Natl. Acad. Sci. U.S.A. 96, 4402–4407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Warshaw D. M., Guilford W. H., Freyzon Y., Krementsova E., Palmiter K. A., Tyska M. J., Baker J. E., Trybus K. M. (2000) The light chain binding domain of expressed smooth muscle heavy meromyosin acts as a mechanical lever. J. Biol. Chem. 275, 37167–37172 [DOI] [PubMed] [Google Scholar]
- 51. Howard J. (2001) Mechanics of Motor Proteins and the Cytoskeleton, Sinauer Associates, Sunderland, MA [Google Scholar]
- 52. Lymn R. W., Taylor E. W. (1971) Mechanism of adenosine triphosphate hydrolysis by actomyosin. Biochemistry 10, 4617–4624 [DOI] [PubMed] [Google Scholar]
- 53. Spudich J. A. (1994) How molecular motors work. Nature 372, 515–518 [DOI] [PubMed] [Google Scholar]
- 54. Harris D. E., Warshaw D. M. (1993) Smooth and skeletal muscle myosin both exhibit low duty cycles at zero load in vitro. J. Biol. Chem. 268, 14764–14768 [PubMed] [Google Scholar]
- 55. Stewart T. J., Jackson D. R., Jr., Smith R. D., Shannon S. F., Cremo C. R., Baker J. E. (2013) Actin sliding velocities are influenced by the driving forces of actin-myosin binding. Cell Mol. Bioeng. 6, 26–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kaya M., Higuchi H. (2010) Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science 329, 686–689 [DOI] [PubMed] [Google Scholar]
- 57. Adamovic I., Mijailovich S. M., Karplus M. (2008) The elastic properties of the structurally characterized myosin II S2 subdomain: a molecular dynamics and normal mode analysis. Biophys. J. 94, 3779–3789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Veigel C., Molloy J. E., Schmitz S., Kendrick-Jones J. (2003) Load-dependent kinetics of force production by smooth muscle myosin measured with optical tweezers. Nat. Cell Biol. 5, 980–986 [DOI] [PubMed] [Google Scholar]
- 59. Lewalle A., Steffen W., Stevenson O., Ouyang Z., Sleep J. (2008) Single-molecule measurement of the stiffness of the rigor myosin head. Biophys. J. 94, 2160–2169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kaya M., Higuchi H. (2013) Stiffness, working stroke, and force of single-myosin molecules in skeletal muscle: elucidation of these mechanical properties via nonlinear elasticity evaluation. Cell Mol. Life Sci. 70, 4275–4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Huxley A. F. (1957) Muscle structure and theories of contraction. Prog. Biophys. Biophys. Chem. 7, 255–315 [PubMed] [Google Scholar]