

Background: Mutations in the G/F domain of the human HSP40 chaperone DNAJB6 cause a protein aggregate myopathy.
Results: Homologous mutations in the yeast HSP40 Sis1 alter prion propagation in a prion strain-dependent manner.
Conclusion: G/F domain mutations affect chaperone-mediated processing of particular client conformers.
Significance: Impaired processing of certain substrate conformations may be one mechanism that contributes to chaperonopathy pathogenesis.
Keywords: Genetic Disease, Heat Shock Protein (HSP), Molecular Chaperone, Muscular Dystrophy, Prion, Protein Aggregation, Yeast
Abstract
The molecular chaperone network protects against the toxic misfolding and aggregation of proteins. Disruption of this network leads to a variety of protein conformational disorders. One such example recently discovered is limb-girdle muscular dystrophy type 1D (LGMD1D), which is caused by mutation of the HSP40 chaperone DNAJB6. All LGMD1D-associated mutations localize to the conserved G/F domain of DNAJB6, but the function of this domain is largely unknown. Here, we exploit the yeast HSP40 Sis1, which has known aggregation-prone client proteins, to gain insight into the role of the G/F domain and its significance in LGMD1D pathogenesis. Strikingly, we demonstrate that LGMD1D mutations in a Sis1-DNAJB6 chimera differentially impair the processing of specific conformers of two yeast prions, [RNQ+] and [PSI+]. Importantly, these differences do not simply correlate to the sensitivity of these prion strains to changes in chaperone levels. Additionally, we analyzed the effect of LGMD1D-associated DNAJB6 mutations on TDP-43, a protein known to form inclusions in LGMD1D. We show that the DNAJB6 G/F domain mutants disrupt the processing of nuclear TDP-43 stress granules in mammalian cells. These data suggest that the G/F domain mediates chaperone-substrate interactions in a manner that extends beyond recognition of a particular client and to a subset of client conformers. We propose that such selective chaperone disruption may lead to the accumulation of toxic aggregate conformers and result in the development of LGMD1D and perhaps other protein conformational disorders.
Introduction
Maintenance of cellular homeostasis requires that proteins adopt a proper tertiary structure. Both genetic and sporadic modifications can impair this process, leading to protein misfolding and the formation of protein aggregates that are present in many neuro- and myodegenerative diseases. Molecular chaperones, such as heat shock proteins (HSPs),3 help combat such aggregation, serving to refold substrates or target them for degradation. Consequently, dysfunction of this protective network of chaperones contributes to a variety of disorders.
Dominantly inherited mutations in the HSP40 chaperone DNAJB6 cause a progressive, late-onset degenerative myopathy called limb-girdle muscular dystrophy type 1D (LGMD1D) (1–4). DNAJB6 is expressed as two isoforms, DNAJB6a and DNAJB6b, with DNAJB6b suggested to be the principal mediator of LGMD1D pathogenesis (2). DNAJB6b has three distinct domains that are conserved in many HSP40 proteins present from yeast to humans. The N-terminal J domain and C-terminal substrate binding domain facilitate the interaction and processing of client proteins in cooperation with HSP70 (5). Between these domains is a poorly understood region called the G/F domain that is rich in glycine and phenylalanine residues. All known LGMD1D-associated mutations localize to an eight-amino acid stretch within the G/F domain. Disruption of the conserved G/F domain of DNAJB6 and similar DNAJ proteins can alter interactions with substrates, thereby highlighting the importance of the G/F domain (2, 6, 7). However, it is unclear how this domain modulates HSP40 function and how mutation of this region can cause disease.
Recently, the RNA-binding protein TDP-43 has emerged as a sensitive and specific marker of myodegeneration and has been shown to form inclusions in LGMD1D (1, 8). Interestingly, TDP-43 has a domain that is rich in glutamine and asparagine residues that is reminiscent of the prion-forming domains of several yeast prion proteins (9, 10). This domain facilitates aggregation of TDP-43 into stress granules during heat shock and can be functionally replaced with the Gln/Asn-rich prion-forming domain of the Rnq1 protein that forms the prion [RNQ+] in the budding yeast Saccharomyces cerevisiae (10, 11).
Yeast prions are self-propagating aggregate structures that template the conversion and recruitment of monomers, acting as epigenetic elements of inheritance to modulate cellular phenotypes (9). This same conformational change underlies the mechanistic basis of prion diseases in mammals and likely several other degenerative disorders (12). One striking feature of prion proteins lies in their innate ability to form a variety of amyloid structures called prion strains, each having a different aggregate conformation that differentially influences cellular and pathological phenotypes (13). This property is not specific to prion proteins, however. In fact, many amyloidogenic proteins exist in a multitude of distinct self-propagating amyloid conformations (14, 15) that likely contribute to the pathological variability observed in many amyloid disorders (16).
Genetic and environmental modifiers likely play a role in the generation and selection of different amyloid conformers, a characteristic we recently described with the [RNQ+] prion (17–20). Like the regulation of proteostasis in humans, molecular chaperones are intimately involved in the propagation of yeast prions. The HSP40 Sis1 binds prion aggregates and, in cooperation with Hsp70 and Hsp104, fragments these aggregates into smaller seeds that can be transmitted to daughter cells (21). Hence, such processing results in and is required for the continued maintenance of the prion state. Interestingly, the DNAJB6 G/F domain shares more homology with the G/F domain of Sis1 than any other yeast HSP40 (1). Moreover, the Sis1 G/F domain is involved in modulating protein aggregation (7). Yet, the role that this domain plays in substrate processing remains unclear.
Here we used the LGMD1D mutations in DNAJB6 with a combination of mammalian and yeast models to investigate how the G/F domain contributes to protein homeostasis. We found that these mutations impaired the chaperone-client interaction in a manner that showed striking dependence on not only the client but also the specific conformation of the substrate. Additionally, G/F domain mutations in another member of the HSP40 family, DNAJB1, resulted in loss of yeast viability, likely due to a loss-of-function in handling essential substrates. Collectively, our data suggest that the G/F domain of DNAJB6 plays a crucial role in recognizing particular client conformers. Such a loss of surveillance activity might lead to the accumulation of a toxic species that contributes to LGMD1D pathogenesis. In addition, the conformer-specific effects may be one factor that underlies the observed variation in disease progression, a hallmark of prion strains and the variability observed in prion diseases.
EXPERIMENTAL PROCEDURES
Yeast Strains and Media
All yeast strains used in this study are derivatives of 74-D694 (ade1-14 his3-Δ200 leu2-3,112 trp1-289 ura3-52). [RNQ+] and [PSI+] yeast strains were kind gifts from S. Liebman (22, 23) and J. Weissman (24). Standard culturing techniques were used. Rich media consisted of YPD (1% yeast extract, 2% peptone, 2% dextrose) or ¼ YPD (0.25% yeast extract, 2% peptone, 2% dextrose) as indicated. Synthetic defined media (0.67% yeast nitrogen base without amino acids, 2% dextrose) lacking the specified nutrients was used to select for plasmids. To make the sis1Δ yeast strains, [rnq−] [psi−] cells were transformed with pRS316-SIS1. SIS1 was then deleted by first amplifying the hphMX4 cassette from pAG32 using oligonucleotides 1415 and 1416 (see the oligonucleotides listed in Table 1), having flanking homology to the SIS1 promoter and terminator. The purified product was then transformed, and cells resistant to hygromycin B were selected. As sis1Δ cells must maintain pRS316-SIS1 because expression of SIS1 is essential, deletion of SIS1 was confirmed by the inability to grow on solid medium containing 1 mg/ml 5-fluoroorotic acid, which selects against cells containing URA3-marked plasmids. These cells were then mated to [PRION+] cells followed by sporulation of the diploids and selection of haploids that were resistant to hygromycin B. Replacement of wild-type (WT) Sis1 with the DNAJB1, SDSS, or Sis1 constructs was then performed by a plasmid shuffle technique using media containing 5-fluoroorotic acid to select against cells containing the URA3-marked SIS1 plasmid.
TABLE 1.
Oligonucleotides used in this study
| Oligonucleotide | Description | Sequence |
|---|---|---|
| 1454 | 5′-Sis1prom-SacI | 5′-CGCGAGCTCGCTAGTCGTTACTTTTC |
| 1455 | 3′-Sis1prom-SpeI | 5′-CGCACTAGTTATTAGTTCTGTATACTATAC |
| 1456 | 5′-Sis1term-ClaI | 5′-CGCATCGATTAGTAATCCTAAGCAAATATAA |
| 1457 | 3′-Sis1term-XhoI | 5′-CGCCTCGAGTTGGACTTACAAAAACTTC |
| 0229 | 5′-Sis1-SpeI | 5′-ATACTAGTATGGTCAAGGAGACAAAAC |
| 0230 | 3′-Sis1-ClaI | 5′-TGATCGATATTTGCTTAGGATTACTATTAAA |
| 1618 | 3′-SDSS | 5′-CTGTCAAAATGACTTCCACCTCCTCCTCCACCATTTAATCCAGGACCACCAGGACCAAAGCTTG |
| 1617 | 5′-SDSS | 5′-CCGTAACCCAGATGATGTCTTCAGGGAATTTTTTGGTGGAAGGGACCCATTCGGTGGTGCTGATGAC |
| 1619 | 5′-SDSS linker | 5′-GGTGGAAGTCATTTTGACAGTCCATTTGAATTTGGCTTCACATTCCGTAACCCAGATGATGTCTTC |
| 1567 | 5′-dnajB6-F89I | 5′-GACAGTCCATTTGAAATTGGCTTCACATTCCGTAAC |
| 1568 | 3′-dnajB6-F89I | 5′-GTTACGGAATGTGAAGCCAATTTCAAATGGACTGTC |
| 1620 | 5′-dnajB6-F93L | 5′-GAATTTGGCTTCACACTCCGTAACCCAGATGATG |
| 1621 | 3′-dnajB6-F93L | 5′-CATCATCTGGGTTACGGAGTGTGAAGCCAAATTC |
| 1569 | 5′-dnajB6-P96R | 5′-GGCTTCACATTCCGTAACCGAGATGATGTCTTCAGGG |
| 1570 | 3′-dnajB6-P96R | 5′-CCCTGAAGACATCATCTCGGTTACGGAATGTGAAGCC |
| 1415 | 5′-Sis1 KO | 5′-CATTCACATCAATATAATAGAGTATAGTATACAGAACTAATACAGCTGAAGCTTCGTACGC |
| 1416 | 3′-Sis1 KO | 5′-GTGATAAATTTATTTGAGTTTATAATTATATTTGCTTAGGATTACTAGCATAGGCCACTAGTGGATCTG |
| 1539 | 5′-dnajb1-F90I | 5′-GTGCCAATGGTACCTCTATCAGCTACACATTCCATG |
| 1540 | 3′-dnajb1-F90I | 5′-CATGGAATGTGTAGCTGATAGAGGTACCATTGGCAC |
| 1492 | 5′-dnajb1-F94L | 5′-CTCTTTCAGCTACACACTCCATGGAGACCCTCATG |
| 1493 | 3′-dnajb1-F94L | 5′-CATGAGGGTCTCCATGGAGTGTGTAGCTGAAAGAG |
| 1541 | 5′-dnajb1-P98R | 5′-CACATTCCATGGAGACCGTCATGCCATGTTTGCTG |
| 1542 | 3′ dnajb1-P98R | 5′-CAGCAAACATGGCATGACGGTCTCCATGGAATGTG |
Plasmid Construction
Plasmids pRS314-sis1-ΔG/F and pRS316CUP1-RNQ1(153–405)-GFP were kind gifts from S. Lindquist (7), and plasmids pTETr-SIS1, pRS316-SIS1, and pRS424GPD-DNAJB1 were kind gifts from E. Craig (25, 26). Oligonucleotides used to clone the yeast plasmids are listed in Table 1. Using pRS316-SIS1 as a template, the endogenous SIS1 promoter was amplified with oligonucleotides 1454 and 1455 and digested with SacI/SpeI, and the endogenous SIS1 terminator was amplified with 1456 and 1457 and digested with ClaI/XhoI. These products were ligated into pRS314 to make an intermediate plasmid used to clone the other constructs. To make pRS314-SIS1, SIS1 was amplified from pRS316-SIS1 with oligonucleotides 0229 and 0230 followed by SpeI/ClaI digestion and ligation. pRS314-SDSS was created by bridge PCR amplifying the N-terminal part with oligonucleotides 0229 and 1618. The C-terminal part of this construct was amplified in two parts using oligonucleotides 1617 and 0230 followed by using this PCR product as a template with oligonucleotides 1619 and 0230 to create the rest of the DNAJB6 G/F domain. The full-length SDSS construct was then amplified using the N-terminal and C-terminal products with oligonucleotides 0229 and 0230 followed by SpeI/ClaI digestion and ligation. Using pRS314-SDSS, the LGMD1D mutations were created by bridge PCR using the following oligonucleotides: 1567 and 1568 (F89I), 1620 and 1621 (F93L), 1569 and 1570 (P96R). The SDSS chimera consists of Sis1 amino acid residues 1–84 and 124–352 flanking the DNAJB6 G/F domain residues 72–108. DNAJB1 mutants were made by bridge PCR using the oligonucleotides 1539 and 1540 (F90I), 1492 and 1493 (F94L), and 1541 and 1542 (P98R) followed by digestion with SpeI/ClaI and ligation with pRS424GPD. Finally, mammalian constructs of DNAJB6b were cloned using site-directed mutagenesis, digested with HindIII/XhoI, and ligated into vector pcDNA3.1 containing a GFP tag. DNAJB6b-ΔG/F consisted of a deletion of amino acid residues 87–97.
Protein Analysis
Sedimentation analysis of Rnq1 was performed as described previously (27) and assessed by SDS-PAGE and Western blot using an αRnq1 antibody. The total and soluble fractions were quantified using ImageJ and used to calculate the percent soluble protein. Semi-denaturing agarose gel electrophoresis (SDD-AGE) for [PSI+] yeast cells was performed as described elsewhere (28) and analyzed by Western blot using an αSup35 antibody. For extraction and immunoblotting of mammalian cells, cells were washed twice with cold PBS, lysed in cold radioimmune precipitation assay buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 1% Nonidet-p40 (IGEPAL), 0.5% sodium deoxycholate, 0.1% SDS, 1 mm phenylmethylsulfonyl fluoride, and protease inhibitor mixture) for 30 min, and centrifuged at 10,000 rpm at 4 °C for 10 min. Equal amounts of total protein (10 μg/sample) were resolved by 10% SDS-PAGE and analyzed by Western blot according to standard procedures. Antibodies used included polyclonal rabbit αTDP-43 (Proteintech, 10782–2-AP, 1:1500), monoclonal mouse αDNAJB6 (Novus Biologicals, H00010049-M01, 1:500), monoclonal mouse αHSP70 (Enzo Life Sciences, ADI-SPA-810, 1:1000), polyclonal rabbit αGAPDH (Cell Signaling Technologies, 2118, 1:1000), and goat αMouse-HRP and goat αRabbit-HRP (Jackson ImmunoResearch Laboratories, 1:5000).
For the TDP-43 sedimentation assay, HeLa cells (5 × 104) were seeded into 6-well plates and incubated in DMEM containing 10% FBS and 1% penicillin/streptomycin Strep at 37 °C and 5% CO2. After an overnight incubation, the cells were co-transfected with Cherry-TDP-43-FL and either WT or mutant versions of DNAJB6b. After a post-transfection period (24 h), the cells were lysed in radioimmune precipitation assay buffer. The lysates were fractionated first with sonication (10 cycles of 30 sec on, 30 sec off) followed by centrifugation at 40,000 rpm for 30 min at 4 °C. The insoluble pellets were dissolved in urea buffer (7 m urea, 2 m thiourea, 4% CHAPS, and 30 mm Tris pH 8.5) and subjected to SDS-PAGE and Western blotting using a polyclonal αTDP-43 antibody. For unknown reasons, the TDP-43-mCherry construct migrates as a doublet between 50–75 kDa, as previously described (29, 30).
Prion Analysis by Fluorescence Microscopy
Imaging of live yeast cells was performed on a Zeiss Axiovert 200 Inverted Microscope equipped with a Zeiss 100×/1.4 NA oil objective. Overnight liquid cultures of cells harboring pRS316CUP1-RNQ1(153–405)-GFP were diluted to an A600 of 0.2. After 1 h of growth, 50 μm CuSO4 was added to cultures, and cells were imaged after ∼2.5 h of induction. Representative images of at least three independent experiments are shown.
Time-course of Prion Loss
Yeast sis1Δ cells expressing pTETr-SIS1 in place of pRS316-SIS1 were used to repress SIS1 as previously described (25). pTETr-SIS1 expresses SIS1 from a tetracycline-repressible promoter, allowing for down-regulation of SIS1 by growing cells in media containing doxycycline, a tetracycline analog. Cultures were maintained in log phase and grown in the presence of 5 μg/ml doxycycline. Samples were subjected to SDD-AGE and Western blot using an αRnq1 antibody following established protocols for the [RNQ+] prion (18), which does not reliably show Rnq1 monomer for unknown reasons. SDS-PAGE and Western blot were also performed using an αSis1 antibody (a gift from E. Craig) and quantified by ImageJ.
Yeast Phenotypic Assays
To monitor cell growth or the [PSI+] status of yeast cells, overnight cultures were normalized by A600, serially diluted 5-fold, and spotted on the indicated media. The [PSI+] status was assessed by colony color on ¼ YPD plates that were incubated for 3 days at 30 °C followed by overnight incubation at 4 °C for additional color development. Representative images of at least three independent experiments are shown.
Mammalian Cell Culture and Heat Shock
HeLa cells were cultured in high glucose formulation of Dulbecco's modified Eagle's medium (high glucose + glutamine, no sodium pyruvate) supplemented with 10% (v/v) fetal bovine serum and penicillin/streptomycin. Primary fibroblast cell lines were derived from patient skin biopsies and grown in FGMTM-2 Fibroblast Growth Medium-2 (Lonza CC-4126) that is developed to support the growth of most primary human fibroblasts.
For heat shock experiments, cells were transfected with Cherry-TDP-43 and either WT or mutant versions of the GFP-DNAJB6b constructs and maintained at 37 °C. 24 h after transfection cells were subjected to heat shock (42 °C, 1 h), then allowed to recover at 37 °C for the indicated times (0–3 h). Imaging of cells was performed as described previously (10).
RESULTS
Homologous LGMD1D Mutations in DNAJB1 Impair Cell Growth and Propagation of the [RNQ+] Prion
To examine the potential mechanism of G/F domain mutations on LGMD1D pathogenesis, we turned to the yeast system, which has provided significant mechanistic insight into many human diseases (31, 32). The yeast HSP40 Sis1 shares homology with DNAJB6 through the G/F domain (Fig. 1A). Although DNAJB6b could not functionally replace the essential role of Sis1 in yeast cells (data not shown), the closest human homolog to Sis1, the HSP40 DNAJB1, can complement for loss of Sis1 (26). In addition to yeast viability, DNAJB1 can complement for the required role that Sis1 plays in the maintenance of the yeast prion [RNQ+] (7, 26). This function in protein aggregate processing requires the Sis1 G/F domain and, in particular, a subdomain of the G/F domain not present in the yeast HSP40 Ydj1 that is comprised of the amino acid stretch containing all three LGMD1D mutant residues in DNAJB6 (26).
FIGURE 1.

Homologous LGMD1D mutations in DNAJB1 impair cell growth and [RNQ+] prion propagation. A, sequence comparison of the G/F domains of DNAJB6, DNAJB1, Sis1, and another yeast HSP40 Ydj1 aligned with ClustalW2. Boxes surround the disease-linked mutations. B, yeast sis1Δ cells propagating m.d. high [RNQ+] and expressing the indicated construct were serially diluted 5-fold and spotted onto rich media (YPD) or on media to select for loss (−Sis1) or co-expression (+Sis1) of WT Sis1. C, m.d. high [RNQ+] cells expressing WT Sis1, WT DNAJB1, DNAJB1-F94L, or DNAJB1-P98R in place of Sis1 were separated by ultracentrifugation into total (T), soluble (S), and insoluble (I) fractions that were then subjected to SDS-PAGE and Western blot using an αRnq1 antibody. Data are representative of n ≥ 3. D, the Rnq1 aggregation pattern in m.d. high [RNQ+] cells expressing the indicated construct and copper-inducible Rnq1-GFP. DIC, differential interference contrast.
Therefore, in taking advantage of the homology in DNAJB1, we created each of the LGMD1D mutations in DNAJB1 to test their effect on [RNQ+] propagation in sis1Δ yeast cells. Strikingly, we found that cells expressing DNAJB1-F90I (homologous to DNAJB6-F89I) were unable to grow without Sis1 expression, similar to the vector control (Fig. 1B). Moreover, cells expressing DNAJB1-P98R (homologous to DNAJB6-P96R) had a slow growth phenotype even when SIS1 was co-expressed. By contrast, cells expressing DNAJB1-F94L (homologous to DNAJB6-F93L) showed no defect in viability.
We then assessed the propagation of the [RNQ+] prion in cells expressing DNAJB1-F94L and DNAJB1-P98R in the absence of wild-type (WT) Sis1 using sedimentation analysis. The [RNQ+] state can be monitored by assessing the amount of soluble versus insoluble Rnq1 after fractionating cell lysates using high speed ultracentrifugation. As most all of the Rnq1 protein is sequestered into aggregates in [RNQ+] cells, Rnq1 accumulates in the pellet fraction (11). In cells expressing WT Sis1 or WT DNAJB1, we found that the [RNQ+] prion was maintained, as most of Rnq1 was in its aggregated, insoluble form (Fig. 1C), in agreement with previous work (26), with only 1.8 ± 1.1 and 1.6 ± 1.3% soluble protein, respectively. In stark contrast, much of the Rnq1 protein was soluble in lysates from cells expressing DNAJB1-F94L (54.4 ± 7.7% soluble protein) and DNAJB1-P98R (31.8 ± 5.2% soluble protein). As a complementary approach to examining [RNQ+] propagation, we used fluorescence microscopy to examine the in vivo aggregate distribution of Rnq1 using a Rnq1-GFP fusion protein. When WT Sis1 or WT DNAJB1 was expressed, we found that cells had many distinct puncta present (Fig. 1D), as previously observed (26). However, cells expressing DNAJB1-F94L exhibited more diffuse fluorescence and fewer puncta if any. Interestingly, expression of DNAJB1-P98R resulted in an abnormal localization pattern, with Rnq1-GFP being neither punctate nor diffuse. Taken together, these data indicate that DNAJB1-F94L and DNAJB1-P98R can functionally replace the essential roles of Sis1 to different extents but are defective in the propagation of [RNQ+].
DNAJB6 Mutations Differentially Impair Prion Propagation of [RNQ+] Strains
Because two of the LGMD1D mutations in DNAJB1 impaired cell growth, we created a chimeric protein to test the effect of all of the disease-linked mutations on [RNQ+] propagation. This chimera had the G/F domain of Sis1 replaced by that of DNAJB6, leaving the other functional domains of Sis1 intact (Fig. 2A). Hereafter, this chimera is referred to as SDSS, denoting the origins of each domain with “S” for Sis1 or “D” for DNAJB6.
FIGURE 2.
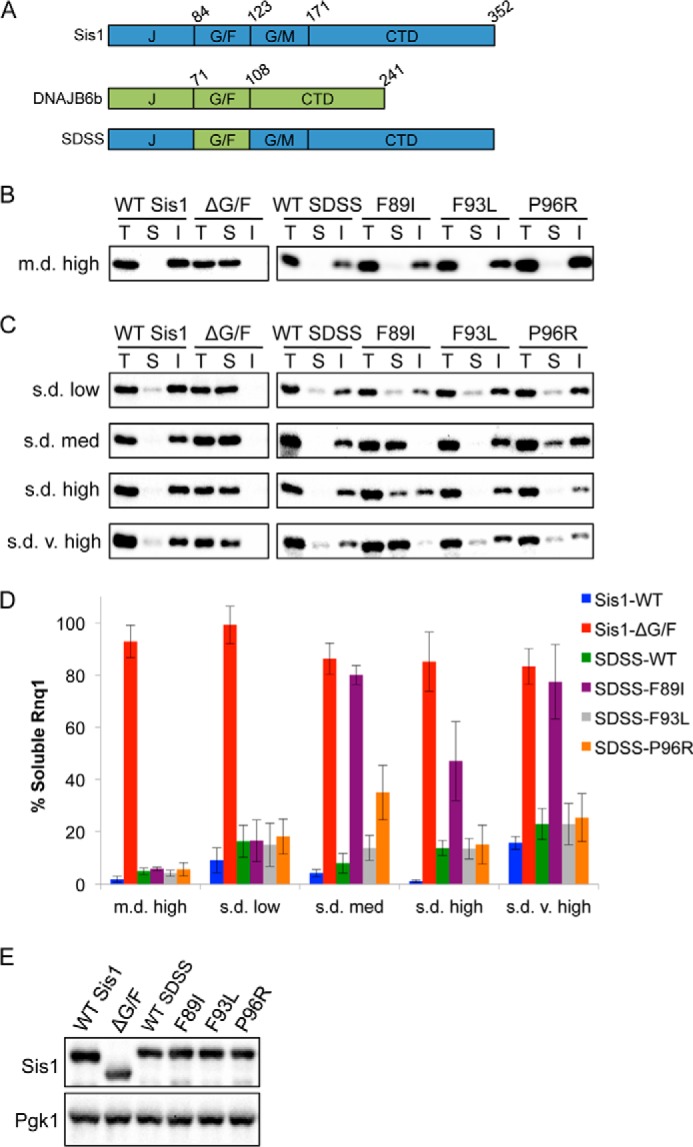
Maintenance of different [RNQ+] strains is differentially impaired by LGMD1D mutations in SDSS. A, domain structures of Sis1, DNAJB6b, and chimera SDSS. B and C, cells propagating m.d. high [RNQ+] (B) or one of the s.d. [RNQ+] strains (C) and expressing WT Sis1, Sis1-ΔG/F, or the indicated SDSS construct were lysed and fractionated as in Fig. 1C followed by SDS-PAGE and Western blot using an αRnq1 antibody. T, total fraction; S, soluble fraction; I, insoluble fraction. The percent of the total protein that was soluble was quantitated and is represented in D as the mean ± S.E. with n ≥ 3. E, Western blot analysis of Sis1 and SDSS mutants in yeast cells. [rnq−] cell lysates harboring the indicated construct were subjected to SDS-PAGE and Western blot with an αSis1 antibody along with an αPgk1 antibody as a loading control.
Using sis1Δ haploid yeast cells, we replaced Sis1 with WT SDSS or LGMD1D mutant SDSS constructs. As controls, we used cells expressing WT Sis1 or Sis1-ΔG/F, which has the G/F domain deleted and is unable to propagate [RNQ+] (7). We then monitored [RNQ+] propagation using sedimentation assays. In contrast to cells expressing WT Sis1, where most of Rnq1 was insoluble, expression of Sis1-ΔG/F resulted in a shift of Rnq1 to the soluble fraction, as expected from earlier studies (7) (Fig. 2B). This suggests that the presence of the G/F domain is required for Sis1 to recognize and maintain the self-propagating Rnq1 aggregate structure. Importantly, expression of WT SDSS phenocopied WT Sis1, indicating that this chimera can functionally replace WT Sis1 in maintaining the [RNQ+] prion. To our surprise, very little soluble Rnq1 was detected in lysates of cells expressing the LGMD1D mutant SDSS constructs. This suggests that the mutant chimera has no defect in the recognition and processing of the Rnq1 aggregates.
However, amyloidogenic proteins, including Rnq1, exist in a variety of different self-propagating structures (18, 19, 33, 34). Prion strains of [RNQ+] have been classified largely on two properties: their ability to induce the formation of the [PSI+] prion (low to very high rates) and the in vivo aggregation pattern observed by expressing Rnq1-GFP (22, 35). Cells propagating a single-dot (s.d.) [RNQ+] strain have predominantly one focus present, whereas multiple-dot (m.d.) [RNQ+] cells exhibit multiple foci. Thus, we hypothesized that the DNAJB6 G/F domain and the LGMD1D mutations might be differentially important in processing distinct aggregate structures. Although we initially analyzed the m.d. high [RNQ+] strain (Fig. 1 and Fig. 2B), we extended our analysis to examine the propagation of four different [RNQ+] strains (Fig. 2, C and D). First, we found that expression of WT SDSS in [RNQ+] cells resulted in the presence of most of the Rnq1 protein in the insoluble fraction (s.d. low and s.d. very high [RNQ+] contain a small pool of soluble Rnq1 that is larger than that of other [RNQ+] strains (22)). Deletion of the G/F domain resulted in accumulation of Rnq1 in the soluble fraction for all [RNQ+] strains, indicating that the G/F domain is universally important for the maintenance of different Rnq1 aggregate structures. Remarkably, however, we found that the LGMD1D mutations differentially impaired the propagation of the [RNQ+] strains despite being expressed at similar levels (Fig. 2E). Like m.d. high [RNQ+], the amount of soluble Rnq1 did not change in s.d. low [RNQ+] cells expressing any of the mutants. In contrast, in s.d. medium [RNQ+] cells, SDSS-F89I phenocopied the deletion of the entire G/F domain. We also observed an accumulation of soluble Rnq1 in s.d. medium [RNQ+] cells expressing SDSS-P96R. Moreover, the F89I mutation eliminated the majority of Rnq1 aggregates in s.d. very high [RNQ+] cells and increased the amount of soluble Rnq1 in s.d. high [RNQ+] cells. However, the P96R mutation did not affect s.d. high or s.d. very high [RNQ+]. These data suggest that LGMD1D mutations impair the role of the G/F domain in processing particular aggregate conformations of Rnq1.
We next examined the effect of the LGMD1D mutations on the prion strain-specific distribution of Rnq1 aggregates using fluorescence microscopy with cells harboring WT or mutant SDSS and propagating either s.d. medium or m.d. high [RNQ+]. We found that expression of SDSS-F89I in s.d. medium [RNQ+] cells caused all of Rnq1-GFP to be diffuse, similar to [rnq−] cells harboring WT SDSS (Fig. 3, A and B). By contrast, the distribution of puncta was unchanged in mutant cells propagating m.d. high [RNQ+] (Fig. 3C), thereby confirming our solubility assay results.
FIGURE 3.

LGMD1D mutations modulate the aggregation pattern of Rnq1 in vivo. The Rnq1 aggregation pattern in [rnq−] cells (A), s.d. medium [RNQ+] cells (B), or m.d. high [RNQ+] cells (C) harboring the indicated SDSS construct and copper-inducible Rnq1-GFP. DIC, differential interference contrast.
[RNQ+] Strains Rely on Sis1 Expression to Different Degrees
As chaperone-mediated substrate processing is an intricate process, changes in chaperone expression have been shown to differentially affect strains of another yeast prion called [PSI+] (36, 37). As such, we hypothesized that the differential effects of the LGMD1D mutations may simply be due to [RNQ+] prion strains having different sensitivities to changes in Sis1 expression. To test this, we down-regulated SIS1 and monitored the rate at which the Rnq1 aggregates were lost using SDD-AGE, a technique that resolves aggregates. We first analyzed the two [RNQ+] strains that had distinctly different sensitivity to the LGMD1D mutations: m.d. high [RNQ+], which was unaffected by the LGMD1D mutations in SDSS, and s.d. medium [RNQ+], which was the most affected by the LGMD1D mutations. Interestingly, we found that m.d. high [RNQ+] cells had a dramatic loss of Rnq1 aggregates by the fifth generation, whereas the loss of Rnq1 aggregates was delayed in s.d. medium [RNQ+] cells (Fig. 4A). Importantly, steady state levels of Sis1 were similarly repressed to 2% of WT by the seventh generation (Fig. 4B). Moreover, the sensitivity to Sis1 expression of the other s.d. [RNQ+] strains, which were variably affected by the LGMD1D mutations, was similar to s.d. medium [RNQ+] (Fig. 4C). These data indicate that the effect of the LGMD1D mutations on the propagation of the [RNQ+] strains does not correlate to the sensitivity of these prion strains to Sis1 expression.
FIGURE 4.

Effect of LGMD1D mutations does not correlate to the sensitivity of [RNQ+] strains to Sis1 expression. A, cells propagating m.d. high or s.d. medium [RNQ+] had endogenous SIS1 replaced by pTETr-SIS1. Cells grown in the presence of doxycycline (+Dox) had samples taken at the indicated number of generations (Gen #) followed by SDD-AGE and Western blot using an αRnq1 antibody. Data are representative of n = 3. B, Western blot analysis of A with an αSis1 antibody. C, as in A for the indicated [RNQ+] strains.
DNAJB6 Mutations Differentially Impair Propagation of [PSI+] Strains
We then asked if the LGMD1D mutations affected another known Sis1 substrate, the [PSI+] prion, which is formed by the translation termination factor Sup35 (23, 38). The presence of [PSI+] modulates translation termination by sequestering Sup35 into aggregates. [PSI+] is easily monitored phenotypically using auxotrophic markers that have a premature termination codon, such as the ade1-14 allele (9). [psi−] cells have soluble Sup35 that faithfully terminates translation at the premature termination codon. Thus, these cells cannot complete the adenine biosynthetic pathway and accumulate a metabolic intermediate that gives [psi−] colonies a red color. In contrast, [PSI+] causes read-through of the premature termination codon (called nonsense suppression), thereby preventing accumulation of the red pigment. Interestingly, different prion strains of [PSI+] are linked to different levels of nonsense suppression: cells propagating stronger [PSI+] strains have more nonsense suppression and are lighter pink as compared with cells propagating weaker [PSI+] strains that are darker pink (23).
To test how the G/F domain and the LGMD1D mutations affect [PSI+] propagation, we used sis1Δ cells propagating four different [PSI+] strains: two weaker [PSI+] strains (Sc37 and weak [PSI+]) and two stronger [PSI+] strains (Sc4 and strong [PSI+]) (23, 24). We first noted that all [PSI+] strains propagated to some degree when Sis1-ΔG/F was expressed, as indicated by a pink or white colony color that was distinct from [psi−] cells (Fig. 5A). As all [RNQ+] strains were eliminated by expression of Sis1-ΔG/F (Fig. 2), this suggests that Sup35 contrasts with Rnq1 as a Sis1 substrate and thereby provides a good means of expanding our analysis. However, unlike previous reports (38), propagation of both weaker [PSI+] strains was impaired, as indicated by cells having a darker pink color on rich media. Propagation of Sc4 and strong [PSI+] was not affected by expression of Sis1-ΔG/F. This indicates that the G/F domain of Sis1 plays a more significant role in propagation of weaker [PSI+] strains as compared with stronger [PSI+] strains, but [RNQ+] has the greatest dependence on this domain.
FIGURE 5.

LGMD1D mutations in SDSS differentially impair propagation of [PSI+] strains. A, equivalent numbers of sis1Δ cells expressing WT Sis1 or Sis1-ΔG/F with the indicated [PSI+] status were serially diluted 5-fold and spotted onto rich media to monitor the [PSI+] phenotype by color. B, as in A with the indicated SDSS construct. Dotted lines represent different parts of the same plate cropped for clarity. C, SDD-AGE analysis of cell lysates expressing the indicated SDSS construct. Data are representative of n = 4.
Next, we analyzed the effect of the LGMD1D mutations in SDSS on [PSI+] propagation. None of the mutations affected the stronger [PSI+] strains (Fig. 5B). However, like the deletion of the G/F domain, the F89I mutation impaired the propagation of Sc37 and weak [PSI+]. Furthermore, as prion strains have different biochemical properties, we hypothesized that the LGMD1D mutations might affect these properties without necessarily affecting the [PSI+] phenotype. Previously, it was shown that one of the major distinguishing features of [PSI+] strains is that weaker [PSI+] strains typically consist of larger Sup35 aggregates as compared with stronger [PSI+] strains and have more soluble Sup35 (39, 40). Therefore, to gain additional insight into how the LGMD1D mutations affect prion propagation, we analyzed aggregate size and soluble Sup35 using SDD-AGE. In agreement with our phenotypic analysis, we found that for Sc37 and weak [PSI+], expression of either SDSS-F89I or Sis1-ΔG/F resulted in a reduction of aggregated Sup35 as well as an increased monomer pool (Fig. 5C and data not shown). Moreover, although expression of SDSS-P96R did not result in any detectable phenotypic change in cells propagating these weaker [PSI+] strains, lysates showed increased soluble Sup35. By contrast, the Sup35 aggregate size and amount of soluble Sup35 of cells propagating Sc4 and strong [PSI+] was not affected by expression of the LGMD1D mutations in SDSS. Therefore, these data show that the G/F domain and the LGMD1D mutants differentially affect propagation of distinct [PSI+] prion strains.
LGMD1D Mutations in DNAJB6 Alter TDP-43 Nuclear Aggregation and Disaggregation
To examine the G/F domain mutations in a mammalian model of LGMD1D, we used TDP-43 as a pathological correlate, as it is known to aggregate in LGMD1D (1). Moreover, TDP-43 contains a prion-like domain that mediates its aggregation (41). In fact, the prion-forming domain of Rnq1 can substitute for the TDP-43 prion-like domain to facilitate aggregation (10), thereby further connecting our yeast studies to a mammalian substrate. To test whether mutations in DNAJB6 affected TDP-43 aggregation, we heat-shocked HeLa cells expressing TDP-43-mCherry and WT or LGMD1D mutant DNAJB6b-GFP, which leads to the formation of nuclear stress bodies of TDP-43 (10). During heat shock and the following recovery, we monitored the presence and co-localization of TDP-43 nuclear stress bodies with DNAJB6b. Interestingly, we found that WT DNAJB6b and all mutant constructs similarly translocated to the nucleus during heat shock and co-localized with TDP-43 nuclear stress bodies (Fig. 6A and data not shown). During recovery, we found that WT DNAJB6b enhanced the dissolution of these stress bodies as compared with GFP controls (Fig. 6, B and C). However, despite no defect in localization to TDP-43 aggregates or differences in expression (Fig. 6D), every DNAJB6b mutant enhanced the formation and persistence of TDP-43 stress bodies (Fig. 6, B and C).
FIGURE 6.

LGMD1D mutations in DNAJB6 alter TDP-43 nuclear aggregation and disaggregation. A, nuclear co-localization of DNAJB6b and TDP-43 upon heat shock. HeLa cells were co-transfected with TDP-43-mCherry and DNAJB6b-GFP (WT or F93L) and subjected to heat shock at 42 °C for 1 h followed by recovery for 2 h at 37 °C. B and C, as in A, TDP-43 nuclear stress body formation (B) and quantitation (C) in HeLa cells transfected with the indicated DNAJB6b construct. Data are represented as the mean ± S.E. with n = 4. D, Western blot analysis of HeLa cell extracts transfected with the indicated constructs as compared with untransfected cells (Control) using antibodies against TDP-43, DNAJB6b, and HSP70 along with GAPDH as a loading control. All samples were run on the same gel but have been cropped for clarity. E, HeLa cells expressing the indicated construct were treated as in A, lysed, and separated into total (T), soluble (S), and insoluble (I) fractions that were then subjected to SDS-PAGE and Western blot using an αTDP-43 antibody.
In addition, we monitored the amount of insoluble TDP-43 using differential detergent extraction and centrifugation. With expression of WT DNAJB6b or our GFP control, we found that TDP-43 entered an insoluble fraction upon heat shock and became soluble upon recovery (Fig. 6E). However, consistent with our immunofluorescence data, when LGMD1D mutants or DNAJB6b-ΔG/F were expressed, more TDP-43 was present in the insoluble fraction under normal conditions and upon heat shock and persisted during the recovery period (Fig. 6E).
We then performed a similar analysis without DNAJB6 overexpression using fibroblasts derived from three independent LGMD1D patients harboring the DNAJB6-F93L mutation, as compared with disease control fibroblasts from three independent patients having sporadic amyotrophic lateral sclerosis. Again, upon heat shock and during recovery, we found that the mutant fibroblasts showed enhanced formation and delayed dissolution of TDP-43 nuclear stress bodies (Fig. 7, A and B). Importantly, these cells expressed DNAJB6b at similar levels (Fig. 7C). This same defect in substrate processing was also seen with endogenous TDP-43 (data not shown). These data indicate that LGMD1D mutations abrogate the ability of DNAJB6b to resolve nuclear TDP-43 stress granules, resulting in the persistence of TDP-43 aggregates.
FIGURE 7.

Dissolution of TDP-43 stress granules is disrupted in patient fibroblasts. A and B, TDP-43 nuclear stress body formation (A) and quantitation (B) in LGMD1D patient (F93L) or disease control fibroblasts. Cells were transfected with TDP-43-mCherry and treated as in Fig. 6A with a 3-h recovery. Data are represented as the mean ± S.E. with n ≥ 4. C, Western blot analysis of patient fibroblasts from three independent disease control patients (C1, C2, C3) having sporadic amyotrophic lateral sclerosis and three different patients from the same family having the DNAJB6-F93L mutation (P1, P2, P3) using antibodies against DNAJB6b and GAPDH. M, marker.
DISCUSSION
Taken together, our study demonstrates that the LGMD1D mutations in the G/F domain of DNAJB6 disrupt client processing in both a substrate- and conformation-specific manner. This defines a novel disease pathomechanism in a protein aggregate disorder, as it is the first time to our knowledge that mutations associated with a chaperonopathy have client conformer-specific effects. Specifically, when placed in the context of Sis1, the DNAJB6 G/F domain can efficiently recognize and process multiple protein aggregate conformers of the same protein (e.g. Rnq1 and Sup35). However, a single disease point mutation in this previously underappreciated region of DNAJ proteins abrogates processing of distinct protein aggregate conformers.
One potential mammalian aggregation-prone protein with specificity to protein aggregate myopathies is TDP-43. We previously established that TDP-43 accumulates in LGMD1D patient muscle and that TDP-43 is a potential DNAJB6 client protein (1, 10). Persistence of TDP-43 aggregates in DNAJB6-expressing cells and patient fibroblasts further demonstrates a DNAJB6-TDP-43 client interaction. In the context of a human protein aggregate disorder, our data now emphasize the expanded and crucial role that the G/F domain plays in protein aggregation by acting as the major regulator of substrate and conformer selectivity. This agrees with previous data with the bacterial DnaJ protein (6). Two possibilities might explain the selectivity: either the G/F domain directly binds substrates or it modulates the neighboring J domain and the interaction of this domain with Hsp70. Future studies will be necessary to distinguish between these two scenarios. In either case, we suggest that LGMD1D mutations in DNAJB6 do not abolish all of its chaperone function but instead confer impaired chaperone surveillance in human skeletal muscle of distinct protein aggregate conformers of TDP-43 or some other important client. Selectively disrupting substrate processing might then lead to the accumulation of these particular aggregate conformers followed by toxicity and ultimately muscle degeneration (Fig. 8).
FIGURE 8.

Schematic model of how mutations in DNAJB6 may contribute to LGMD1D pathogenesis. In skeletal muscle, various insults may lead to the misfolding of a substrate into a range of distinct aggregate conformers. A, WT DNAJB6 is capable of properly processing these misfolded conformers, which might include refolding the substrate into its native conformation. B, in the case of LGMD1D, mutations in DNAJB6 impair the processing of particular conformers. This leads to the accumulation of this specific aggregate conformer and ultimately to cellular toxicity and myocyte degeneration.
DNAJB6 has previously been implicated in modulating the turnover of a number of different proteins (10, 42–44). Interestingly, many proteins can populate a variety of different aggregated structures or different folding intermediates (13, 45). Hence, the chaperone network is charged with the task of handling a large spectrum of substrate conformations to maintain cellular homeostasis. Moreover, although there are some common pathogenic mechanisms involved in protein misfolding disorders, there is also tremendous tissue specificity. DNAJB6 is one of the more abundant HSP40s in skeletal muscle, yet it is ubiquitously expressed and is more abundant in other tissues (46). Therefore, it is intriguing to speculate that certain tissue-specific protein conformers, whether aggregated or simply unfolded, are more refractory to dissolution and cannot be resolved by DNAJB6 G/F domain mutants.
By analyzing the LGMD1D mutations in a yeast model, we found striking differences between these mutations on phenotypic consequences. Expression of the homologous mutation of F89I showed the greatest effect in terms of cell viability using DNAJB1 and propagation of [RNQ+] and [PSI+] using SDSS. Indeed, previous work in zebrafish similarly showed that the F89I mutation caused more severe muscle degeneration than the F93L mutation (2). This is also consistent with human LGMD1D; although the disease is typically late onset, patients with the F89I mutation have been reported to present with childhood onset weakness (47). Curiously, we found that the homologous F89I mutation in DNAJB1 was inviable in yeast, but its effect was not dominant. By contrast, the homologous P96R mutation in DNAJB1 maintained yeast viability but was dominantly toxic. This surprising difference suggests that the effect of these mutations is distinct, with the homologous F89I mutation possibly abolishing all chaperone activity and preventing polymerization of HSP40, whereas the homologous P96R mutation might influence the chaperone activity in the HSP40 polymeric state. In addition, it is important to note that even though there was no effect of SDSS-F93L on propagation of the m.d. high [RNQ+] strain, the homologous F94L mutation present in DNAJB1 impaired propagation. Therefore, we propose that different mutations may be associated with variation in disease severity by impairing HSP40 function to different degrees.
Mutations in other DNAJ proteins have also been shown to cause disease, and the involvement of these proteins has been implicated in many other disorders (48). As this family of chaperones has more than 40 members in humans, there likely is a high level of specificity in cellular functions (5). A variety of factors can influence this functional specificity. Here, we show that the HSP40 G/F domain plays a major role in substrate conformer selectivity. Hence, when examining the mechanistic basis of chaperonopathies, our data suggest that substrate conformation is another important variable that may contribute to disease progression.
Acknowledgments
We thank R. Stewart and R. Wilkinson for microscopy assistance. For reagents, we are grateful to S. Liebman, S. Lindquist, E. Craig, and J. Weissman. We thank A. Cashikar, D. Summers, and L. Westergard for helpful comments on the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grants F31AG040899 and T32GM007067 (to K. C. S.), GM072778 (to H. L. T.), and AG031867 and AG042095 (to C. C. W.). This work was also supported by the Muscular Dystrophy Association (to C. C. W.) and the Hope Center for Neurological Disorders (to C. C. W., M. B. H., and H. L. T.).
- HSP
- heat shock protein
- LGMD1D
- limb-girdle muscular dystrophy type 1D
- m.d.
- multiple-dot
- s.d.
- single-dot
- SDD-AGE
- semi-denaturing detergent agarose gel electrophoresis.
REFERENCES
- 1. Harms M. B., Sommerville R. B., Allred P., Bell S., Ma D., Cooper P., Lopate G., Pestronk A., Weihl C. C., Baloh R. H. (2012) Exome sequencing reveals DNAJB6 mutations in dominantly inherited myopathy. Ann. Neurol. 71, 407–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sarparanta J., Jonson P. H., Golzio C., Sandell S., Luque H., Screen M., McDonald K., Stajich J. M., Mahjneh I., Vihola A., Raheem O., Penttilä S., Lehtinen S., Huovinen S., Palmio J., Tasca G., Ricci E., Hackman P., Hauser M., Katsanis N., Udd B. (2012) Mutations affecting the cytoplasmic functions of the co-chaperone DNAJB6 cause limb-girdle muscular dystrophy. Nat. Genet. 44, 450–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sato T., Hayashi Y. K., Oya Y., Kondo T., Sugie K., Kaneda D., Houzen H., Yabe I., Sasaki H., Noguchi S., Nonaka I., Osawa M., Nishino I. (2013) DNAJB6 myopathy in an Asian cohort and cytoplasmic/nuclear inclusions. Neuromuscul. Disord. 23, 269–276 [DOI] [PubMed] [Google Scholar]
- 4. Couthouis J., Raphael A. R., Siskind C., Findlay A. R., Buenrostro J. D., Greenleaf W. J., Vogel H., Day J. W., Flanigan K. M., Gitler A. D. (2014) Exome sequencing identifies a DNAJB6 mutation in a family with dominantly inherited limb-girdle muscular dystrophy. Neuromuscul. Disord. 24, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kampinga H. H., Craig E. A. (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat. Rev. Mol. Cell Biol. 11, 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Perales-Calvo J., Muga A., Moro F. (2010) Role of DnaJ G/F-rich domain in conformational recognition and binding of protein substrates. J. Biol. Chem. 285, 34231–34239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sondheimer N., Lopez N., Craig E. A., Lindquist S. (2001) The role of Sis1 in the maintenance of the [RNQ+] prion. EMBO J. 20, 2435–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Udan M., Baloh R. H. (2011) Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 5, 1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liebman S. W., Chernoff Y. O. (2012) Prions in yeast. Genetics 191, 1041–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Udan-Johns M., Bengoechea R., Bell S., Shao J., Diamond M. I., True H. L., Weihl C. C., Baloh R. H. (2014) Prion-like nuclear aggregation of TDP-43 during heat shock is regulated by HSP40/70 chaperones. Hum. Mol. Genet. 23, 157–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sondheimer N., Lindquist S. (2000) Rnq1: an epigenetic modifier of protein function in yeast. Mol. Cell 5, 163–172 [DOI] [PubMed] [Google Scholar]
- 12. Chiti F., Dobson C. M. (2006) Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75, 333–366 [DOI] [PubMed] [Google Scholar]
- 13. Toyama B. H., Weissman J. S. (2011) Amyloid structure: conformational diversity and consequences. Annu. Rev. Biochem. 80, 557–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sawaya M. R., Sambashivan S., Nelson R., Ivanova M. I., Sievers S. A., Apostol M. I., Thompson M. J., Balbirnie M., Wiltzius J. J., McFarlane H. T., Madsen A. Ø., Riekel C., Eisenberg D. (2007) Atomic structures of amyloid cross-β spines reveal varied steric zippers. Nature 447, 453–457 [DOI] [PubMed] [Google Scholar]
- 15. Wiltzius J. J., Landau M., Nelson R., Sawaya M. R., Apostol M. I., Goldschmidt L., Soriaga A. B., Cascio D., Rajashankar K., Eisenberg D. (2009) Molecular mechanisms for protein-encoded inheritance. Nat. Struct. Mol. Biol. 16, 973–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kikis E. A., Gidalevitz T., Morimoto R. I. (2010) Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 694, 138–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stein K. C., True H. L. (2014) Extensive diversity of prion strains is defined by differential chaperone interactions and distinct amyloidogenic regions. PLoS Genet. 10, e1004337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Huang V. J., Stein K. C., True H. L. (2013) Spontaneous variants of the [RNQ+] prion in yeast demonstrate the extensive conformational diversity possible with prion proteins. PLoS ONE 8, e79582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Westergard L., True H. L. (2014) Wild yeast harbour a variety of distinct amyloid structures with strong prion-inducing capabilities. Mol. Microbiol. 92, 183–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Westergard L., True H. L. (2014) Extracellular environment modulates the formation and propagation of particular amyloid structures. Mol. Microbiol. 92, 698–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. True H. L. (2006) The battle of the fold: chaperones take on prions. Trends Genet. 22, 110–117 [DOI] [PubMed] [Google Scholar]
- 22. Bradley M. E., Edskes H. K., Hong J. Y., Wickner R. B., Liebman S. W. (2002) Interactions among prions and prion “strains” in yeast. Proc. Natl. Acad. Sci. U.S.A. 99, 16392–16399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Derkatch I. L., Chernoff Y. O., Kushnirov V. V., Inge-Vechtomov S. G., Liebman S. W. (1996) Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics 144, 1375–1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tanaka M., Collins S. R., Toyama B. H., Weissman J. S. (2006) The physical basis of how prion conformations determine strain phenotypes. Nature 442, 585–589 [DOI] [PubMed] [Google Scholar]
- 25. Aron R., Higurashi T., Sahi C., Craig E. A. (2007) J-protein co-chaperone Sis1 required for generation of [RNQ+] seeds necessary for prion propagation. EMBO J. 26, 3794–3803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lopez N., Aron R., Craig E. A. (2003) Specificity of class II Hsp40 Sis1 in maintenance of yeast prion [RNQ+]. Mol. Biol. Cell 14, 1172–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bardill J. P., Dulle J. E., Fisher J. R., True H. L. (2009) Requirements of Hsp104p activity and Sis1p binding for propagation of the [RNQ(+)] prion. Prion 3, 151–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dulle J. E., Stein K. C., True H. L. (2014) Regulation of the Hsp104 middle domain activity is critical for yeast prion propagation. PLoS ONE 9, e87521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ju J.-S., Fuentealba R. A., Miller S. E., Jackson E., Piwnica-Worms D., Baloh R. H., Weihl C. C. (2009) Valosin-containing protein (VCP) is required for autophagy and is disrupted in VCP disease. J. Cell Biol. 187, 875–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Walker A. K., Soo K. Y., Sundaramoorthy V., Parakh S., Ma Y., Farg M. A., Wallace R. H., Crouch P. J., Turner B. J., Horne M. K., Atkin J. D. (2013) ALS-associated TDP-43 induces endoplasmic reticulum stress, which drives cytoplasmic TDP-43 accumulation and stress granule formation. PLoS ONE 8, e81170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tenreiro S., Munder M. C., Alberti S., Outeiro T. F. (2013) Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 127, 438–452 [DOI] [PubMed] [Google Scholar]
- 32. Pereira C., Coutinho I., Soares J., Bessa C., Leão M., Saraiva L. (2012) New insights into cancer-related proteins provided by the yeast model. FEBS J. 279, 697–712 [DOI] [PubMed] [Google Scholar]
- 33. Eisenberg D., Jucker M. (2012) The amyloid state of proteins in human diseases. Cell 148, 1188–1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stein K. C., True H. L. (2011) The [RNQ+] prion: a model of both functional and pathological amyloid. Prion 5, 291–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bradley M. E., Liebman S. W. (2003) Destabilizing interactions among [PSI(+)] and [PIN(+)] yeast prion variants. Genetics 165, 1675–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hines J. K., Higurashi T., Srinivasan M., Craig E. A. (2011) Influence of prion variant and yeast strain variation on prion-molecular chaperone requirements. Prion 5, 238–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chernoff Y. O., Lindquist S. L., Ono B., Inge-Vechtomov S. G., Liebman S. W. (1995) Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science 268, 880–884 [DOI] [PubMed] [Google Scholar]
- 38. Higurashi T., Hines J. K., Sahi C., Aron R., Craig E. A. (2008) Specificity of the J-protein Sis1 in the propagation of 3 yeast prions. Proc. Natl. Acad. Sci. U.S.A. 105, 16596–16601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kryndushkin D. S., Alexandrov I. M., Ter-Avanesyan M. D., Kushnirov V. V. (2003) Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643 [DOI] [PubMed] [Google Scholar]
- 40. Uptain S. M., Sawicki G. J., Caughey B., Lindquist S. (2001) Strains of [PSI(+)] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J. 20, 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fuentealba R. A., Udan M., Bell S., Wegorzewska I., Shao J., Diamond M. I., Weihl C. C., Baloh R. H. (2010) Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 285, 26304–26314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Durrenberger P. F., Filiou M. D., Moran L. B., Michael G. J., Novoselov S., Cheetham M. E., Clark P., Pearce R. K., Graeber M. B. (2009) DnaJB6 is present in the core of Lewy bodies and is highly up-regulated in parkinsonian astrocytes. J. Neurosci. Res. 87, 238–245 [DOI] [PubMed] [Google Scholar]
- 43. Hageman J., Rujano M. A., van Waarde M. A., Kakkar V., Dirks R. P., Govorukhina N., Oosterveld-Hut H. M., Lubsen N. H., Kampinga H. H. (2010) A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol. Cell 37, 355–369 [DOI] [PubMed] [Google Scholar]
- 44. Watson E. D., Geary-Joo C., Hughes M., Cross J. C. (2007) The Mrj co-chaperone mediates keratin turnover and prevents the formation of toxic inclusion bodies in trophoblast cells of the placenta. Development 134, 1809–1817 [DOI] [PubMed] [Google Scholar]
- 45. Jahn T. R., Radford S. E. (2008) Folding versus aggregation: polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 469, 100–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hageman J., Kampinga H. H. (2009) Computational analysis of the human HSPH/HSPA/DNAJ family and cloning of a human HSPH/HSPA/DNAJ expression library. Cell Stress Chaperones 14, 1–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Suarez-Cedeno G., Winder T., Milone M. (2014) DNAJB6 myopathy: a vacuolar myopathy with childhood onset. Muscle Nerve 49, 607–610 [DOI] [PubMed] [Google Scholar]
- 48. Kakkar V., Prins L. C., Kampinga H. H. (2012) DNAJ proteins and protein aggregation diseases. Curr. Top. Med. Chem. 12, 2479–2490 [DOI] [PubMed] [Google Scholar]