

Background: HEXIM1 and LaRP7 bind to 7SK snRNA.
Results: HEXIM1 and LaRP7 activation domain chimeras activated plasmid targets via defined 7SK snRNA motifs in cells.
Conclusion: Specific RNA targets of HEXIM1 and LaRP7 and inhibition of P-TEFb were dissected genetically in vivo.
Significance: This system facilitates studies of 7SK snRNP in cells.
Keywords: Gene Expression, RNA Binding Protein, RNA Folding, RNA-Protein Interaction, Transcription Elongation Factor
Abstract
The positive transcription elongation factor b (P-TEFb), comprised of cyclin-dependent kinase 9 (CDK9) and cyclins T1 (CycT1) or T2 (CycT2), activates eukaryotic transcription elongation. In growing cells, P-TEFb exists in active and inactive forms. In the latter, it is incorporated into the 7SK small nuclear ribonucleoprotein, which contains hexamethylene bisacetamide-induced proteins (HEXIM) 1 or 2, La-related protein 7 (LaRP7), methyl phosphate capping enzyme, and 7SK small nuclear RNA (7SK). HEXIM1 inhibits the kinase activity of CDK9 via interactions between 7SK, HEXIM1, and CycT1. LaRP7 and methyl phosphate capping enzyme interact with 7SK independently of HEXIM1 and P-TEFb. To analyze genetic interactions between HEXIM1 and/or LaRP7 and 7SK using a cell-based system, we established artificial heterologous RNA tethering assays in which reporter gene expression depended on interactions between selected regions of 7SK and its cognate binding partners fused to a strong activator. This system enabled us to map the HEXIM1- and LaRP7- binding regions of 7SK. Assays with various mutant 7SK plasmid targets revealed that the 5′ U-U bulge and central loop of stem-loop I or RNA motif 3 of 7SK are required for transactivation, suggesting that HEXIM1 and CycT1 form a combinatorial binding surface for 7SK. Moreover, a region in HEXIM1 C-terminal to its previously mapped RNA-binding motif was also required for interactions between HEXIM1 and 7SK. Finally, a tyrosine-to-alanine mutation in HEXIM1, which is critical for its inhibitory effect on CDK9, changed HEXIM1 into an activator. These cell-based assays elucidate this important aspect of transcription elongation in vivo.
Introduction
Eukaryotic transcription by RNA polymerase II (RNAPII)2 is regulated at multiple steps, including initiation, promoter clearance, elongation, and cotranscriptional processing of nascent transcripts (1). A recent genome-wide analysis revealed that elongation is a critical step of transcription (2–4). P-TEFb, which contains CycT1, CycT2 (collectively, CycT), and CDK9, plays a major role in this process. P-TEFb phosphorylates serines at position 2 (Ser-2) in the C-terminal domain of RNAPII as well as those in the 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole sensitivity-inducing factor and the negative elongation factor (5). In cells, P-TEFb exists in two major forms (6). The catalytically active P-TEFb binds bromodomain-containing protein 4 (BRD4), subunits of the superelongation complex, or other DNA- or RNA-bound activators (7–10). In contrast, P-TEFb in the 7SK small nuclear ribonucleoprotein (snRNP) complex is inactive. It contains 7SK, HEXIM1 or HEXIM2, LaRP7, and methyl phosphate capping enzyme (11). In the 7SK snRNP, HEXIM proteins inhibit the kinase activity of CDK9. This P-TEFb equilibrium determines the state of cellular activation, proliferation, and differentiation. Many stresses, such as UV light, heat, inhibition of transcription by Actinomycin D, 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole or flavopiridol, histone deacetylase inhibitors such as trichostatin A suberoylanilide hydroxamic acid, and specific intracellular signaling cascades, release P-TEFb from 7SK snRNP (3, 12–16).
7SK (330–332 nucleotides, depending on posttranscriptional nuclease digestion) is transcribed abundantly by RNAPIII in cells (17). There, 7SK partitions between RNA complexes with the heterogeneous ribonucleoproteins A1, A2, Q1, and R (18) or 7SK snRNP (19). HEXIM1 binds to the upper stem-loop I (short SLI) or RNA motif 3 (M3) (positions 24–87) of 7SK in vitro (20). M3 resembles the transactivation response (TAR) RNA hairpin from HIV type 1, in which the viral transactivator of transcription (Tat) and P-TEFb bind to its 5′ U bulge and the central loop, respectively, to stimulate HIV transcription (21). Indeed, Tat also binds to M3 in vitro and releases P-TEFb from the 7SK snRNP (21–23). In turn, methyl phosphate capping enzyme and LaRP7 bind to the lower 5′ SLI and the U-rich SLIV/M8 RNA hairpin in vitro, respectively (19, 24). Of note, interactions between LaRP7 and 7SK are required for the recruitment of P-TEFb into the 7SK snRNP (24).
HEXIM1 contains 359 residues in four functional domains (25, 26). A variable N-terminal region (residues 1–149) forms a self-inhibitory domain (27). The central basic domain (residues 150–177) forms a nuclear localization signal and an RNA-binding domain (27). Interestingly, the sequence of the HEXIM1 nuclear localization signal is almost identical to the arginine-rich motif of Tat (26). In vitro studies indicated that HEXIM1 binds to double-stranded RNA in a relatively sequence-independent manner (28). Indeed, the same study demonstrated that HEXIM1 also binds to microRNAs (miR16) (28). Immediately C-terminal to this region (residues 178–220) are found acidic amino acids, the PYNT motif (residues 203–206) and the evolutionally conserved phenylalanine at position 208 (Phe-208), which interact with CycT1 and help to inhibit the kinase activity of CDK9 (26, 29, 30). This acidic region also binds to the basic region of HEXIM1 in the absence of 7SK and, in its presence, improves these RNA-binding interactions (31). Finally, the C-terminal coiled-coil domain (residues 250–359) interacts with CycT1 and supports the formation of HEXIM1 dimers (29, 32, 33). This region also contains an additional tyrosine residue at position 271 (Tyr-271) that is critical for the inhibition of CDK9 (30).
In this study, we established an experimental approach to analyze genetic interactions between HEXIM1, LaRP7, and 7SK using a cell-based system. We adopted an artificial heterologous RNA tethering reporter assay (34–37) and constructed new plasmid targets by replacing the TAR of HIVSCAT with various regions of 7SK. By recruiting P-TEFb via hybrid proteins that also contained the transcriptional activation domain of Tat (residues 1–48), the HEXIM1·Tat48 and LaRP7·Tat48 chimeras stimulated plasmid targets that contained M3 and M8, respectively. The central region of HEXIM1 (residues 150–220) was sufficient to bind to M3. Deletion of U40/U41 or disruption of the GAUC motif abolished this activation, supporting previous binding studies in vitro (38). In addition, the central loop (U57/58) of M3 was also required for transactivation, suggesting that HEXIM1 and CycT1 form a combinatorial binding surface for 7SK, which resembles interactions between Tat, CycT1, and TAR. Finally, mutating Tyr-271 of HEXIM1 to alanine turned HEXIM1 into an activator on M3. Therefore, we established a reliable cell-based assay to analyze genetic interactions between HEXIM1, 7SK, and P-TEFb.
EXPERIMENTAL PROCEDURES
Cell Lines and Antibodies
HeLa or HEK293T cells were grown in DMEM containing 10% FCS at 37 °C. Rabbit anti-cMyc (catalog no. ab9106) and anti-β tubulin (catalog no. ab6046) antibodies were purchased from Abcam. Rabbit anti-CDK9 (catalog no. SC-484) was purchased from Santa Cruz Biotechnology.
Plasmids
Chloramphenicol acetyltransferase (CAT) plasmid targets were constructed by inserting double-stranded DNA into pHIVSCAT using BglII and SacI restriction enzymes (34, 38). The cDNAs of the RNA sequences from 7SK snRNA and their negative strand DNAs were chemically synthesized (IDTDNA) and annealed to form double-strand DNA inserts. Both the 5′ and 3′ ends of each cDNA and the negative strand contain unique linkers to create the adhesive ends of the restriction enzymes and to maintain the Watson-Crick base pairing that forms an RNA stem structure as in the TAR region of the original HIVSCAT. The linker sequences were as follows: cDNA strand, 5′-AGATCT (cDNA) GAGCT-3′; negative strand, 5′-C (negative strand DNA) A-3′. The actual DNA sequences of each reporter are available upon request. Full-length or truncated HEXIM1 and the HEXIM1 chimera were constructed in the pEF-Bos mammalian expression plasmid. All proteins contained a cMyc antibody epitope tag at the N terminus, and their expression in cells was confirmed by Western blot analysis using an anti-cMyc antibody. The CDK9·CycT1 chimera was created as described previously, except that the full-length CycT1 was fused to CDK9 (39). As described previously, the CDK9·CycT1 chimera forms an active P-TEFb complex when expressed ectopically in cells (39). Point mutations were created by using the QuikChange site-directed mutagenesis kit (Agilent Technologies).
RNA Immunoprecipitation
HEXIM1 (Figs. 3B and 4) or Hex·Tat48 fusion proteins (Fig. 4) were expressed in HEK293T cells (4 × 106) by transient transfection using Lipofectamine 2000 (Invitrogen) in the presence (Fig. 3B) or absence (Fig. 4) of the WT or mutant M3 plasmid reporters (0.5 μg). The cells were lysed in buffer A (20 mm HEPES-KOH (pH 7.8), 0.1 m KCl, 0.1% Nonidet P-40, and 0.2 mm EDTA) on ice for 10 min. Cell lysates were centrifuged at 14,000 rpm for 5 min at 4 °C, and supernatants were collected. Cell lysates were then precleared with protein A-Sepharose beads (Invitrogen) and divided into two aliquots. Each aliquot was incubated with 1 μg of normal-rabbit IgG or anti-cMyc antibodies overnight at 4 °C. Then, 20 μl of protein-A-Sepharose beads precoated with BSA and yeast tRNA were added to each aliquot for an additional 2 h at 4 °C. Beads were washed five times with buffer A. RNA was then extracted with TRIzol (Invitrogen), followed by DNase I treatment (Ambion). Reverse transcription quantitative PCR (RT-qPCR) analyses were performed to quantify endogenous 7SK (Fig. 4) or RNA expressed from the plasmid reporters (Fig. 3B) enriched in the immunoprecipitates. The same sets of qPCR analyses using the samples without reverse transcription confirmed that the DNA contamination from transfected plasmid reporters was negligible (data not shown). The specific primers for endogenous 7SK or RNA from the reporter plasmids were as follows: 7SK, GAGGGCGATCTGGCTGCGACAT (forward) and ACATGGAGCGGTGAGGGAGGAA (reverse); 7SK-CAT, TTTCCCAGGAAGA TCCTGAC (forward) and ACCTTGGTGAGATCGAATGG (reverse). Data were normalized to input amounts of 7SK and calculated as values relative to the amount obtained with IgG control of untransfected cells (set to 1).
FIGURE 3.

HEXIM1(178–220) determines the target specificity of HEXIM1. A, the RNA sequence and putative secondary structure of plasmid effectors are presented above the CAT data. 7SK plasmid targets (indicated in each panel of CAT data) were coexpressed with effectors, which are presented below the CAT data, in HeLa cells. CAT data are presented as fold activation by calculating relative CAT values obtained with Tat48 fusion proteins over those obtained with the empty plasmid (columns 1, 5, and 9). Error bars are as in Fig. 2. The expression of effectors was determined by Western blotting with anti-cMyc antibodies with tubulin as the loading control (bottom panel). B, HEXIM1 interacts with the WT M3 but not the mutant M3ΔU40/41 or M3A43G/C44U plasmid targets. 7SK plasmid targets (indicated below the graph) were coexpressed with HEXIM1 in HeLa cells. HEXIM1 (Hex, cMyc epitope-tagged) was immunoprecipitated by anti-cMyc antibodies, and associated RNA was subjected to RT-qPCR using primer sets specific to these plasmid targets. Data are presented as fold RNA enrichment by calculating relative amounts of cDNA over the value obtained with non-transfected cells (column 1 set at 1). Immunoprecipitations (IP) with IgG from cells expressing HEXIM1 and each plasmid target were performed as negative controls (C) and confirmed that the signals are specific to HEXIM1 immunoprecipitations (data not shown).
FIGURE 4.
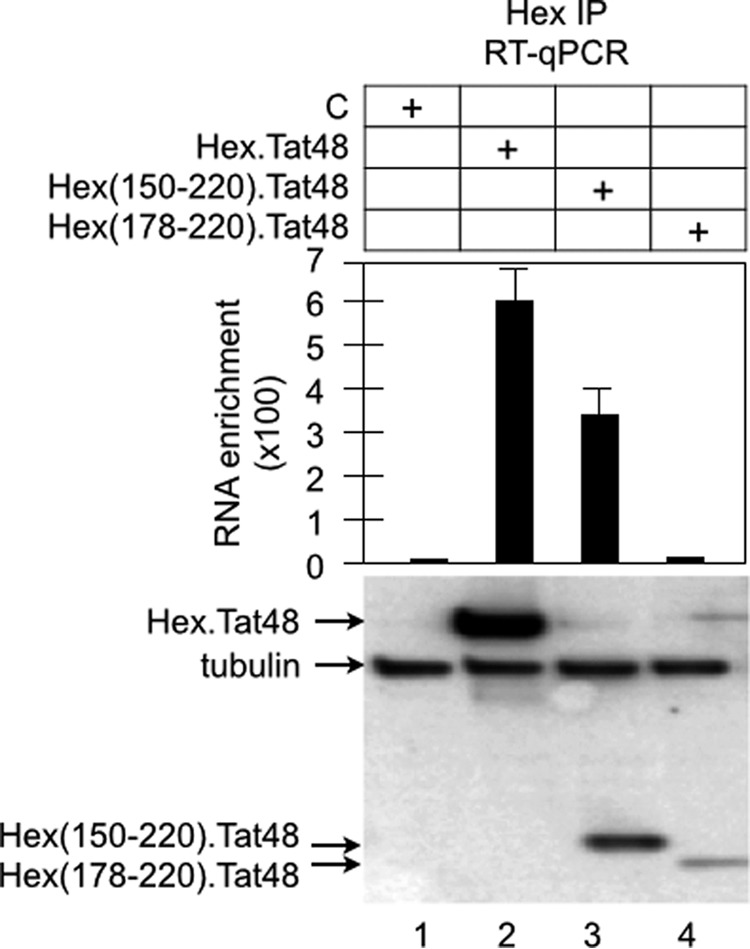
The Hex·Tat48 and Hex(150–220)· Tat48 chimeras, but not the Hex(178–220)·Tat48 chimera, interact with 7SK in cells. The Hex·Tat48 chimeras (indicated at the top of the graph) were expressed in HEK293T cells and immunoprecipitated (IP) by anti-cMyc antibodies. To quantify 7SK, RNA in immunoprecipitations was subjected to RT-qPCR by using a set of specific primers. Data are presented as fold RNA enrichment by calculating relative amounts of cDNA over values obtained with untransfected cells (column 1 set at 1). Immunoprecipitation with IgG was performed as a negative control (C) and confirmed that the signals were specific to HEXIM1 immunoprecipitations (data not shown). Note that Hex(150–220)·Tat48 and Hex(178–220)·Tat48 chimeras (118 and 70 residues, respectively) were not retained quantitatively on nitrocellulose filters because of their small sizes.
Transient Transfection and CAT Assay
HeLa cells were seeded into 24-well plates approximately 12 h prior to cotransfection of the plasmid reporters (0.1 μg) and plasmids encoding the effectors (0.3 μg for Hex and Hex chimeras or 0.1 μg for LaRP7 and the LaRP7 chimera) using Xtreme HP (Roche). Forty-eight hours after transfection, cells were lysed in and subjected to CAT enzymatic assays as described previously (36). The remaining lysates were subjected to Western blot analysis using anti-cMyc antibody and anti-tubulin antibody as a loading control.
RESULTS
HEXIM1 and LaRP7 Bind to M3 and M8, Respectively, in Cells
Wassarman and Steitz (40) originally predicted the secondary structure of 7SK. Their model contains four RNA stem-loops (Fig. 1A, SLI-SLIV). Recently, Bensaude and colleagues (20) published a modified structure that contains eight evolutionally conserved motifs (Fig. 1B, M1-M8). In vitro binding studies indicated that sequences from positions 24–87 of 7SK, which correspond to M3 or short SLI, interact with HEXIM1. SLIV/M8 interacts with LaRP7 (24, 41–43). To study genetic interactions between HEXIM1 or LaRP7 and 7SK, we modified an artificial heterologous RNA tethering system (34). In these systems, the TAR in the HIV LTR is replaced by an RNA sequence of interest (34), and RNA-binding proteins that interact with it are used as fusion partners of specific effectors. One well studied plasmid target contains the stem-loop IIB (SLIIB) from the HIV Rev response element and Rev proteins fused to activators of transcription elongation (35, 37, 44). In particular, CycT1 from P-TEFb is a strong activator when fused to Rev, as are the transcriptional activation domain of Tat (residues 1–48, TAD48) and RelA, a subunit of nuclear factor κB (44, 45).
FIGURE 1.

Schematic of proposed secondary structures of 7SK snRNA. A, the structure proposed by Wasserman and Steitz (40) contains four stem-loops (SLI-SLIV). B, Bensaude and coworkers (20) proposed a modified structure of 7SK snRNA that contains eight motifs (M1∼M8).
We first created chimeras between HEXIM1 or LaRP7 and Tat48 (Fig. 2). Because Tat48 binds to CycT1 and not to RNA (46), our chimeras could only activate these plasmid targets when their fusion partners bound to their cognate RNA sequences. Indeed, Tat48 did not activate plasmid reporters used in this study (see below). Next, we constructed plasmid targets with nucleotides forming the proposed secondary structures, which are presented in Fig. 1A. They are short SLI/M3, SLII, SLIII/M7, and SLIV/M8. HEXIM1 (Hex), Hex·Tat48, LaRP7, or LaRP7·Tat48 chimeras were coexpressed with these plasmid targets by transient transfection in HeLa cells. Forty-eight hours after the transfection, CAT enzymatic activities were measured and calculated as fold activation over the values obtained with non-fused proteins. As presented in Fig. 2, the Hex·Tat48 chimera activated M3 7-fold over Hex alone (Fig. 2, columns 1 and 2) but not other plasmid targets (Fig. 2, columns 6, 10, and 14). On the other hand, the LaRP7·Tat48 chimera activated M8 5-fold over LaRP7 alone (Fig. 2, columns 15 and 16). It also failed to activate other plasmid targets (Fig. 2, columns 4, 8, and 12). Western blotting of cell lysates revealed amounts of ectopically expressed proteins. We conclude that HEXIM1 and LaRP7 bind to M3 and M8, respectively.
FIGURE 2.

HEXIM1 and LaRP7 bind to M3 and M8, respectively, in cells. CAT plasmid targets were constructed by replacing TAR in HIVSCAT with the sequence of 7SK. The RNA sequence and putative secondary structure of each reporter are presented above the CAT data. A schematic of plasmid targets and effectors is shown left of the CAT data. The 7SK plasmid targets (indicated in each panel of CAT data) were coexpressed with effectors, which are shown below the CAT data, in HeLa cells. CAT data are presented as fold activation by calculating relative CAT values obtained with Tat48 fusion proteins (columns with even numbers) over the cognate non-fusion protein (columns with odd numbers) on each reporter. Error bars represent mean ± S.E. of experiments performed in triplicate. The expression of the effector proteins was determined by Western blotting with anti-cMyc antibodies and tubulin as the loading control (bottom panel). m, cMyc epitope tag. pA, polyadenylation site.
Residues from Positions 178–220 Determine the Target Specificity of HEXIM1
Although HEXIM1 binds to CycT1, it failed to activate M3 (Fig. 2), presumably because it inhibits the kinase activity of CDK9. Next, we sought to map the minimal region of HEXIM1 that binds to 7SK by fusing various regions of HEXIM1 to Tat48. The RNA-binding domain of HEXIM1 was mapped to its central basic residues from positions 150–177 in vitro (27). Rana and co-workers (41) demonstrated that its neighboring C-terminal residues from positions 178–220 also contribute to this binding. Therefore, we investigated whether the truncated Hex(150–177)·Tat48 or Hex(150–220)·Tat48 chimeras also activate M3 (Fig. 3A). In addition, because Tat can recruit P-TEFb from 7SK snRNP by competing with HEXIM1 for binding to 7SK and to P-TEFb, we wondered whether the full-length Tat protein also activates M3 (21–23). Indeed, the Tat, Hex(150–220)·Tat48, and Hex(150–177) chimeras activated M3 to similar levels (Fig. 3A, columns 2–4). Neither Tat48 (Fig. 3A, column 1) nor the Hex(178–220)·Tat48 chimera (data not shown) activated this plasmid target. These results indicate that Tat and HEXIM1 bind to M3. Indeed, RNA immunoprecipitations indicated that Hex·Tat48 and Hex(150–177)·Tat48, but not the Hex(178–220)·Tat48 chimera, interact with 7SK (Fig. 4, top panel, second through fourth columns). Because they measured less than 10 kDa, smaller fusion proteins were not retained quantitatively on the nitrocellulose filter (Fig. 4, bottom panel, Western blot analysis, lanes 3 and 4).
An extensive in vitro study conducted by Dock-Bregeon and co-workers (43) indicated that the U-U bulge (U40/U41) as well as the GAUC:CUAG stem adjacent to the U-U bulge (positions 42–45 and 64–67, respectively) are critical for interactions between HEXIM1 and 7SK. To examine the importance of these motifs in our CAT reporter system, we created mutant M3 plasmid targets in which the U-U bulge was removed (M3Δ40/41) or the GUAC motif was mutated (M3A43G/U44C) (Fig. 3A). Surprisingly, although Tat and the Hex(150–177)·Tat48 chimera retained a lower but significant activity on these mutant plasmid targets (Fig. 3A, columns 6, 8, 10, and 12), the Hex(150–220)·Tat48 chimera failed completely to activate them (Fig. 3A, columns 7 and 11). The full-length Hex·Tat48 chimera also failed to activate these plasmid targets (data not shown). To determine whether RNAs expressed from the WT or mutant M3 plasmid targets interact with HEXIM1, we performed additional RNA immunoprecipitations. HEXIM1 was coexpressed with WT and mutant plasmid targets, followed by immunoprecipitations with anti-cMyc antibodies. RNAs associated with HEXIM1 were extracted, treated with DNase I, and quantified by RT-qPCR using primer sets specific to the 3′ region of M3 and the 5′ proximal region of the CAT gene. A parallel qPCR analysis of the same RNA samples without RT reaction confirmed that the RT-qPCR signals were not from the plasmid DNA (data not shown). As presented in Fig. 3B, although the WT M3 RNA was enriched (∼100-fold over the IgG control) in HEXIM1 immunoprecipitations, it was not observed with mutant M3Δ40/41 and M3A43G/U44C RNAs (Fig. 3B, columns 2–4). From these results, we conclude that HEXIM1 interacts specifically with M3 and that residues from positions 178–220 of HEXIM1 are critical for interactions between HEXIM1 and 7SK.
U Residues in the Central Loop of M3 Are Also Important for Interactions with HEXIM1
In vitro binding studies indicated additional sequences in M3 that might be involved in interactions between HEXIM1 and 7SK snRNP. For instance, deletion of the uridine at position 63 or the C-U bulge at positions 71 and 72 reduce this binding affinity (43). In addition, there is another bulge (from positions 75–77) containing a uridine residue that could interact with HEXIM1. Finally, 7SK contains a central loop (from positions 50–59) that resembles the central loop of TAR where CycT1 binds to form a stable complex between Tat, P-TEFb, and TAR (21, 47, 48). Therefore, we constructed a series of mutant M3 plasmid targets to identify other residues critical for these interactions. Next, we determined whether the Hex(150–220)·Tat48 chimera can activate these plasmid targets (Fig. 5). As presented in Fig. 5, the mutant M3ΔU63, M3ΔC34/C35, M3ΔC71/U72, and M3ΔC75/U77 plasmid targets were activated by the Hex(150–220)·Tat48 chimera to levels similar to the WT M3 plasmid target, indicating that they still interact with HEXIM1 in cells (Fig. 5, bars 1 and 6–9). In contrast, the Hex(150–220)·Tat48 chimera activated the mutant ΔU57/U58 plasmid target to a lesser extent (Fig. 5, bars 4 and 5). This finding and the previous data on residues from positions 178–220 of HEXIM1, which contribute to its binding specificity (Fig. 3), support the model where RNA-binding residues from positions 150–177 and those from positions 178–220 of HEXIM1 form a tripartite HEXIM1-CycT1–7SK complex that resembles the Tat-CycT1-TAR complex (21, 47, 48).
FIGURE 5.

Mapping of the residues in M3 that are important for HEXIM1 interactions. Plasmid targets are presented to the left of the CAT data. The Hex(150–220)·Tat48 chimera (0.3 μg) was coexpressed with each plasmid target (0.1 μg) in HeLa cells. CAT data are presented as fold activation by calculating relative CAT values obtained with the Hex(150–220)·Tat48 chimera over the values obtained with the empty plasmid (set to 1). Error bars are as in Fig. 2. m, cMyc epitope tag. pA, polyadenylation site.
No Other Proposed Secondary Structures in 7SK Interact with HEXIM1
In the secondary structure of 7SK proposed by Bensaude and co-workers (Ref. 20 and Fig. 1B), there are eight evolutionally conserved motifs (M1-M8). We next examined whether HEXIM 1 binds to other motifs in cells. In addition, it has been demonstrated that the uridine at position 30 contacts HEXIM1 (41). Therefore, we constructed a plasmid target with a truncated M3 (short M3) from positions 31–75. As presented in Fig. 6, although the M4, M5, M6, and M7 plasmid targets were not activated by the Hex(150–220)·Tat48 chimera (Fig. 6, bars 5–7), short M3 was activated to a level similar to M3 by the Hex(150–220)·Tat48 chimera (Fig. 6, bars 3 and 4). Although it has been demonstrated that HEXIM1 interacts with 7SK(10–48) in vitro (28), 7SK(10–48) was not activated by the Hex(150–220)·Tat48 chimera (Fig. 6, bar 1). Finally, HEXIM1 did not bind to the lower stem of SLI (Fig. 6, bar 2, lsSLI).
FIGURE 6.
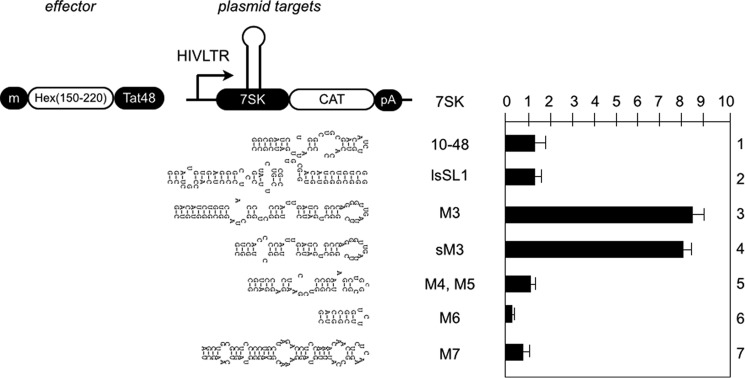
No other proposed secondary structures in 7SK interact with HEXIM1. Reporters are presented next to the CAT data. The Hex(150–220)·Tat48 chimera (0.3 μg) was coexpressed with each reporter (0.1 μg) in HeLa cells. CAT data are presented as fold activation by calculating relative CAT values over those obtained with empty plasmids. Error bars are as in Fig. 2. m, cMyc epitope tag; lsSLI, lower stem of SLI; sM3, short M3. pA, polyadenylation site.
Mutating the Tyrosine at Position 271 to Alanine Transforms HEXIM1 into an Activator
Previous results indicated that HEXIM1 activates M3 only when the Hex·Tat48 chimera recruits P-TEFb. Because Tat can compete with HEXIM1 for binding to CycT1 (22, 23), P-TEFb recruited via Tat48 is also not inhibited by HEXIM1 in the context of the Hex·Tat48 chimera. However, HEXIM1 alone inhibited M3 (Fig. 7, bar 1), indicating that interactions between HEXIM1 and M3 are sufficient to inactivate P-TEFb. Mutating a tyrosine at position 271 to alanine (Y271A) abolishes this inhibitory effect of HEXIM1 without affecting its ability to interact with 7SK and CycT1 (30). Indeed, the mutant Hex(Y271A) proteins exhibited a positive effect (∼4-fold) on reporter M3 (Fig. 7, bar 3). The mutant Hex(Y271A) proteins did not activate other plasmid targets, such as HIVSCAT (data not shown). Therefore, HEXIM1 can activate plasmid targets via 7SK when its ability to inhibit P-TEFb is diminished.
FIGURE 7.
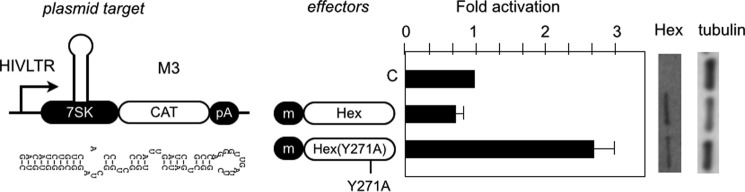
Mutation of tyrosine 271 in HEXIM1 changes HEXIM1 into an activator. WT HEXIM1 or mutant Hex(Y271A) proteins were coexpressed with M3 in HeLa cells. CAT data are presented as fold activation by calculating relative CAT values over those obtained with empty plasmids. Error bars are as in Fig. 2. The expression of the effectors was determined by Western blotting as in Fig. 2. m, cMyc epitope tag; C, control. pA, polyadenylation site.
Recruitment of Active (Free) P-TEFb Is Required for Optimal Activation by the Mutant Hex(Y271A) Protein
Although a Tyr-to-Ala mutation at position 271 changed HEXIM1 into an activator (Fig. 7), its effects on M3 were small (4-fold, Fig. 7). Because the majority of P-TEFb is found in the inactive 7SK snRNP in HeLa cells and because ectopically expressed HEXIM1 proteins are inefficiently incorporated into the 7SK snRNP (Ref. 17 and data not shown), the lower activation by the mutant Hex(Y271A) protein could be due to limited amounts of active P-TEFb in cells. To examine this possibility, we constructed the Hex·CycT1 chimera that contained WT HEXIM1 or mutant Hex(Y271A) proteins and determined its ability to activate M3 (Fig. 8A). Although the WT Hex·CycT1 chimera weakly activated this plasmid target (1.5-fold), the mutant Hex(Y271A)·CycT1 chimera exhibited a strong effect (12-fold, Fig. 8A, columns 2 and 3). The Hex·CycT1 and the mutant Hex(Y271A)·CycT1 chimeras were expressed at similar levels (Fig. 8A). Similarly, the mutant Hex(Y271A) protein activated M3 when the active P-TEFb complex (CDK9·CycT1 chimera) was provided in trans by transient coexpression (8-fold, Fig. 8B, columns 2 and 4). We conclude that the optimal activation of M3 by the mutant Hex(Y271A) protein requires the active P-TEFb in cis or in trans.
FIGURE 8.
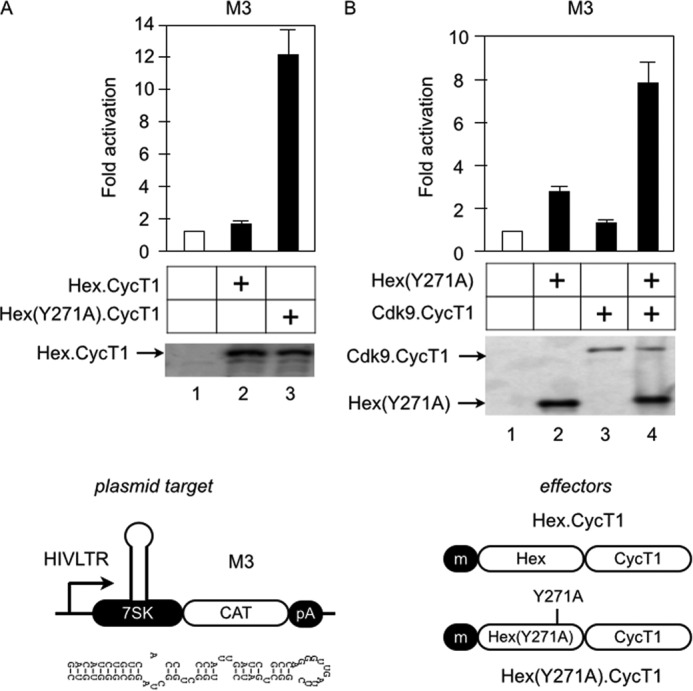
Recruitment of active (free) P-TEFb is required for optimal transactivation by Hex(Y271A). A, the WT Hex·CycT1 (column 2) or mutant Hex(Y271A)·CycT1 (column 3) chimeras were coexpressed with the M3 reporter in HeLa cells. CAT data are presented as fold activation by calculating the relative CAT values over the values obtained with empty plasmid (column 1 set to 1). Error bars are as in Fig. 2. The expression of effectors was confirmed by Western blotting. B, the mutant Hex(Y271A) protein was coexpressed with M3 in HeLa cells with (columns 3 and 4) or without (columns 1 and 2) the CDK9·CycT1 chimera. CAT data are as in A (column 1 set to 1). Error bars are as in Fig. 2. The expression of the effector proteins and the CDK9·CycT1 chimera was detected by Western blotting. m, cMyc epitope tag. pA, polyadenylation site.
DISCUSSION
In this study, we established a reporter-based system to measure genetic interactions between 7SK and HEXIM1 or LaRP7. In agreement with previous studies, HEXIM1 bound to M3, and LaRP7 bound to M8. The former interaction was mediated not only by the U-U bulge and GUAC motif of 7SK but also U residues in its central loop. Finally, a mutation at Tyr-271 to Ala transformed HEXIM1 into an activator.
In our heterologous RNA tethering system to study interactions between 7SK and HEXIM1, Tat48 was used as the effector. Maximal activities obtained with the Hex·Tat48 or LaRP7·Tat48 chimeras were somewhat lower (5- to 8-fold) than those obtained with other systems, such as Rev and SLIIB (20- to 30-fold) (34, 35, 37, 44). This difference could reflect the ability of Rev to form higher-order oligomers that recruit more activators to the elongation complex. Although HEXIM1 forms dimers on 7SK (32, 49), only one HEXIM1-binding site was identified in our study. Also, because of high levels of the endogenous HEXIM1 protein, it was not clear whether our Hex·Tat48 chimera bound to the reporter as a monomer or a dimer with itself or the endogenous HEXIM1 protein. Finally, WT 7SK contains seven AUG codons that reduced the efficiency CAT protein expression using the endogenous in-frame start codon. However, RT-qPCR data supported our CAT data, i.e. that transcription of the longer reporter genes that contained the full-length 7SK were activated 3- to 5-fold by the Hex·Tat48 chimera (data not shown).
In the absence of Tat, RNAPII transcribes up to 150 nucleotides past the transcriptional start site on the HIV LTR (50). Supporting this observation, recent genome-wide analyses revealed that pausing sites distribute over an extended distance after initiation (51). These findings suggest that stable RNA structures are only surrogate markers for pausing rather than stalling RNAPII by themselves. Therefore, transcription from our 7SK plasmid targets should exhibit patterns of RNAPII pausing similar to those on the parental HIV LTR. To this end, when TAR was substituted with the stem-loop IIB (SL((B) from the Rev response element, the Rev·CycT1 chimera activated HIV transcription indistinguishably from Tat and TAR (35).
HEXIM1 can be recruited to promoter and 5′ proximal regions in genes directly or via RNA-binding proteins such as SC35, presumably for loading of P-TEFb (52, 53). Although chromatin immunoprecipitations revealed HEXIM1 and LaRP7 on DNA, they did not demonstrate the presence of 7SK or the entire 7SK snRNP. Of interest, we also observed tight associations between HEXIM1 and chromatin but only when it was phosphorylated on serine at position 158 (54). Thus modified, the phosphorylated HEXIM1 protein neither bound 7SK nor inhibited P-TEFb (54). Therefore, it would be of interests to determine whether the chromatin-bound HEXIM1 is capable of interacting with 7SK. Nevertheless, we observed effects of HEXIM1 only in the presence of M3 and not without its specific RNA target.
Our results indicated that the minimal HEXIM1-binding region of 7SK contains the U-U bulge (positions 40 and 41), the GUAC motif, and the central loop. No other predicted secondary structures were found to bind to HEXIM1. This region also binds to Tat, which is consistent with previous studies in vitro (21). Although those binding studies demonstrated the importance of the U40-U41 bulge and the GUAC motif for the interaction between HEXIM1 and 7SK (43), the importance of the central loop for assembling the 7SK snRNP had not been appreciated. In contrast, our results revealed that the uridine residues at positions 57 and 58 in the central loop are also required. It is to be noted that, in addition to the basic region of HEXIM1 (from positions 150–177), its immediate C-terminal residues (from positions 178–220) support specific interactions between HEXIM1 and 7SK (26). This region contains the PYNT motif (from positions 203–206) and the phenylalanine at position 208, which also help to inhibit the kinase activity of CDK9 (26, 29, 30). These RNA-protein interactions cause a conformational change to create a combinatorial binding surface for CycT1, which resembles those between Tat, TAR, and CycT1 (23, 31) where the central loop of TAR is required for binding to CycT1 (46). Our results provide a genetic and functional support for this model (Fig. 9). Because cooperative interactions between HEXIM1 and P-TEFb require the proper spatial arrangement between the 5′ U-U bulge and the central loop in M3, which is not maintained in TAR, HEXIM(Y271A) did not work on TAR. On the other hand, the LaRP7-binding region of 7SK was mapped to M8 (Fig. 2).
FIGURE 9.
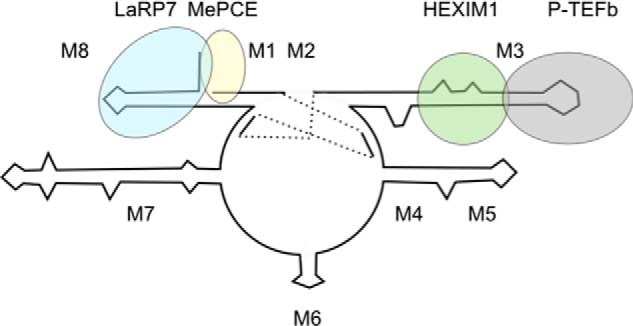
A model of 7SK snRNP. Although HEXIM1 and P-TEFb bind cooperatively to M3, LaRP7 binds to M8 of 7SK.
The maintenance of the P-TEFb equilibrium is a critical step in transcription elongation. At homeostasis, the amount of active P-TEFb is limited because it is mostly sequestered in the 7SK snRNP. Therefore, the release of P-TEFb from 7SK snRNP by various stimuli or stresses is a prerequisite step for activation of transcription and for cellular growth. However, Tat can recruit P-TEFb directly from the 7SK snRNP (22, 23). Not only does Tat contain an RNA-binding region almost identical to that in HEXIM1, but TAR folds into a stem-loop similar to the minimal HEXIM1-binding region of 7SK. As such, Tat can compete with HEXIM1 and 7SK for binding to CycT1, which forms a stable tripartite complex with TAR (22). Therefore, it is intriguing to speculate that HIV evolved by modifying HEXIM1 and removing its P-TEFb inhibitory sequences and adjusting its RNA-protein interaction to obtain the simplest unit for transcription elongation of HIV.
In conclusion, our reporter-based assays provide a quantitative measurement of RNA-protein interactions that assemble 7SK snRNP. They facilitate the analysis of these interactions, which could be used to measure efficiencies of compounds that interfere with and to develop high throughput screening systems for inhibitors that block these RNA-protein interactions in cells.
Acknowledgments
We thank the members of the Peterlin laboratory for helpful discussions.
This work was supported, in whole or in part, by National Institute of Health CARE Center Grant U19 AI076113, HARC Center Grant P50 GM082250, and Grant AI076113 (to B. M. P.). This work was also supported by American Foundation of AIDS Research Grant 108241-51-RGRL (to K. F.).
- RNAPII
- RNA polymerase II
- P-TEFb
- positive transcription elongation factor b
- snRNP
- small nuclear ribonucleoprotein
- HEXIM
- hexamethylene bisacetamide-induced protein
- TAR
- transactivation response element
- SL
- stem-loop
- CAT
- chloramphenicol acetyltransferase
- RT-qPCR
- quantitative RT-PCR.
REFERENCES
- 1. Shilatifard A., Conaway R. C., Conaway J. W. (2003) The RNA polymerase II elongation complex. Annu. Rev. Biochem. 72, 693–715 [DOI] [PubMed] [Google Scholar]
- 2. Fuda N. J., Ardehali M. B., Lis J. T. (2009) Defining mechanisms that regulate RNA polymerase II transcription in vivo. Nature 461, 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peterlin B. M. (2010) Transcription elongation takes central stage: the P-TEFb connection. Cell Cycle 9, 2933–2934 [DOI] [PubMed] [Google Scholar]
- 4. Price D. H. (2008) Poised polymerases: on your mark, get set, go! Mol. Cell 30, 7–10 [DOI] [PubMed] [Google Scholar]
- 5. Peterlin B. M., Price D. H. (2006) Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 23, 297–305 [DOI] [PubMed] [Google Scholar]
- 6. Zhou Q., Yik J. H. (2006) The Yin and Yang of P-TEFb regulation: implications for human immunodeficiency virus gene expression and global control of cell growth and differentiation. Microbiol. Mol. Biol. Rev. 70, 646–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. He N., Liu M., Hsu J., Xue Y., Chou S., Burlingame A., Krogan N. J., Alber T., Zhou Q. (2010) HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 38, 428–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sobhian B., Laguette N., Yatim A., Nakamura M., Levy Y., Kiernan R., Benkirane M. (2010) HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 38, 439–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jang M. K., Mochizuki K., Zhou M., Jeong H. S., Brady J. N., Ozato K. (2005) The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19, 523–534 [DOI] [PubMed] [Google Scholar]
- 10. Yang Z., Yik J. H., Chen R., He N., Jang M. K., Ozato K., Zhou Q. (2005) Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 19, 535–545 [DOI] [PubMed] [Google Scholar]
- 11. He N., Zhou Q. (2011) New insights into the control of HIV-1 transcription: when Tat meets the 7SK snRNP and super elongation complex (SEC). J. Neuroimmune Pharmacol. 6, 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen R., Liu M., Li H., Xue Y., Ramey W. N., He N., Ai N., Luo H., Zhu Y., Zhou N., Zhou Q. (2008) PP2B and PP1α cooperatively disrupt 7SK snRNP to release P-TEFb for transcription in response to Ca2+ signaling. Genes Dev. 22, 1356–1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. He N., Pezda A. C., Zhou Q. (2006) Modulation of a P-TEFb functional equilibrium for the global control of cell growth and differentiation. Mol. Cell Biol. 26, 7068–7076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartholomeeusen K., Fujinaga K., Xiang Y., Peterlin B. M. (2013) HDAC inhibitors that release positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. J. Biol. Chem. 288, 14400–14407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bartholomeeusen K., Xiang Y., Fujinaga K., Peterlin B. M. (2012) BET bromodomain inhibition activates transcription via a transient release of P-TEFb from 7SK snRNP. J. Biol. Chem. 287, 36609–36616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Contreras X., Barboric M., Lenasi T., Peterlin B. M. (2007) HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 3, 1459–1469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Peterlin B. M., Brogie J. E., Price D. H. (2012) 7SK snRNA: a noncoding RNA that plays a major role in regulating eukaryotic transcription. Wiley Interdiscip. Rev. RNA 3, 92–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barrandon C., Bonnet F., Nguyen V. T., Labas V., Bensaude O. (2007) The transcription-dependent dissociation of P-TEFb-HEXIM1–7SK RNA relies upon formation of hnRNP-7SK RNA complexes. Mol. Cell Biol. 27, 6996–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Krueger B. J., Jeronimo C., Roy B. B., Bouchard A., Barrandon C., Byers S. A., Searcey C. E., Cooper J. J., Bensaude O., Cohen E. A., Coulombe B., Price D. H. (2008) LARP7 is a stable component of the 7SK snRNP while P-TEFb, HEXIM1 and hnRNP A1 are reversibly associated. Nucleic Acids Res. 36, 2219–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marz M., Donath A., Verstraete N., Nguyen V. T., Stadler P. F., Bensaude O. (2009) Evolution of 7SK RNA and its protein partners in metazoa. Mol. Biol. Evol. 26, 2821–2830 [DOI] [PubMed] [Google Scholar]
- 21. Muniz L., Egloff S., Ughy B., Jády B. E., Kiss T. (2010) Controlling cellular P-TEFb activity by the HIV-1 transcriptional transactivator Tat. PLoS Pathog. 6, e1001152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barboric M., Yik J. H., Czudnochowski N., Yang Z., Chen R., Contreras X., Geyer M., Peterlin B. M., Zhou Q. (2007) Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 35, 2003–2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sedore S. C., Byers S. A., Biglione S., Price J. P., Maury W. J., Price D. H. (2007) Manipulation of P-TEFb control machinery by HIV: recruitment of P-TEFb from the large form by Tat and binding of HEXIM1 to TAR. Nucleic Acids Res. 35, 4347–4358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muniz L., Egloff S., Kiss T. (2013) RNA elements directing in vivo assembly of the 7SK/MePCE/Larp7 transcriptional regulatory snRNP. Nucleic Acids Res. 41, 4686–4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yik J. H., Chen R., Nishimura R., Jennings J. L., Link A. J., Zhou Q. (2003) Inhibition of P-TEFb (CDK9/Cyclin T) kinase and RNA polymerase II transcription by the coordinated actions of HEXIM1 and 7SK snRNA. Mol. Cell 12, 971–982 [DOI] [PubMed] [Google Scholar]
- 26. Michels A. A., Nguyen V. T., Fraldi A., Labas V., Edwards M., Bonnet F., Lania L., Bensaude O. (2003) MAQ1 and 7SK RNA interact with CDK9/cyclin T complexes in a transcription-dependent manner. Mol. Cell Biol. 23, 4859–4869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yik J. H., Chen R., Pezda A. C., Samford C. S., Zhou Q. (2004) A human immunodeficiency virus type 1 Tat-like arginine-rich RNA-binding domain is essential for HEXIM1 to inhibit RNA polymerase II transcription through 7SK snRNA-mediated inactivation of P-TEFb. Mol. Cell Biol. 24, 5094–5105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li Q., Cooper J. J., Altwerger G. H., Feldkamp M. D., Shea M. A., Price D. H. (2007) HEXIM1 is a promiscuous double-stranded RNA-binding protein and interacts with RNAs in addition to 7SK in cultured cells. Nucleic Acids Res. 35, 2503–2512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Michels A. A., Fraldi A., Li Q., Adamson T. E., Bonnet F., Nguyen V. T., Sedore S. C., Price J. P., Price D. H., Lania L., Bensaude O. (2004) Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 23, 2608–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li Q., Price J. P., Byers S. A., Cheng D., Peng J., Price D. H. (2005) Analysis of the large inactive P-TEFb complex indicates that it contains one 7SK molecule, a dimer of HEXIM1 or HEXIM2, and two P-TEFb molecules containing Cdk9 phosphorylated at threonine 186. J. Biol. Chem. 280, 28819–28826 [DOI] [PubMed] [Google Scholar]
- 31. Barboric M., Kohoutek J., Price J. P., Blazek D., Price D. H., Peterlin B. M. (2005) Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 24, 4291–4303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blazek D., Barboric M., Kohoutek J., Oven I., Peterlin B. M. (2005) Oligomerization of HEXIM1 via 7SK snRNA and coiled-coil region directs the inhibition of P-TEFb. Nucleic Acids Res. 33, 7000–7010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bigalke J. M., Dames S. A., Blankenfeldt W., Grzesiek S., Geyer M. (2011) Structure and dynamics of a stabilized coiled-coil domain in the P-TEFb regulator Hexim1. J. Mol. Biol. 414, 639–653 [DOI] [PubMed] [Google Scholar]
- 34. Tiley L. S., Madore S. J., Malim M. H., Cullen B. R. (1992) The VP16 transcription activation domain is functional when targeted to a promoter-proximal RNA sequence. Genes Dev. 6, 2077–2087 [DOI] [PubMed] [Google Scholar]
- 35. Bieniasz P. D., Grdina T. A., Bogerd H. P., Cullen B. R. (1999) Recruitment of cyclin T1/P-TEFb to an HIV type 1 long terminal repeat promoter proximal RNA target is both necessary and sufficient for full activation of transcription. Proc. Natl. Acad. Sci. U.S.A. 96, 7791–7796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fujinaga K., Cujec T. P., Peng J., Garriga J., Price D. H., Graña X., Peterlin B. M. (1998) The ability of positive transcription elongation factor B to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J. Virol. 72, 7154–7159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fujinaga K., Taube R., Wimmer J., Cujec T. P., Peterlin B. M. (1999) Interactions between human cyclin T, Tat, and the transactivation response element (TAR) are disrupted by a cysteine to tyrosine substitution found in mouse cyclin T. Proc. Natl. Acad. Sci. U.S.A. 96, 1285–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lu X., Welsh T. M., Peterlin B. M. (1993) The human immunodeficiency virus type 1 long terminal repeat specifies two different transcription complexes, only one of which is regulated by Tat. J. Virol. 67, 1752–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Fujinaga K., Irwin D., Geyer M., Peterlin B. M. (2002) Optimized chimeras between kinase-inactive mutant Cdk9 and truncated cyclin T1 proteins efficiently inhibit Tat transactivation and human immunodeficiency virus gene expression. J. Virol. 76, 10873–10881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wassarman D. A., Steitz J. A. (1991) Structural analyses of the 7SK ribonucleoprotein (RNP), the most abundant human small RNP of unknown function. Mol. Cell Biol. 11, 3432–3445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bélanger F., Baigude H., Rana T. M. (2009) U30 of 7SK RNA forms a specific photo-cross-link with Hexim1 in the context of both a minimal RNA-binding site and a fully reconstituted 7SK/Hexim1/P-TEFb ribonucleoprotein complex. J. Mol. Biol. 386, 1094–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Egloff S., Van Herreweghe E., Kiss T. (2006) Regulation of polymerase II transcription by 7SK snRNA: two distinct RNA elements direct P-TEFb and HEXIM1 binding. Mol. Cell Biol. 26, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lebars I., Martinez-Zapien D., Durand A., Coutant J., Kieffer B., Dock-Bregeon A. C. (2010) HEXIM1 targets a repeated GAUC motif in the riboregulator of transcription 7SK and promotes base pair rearrangements. Nucleic Acids Res. 38, 7749–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Barboric M., Nissen R. M., Kanazawa S., Jabrane-Ferrat N., Peterlin B. M. (2001) NF-κB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol. Cell 8, 327–337 [DOI] [PubMed] [Google Scholar]
- 45. Southgate C., Zapp M. L., Green M. R. (1990) Activation of transcription by HIV-1 Tat protein tethered to nascent RNA through another protein. Nature 345, 640–642 [DOI] [PubMed] [Google Scholar]
- 46. Wei P., Garber M. E., Fang S. M., Fischer W. H., Jones K. A. (1998) A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 92, 451–462 [DOI] [PubMed] [Google Scholar]
- 47. Richter S., Cao H., Rana T. M. (2002) Specific HIV-1 TAR RNA loop sequence and functional groups are required for human cyclin T1-Tat-TAR ternary complex formation. Biochemistry 41, 6391–6397 [DOI] [PubMed] [Google Scholar]
- 48. Richter S., Ping Y. H., Rana T. M. (2002) TAR RNA loop: a scaffold for the assembly of a regulatory switch in HIV replication. Proc. Natl. Acad. Sci. U.S.A. 99, 7928–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Byers S. A., Price J. P., Cooper J. J., Li Q., Price D. H. (2005) HEXIM2, a HEXIM1-related protein, regulates positive transcription elongation factor b through association with 7SK. J. Biol. Chem. 280, 16360–16367 [DOI] [PubMed] [Google Scholar]
- 50. Selby M. J., Bain E. S., Luciw P. A., Peterlin B. M. (1989) Structure, sequence, and position of the stem-loop in tar determine transcriptional elongation by tat through the HIV-1 long terminal repeat. Genes Dev. 3, 547–558 [DOI] [PubMed] [Google Scholar]
- 51. Kwak H., Fuda N. J., Core L. J., Lis J. T. (2013) Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339, 950–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ji X., Zhou Y., Pandit S., Huang J., Li H., Lin C. Y., Xiao R., Burge C. B., Fu X. D. (2013) SR proteins collaborate with 7SK and promoter-associated nascent RNA to release paused polymerase. Cell 153, 855–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McNamara R. P., McCann J. L., Gudipaty S. A., D'Orso I. (2013) Transcription factors mediate the enzymatic disassembly of promoter-bound 7SK snRNP to locally recruit P-TEFb for transcription elongation. Cell Rep. 5, 1256–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fujinaga K., Barboric M., Li Q., Luo Z., Price D. H., Peterlin B. M. (2012) PKC phosphorylates HEXIM1 and regulates P-TEFb activity. Nucleic Acids Res. 40, 9160–9170 [DOI] [PMC free article] [PubMed] [Google Scholar]