

Background: Chemotherapy-induced peripheral neuropathy (CIPN) is a critical dose-limiting side effect of many chemotherapeutic agents, including paclitaxel.
Results: Spinal activation of the S1P-to-S1PR1 axis contributes to the development and maintenance of paclitaxel-induced neuropathic pain through enhanced neuroinflammatory processes.
Conclusion: Inhibition of S1PR1 blocks and reverses paclitaxel-induced neuropathic pain without interfering with anticancer effects.
Significance: Targeting the S1PR1 signaling pathway could be an effective approach for the treatment of CIPN.
Keywords: Drug Action, Neuroinflammation, Pain, Pharmacology, Rat, Sphingosine 1-Phosphate (S1P), CIPN, FTY720, Oxaliplatin, Paclitaxel
Abstract
The ceramide-sphingosine 1-phosphate (S1P) rheostat is important in regulating cell fate. Several chemotherapeutic agents, including paclitaxel (Taxol), involve pro-apoptotic ceramide in their anticancer effects. The ceramide-to-S1P pathway is also implicated in the development of pain, raising the intriguing possibility that these sphingolipids may contribute to chemotherapy-induced painful peripheral neuropathy, which can be a critical dose-limiting side effect of many widely used chemotherapeutic agents. We demonstrate that the development of paclitaxel-induced neuropathic pain was associated with ceramide and S1P formation in the spinal dorsal horn that corresponded with the engagement of S1P receptor subtype 1 (S1PR1)-dependent neuroinflammatory processes as follows: activation of redox-sensitive transcription factors (NFκB) and MAPKs (ERK and p38) as well as enhanced formation of pro-inflammatory and neuroexcitatory cytokines (TNF-α and IL-1β). Intrathecal delivery of the S1PR1 antagonist W146 reduced these neuroinflammatory processes but increased IL-10 and IL-4, potent anti-inflammatory/neuroprotective cytokines. Additionally, spinal W146 reversed established neuropathic pain. Noteworthy, systemic administration of the S1PR1 modulator FTY720 (Food and Drug Administration-approved for multiple sclerosis) attenuated the activation of these neuroinflammatory processes and abrogated neuropathic pain without altering anticancer properties of paclitaxel and with beneficial effects extended to oxaliplatin. Similar effects were observed with other structurally and chemically unrelated S1PR1 modulators (ponesimod and CYM-5442) and S1PR1 antagonists (NIBR-14/15) but not S1PR1 agonists (SEW2871). Our findings identify for the first time the S1P/S1PR1 axis as a promising molecular and therapeutic target in chemotherapy-induced painful peripheral neuropathy, establish a mechanistic insight into the biomolecular signaling pathways, and provide the rationale for the clinical evaluation of FTY720 in chronic pain patients.
Introduction
Paclitaxel (Taxol®) is a widely used chemotherapeutic agent indicated for treating breast, ovarian, non-small cell lung carcinomas, and Kaposi sarcoma. Unfortunately, the dose-limiting side effect and leading cause of discontinuation of this highly efficacious anticancer drug is peripheral neuropathy accompanied by chronic neuropathic pain (CIPN)2 that may resolve within weeks or could persist for years after drug termination (1). The clinical management of these patients becomes difficult as current pain drugs are marginally effective and display unacceptable side effects (1). Identification of target-directed therapeutic approaches based on mechanistic insight is of paramount significance as the estimated incidence of CIPN is 30–90% of patients treated with taxanes and combinational chemotherapies (1).
As a desirable anticancer mechanism, paclitaxel can activate the sphingolipid pathway to produce the potent pro-apoptotic ceramide, which can induce apoptosis in a variety of neoplasms (2). Ceramide is then hydrolyzed by ceramidases to sphingosine and then phosphorylated by sphingosine kinases (SphK1 and -2) to produce sphingosine 1-phosphate (S1P) (3). S1P levels are further regulated by its dephosphorylation by S1P phosphatases and lipid phosphate phosphatases or cleaved by S1P lyase (3). Once released, S1P initiates signaling through a family of five cognate G protein-coupled receptors (S1PR1–5) leading to various cellular responses (3, 4). In contrast to ceramide, S1P is a potent anti-apoptotic sphingolipid; it is hypothesized that the ceramide/S1P rheostat plays a critical role in regulating cancer cell fate with elevated levels of ceramide inducing cell death, whereas elevated levels of S1P lead to survival and proliferation (2). To this end, therapeutic strategies to treat various types of cancer by increasing the levels of ceramide (e.g. with ceramidase inhibitors), reducing S1P bioavailability (e.g. with SphK inhibitors), or attenuating S1P/S1PR1 signaling with anti-S1P antibodies or S1P1 modulators, such as FTY720, are an active area of investigation and are moving forward as novel anticancer agents (2, 5). FTY720 (fingolimod/Gilenya®) is the first orally available agent approved by the Food and Drug Administration for the treatment of relapsing-remitting multiple sclerosis (MS) (6), an autoimmune disorder characterized by neuroinflammation in the central nervous system (CNS), demyelination, and neurodegeneration. In addition to their well established roles in inflammation and cancer, ceramide and S1P are emerging as important modulators in the development of peripheral and central sensitization associated with enhanced pain processing (7, 8). For example, peripheral ceramide and S1P (acting via S1PR1) increase the excitability of small diameter sensory neurons and contribute to nerve growth factor-induced sensitization of sensory neurons (9–13). Intraplantar injection of ceramide (14–16), S1P, or S1PR1 agonists (15, 17) in rats or mice evoke profound mechano-hypersensitivity via activation of the S1P1 receptors and subsequent formation of a peripheral inflammatory response (14, 15, 18). In the CNS, these sphingolipids also appear to be important mediators in the development of spinal sensitization associated with increased nociceptive input. For example, ceramide/S1P levels are elevated in the spinal dorsal horn of neuropathic animals (19) and in morphine-tolerant rats where they contribute to the development of central sensitization by hyperactivating glial cells and increasing the production of pro-inflammatory/neuroexcitatory cytokines and nitro-oxidative species (20, 21). Furthermore, Yan and Weng (22) recently reported that IL-1β generated in the spinal cord of neuropathic rats contributes to central sensitization; the activity of presynaptic NMDA receptors is enhanced by activation of the sphingomyelinase/ceramide signaling pathway that results in increased glutamate release from the primary afferent terminals. Whereas the underlying causative mechanisms of CIPN following paclitaxel are multifactorial and include neuropathological changes in the periphery (23), prominent neuropathological changes in the CNS have been documented to contribute through the development of neuroinflammation and dysregulation of neuroglia communication in the spinal cord (24). We hypothesize that if paclitaxel-induced neuropathic pain is dependent on the activation of the S1P/S1PR1 axis, then anti-S1PR1-targeted approaches should provide an effective means to mitigate CIPN without interfering with anticancer effects. Indeed, our results identify for the first time S1PR1 as a promising molecular target in CIPN, establish a mechanistic link into potential biomolecular signaling pathways, and provide the foundation to consider “fast-track” clinical use of FTY720 as a therapeutic agent in CIPN patients.
EXPERIMENTAL PROCEDURES
Experimental Animals
Adult male Sprague-Dawley rats (200–220 g starting weight) from Harlan Laboratories (Nossan, Milan, Italy, and Indianapolis, IN; Frederick, MD breeding colony) were housed 3–4 per cage in a controlled environment (12-h light/dark cycle) with food and water available ad libitum. All experiments were performed in accordance with the International Association for the Study of Pain and the National Institutes of Health guidelines on laboratory animal welfare and were approved by the Saint Louis University Institutional Animal Care and Use Committee and the University of Messina Review Board for the care of animals. All animal experiments complied with regulations in Italy (DM 116192) as well as European Union regulations (Official Journal of the European Union of the European Council L 358/112/18/1986). All experiments were conducted with the experimenters blinded to treatment conditions.
Animal Models of Chemotherapy-induced Neuropathic Pain
Paclitaxel (Parenta Pharmaceuticals, Yardley, PA) or its vehicle (Cremophor EL and 95% EtOH in 1:1 ratio, Sigma) was injected intraperitoneally in rats on 4 alternate days (D0, 2, 4, and 6; cumulative dose of 8 mg/kg) (25). Oxaliplatin (Oncology Supply, Dothan, AL) or its vehicle (5% dextrose) was injected intraperitoneally in rats on 5 consecutive days (D0–4) for a final cumulative dose of 10 mg/kg. This low dose paradigm does not cause kidney injury (26).
S1PR1-induced Mechano-hypersensitivity
Chronic intrathecal cannulae were placed in rats using the L5/L6 lumbar approach, as described previously (27). To induce mechano-allodynia and mechano-hyperalgesia, SEW2871 (10 μl) or its vehicle (2% DMSO in saline) was administered via these cannulae and flushed with sterile physiological saline (10 μl). Behavior was assessed before any treatment (baseline) and then periodically after drug administration for 2–5 h.
Test Compounds
In the prophylactic paradigms, all test compounds were given 30 min prior to chemotherapeutic agents or SEW2871. In some experiments, W146 (Cayman Chemical), W140 (Cayman Chemical), FTY720 (Fingolimod, Cayman Chemical), CYM-5442 (Tocris Biosciences, Bristol, UK), SN-50 (Millipore), SB20350 (Millipore), U0126 (Millipore), SK-I (2-(p-hydroxyanilino)-4-(p-chlorophenyl) thiazole) (Millipore), or their vehicle (0.25–2% DMSO in saline) were given i.th. via chronically implanted i.th. cannulae as described above. In other experiments, FTY720 or its vehicle (2% DMSO in saline) was delivered systemically by injection (intraperitoneal) or oral gavage. NIBR-14, CYM-5442, ponesimod, or their vehicle (5% DMSO in saline) was given by oral gavage. Ponesimod (ACT-128800, (Z,Z)-5-[3-chloro-4-((2R)-2,3-dihydroxypropoxy)-benzylidene]-2-propylimino-3-o-tolylthiazolidin-4-one) a selective, reversible, and orally active S1P1 receptor agonist was prepared by Shanghai ChemPartner Co. according to the method of Bolli et al. (28). The final product was purified by preparative HPLC (purity >97% by LC/MS).
Osmotic Pump Implantation
On D16, rats were lightly anesthetized with isoflurane (3% in 100% O2), and their backs were shaved and scrubbed with Nolvasan. An incision was made in the interscapular region for subcutaneous implantation of primed osmotic minipumps (Alzet 2001; Alza) that infused 1 μl/h over a 7-day period. Minipumps were filled according to the manufacturer's specifications with FTY720, NIBR-14, CYM5442, or their vehicle, 100% DMSO. Immediately following surgery, FTY720-treated rats were injected with a loading intraperitoneal dose of 0.03 mg/kg FTY720 or its vehicle (2% DMSO), and the minipump was allowed to deliver FTY720 (0.03 and 0.1 mg/kg/day) or vehicle for 6 days. Likewise, NIBR-14-treated rats were given a loading intraperitoneal dose of 0.3 mg/kg/day NIBR-14 or vehicle (2% DMSO), and the minipump was allowed to deliver NIBR-14 (1 or 3 mg/kg/day) or vehicle for 6 days. For CYM-5442-treated rats, a loading dose of 3 mg/kg CYM-5442 or vehicle (5% DMSO) was given intraperitoneally, and the minipump was allowed to deliver CYM-5442 (3 mg/kg/day) or vehicle for 6 days.
Behavior Testing
Assessment of behavior was always started in the morning. Rats were randomized into treatment groups before any behavioral assessment was performed. Behavioral testing was done prior to any drug administration (D0 or baseline) and then subsequently at selected time points. For mechano-allodynia, rats were placed in elevated Plexiglas chambers (28X40X35-cm) upon a wire mesh floor and allowed to acclimate for 15 min prior to measuring the mechanical paw withdrawal thresholds, grams (PWT (g)), using calibrated von Frey filaments (Stoelting, Wood Dale, IL, ranging from 3.61 (0.407 g) to 5.46 (26 g) bending force) according to the “up-and-down” method (29) or with an electronic version of the von Frey test (dynamic plantar aesthesiometer, model 37450, Ugo Basile, Geminio, Italy) with a cutoff set at 50 g. The development of mechano-allodynia is evidenced by a significant (p < 0.05) reduction in mechanical mean absolute PWT (g) at forces that failed to elicit withdrawal responses before treatment (baseline or D0). For mechanical hyperalgesia, PWTs (g) were measured by the Randall and Sellitto paw pressure test (30) using a Ugo-Basile analgesiometer (model 37215) that applies a linearly increasing mechanical force to the dorsum of the rat's hind paw. The nociceptive threshold was defined as the force (gram) at which the rat withdrew its paw (cutoff set at 250 g). Animals receiving chemotherapeutic agents in the presence or absence of the experimental test substances did not display signs of toxicity, i.e. they exhibited normal posture, grooming, locomotor behavior, hair coat was normal, no signs of piloerection or ocular porphyrin discharge, and they gained body weight normally and comparable with vehicle-treated rats. If testing coincided with a day when rats received test substance, behavioral measurements were always taken before the injection of the test substance.
Tail Flick
The tail flick test, which measures the latency (in seconds) of tail withdrawal from a noxious radiant heat source (Ugo Basile; model number 37360), was used to measure acute thermal nociceptive sensitivity in rats with baseline latencies of 2–5 s and a cutoff time of 15 s to prevent tissue injury (31). Tail flick latencies were taken before and at 15, 30, 60, and 90 min after injection of test compounds.
Tissue for Biochemical Analysis
For all biochemical analyses, rats were sacrificed at a time of peak mechano-hypersensitivity (D16). The lower lumbar enlargement of the spinal cord (L4 to L6) was harvested, flash-frozen in liquid nitrogen, and kept at −80 °C until the appropriate assay could be performed.
Sphingomyelinase Activity
Spinal cord tissues were homogenized in specific buffers, as described previously (32). Following homogenization, samples were preincubated with sphingomyelin for 30 min at 37 °C before the sphingomyelinase activity was measured for 1.5 h using an Amplex® red sphingomyelinase assay kit (Molecular Probes, Eugene, OR) in a fluorescence microplate reader. The sphingomyelinase activity was expressed as milliunits/s and normalized by protein concentration (μg/ml). Hydrogen peroxide and purified sphingomyelinase were used as positive controls.
Serine Palmitoyltransferase (SPT) Activity
SPT activity was determined by measuring the incorporation of [3H]serine into 3-ketosphinganine following a previously described method (33). SPT activity was measured by number of counts/min and normalized by protein concentration (μg/ml).
Sphingosine Kinase Activity
Spinal cord tissue was homogenized in SK1 buffer (containing 20 mm Tris-HCl, pH 7.4, 1 mm EDTA, 0.5 mm deoxypyridoxine, 15 mm NaF, 1 mm β-mercaptoethanol, 1 mm sodium orthovanadate, 40 mm β-glycerophosphate, 0.4 mm phenylmethylsulfonyl fluoride, 10% glycerol, 0.5% Triton X-100, and a complete protease inhibitor mixture) using a rotor homogenizer (61). After brief sonication and quantitation of protein concentration by the BCA method, 30 μg of protein was incubated in 90 μl of reaction mixture containing sphingosine (50 μm, delivered in 4 mg/ml fatty acid-free bovine serum albumin), [γ-32P]ATP (5 μCi, 1 mm dissolved in 10 mm MgCl2), and SK1 buffer for 30 min at 37 °C. The reaction was terminated by the addition of 10 μl of 1 n HCl and 400 μl of chloroform/methanol/HCl (100:200:1, v/v/v). Subsequently, 120 μl of chloroform and 120 μl of 2 m KCl were added, and samples were centrifuged at 3000 × g for 5 min. The organic phase (200 μl) was transferred to new glass tubes and dried. Samples were resuspended in chloroform/methanol/HCl (100: 100:1, v/v/v). Lipids were then resolved on silica thin layer chromatography plates using 1-butanol/methanol/acetic acid/water (8:2:1:2, v/v/v/v) as the solvent system and visualized by autoradiography. The radioactive spots corresponding to S1P were scraped from the plates and counted for radioactivity. Background values were determined in negative controls in which sphingosine was not added to the reaction mixture.
Immunofluorescence
After behavioral measurements, rats were anesthetized with ketamine/xylazine and transcardially perfused with phosphate-buffered saline (PBS, pH 7.4) followed by 10% buffered neutral formalin. Spinal cord was harvested and post-fixed in the same fixative for 16 h at 4 °C. After rinsing in PBS, the tissue was infiltrated with 30% (w/v) sucrose in PBS at 4 °C for 48 h, rinsed again in PBS, transferred to OCT, and frozen on dry ice. Transverse sections (20 μm) were cut in a cryostat, collected on gelatin-coated glass microscope slides, air-dried overnight, and stored at −20 °C. Spinal cord sections were blocked (10% normal goat serum, 2% bovine serum albumin, 0.2% Triton X-100 in PBS) and then immunolabeled as described previously (20, 34, 35) using a well characterized primary antibody, rabbit IgG polyclonal anti-ceramide (36) (1:50, incubated 18 h at 4 °C), followed by several PBS washes and incubation (2 h room temperature in the dark) with a goat anti-rabbit IgG antibody conjugated to Alexa Fluor 568 (1:250, Invitrogen). The coverslips were mounted with Fluorogel II containing DAPI (Electron Microscopy Sciences, Hatfield, PA) and photographed with an Olympus FV1000 MPE confocal microscope (multiline argon lasers with excitation at 405 and 543 nm) using a 10× objective (UPLSAPO; 0.4 NA) for regional fluorescence intensity image analysis and with 60× oil immersion objective (PLAPON; 1.42 NA) and 2.4× optical zoom (0.1 μm pixel dimensions in the X-Y plane and the pinhole set at 1 Airy unit) for higher magnification images. Images were acquired within the dynamic range of the microscope (i.e. no pixel intensity values of 0 or 255 in an 8-bit image). Sections treated with rabbit IgG at equivalent concentrations to primary antibodies were used as controls yielding only nonspecific background fluorescence. The fluorescence intensity of immunolabeled ceramide was calculated as reported previously (34, 35, 37) in selected regions of the lumbar spinal cord. The mean fluorescence intensity (MFI) was calculated by Equation 1,
where i is the mean gray value; pp is the positive pixel area, and p is total pixel area. Image analysis was performed using the freeware program ImageJ (version 1.43, National Institutes of Health) (38). Images received background threshold corrections prior to analysis using the automated ImageJ intermode histogram-based threshold function. The superficial dorsal horns (laminae I and II), dorsal horns, and ventral horns at the L4, L5, and L6 levels were outlined on images bilaterally using the ImageJ region of interest tool. The borders of these regions were determined and confirmed using cresyl violet-stained sections of regions adjacent to immunolabeled sections and an atlas (39). There were no significant differences bilaterally, so MFI was calculated as a combined value and reported as fold change compared with the vehicle group. Data are expressed as arbitrary units.
Preparation of Cytosolic and Nuclear Extracts and Western Blot Analysis
Western blot analyses were performed as described previously (93) with slight modifications. Briefly, samples were suspended in extraction buffer A (containing 0.2 mm phenylmethylsulfonyl fluoride (PMSF), 0.15 mm pepstatin A, 20 mm leupeptin, 1 mm sodium orthovanadate), homogenized at the highest setting for 2 min, and centrifuged for 15 min at 4 °C. Supernatants (cytosolic fractions) were collected, and the pellets (nuclear fractions) were lysed in buffer B containing 1% Triton X-100, 150 mm NaCl, 10 mm Tris-HCl, pH 7.4, 1 mm EGTA, 1 mm EDTA, 0.2 mm PMSF, 20 μm leupeptin, 0.2 mm sodium orthovanadate. The pellet lysates were clarified by centrifugation (30 min at 15,000 × g, 4 °C), and the supernatants containing the nuclear protein were stored at −80 °C. The levels of cytosolic NFκB p65, p-ERK, ERK, p-p38, and p38 were measured using the cytosolic fractions, whereas nuclear NFκB p65 levels were measured in the nuclear fraction. Protein concentrations were determined using the Bio-Rad protein assay kit. Proteins (40 μg) were reduced in Laemmli Sample Buffer by boiling for 5 min. The proteins were resolved by SDS-PAGE (10 or 12% polyacrylamide) and transferred to nitrocellulose membranes. The membranes were blocked with 5% (w/v) nonfat dried milk in phosphate-buffered saline for 45 min at room temperature and subsequently probed with specific antibodies as follows: anti-pERK (1:500; Santa Cruz Biotechnology), anti-ERK (1:500; Santa Cruz Biotechnology), anti-phospho-38 (1:500; Santa Cruz Biotechnology), anti-p38 (1:500; Santa Cruz Biotechnology), anti-phospho-NFκB p65 (Ser-536) (1:500; Santa Cruz Biotechnology), or anti-NFκB p65 (1:1000; Cell Signaling) in 1× PBS, 5% w/v nonfat dried milk, and 0.1% Tween 20 (PMT) at 4 °C overnight. The bound antibodies were visualized following incubation with peroxidase-conjugated bovine anti-mouse immunoglobulin G (IgG) secondary antibody or peroxidase-conjugated goat anti-rabbit IgG (1:2000; Jackson ImmunoResearch) for 1 h at room temperature using enhanced chemiluminescence detection system reagent, according to the manufacturer's instructions (SuperSignal West Pico Chemiluminescent Substrate, Thermo Fisher Scientific Inc.). Relative protein expression was quantified by band densitometry with Chemidoc XRS+ Documentation System Bio-Rad software. Each membrane was subsequently probed for β-actin (for cytosolic extract) or laminin B1 (for nuclear extract) proteins (1:10,000; Sigma) for use as endogenous loading controls. All densitometry data were normalized to β-actin or laminin B1 bands.
ELISA or Multiplex Assay for Cytokines
The levels of cytokines in spinal cord lysates were either assessed using commercially available ELISA kits (R&D Systems) or by using a commercially available magnetic multiplex cytokine kit (Bio-Rad). Samples were processed according to the manufacturer's protocol.
Anticancer Activity
The effects of FTY720 on the anticancer activity of paclitaxel on human breast adenocarcinoma cells (SKBr3) (41) or of oxaliplatin on human colon carcinoma cells (SW480) (42) were assessed using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay adapted from a previously described assay (43, 44). Cells were cultured and assayed in DMEM (SKBr3, Mediatech) or L15 (SW480, Sigma) media supplemented with 10% FBS (Thermo Fisher Scientific) and penicillin/streptomycin (Invitrogen). Cells (3.13 × 104 cells/cm2) were cultured overnight in 24-well plates (BD Biosciences) in complete media. This plating regimen yielded 60% confluent plate cultures for testing. After equilibrating in fresh media (5 h), cells were treated for 48 h with FTY720 (5 μm) or its vehicle (PBS), chemotherapeutic paclitaxel (1–100 nm final concentration) or its vehicle (1.1% Cremophor EL, 0.9% ethanol, final concentration, Sigma), and oxaliplatin (0–30 μm) or its vehicle (0.01% DMSO, final concentration). Two naive control wells (media only) were included as a control for the anticancer effects of FTY720 alone. Cell survival was assessed by incubating cells with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (500 μg/ml; Sigma) for 75 min, removing media, and dissolving the resulting tetrazolium crystals in isopropyl alcohol. The tetrazolium absorption (A560–570 nm) was measured from paclitaxel experiments using a Unicam UV1 spectrophotometer (Thermo Fisher Scientific) and from oxaliplatin experiments using a Glomax® multidetection system (Promega). The antitumor effects of FTY720 alone were determined by Equation 2.
 |
The LD50 of each chemotherapeutic agent + FTY720 or its vehicle was calculated using three-parameter nonlinear analysis using Equation 3.
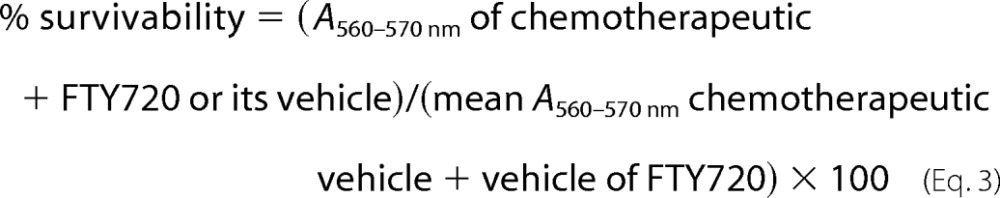 |
The top and bottom plateaus were constrained using GraphPad Prism Version 5.03 (GraphPad Software, Inc.).
S1P ELISA
Spinal cord tissue was homogenized. The level of S1P was measured in spinal cord lysates using commercially available ELISA kits (Echelon Biosciences, Salt Lake City, UT) according to the manufacturer's protocol.
Leukocyte Counts
Six days following osmotic pump implantation, whole blood was collected via intracardiac puncture into K3EDTA Vacutainer tubes and sent to Advanced Veterinary Labs (St. Louis) for a complete blood count panel with white blood cell differential.
Synthesis of S1PR1 Competitive Antagonist NIBR-14, Methyl Ester Pro-drug
The Novartis competitive antagonist (S)-2-{[3′-(4-chloro-2,5-dimethylphenylsulfonylamino)-3,5-dimethylbiphenyl-4-carbonyl]-methylamino}-4-dimethylaminobutyric acid methyl ester 14 (NIBR-14; MW = 600.17) was prepared as described in the literature (40). LC/MS (50–95% acetonitrile in 0.05% TFA over 6 min) with retention time = 7.15 min, C31H38ClN3O5S, m/z = 600 ([M + H+]), using a Waters Alliance-SQ 3100 system (positive ion electrospray mode) was performed with an Agilent Eclipse (XDB-C18, 4.6 × 150 mm, 5-μm) column.
Synthesis of S1PR1 Competitive Antagonist NIBR-15
The methyl ester prodrug NIBR-14 (90 mg, 0.15 mmol) was dissolved in acetic acid (6 ml) and treated with concentrated HCl (1.5 ml). This mixture was stirred at 50 °C for 12 h and concentrated. Recrystallization from acetonitrile afforded 45 mg (48% yield) of NIBR-15 HCl salt (Mr = 622.60). LC/MS (15–95% acetonitrile in 0.05% TFA over 6 min) was performed with the following parameters: retention time = 6.97 min, C30H37Cl2N3O5S, m/z = 586 ([M + H+ − Cl]+).
Determining ED50 Values
The ED50 values and the corresponding 95% confidence interval (95% CI) were determined by curve-fitting the % prevention (Equation 4) of mechano-allodynia or mechano-hyperalgesia using the least sum of squares method by a normalized three-parameter, nonlinear analysis (Hill slope = 1) using GraphPad Prism (Release 5.03, GraphPad Software, Inc.),
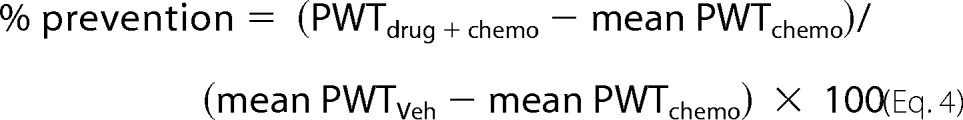 |
where drug = W146 or FTY720, chemo = paclitaxel or oxaliplatin, and Veh = vehicle.
Statistical Analysis
Data are expressed as mean ± S.D. for n animals. Differences in behavioral data from the full time course studies were analyzed by two-way repeated measures ANOVA with Bonferroni comparisons. All other biochemical data were analyzed by one-way ANOVA with Dunnett's comparisons. Differences in fluorescence intensity and single comparison behavioral tests as noted were analyzed using the unpaired Student's t test. Significant differences were defined at p < 0.05.
RESULTS
S1P/S1PR1 Signaling Pathway Is Activated in Spinal Cord Dorsal Horn in Response to Paclitaxel
Consistent with previous studies (45, 46), paclitaxel produced a time-dependent development of mechano-allodynia and mechano-hyperalgesia (i.e. mechano-hypersensitivity) (Fig. 1, B and C) as evidenced by a decrease in PWTs (g) by D12 (onset), which peaked by D16, and plateaued throughout our observation period (D25; times relative to first injection). The delay between the last exposure to paclitaxel and onset of mechano-hypersensitivity mimics the clinical “coasting” phenomenon described in patients (1). Ceramide is generated by enzymatic hydrolysis of sphingomyelin by sphingomyelinases (sphingomyelin pathway) and from de novo synthesis by SPT and ceramide synthase (de novo pathway) (47). Ceramide, in turn, is hydrolyzed to sphingosine, which is phosphorylated by SphK1 and -2 to form S1P (47). SphK1 activity is the major determinant of S1P levels in inflammatory diseases and is known to be activated by several cytokines, in particular TNF-α (48). As shown in Fig. 2, the peak of paclitaxel-induced mechano-hypersensitivity was associated with increased SPT and neutral/acidic sphingomyelinase activities in lumbar spinal cord (L4–6) (Fig. 2, A and B) as well as enhanced bilateral ceramide immunolabeling within laminae I and II of the L4–6 superficial dorsal horn (discrete pain-processing region that undergoes neuroinflammatory changes during chronic pain Fig. 2, C–E) (49, 50)). The intensity of spinal ceramide immunofluorescence (MFI) was compared between vehicle and paclitaxel groups using automated image analysis (38) of confocal microscopy images by calculating the product of the mean gray value and percentage of positive pixel area for each region of interest (e.g. superficial dorsal horn). Compared with vehicle, paclitaxel was associated with increased MFI in the superficial dorsal horn (Fig. 2C) but not the ventral horn (data not shown). In addition to the activation of the enzymes involved in the biosynthesis of ceramide, paclitaxel also led to increased SphK1 activity and S1P formation in spinal cord (Fig. 3, A and B). Daily (D0–D15) intrathecal injections of a commonly used and well characterized SphK1/2 inhibitor, SK-I (0.3 μm/d) (51), given at a dose previously shown to block increased spinal production of S1P during central sensitization evoked by prolonged use of opioids (20) blocked the following: 1) the increased activation of SphK1 (Fig. 3A); 2) the increased production of S1P (Fig. 3B) in spinal cord; and 3) the development of mechano-allodynia and mechano-hyperalgesia (Fig. 3, C and D). These results establish the role of the sphingosine kinase to S1P signaling pathway in spinal cord during CIPN (Fig. 3E).
FIGURE 1.

Spinal administration of an S1PR1 antagonist blocks the development of paclitaxel-induced neuropathic pain. Treatment with paclitaxel (P-V, ●), but not its vehicle (V-V, ▵), led to a time-dependent development of mechano-allodynia (B) and mechano-hyperalgesia (C). Daily (D0–15) W146 (A; i.th.; 0.2, ▴; 0.7, ▾; or 2.2 nmol/day, ■) but not its inactive S-enantiomer, W140 (i.th., 2.2 nmol/day, ♢), blocked mechano-hypersensitivity (B and C) in a dose-dependent manner. W146 (i.th.; 2.2 nmol/day, ○) alone had no effect on PWTs in vehicle group (B and C). Results are expressed as mean ± S.D. for n = 7 rats analyzed by two-way repeated measures ANOVA with Bonferroni comparisons. *, p < 0.05 versus D0; †, p < 0.05 versus P-V.
FIGURE 2.

Spinal ceramide metabolic pathway is up-regulated during paclitaxel-induced neuropathic pain. When compared with vehicle (V, open bars) on D16, paclitaxel (P, black bars) activated ceramide metabolic pathways by enhancing SPT (A) and sphingomyelinase (SMase) (B) activities, which correlated with increased ceramide levels (D and E). D, ceramide immunolabeling (red) in laminae I and II (yellow border) of the L6 dorsal horn (white border) and merged with DAPI (blue). Negative controls exhibited low levels of background fluorescence (not shown). E, high magnification representative images (orange boxes) of the dorsal horn. Micrographs represent n = 3 animals/group (three sections/animal). Scale bars: white, 250 μm; green, 20 μm. Results are expressed as mean ± S.D. for n = 4–5 rats analyzed by unpaired Student's t test. *, p < 0.05 versus V.
FIGURE 3.
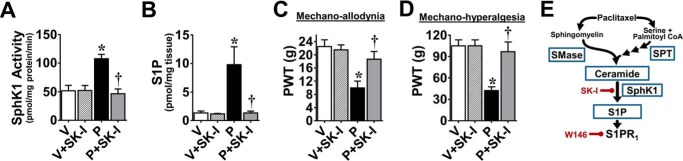
Spinal administration of SK-I blocks ceramide metabolic pathway and paclitaxel-induced neuropathic pain. When compared with vehicle (V, open bars) on D16, paclitaxel (P, black bars) activated ceramide metabolic pathways by increasing SphK1 activity (A) and S1P formation (B). C, daily (D0–15) treatment with SK-I (i.th., gray bars) prevented the increased SphK1 activity (A) and S1P formation (B) as well as the development of mechano-allodynia (C) and -hyperalgesia (D). SK-I had no effect on its own (V+SK-I, hatched bars). E, summary schematic. Results are expressed as mean ± S.D. for n = 6 rats analyzed by unpaired Student's t test. *, p < 0.05 versus V; †, p < 0.05 versus P.
Spinal Administration of S1PR1 Antagonists Block the Development of Paclitaxel-induced Neuropathic Pain
Daily spinal (D0–D15) delivery of the selective S1PR1 antagonist W146 (0.2–2.2 nmol/day; Fig. 1A) but not its inactive S-enantiomer W140 (2.2 nmol/day) (52), in rats with previously implanted i.th. catheters, attenuated paclitaxel-induced mechano-allodynia and mechano-hyperalgesia (Fig. 1, B and C) in a dose-dependent manner; doses were chosen from previous studies (15). Although chemotherapy is completed within a few days, we continued dosing until peak mechano-allodynia and mechano-hyperalgesia were achieved because the delay to symptom onset introduces uncertainty regarding the exact initiation of relevant pathological processes making continued treatment prudent. When W146 treatment was discontinued on D15, mechano-hypersensitivity did not emerge through D25 (Fig. 1, B and C). The ED50 (effective dose providing 50% effect) of W146 for mechano-allodynia and mechano-hyperalgesia at D25 was 0.4 nmol/day (95% CI: 0.2–0.7) and 0.8 nmol/day (95% CI: 0.5–1.3).
Because all drugs tested had comparable effects on mechano-allodynia and mechano-hyperalgesia, we will only show the latter for simplicity in the subsequent figures, and we refer to it as mechano-hypersensitivity in the text.
Intrathecal Injection of Selective S1PR1 Agonist SEW2871 Causes Mechano-hypersensitivity
Taken together, these results suggest that the formation of S1P in spinal cord in response to paclitaxel engages the S1P1 receptor to evoke mechano-hypersensitivity. Therefore, does spinal activation of S1PR1 with exogenous application of an S1PR1 agonist mimic these behavioral outcomes? In normal rats with previously implanted i.th. catheters, injection of the selective S1PR1 agonist, SEW2871 (0.8 nmol) (53), led to a time-dependent development of mechano-hypersensitivity that peaked by 2 h (Fig. 4). These effects were blocked with W146, but not W140 (2.2 nmol, i.th.; given 30 min before SEW2871 treatment; Fig. 4A), confirming an S1PR1 mechanism of action. These results suggest that activation of S1PR1 in spinal cord by an exogenous S1PR1 agonist can recapitulate behavioral features associated with paclitaxel administration
FIGURE 4.

Inhibition of SEW2871-induced pain by S1PR1 antagonists and S1PR1 modulators. Intrathecal injection of SEW2871 (V-SEW, black bars) but not its vehicle (V-V, open bars) led to the development of mechano-hypersensitivity with peak effect at 2 h (A–D). When given 30 min before SEW2871, intrathecal administration of W146 (A; 2.2 nmol), FTY720 (B; 0.8 nmol), or CYM-5442 (C; 0.8 nmol) but not W140 (A; 2.2 nmol) blocked mechano-hypersensitivity. Likewise, pretreatment with oral FTY720 (B; 0.1 mg/kg), CYM-5442 (C; 0.3 mg/kg), or NIBR-14 (D; 3 mg/kg) prevented the development of SEW2871-induced mechano-hypersensitivity. Results are expressed as mean ± S.D. for n = 4–7 rats and analyzed by one-way ANOVA with Bonferroni comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus V-SEW.
Intrathecal Injection of S1PR1 Modulators Block SEW2871-induced Mechano-hypersensitivity
Binding of SEW2871 (or the endogenous ligand S1P) to the S1P1 receptor (53) allows S1PR1 recycling back to the plasma membrane after internalization. In contrast, other S1PR1 agonists potently induce irreversible down-regulation of S1PR1 resulting in the ubiquitinylation and proteosomal degradation of the receptor, yielding a net decrease in S1PR1 expression at the plasma membrane (54). S1PR1 agonists in this class referred to as S1PR1 modulators include the following: FTY720 (a sphingosine analog that is phosphorylated in vivo by SphK2 to produce its bioactive S-isomeric monophosphate ester FTY720-P, which is an S1P mimetic capable of binding to all S1PRs except S1PR2 (6)); CYM-5442 (55); and ponesimod (28). These S1PR1 modulators therefore act as functional antagonists (unlike SEW2871 or S1P) at S1PR1 to block S1P/S1PR1 signaling and exert effects similar to those observed with other S1PR1 antagonists (56). This raises the following question. Do these S1PR1 modulators block the sensitizing effects of SEW2871? Intrathecal injection of FTY720 or CYM-5442 (0.8 nmol) 30 min before SEW2871 blocked its ability to elicit mechano-hyperalgesia and mechano-allodynia (Fig. 4, B and C). Similar results were obtained when FTY720 or CYM-5442 was given systemically consistent with their high CNS permeability (57, 58) and were corroborated with NIBR-14 (3 mg/kg; Fig. 4D). NIBR-14 is a methyl ester pro-drug that is rapidly hydrolyzed in vivo to its corresponding carboxylic acid (NIBR-15) that acts as a potent and selective S1PR1 antagonist (40). The suppressive effects of FTY720 and CYM-5442 therefore mimic those observed with other S1PR1 antagonists and support their role as functional antagonists for their mechanism of action at S1PR1.
Systemic Administration of S1PR1 Modulators and S1PR1 Antagonists Block the Development of Paclitaxel-induced Neuropathic Pain
Results gathered so far define the important contribution of the S1P/S1PR1 axis in the spinal cord to the development of CIPN; blocking this signaling pathway mitigates CIPN. This raises the question as to whether these anti-S1PR1 agents would be effective when given systemically, thereby paralleling potential clinical conditions. Systemic FTY720 (0.003, 0.01, and 0.03 mg/kg/day; i.p), CYM-5442, ponesimod, or NIBR-14 (all at 0.3, 1, and 3 mg/kg/day; p.o.) blocked mechano-hypersensitivity in a dose-dependent fashion (Fig. 5, A–D). ED50 values for mechano-allodynia and mechano-hyperalgesia on D25 for each agent are reported in Table 1. We also examined whether restricting the dosing regimen of the anti-S1PR1 agents to coincide with only the paclitaxel treatment would afford protection as dosing patients only when they receive the chemotherapeutic agent would be a preferred regimen. FTY720 (0.03 mg/kg/day), CYM-5442, ponesimod, or NIBR-14/-15 (all at 3 mg/kg/day), when given orally at the same time as paclitaxel (D0, -2, -4, and -6), blocked neuropathic pain through at least D25 (data not shown).
FIGURE 5.

Systemic delivery of S1PR1 modulators and S1PR1 antagonists blocks paclitaxel-induced neuropathic pain. When compared with vehicle (V-V, ▵), treatment with paclitaxel (P-V, ●) led to the development of mechano-hypersensitivity by D25 (A–D). Daily (D0–15) treatment with FTY720 (A; 0.003, ▴; 0.01, ▾; or 0.03 mg/kg/day, ■), CYM-5442 (B; 0.03, ▴; 0.1, ▾; or 0.3 mg/kg/day, ■), ponesimod (C; 0.3, ▴; 1, ▾; or 3 mg/kg/day, ■), or NIBR-14 (D; 0.3, ▴; 1, ▾; or 3 mg/kg/day, ■) blocked the development of mechano-hypersensitivity in a dose-dependent manner. Results are expressed as mean ± S.D. for n = 7 rats and analyzed by one-way ANOVA Dunnett's comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus P-V.
TABLE 1.
ED50 values for drugs tested in models of chemotherapy-induced neuropathic pain
| Treatment | Allodynia |
Hyperalgesia |
||
|---|---|---|---|---|
| ED50 | 95% CI | ED50 | 95% CI | |
| mg/kg/day | mg/kg/day | |||
| P-FTY720 | 0.007 | 0.0044–0.012 | 0.01 | 0.0066–0.014 |
| P-CYM-5442 | 0.3 | 0.17–0.41 | 0.3 | 0.25–0.46 |
| P-Ponesimod | 0.3 | 0.2–0.37 | 0.3 | 0.23–0.52 |
| P-NIBR-14 | 0.3 | 0.22–0.36 | 0.5 | 0.32–0.67 |
| Ox-FTY720 | 0.008 | 0.0047–0.012 | 0.007 | 0.0043–0.010 |
Contribution of Spinal NFκB and MAPKs to Paclitaxel-induced Neuropathic Pain
We previously reported that the development of paclitaxel-induced neuropathic pain is associated with increased formation of TNF-α and IL-1β in the spinal cord and that inhibiting these pro-inflammatory cytokines blocks CIPN (45). We now extend these findings and demonstrate that at peak mechano-hypersensitivity on D16, additional canonical pro-inflammatory signaling pathways are activated. These include the activation of the NFκB signaling pathway as indicated by p65 phosphorylation at serine 536 (Fig. 6A), nuclear translocation of the p65 subunit of NFκB (Fig. 6B), and the activation of the MAPK signaling pathway as evidenced by the increased phosphorylation of ERK1/2 (Fig. 6C) and p38 (Fig. 6D). Because the function of each of these signaling pathways in the context of CIPN is not known, selective inhibitors for each of these pathways were used to establish their possible contribution. The selective inhibitor of NFκB, SN50, is a small peptide that binds to and prevents the translocation of NFκB to the nucleus (59), whereas the MAPK inhibitors U0126 and SB203580 prevent MAPK/ERK1/2 (MEK1/2) phosphorylation of ERK (60) and phospho-p38 catalytic activity (61), respectively. As can be seen in Fig. 6, i.th. delivery of SN50 (2 ng/kg/day) (62), U0126 (1 μg/kg/day) (63), and SB203580 (10 μg/kg/day) (62, 64) attenuated their respective pathways (Fig. 6, A–D), as well as the development of mechano-hypersensitivity (Fig. 6, E–G).
FIGURE 6.

Activation of NFκB and MAPK activation in spinal cord following paclitaxel treatment. On D16 and when compared with vehicle (V-V, open bars), administration of paclitaxel (P-V, black bars) increased the phosphorylation of cytosolic p65 (A), nuclear translocation of NFκB p65 (B), and the phosphorylation of ERK1/2 (C) and p38 (D). Daily i.th. (D0–15) delivery of selective inhibitors (gray bars) of NFκB (A and B: SN50, 2 ng/kg/day), MEK1/2 (C, U0126, 1 μg/kg/day), and p38 (D, SB203580, 10 μg/kg/day) blocked their respective pathway (A–D) and mechano-hypersensitivity as tested on D16 (E–G). Results are expressed as mean ± S.D. for n = 6–7 rats and analyzed by one-way ANOVA with Dunnett's comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus P-V.
Spinal Neuroinflammation Is Blocked by Anti-S1PR1 Approaches
Inhibition of CIPN by W146 (i.th.; 2.2 nmol/day) at peak mechano-hypersensitivity was associated with inhibition of the activation of NFκB (p65 phosphorylation at serine 536 and nuclear translocation of the p65 subunit of NFκB; Fig. 7, A and B) and MAPKs (reflected as phosphorylation of ERK1/2 and p38 (Fig. 7, D and E) as well as the overproduction of TNF-α and IL-1β (Fig. 7F). Conversely, W146 increased formation of anti-inflammatory cytokines (IL-10 and IL-4) (Fig. 7F). Supporting a common anti-S1PR1 mechanism of action in the modulation of spinal neuroinflammation in the genesis of CIPN, systemic FTY720, CYM-5442, ponesimod, or NIBR-14 given at the highest dose attenuated TNF-α and IL-1β but increased IL-10 and IL-4 (Fig. 8, A–D).
FIGURE 7.

Inhibition of spinal neuroinflammation by the S1PR1 antagonist W146. On D16 and when compared with vehicle (V-V, open bars), administration of paclitaxel (P-V, black bars) increases cytosolic phosphorylation of NFκB p65 (A), nuclear translocation of NFκB p65 (B), and phosphorylation of ERK1/2 (C) and p38 (D). These events were blocked by daily i.th. delivery (D0–15) of W146 (2.2 nmol/day, gray bars). E, when compared with vehicle on D16, paclitaxel treatment also increased TNF-α and IL-1β production, which was blocked by W146; W146 increased the levels of IL-4 and IL-10. W146 alone had no effect in the spinal cord of vehicle-treated rats (light gray bars). Results are expressed as mean ± S.D. for n = 5–6 rats and analyzed by one-way ANOVA with Dunnett's comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus P-V.
FIGURE 8.

Systemic delivery of S1PR1 modulators and S1PR1 antagonists blocks paclitaxel-induced pro-inflammatory cytokine production in spinal cord. As compared with vehicle (V-V, open bars), paclitaxel (P-V, black bars)-induced increases in the production of proinflammatory cytokines TNF-α and IL-1β on D16 were blocked by daily administration at the highest effective dose of FTY720 (A), CYM-5442 (B), ponesimod (C), or NIBR-14 (D). Administration of these compounds in paclitaxel-treated rats increased the production of IL-4 and IL-10. Results are expressed as mean ± S.D. for n = 6 rats and analyzed by one-way ANOVA Dunnett's comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus P-V.
S1PR1 Modulators and S1PR1 Antagonists, but Not S1PR1 Agonists, Reverse Established Paclitaxel-induced Neuropathic Pain in a Naloxone-independent Manner
We next sought to define whether activation of spinal S1PR1 contributes to the maintenance of CIPN. Intrathecal administration of W146 on D25, but not W140, caused a rapid reversal of mechano-hypersensitivity, which was maximal within 2 h (Fig. 9A). In contrast, SEW2871 (i.th.; 0.8 nmol; Fig. 9A) had no effect, reinforcing the notion that S1PR1 antagonism and not agonism provides antinociception. In added support, the administration of a single oral dose of FTY720 (0.1 mg/kg) or NIBR-14 (3 mg/kg) on D25 reversed mechano-hypersensitivity and this was not blocked by a high dose (intraperitoneal) of the nonselective opioid receptor antagonist naloxone (2 mg/kg), thus excluding the contribution of an endogenous opioid pathway (Fig. 9, B and C). It is noteworthy that a subcutaneous mini-pump infusion of FYT720 (0.1 mg/kg/day for 6 days), CYM-5442, or NIBR-14 (both 3 mg/kg/day for 6 days) following an intraperitoneal loading dose completely reversed mechano-hypersensitivity (Fig. 10A) without any evident analgesic tolerance or alteration to the peripheral blood leukocyte differential (Table 2).
FIGURE 9.

W146, FTY720, and NIBR-14 reverse-established paclitaxel-induced neuropathic pain. Intrathecal administration of W146 (P-W146, gray hatched bars), but not W140 (P-W140, light gray bars) or SEW2871 (2.2 nmol, P-SEW2871, dark gray bars), at the time of peak plateau development of paclitaxel (P-V, black bars)-induced mechano-hypersensitivity as compared with vehicle (V-V, open bars) reversed hypersensitivity with peak effects at 2 h post-injection (A). Similar results were obtained with oral administration of FTY720 (B; 0.1 mg/kg, P-FTY720, dark gray bar) or NIBR-14 (iC; 3 mg/kg, P-NIBR-14, dark gray bar). The effects of FTY720 and NIBR-14 were not affected by pretreatment with naloxone (B and C; 2 mg/kg, i.p., light gray bars). Results are expressed as mean ± S.D. for n = 3–6 animals and analyzed by one-way ANOVA with Dunnett's comparisons. *, p < 0.05 versus V-V; †, p < 0.05 versus P-V.
FIGURE 10.

FTY720, CYM5442, and NIBR-14 reverse established paclitaxel-induced neuropathic pain without altering acute pain. A, miniosmotic pumps implanted subcutaneously on D16 followed by a single loading dose of either FTY720 (P-FTY720, 0.1 mg/kg/day, ■), CYM5442 (P-CYM5442, 3 mg/kg/day, ▾) or NIBR-14 (P-NIBR-14, 3 mg/kg/day, ♦) significantly attenuated mechano-hypersensitivity induced by paclitaxel (P-V, ●) treatment and maintained its efficacy over the 6 days tested. B, treatment with morphine (subcutaneously, 5 mg/kg, ○) led to a time-dependent increase in tail flick latency(ies), whereas similar subcutaneous injections of FTY720 (3 mg/kg, ■), NIBR-14 (3 mg/kg, ▴), ponesimod (3 mg/kg, ▾), or CYM-5442 (3 mg/kg, ♦) had no effect on latency throughout the time tested. C, summary schematic. Results are expressed as mean ± S.D. for n = 3–5 animals and analyzed by one-way ANOVA with Dunnett's comparisons. *, p < 0.05 versus D0/0 min.
TABLE 2.
Complete blood cells counts
No basophils were observed in any group.
| Treatment | Neutrophils |
Lymphocytes |
Monocytes |
Eosinophils |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | S.D. | N | Mean | S.D. | N | Mean | S.D. | N | Mean | S.D. | N | |
| V | 738 | 156 | 4 | 6720 | 2939 | 4 | 153 | 106 | 4 | 9.8 | 7.1 | 4 |
| V-FTY720 | 1061 | 393 | 4 | 3877 | 2875 | 4 | 120 | 26 | 4 | 6.5 | 9.3 | 4 |
| P | 522 | 293 | 4 | 4475 | 1417 | 4 | 110 | 71 | 4 | 4.8 | 4.9 | 4 |
| P-FTY720 | 1141 | 853 | 4 | 2572 | 1663 | 4 | 241 | 157 | 4 | 11.3 | 14.3 | 4 |
FTY720 Blocks Oxaliplatin-induced Neuropathic Pain
To examine whether the beneficial effects of an anti-S1PR1 strategy can be extended to neuropathic pain resulting from other chemotherapeutic agents with different mechanisms of anticancer action, we used oxaliplatin, which is widely used for metastatic colon cancer and other gastrointestinal cancers. We decided to examine this question with FTY720 only because this drug is already used clinically and therefore provides an advantage over the other S1PR1 antagonists/modulators that are in preclinical discovery or clinical trials. Confirming our previous studies (65), oxaliplatin-induced neuropathic pain was significant by D11 (onset), peaked by D17, and plateaued throughout our observation period (D25). Mechano-hypersensitivity was attenuated in a dose-dependent manner by FTY720 with ED50 values reported in Table 1. Furthermore, and mimicking effects seen with paclitaxel, at the highest dose tested, FTY720 (0.01 mg/kg/day) blocked the increased production of TNF-α and IL-1β, whereas the levels of anti-inflammatory cytokines IL-10 and IL-4 increased significantly (data not shown).
Lack of Effects on Acute Nociception
In contrast to morphine, which was used as a positive control at 5 mg/kg, the highest doses of FTY720 (3 mg/kg, intraperitoneally), CYM-5442, ponesimod, or NIBR-14 (3 mg/kg, oral gavage) tested had no effects on acute nociception as measured by tail flick latency (Fig. 10B).
FTY720 Does Not Alter Anticancer Activity in Vitro
The anticancer effects of FTY720 and other anti-S1P/S1PR1 agents are well documented (2, 5) and are therefore not expected to interfere with the chemotherapeutic effectiveness of paclitaxel or oxaliplatin. Based upon the reported pharmacokinetic studies in rats (6), the anticipated plasma levels of FTY720 at doses providing maximal blockade of CIPN are about 0.5–0.7 nm. When tested at doses at least 10-fold higher, FTY720 (10 nm) did not diminish the in vitro anticancer effects of paclitaxel (1–100 nm) or oxaliplatin (0.3–30 μm) in human breast adenocarcinoma (SKBr3) or colon carcinoma (SW480) cells, respectively. FTY720 (10 nm) alone caused a 3–20% decrease in anticancer cell survival, which is not surprising (Table 3).
TABLE 3.
FTY720 does not interfere with the in vitro anticancer activity of paclitaxel or oxaliplatin in vitro
| Treatment | LD50 | 95% CI | N |
|---|---|---|---|
| SKBr3 cells | |||
| Paclitaxel | 7.3 nM | 3.1–17 nM | 3 |
| P-FTY720 (10 nM) | 7.1 nM | 3.1–16 nM | 3 |
| SW480 cells | |||
| Oxaliplatin | 0.6 μm | 0.03–10.7 μm | 4 |
| Ox-FTY720 (10 nm) | 0.1 μm | 0.01–1.3 μm | 4 |
DISCUSSION
Despite extensive research efforts, chronic neuropathic pain remains a largely unmet medical need, and novel therapies are needed. Several important findings have emerged from our study.
First, our results establish that the S1P-S1PR1 axis is a critical determinant in the development and maintenance of paclitaxel-induced neuropathic pain and identify the following two viable therapeutics with potential for fast translational impact to patient care: FTY720, which is Food and Drug Administration-approved (6), and ponesimod, which is in phase 2 clinical trials (66). In addition to the clinically feasible prevention treatment paradigm, the fact that anti-S1PR1-targeted approaches can be used in an intervention paradigm allows therapy to be extended to those patients who have developed the neuropathy. It is reasonable to hypothesize that anti-S1PR1 approaches may provide added therapeutic opportunities because these are proving useful in anticancer therapies as stand-alone drugs or as adjuncts to chemotherapeutic agents (2, 5). Thus, drugs, such as FTY720, may enhance the anticancer effects of chemotherapeutic agents while minimizing the major dose-limiting toxicity neuropathic pain as shown in this study. Noteworthy, these beneficial effects were not restricted to paclitaxel only but also extended to other chemotherapeutic agents, raising the exciting possibility of a more widespread impact to mitigate CIPN. Effects were naloxone-independent and therefore are not mediated by activation of an endogenous opioid system reducing the potential for the development of analgesic tolerance. Because the S1P/S1PR1 axis is critically involved in the egress of T and B cells from lymphoid tissues (67), it was initially thought that the efficacy of FTY720 and other S1PR1 functional antagonists was mediated by their ability to block the infiltration of autoreactive T cells into the CNS where they play an important role in the progression of MS (10). However, phase II clinical trials revealed that the peak effectiveness of FTY720 in the treatment of MS occurs at doses that are suboptimal for the induction of lymphopenia (6). These paradoxical human clinical data, coupled with the findings obtained in the CNS that S1P is produced by both neurons and glia expressing S1PR1, suggest that the beneficial effects of these agents may also be exerted by nonimmunocompetent cells in the CNS (68). It is known that FTY720 efficiently crosses the blood-brain barrier where it is phosphorylated by SphK2 (predominantly expressed in neurons and glial cells) (69) to yield FTY720-P (70). The CNS is therefore an important site for mechanism of action of not only FTY720 but also of other S1PR1 modulators, including CYM-5442 (6, 57, 71). In our studies, the maximal efficacy regarding the reduction of mechano-hypersensitivity was detected with doses of FTY720 at least 10-fold lower than those shown to cause lymphopenia (6) or bradycardia (72) and for NIBR-14/15 to cause pulmonary vascular leakage (40). Furthermore, we did not observe any changes in differential white blood cell counts following a 6-day infusion of FTY720 that provided near-to-maximal inhibition of CIPN. Similar findings were reported by Coste et al. (73), where they demonstrated that FTY720 at doses devoid of immunosuppressive effects was capable of reversing the neuropathic pain produced by a chronic constriction injury of the sciatic nerve.
Second, our results identify functional antagonism and not agonism as the mechanism of action of the S1PR1 modulators in blocking pain. To this end, intrathecal delivery of a selective S1PR1 agonist, SEW2871, evoked pronociceptive and not antinociceptive behavioral outcomes that were blocked by pretreatment with FTY720, CYM-5442, and ponesimod as well as by S1PR1 antagonists. In addition, S1PR1 modulators and S1PR1 antagonists, but not the S1PR1 agonist SEW2871, blocked and reversed CIPN. In a previous study, intrathecal injection of SEW2871 had no effect on mechanical hypersensitivity resulting from the injection of formalin into the rat's hind paw (74). This lack of effect is consistent with the fact that SEW2871 is not a functional antagonist and agrees with our findings that SEW2871 does not reverse CIPN. In addition, our results support previous findings in various animal models for transplantation and autoimmune diseases, including MS and arthritis, where functional antagonism of S1PR1 rather than persistent agonism/signaling at S1PR1 was identified as the mechanism of action (56).
Third, our results identify the spinal cord as one site of action for anti-S1PR1 agents and inhibition of spinal neuroinflammation as a molecular target in their pharmacological activity. This parallels the documented CNS mechanisms of action of anti-S1PR1 therapies in MS (71). Initial studies using FTY720 in animal models of MS revealed the importance of the S1P/S1PR1 axis to neuroinflammatory processes in the CNS, as well as the protective actions of various S1PR1 functional antagonists or selective S1PR1 antagonists (56). In a rodent model of MS, FTY720 (75) or CYM-5442 (57) inhibits the S1P/S1PR1 activation and signaling in astrocytes and the downstream formation of pro-inflammatory cytokines, including IL-1β and IL-6. Pharmacological and genetic manipulations reveal that these beneficial effects are exerted through S1PR1 functional antagonism in astrocytes and neurons (56). Likewise, accumulating evidence implicates neuroinflammatory processes in the alteration of glia-neuronal communication in the dorsal horn of the spinal cord that is associated with paclitaxel-induced neuropathic pain. Specifically, hyperactivation of glial cells (45, 76, 77), enhanced TNF-α and IL-1β (45, 78, 79), and nitroxidative species production (45) as well as dysregulation of glutamate homeostasis (45, 80, 81) have been documented. A recent study revealed that increased activation of GSK-3β in spinal cord contributes to the development and maintenance of paclitaxel-induced neuropathic pain by activating astrocytes and causing the overproduction of IL-1β (79). It is, however, known that in addition to GSK-3β, redox-activated transcription factors such as NFκB and MAPKs regulate the levels of TNF-α and IL-1β (and vice versa) and contribute to the development of several different neuropathic pain states (82). However, to date their functional contribution to CIPN was not documented. Our results now reveal that these signaling pathways are activated in response to paclitaxel and contribute to CIPN because their inhibition attenuates mechano-hypersensitivity. Moreover, our findings that S1PR1 antagonism blocks the activation of NFκB and MAPKs and shifts the cytokine environment in the spinal cord from pro-inflammatory to anti-inflammatory suggest that inhibition of spinal neuroinflammation is an important component in their beneficial armamentarium. Noteworthy, similar enhancement in the expression of anti-inflammatory IL-10 following treatment with FTY720 has been described with activated dendritic cells (83) and in colitis (84). In addition to inhibition of neuroinflammatory processes in spinal cord, it is important to recognize that blocking neuronal S1PR1 signaling may also contribute to the beneficial effects of anti-S1PR1 approaches (8). For example, blocking S1PR1 may also attenuate the increased neuronal excitability in spinal dorsal horn observed during paclitaxel-induced neuropathic pain (85).
It has been previously reported in other animal models that cytokines and nitroxidative species for example can activate SphK1 (48) and sphingomyelinases (47) as well as regulate the enzymatic activities of SPT, sphingomyelinases, and ceramidases (86–88). Reciprocally, ceramide and S1P can increase the formation of cytokines, and nitroxidative species from glial cells (89) and can also regulate cellular redox homeostasis (87). Therefore, the ceramide/S1P pathway may not only be a target for the action of neuroinflammatory species, it may also serve as a trigger for their formation. Together, these nitroxidative- and cytokine-rich environments may synergize with S1P signaling to prolong and exacerbate CIPN by instituting feedback loops that sustain local neuroinflammatory processes in spinal cord.
Although our studies focused on changes in the spinal cord, we cannot exclude the likely contributions of S1P/S1PR1 in the periphery. For example, S1PR1 activation increases the excitability of rat sensory neurons, and blocking S1P/S1PR1 signaling reduces their excitability to inflammatory stimuli (90, 91). Therefore, further understanding of the S1P/S1PR1 axis could provide the framework for clinical development of S1PR1 antagonists/modulators in CIPN and perhaps other chronic neuropathic pain states such as diabetic neuropathy, whose underlying pathology shares commonalities with CIPN (92).
Acknowledgments
We thank Dr Gary J. Bennett (McGill University, Montreal, Canada) for pertinent insight, guidance, and helpful suggestions with the writing of this paper. The studies performed in GDN's laboratory were conducted in a facility constructed with support from Research Facilities Improvement Program Grant C06 RR015481-01 from the National Center for Research Resources, National Institutes of Health.
This work was supported by the Leukemia and Lymphoma Society Translational Research Program and the Mayday Fund and in part by the Saint Louis Cancer Center. These studies were also supported by the Ralph W. and Grace M. Showalter Trust (to G. D. N.).
- CIPN
- chemotherapy-induced neuropathic pain
- S1P
- sphingosine 1-phosphate
- SphK
- sphingosine kinase
- MS
- multiple sclerosis
- i.th.
- intrathecal
- PWT
- paw withdrawal threshold
- SPT
- serine palmitoyltransferase
- MFI
- mean fluorescence intensity
- S1PR1
- S1P receptor subtype 1
- ANOVA
- analysis of variance
- CI
- confidence interval.
REFERENCES
- 1. Farquhar-Smith P. (2011) Chemotherapy-induced neuropathic pain. Curr. Opin. Support Palliat. Care 5, 1–7 [DOI] [PubMed] [Google Scholar]
- 2. Ogretmen B., Hannun Y. A. (2004) Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 4, 604–616 [DOI] [PubMed] [Google Scholar]
- 3. Spiegel S., Milstien S. (2011) The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 11, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rosen H., Goetzl E. J. (2005) Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat. Rev. Immunol. 5, 560–570 [DOI] [PubMed] [Google Scholar]
- 5. Adan-Gokbulut A., Kartal-Yandim M., Iskender G., Baran Y. (2013) Novel agents targeting bioactive sphingolipids for the treatment of cancer. Curr. Med. Chem. 20, 108–122 [PubMed] [Google Scholar]
- 6. Brinkmann V., Billich A., Baumruker T., Heining P., Schmouder R., Francis G., Aradhye S., Burtin P. (2010) Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat. Rev. Drug Discov. 9, 883–897 [DOI] [PubMed] [Google Scholar]
- 7. Welch S. P., Sim-Selley L. J., Selley D. E. (2012) Sphingosine-1-phosphate receptors as emerging targets for treatment of pain. Biochem. Pharmacol. 84, 1551–1562 [DOI] [PubMed] [Google Scholar]
- 8. Salvemini D., Doyle T., Kress M., Nicol G. (2013) Therapeutic targeting of the ceramide-to-sphingosine 1-phosphate pathway in pain. Trends Pharmacol. Sci. 34, 110–118 [DOI] [PubMed] [Google Scholar]
- 9. Zhang Y. H., Nicol G. D. (2004) NGF-mediated sensitization of the excitability of rat sensory neurons is prevented by a blocking antibody to the p75 neurotrophin receptor. Neurosci. Lett. 366, 187–192 [DOI] [PubMed] [Google Scholar]
- 10. Zhang Y. H., Vasko M. R., Nicol G. D. (2002) Ceramide, a putative second messenger for nerve growth factor, modulates the TTX-resistant Na+ current and delayed rectifier K+ current in rat sensory neurons. J. Physiol. 544, 385–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang Y. H., Vasko M. R., Nicol G. D. (2006) Intracellular sphingosine 1-phosphate mediates the increased excitability produced by nerve growth factor in rat sensory neurons. J. Physiol. 575, 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nicol G. D. (2008) Nerve growth factor, sphingomyelins, and sensitization in sensory neurons. Sheng Li Xue Bao 60, 603–604 [PubMed] [Google Scholar]
- 13. Choi J. I., Svensson C. I., Koehrn F. J., Bhuskute A., Sorkin L. S. (2010) Peripheral inflammation induces tumor necrosis factor dependent AMPA receptor trafficking and Akt phosphorylation in spinal cord in addition to pain behavior. Pain 149, 243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Doyle T., Chen Z., Muscoli C., Obeid L. M., Salvemini D. (2011) Intraplantar-injected ceramide in rats induces hyperalgesia through an NF-κB- and p38 kinase-dependent cyclooxygenase 2/prostaglandin E2 pathway. FASEB J. 25, 2782–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Doyle T., Chen Z., Obeid L. M., Salvemini D. (2011) Sphingosine-1-phosphate acting via the S1P(1) receptor is a downstream signaling pathway in ceramide-induced hyperalgesia. Neurosci. Lett. 499, 4–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joseph E. K., Levine J. D. (2004) Caspase signalling in neuropathic and inflammatory pain in the rat. Eur. J. Neurosci. 20, 2896–2902 [DOI] [PubMed] [Google Scholar]
- 17. Mair N., Benetti C., Andratsch M., Leitner M. G., Constantin C. E., Camprubí-Robles M., Quarta S., Biasio W., Kuner R., Gibbins I. L., Kress M., Haberberger R. V. (2011) Genetic evidence for involvement of neuronally expressed S1P receptor in nociceptor sensitization and inflammatory pain. PLoS One 6, e17268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finley A., Chen Z., Esposito E., Cuzzocrea S., Sabbadini R., Salvemini D. (2013) Sphingosine 1-phosphate mediates hyperalgesia via a neutrophil-dependent mechanism. PLoS One 8, e55255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Patti G. J., Yanes O., Shriver L. P., Courade J. P., Tautenhahn R., Manchester M., Siuzdak G. (2012) Metabolomics implicates altered sphingolipids in chronic pain of neuropathic origin. Nat. Chem. Biol. 8, 232–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Muscoli C., Doyle T., Dagostino C., Bryant L., Chen Z., Watkins L. R., Ryerse J., Bieberich E., Neumman W., Salvemini D. (2010) Counter-regulation of opioid analgesia by glial-derived bioactive sphingolipids. J. Neurosci. 30, 15400–15408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ndengele M. M., Cuzzocrea S., Masini E., Vinci M. C., Esposito E., Muscoli C., Petrusca D. N., Mollace V., Mazzon E., Li D., Petrache I., Matuschak G. M., Salvemini D. (2009) Spinal ceramide modulates the development of morphine antinociceptive tolerance via peroxynitrite-mediated nitroxidative stress and neuroimmune activation. J. Pharmacol. Exp. Ther. 329, 64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yan X., Weng H. R. (2013) Endogenous interleukin-1β in neuropathic rats enhances glutamate release from the primary afferents in the spinal dorsal horn through coupling with presynaptic N-methyl-d-aspartic acid receptors. J. Biol. Chem. 288, 30544–30557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrier J., Pereira V., Busserolles J., Authier N., Balayssac D. (2013) Emerging trends in understanding chemotherapy-induced peripheral neuropathy. Curr. Pain Headache Rep. 17, 364. [DOI] [PubMed] [Google Scholar]
- 24. Wang X. M., Lehky T. J., Brell J. M., Dorsey S. G. (2012) Discovering cytokines as targets for chemotherapy-induced painful peripheral neuropathy. Cytokine 59, 3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Polomano R. C., Mannes A. J., Clark U. S., Bennett G. J. (2001) A painful peripheral neuropathy in the rat produced by the chemotherapeutic drug, paclitaxel. Pain 94, 293–304 [DOI] [PubMed] [Google Scholar]
- 26. Xiao W. H., Zheng H., Bennett G. J. (2012) Characterization of oxaliplatin-induced chronic painful peripheral neuropathy in the rat and comparison with the neuropathy induced by paclitaxel. Neuroscience 203, 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Størkson R. V., Kjørsvik A., Tjølsen A., Hole K. (1996) Lumbar catheterization of the spinal subarachnoid space in the rat. J. Neurosci. Methods 65, 167–172 [DOI] [PubMed] [Google Scholar]
- 28. Bolli M. H., Abele S., Binkert C., Bravo R., Buchmann S., Bur D., Gatfield J., Hess P., Kohl C., Mangold C., Mathys B., Menyhart K., Müller C., Nayler O., Scherz M., Schmidt G., Sippel V., Steiner B., Strasser D., Treiber A., Weller T. (2010) 2-Imino-thiazolidin-4-one derivatives as potent, orally active S1P1 receptor agonists. J. Med. Chem. 53, 4198–4211 [DOI] [PubMed] [Google Scholar]
- 29. Dixon W. J. (1980) Efficient analysis of experimental observations. Annu. Rev. Pharmacol. Toxicol. 20, 441–462 [DOI] [PubMed] [Google Scholar]
- 30. Randall L. O., Selitto J. J. (1957) A method for measurement of analgesic activity on inflamed tissue. Arch. Int. Pharmacodyn. Ther. 111, 409–419 [PubMed] [Google Scholar]
- 31. D'Amour F. E., Smith D. L. (1941) A method for determining loss of pain sensation. J. Pharmacol. Exp Ther. 72, 74–79 [Google Scholar]
- 32. Dobrowsky R. T., Kolesnick R. N. (2001) Analysis of sphingomyelin and ceramide levels and the enzymes regulating their metabolism in response to cell stress. Methods Cell Biol. 66, 135–165 [DOI] [PubMed] [Google Scholar]
- 33. Williams R. D., Wang E., Merrill A. H., Jr. (1984) Enzymology of long-chain base synthesis by liver: characterization of serine palmitoyltransferase in rat liver microsomes. Arch. Biochem. Biophys. 228, 282–291 [DOI] [PubMed] [Google Scholar]
- 34. Little J. W., Chen Z., Doyle T., Porreca F., Ghaffari M., Bryant L., Neumann W. L., Salvemini D. (2012) Supraspinal peroxynitrite modulates pain signaling by suppressing the endogenous opioid pathway. J. Neurosci. 32, 10797–10808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Youle M., Osio M., and ALCAR Study Group (2007) A double-blind, parallel-group, placebo-controlled, multicentre study of acetyl l-carnitine in the symptomatic treatment of antiretroviral toxic neuropathy in patients with HIV-1 infection. HIV Med. 8, 241–250 [DOI] [PubMed] [Google Scholar]
- 36. Krishnamurthy K., Dasgupta S., Bieberich E. (2007) Development and characterization of a novel anti-ceramide antibody. J. Lipid Res. 48, 968–975 [DOI] [PubMed] [Google Scholar]
- 37. Marvizón J. C., Chen W., Murphy N. (2009) Enkephalins, dynorphins, and β-endorphin in the rat dorsal horn: an immunofluorescence colocalization study. J. Comp. Neurol. 517, 51–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Schneider C. A., Rasband W. S., Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Meth. 9, 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Paxinos G., Watson C. (1998) The Rat Brain in Stereotaxic Coordinates, 4th Ed., Academic Press, San Diego [Google Scholar]
- 40. Angst D., Janser P., Quancard J., Buehlmayer P., Berst F., Oberer L., Beerli C., Streiff M., Pally C., Hersperger R., Bruns C., Bassilana F., Bollbuck B. (2012) An oral sphingosine 1-phosphate receptor 1 (S1P(1)) antagonist prodrug with efficacy in vivo: discovery, synthesis, and evaluation. J. Med. Chem. 55, 9722–9734 [DOI] [PubMed] [Google Scholar]
- 41. Itamochi H., Oishi T., Shimada M., Sato S., Uegaki K., Naniwa J., Sato S., Nonaka M., Terakawa N., Kigawa J., Harada T. (2011) Inhibiting the mTOR pathway synergistically enhances cytotoxicity in ovarian cancer cells induced by etoposide through up-regulation of c-Jun. Clin. Cancer Res. 17, 4742–4750 [DOI] [PubMed] [Google Scholar]
- 42. Dahan L., Sadok A., Formento J. L., Seitz J. F., Kovacic H. (2009) Modulation of cellular redox state underlies antagonism between oxaliplatin and cetuximab in human colorectal cancer cell lines. Br. J. Pharmacol. 158, 610–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shah M. R., Kriedt C. L., Lents N. H., Hoyer M. K., Jamaluddin N., Klein C., Baldassare J. (2009) Direct intra-tumoral injection of zinc-acetate halts tumor growth in a xenograft model of prostate cancer. J. Exp. Clin. Cancer Res. 28, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kriedt C. L., Baldassare J., Shah M., Klein C. (2010) Zinc functions as a cytotoxic agent for prostate cancer cells independent of culture and growth conditions. J. Exp. Ther. Oncol. 8, 287–295 [PubMed] [Google Scholar]
- 45. Doyle T., Chen Z., Muscoli C., Bryant L., Esposito E., Cuzzocrea S., Dagostino C., Ryerse J., Rausaria S., Kamadulski A., Neumann W. L., Salvemini D. (2012) Targeting the overproduction of peroxynitrite for the prevention and reversal of paclitaxel-induced neuropathic pain. J. Neurosci. 32, 6149–6160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Flatters S. J., Bennett G. J. (2006) Studies of peripheral sensory nerves in paclitaxel-induced painful peripheral neuropathy: evidence for mitochondrial dysfunction. Pain 122, 245–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hannun Y. A., Obeid L. M. (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 9, 139–150 [DOI] [PubMed] [Google Scholar]
- 48. Snider A. J., Orr Gandy K. A., Obeid L. M. (2010) Sphingosine kinase: role in regulation of bioactive sphingolipid mediators in inflammation. Biochimie 92, 707–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang H., Yoon S. Y., Dougherty P. M. (2012) Evidence that spinal astrocytes but not microglia contribute to the pathogenesis of paclitaxel-induced painful neuropathy. J. Pain 3, 239–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zheng F. Y., Xiao W. H., Bennett G. J. (2011) The response of spinal microglia to chemotherapy-evoked painful peripheral neuropathies is distinct from that evoked by traumatic nerve injuries. Neuroscience 176, 447–454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Delgado A., Casas J., Llebaria A., Abad J. L., Fabrias G. (2006) Inhibitors of sphingolipid metabolism enzymes. Biochim. Biophys. Acta 1758, 1957–1977 [DOI] [PubMed] [Google Scholar]
- 52. Sanna M. G., Wang S. K., Gonzalez-Cabrera P. J., Don A., Marsolais D., Matheu M. P., Wei S. H., Parker I., Jo E., Cheng W. C., Cahalan M. D., Wong C. H., Rosen H. (2006) Enhancement of capillary leakage and restoration of lymphocyte egress by a chiral S1P1 antagonist in vivo. Nat. Chem. Biol. 2, 434–441 [DOI] [PubMed] [Google Scholar]
- 53. Sanna M. G., Liao J., Jo E., Alfonso C., Ahn M. Y., Peterson M. S., Webb B., Lefebvre S., Chun J., Gray N., Rosen H. (2004) Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 279, 13839–13848 [DOI] [PubMed] [Google Scholar]
- 54. Verzijl D., Peters S. L., Alewijnse A. E. (2010) Sphingosine-1-phosphate receptors: zooming in on ligand-induced intracellular trafficking and its functional implications. Mol. Cells 29, 99–104 [DOI] [PubMed] [Google Scholar]
- 55. Gonzalez-Cabrera P. J., Jo E., Sanna M. G., Brown S., Leaf N., Marsolais D., Schaeffer M. T., Chapman J., Cameron M., Guerrero M., Roberts E., Rosen H. (2008) Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol. Pharmacol. 74, 1308–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bigaud M., Guerini D., Billich A., Bassilana F., Brinkmann V. (2013) Second generation S1P pathway modulators: research strategies and clinical developments. Biochim. Biophys. Acta 1841, 745–758 [DOI] [PubMed] [Google Scholar]
- 57. Gonzalez-Cabrera P. J., Cahalan S. M., Nguyen N., Sarkisyan G., Leaf N. B., Cameron M. D., Kago T., Rosen H. (2012) S1P(1) receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol. Pharmacol. 81, 166–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Meno-Tetang G. M., Li H., Mis S., Pyszczynski N., Heining P., Lowe P., Jusko W. J. (2006) Physiologically based pharmacokinetic modeling of FTY720 (2-amino-2[2-(-4-octylphenyl)ethyl]propane-1,3-diol hydrochloride) in rats after oral and intravenous doses. Drug Metab. Dispos. 34, 1480–1487 [DOI] [PubMed] [Google Scholar]
- 59. Lin Y.-Z., Yao S. Y., Veach R. A., Torgerson T. R., Hawiger J. (1995) Inhibition of nuclear translocation of transcription factor NF-κB by a synthetic peptide containing a cell membrane-permeable motif and nuclear localization sequence. J. Biol. Chem. 270, 14255–14258 [DOI] [PubMed] [Google Scholar]
- 60. Favata M. F., Horiuchi K. Y., Manos E. J., Daulerio A. J., Stradley D. A., Feeser W. S., Van Dyk D. E., Pitts W. J., Earl R. A., Hobbs F., Copeland R. A., Magolda R. L., Scherle P. A., Trzaskos J. M. (1998) Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 273, 18623–18632 [DOI] [PubMed] [Google Scholar]
- 61. Cuenda A., Rouse J., Doza Y. N., Meier R., Cohen P., Gallagher T. F., Young P. R., Lee J. C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233 [DOI] [PubMed] [Google Scholar]
- 62. Lee M. K., Han S. R., Park M. K., Kim M. J., Bae Y. C., Kim S. K., Park J. S., Ahn D. K. (2011) Behavioral evidence for the differential regulation of p-p38 MAPK and p-NF-κB in rats with trigeminal neuropathic pain. Mol. Pain 7, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Daulhac L., Mallet C., Courteix C., Etienne M., Duroux E., Privat A. M., Eschalier A., Fialip J. (2006) Diabetes-induced mechanical hyperalgesia involves spinal mitogen-activated protein kinase activation in neurons and microglia via N-methyl-d-aspartate-dependent mechanisms. Mol. Pharmacol. 70, 1246–1254 [DOI] [PubMed] [Google Scholar]
- 64. Jin S. X., Zhuang Z. Y., Woolf C. J., Ji R. R. (2003) p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J. Neurosci. 23, 4017–4022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Z., Janes K., Chen C., Doyle T., Bryant L., Tosh D. K., Jacobson K. A., Salvemini D. (2012) Controlling murine and rat chronic pain through A3 adenosine receptor activation. FASEB J. 26, 1855–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Brossard P., Derendorf H., Xu J., Maatouk H., Halabi A., Dingemanse J. (2013) Pharmacokinetics and pharmacodynamics of ponesimod, a selective S1P1 receptor modulator, in the first-in-human study. Br. J. Clin. Pharmacol. 76, 888–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Matloubian M., Lo C. G., Cinamon G., Lesneski M. J., Xu Y., Brinkmann V., Allende M. L., Proia R. L., Cyster J. G. (2004) Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature 427, 355–360 [DOI] [PubMed] [Google Scholar]
- 68. Jackson S. J., Giovannoni G., Baker D. (2011) Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflammation 8, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Blondeau N., Lai Y., Tyndall S., Popolo M., Topalkara K., Pru J. K., Zhang L., Kim H., Liao J. K., Ding K., Waeber C. (2007) Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 103, 509–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Foster C. A., Howard L. M., Schweitzer A., Persohn E., Hiestand P. C., Balatoni B., Reuschel R., Beerli C., Schwartz M., Billich A. (2007) Brain penetration of the oral immunomodulatory drug FTY720 and its phosphorylation in the central nervous system during experimental autoimmune encephalomyelitis: consequences for mode of action in multiple sclerosis. J. Pharmacol. Exp. Ther. 323, 469–475 [DOI] [PubMed] [Google Scholar]
- 71. Groves A., Kihara Y., Chun J. (2013) Fingolimod: direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 328, 9–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Fryer R. M., Muthukumarana A., Harrison P. C., Nodop Mazurek S., Chen R. R., Harrington K. E., Dinallo R. M., Horan J. C., Patnaude L., Modis L. K., Reinhart G. A. (2012) The clinically-tested S1P receptor agonists, FTY720 and BAF312, demonstrate subtype-specific bradycardia (S1P(1)) and hypertension (S1P(3)) in rat. PLoS One 7, e52985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Coste O., Brenneis C., Linke B., Pierre S., Maeurer C., Becker W., Schmidt H., Gao W., Geisslinger G., Scholich K. (2008) Sphingosine 1-phosphate modulates spinal nociceptive processing. J. Biol. Chem. 283, 32442–32451 [DOI] [PubMed] [Google Scholar]
- 74. Coste O., Pierre S., Marian C., Brenneis C., Angioni C., Schmidt H., Popp L., Geisslinger G., Scholich K. (2008) Antinociceptive activity of the S1P-receptor agonist FTY720. J. Cell. Mol. Med. 12, 995–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Choi J. W., Gardell S. E., Herr D. R., Rivera R., Lee C. W., Noguchi K., Teo S. T., Yung Y. C., Lu M., Kennedy G., Chun J. (2011) FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. U.S.A. 108, 751–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Peters C. M., Jimenez-Andrade J. M., Jonas B. M., Sevcik M. A., Koewler N. J., Ghilardi J. R., Wong G. Y., Mantyh P. W. (2007) Intravenous paclitaxel administration in the rat induces a peripheral sensory neuropathy characterized by macrophage infiltration and injury to sensory neurons and their supporting cells. Exp. Neurol. 203, 42–54 [DOI] [PubMed] [Google Scholar]
- 77. Warwick R. A., Hanani M. (2013) The contribution of satellite glial cells to chemotherapy-induced neuropathic pain. Eur. J. Pain 17, 571–580 [DOI] [PubMed] [Google Scholar]
- 78. Burgos E., Gómez-Nicola D., Pascual D., Martín M. I., Nieto-Sampedro M., Goicoechea C. (2012) Cannabinoid agonist WIN 55,212-2 prevents the development of paclitaxel-induced peripheral neuropathy in rats. Possible involvement of spinal glial cells. Eur. J. Pharmacol. 682, 62–72 [DOI] [PubMed] [Google Scholar]
- 79. Gao M., Yan X., Weng H. R. (2013) Inhibition of glycogen synthase kinase 3β activity with lithium prevents and attenuates paclitaxel-induced neuropathic pain. Neuroscience 254, 301–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Cata J. P., Weng H. R., Lee B. N., Reuben J. M., Dougherty P. M. (2006) Clinical and experimental findings in humans and animals with chemotherapy-induced peripheral neuropathy. Minerva Anestesiol. 72, 151–169 [PubMed] [Google Scholar]
- 81. Weng H. R., Aravindan N., Cata J. P., Chen J. H., Shaw A. D., Dougherty P. M. (2005) Spinal glial glutamate transporters downregulate in rats with taxol-induced hyperalgesia. Neurosci. Lett. 386, 18–22 [DOI] [PubMed] [Google Scholar]
- 82. Ji R. R., Gereau R. W., 4th, Malcangio M., Strichartz G. R. (2009) MAP kinase and pain. Brain Res. Rev. 60, 135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Durafourt B. A., Lambert C., Johnson T. A., Blain M., Bar-Or A., Antel J. P. (2011) Differential responses of human microglia and blood-derived myeloid cells to FTY720. J. Neuroimmunol. 230, 10–16 [DOI] [PubMed] [Google Scholar]
- 84. Daniel C., Sartory N., Zahn N., Geisslinger G., Radeke H. H., Stein J. M. (2007) FTY720 ameliorates Th1-mediated colitis in mice by directly affecting the functional activity of CD4+CD25+ regulatory T cells. J. Immunol. 178, 2458–2468 [DOI] [PubMed] [Google Scholar]
- 85. Cata J. P., Weng H. R., Chen J. H., Dougherty P. M. (2006) Altered discharges of spinal wide dynamic range neurons and down-regulation of glutamate transporter expression in rats with paclitaxel-induced hyperalgesia. Neuroscience 138, 329–338 [DOI] [PubMed] [Google Scholar]
- 86. Perrotta C., Clementi E. (2010) Biological roles of acid and neutral sphingomyelinases and their regulation by nitric oxide. Physiology 25, 64–71 [DOI] [PubMed] [Google Scholar]
- 87. Won J. S., Singh I. (2006) Sphingolipid signaling and redox regulation. Free Radic. Biol. Med. 40, 1875–1888 [DOI] [PubMed] [Google Scholar]
- 88. Huwiler A., Pfeilschifter J. (2003) Nitric oxide signalling with a special focus on lipid-derived mediators. Biol. Chem. 384, 1379–1389 [DOI] [PubMed] [Google Scholar]
- 89. Nayak D., Huo Y., Kwang W. X., Pushparaj P. N., Kumar S. D., Ling E. A., Dheen S. T. (2010) Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 166, 132–144 [DOI] [PubMed] [Google Scholar]
- 90. Chi X. X., Nicol G. D. (2010) The sphingosine 1-phosphate receptor, S1PR(1), plays a prominent but not exclusive role in enhancing the excitability of sensory neurons. J. Neurophysiol. 104, 2741–2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Mair N., Benetti C., Andratsch M., Leitner M. G., Constantin C. E., Camprubí-Robles M., Quarta S., Biasio W., Kuner R., Gibbins I. L., Kress M., Haberberger R. V. (2011) Genetic evidence for involvement of neuronally expressed S1P(1) receptor in nociceptor sensitization and inflammatory pain. PLoS One 6, e17268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bennett G. J. (2010) Pathophysiology and animal models of cancer-related painful peripheral neuropathy. Oncologist 15, 9–12 [DOI] [PubMed] [Google Scholar]
- 93. Paterniti I., Mazzon E., Riccardi L., Galuppo M., Impellizzeri D., Esposito E., Bramanti P., Cappellani A., Cuzzocrea S. (2012) Peroxisome proliferator-activated receptor beta/sigma agonist GW0742 ameliorates cerulein- and taurocholate-induced acute pancreatitis in mice. Surgery 152, 90–106 [DOI] [PubMed] [Google Scholar]