

Abstract
Hepatocerebral mitochondrial DNA depletion syndromes are classically considered diseases of early childhood, typically affecting the liver, peripheral, and central nervous systems with a rapidly progressive course. Evidence is emerging that initial symptom onset can extend into adulthood, though few such cases have been reported. We describe a 25-year-old woman who presented initially with secondary amenorrhea, followed by a megaloblastic anemia, lactic acidosis, leukoencephalopathy, progressive peripheral neuropathy, and liver cirrhosis. An apparently homozygous P98L mutation was identified in MPV17, a gene associated with a lethal infantile neurohepatopathy. Homozygosity for the same allele was recently reported in a man with a similar hepatic and neurologic phenotype. This is the first clinical report of an adult female with this disorder, and the first to describe amenorrhea and megaloblastic anemia as likely associated symptoms.
Mitochondrial DNA depletion syndromes are a heterogeneous group of diseases associated with reduced copy number of mitochondrial DNA due to defects in the replication or maintenance of the mitochondrial genome. Frequent manifestations include neuropathy, leukoencephalopathy, myopathy, and hepatic disease. Though depletion of the circular mitochondrial genome and subsequent mitochondrial dysfunction and energy failure are thought to underlie the pathophysiology of these disorders, they are caused by mutations in nuclear-encoded mitochondrial proteins; the inheritance pattern is usually autosomal recessive. These syndromes generally present in infancy, and death often occurs within the first year of life.
Located on chromosome 2, MPV17 encodes an inner membrane-associated mitochondrial protein of unclear function (El-Hattab and Scaglia 2013). Mutations in MPV17 are known to cause Navajo neurohepatopathy (OMIM #256810), a recessive, rapidly progressive mitochondrial DNA depletion syndrome of liver failure and neurologic deterioration with onset in the first year of life (Karadimas et al. 2006; El-Hattab et al. 2010; AlSaman et al. 2012). More recently, mutations in this gene have been identified in an older child (El-Hattab et al. 2010) and two adults (Blakely et al. 2012; Garone et al. 2012) with subacute onset of many of the same symptoms seen in the infantile form, including peripheral neuropathy, liver dysfunction or failure, progressive white matter disease, and lactic acidosis.
The MPV17 mutation P98L was identified in the heterozygous state in a child who survived to 2.5 years without liver transplantation, and was hypothesized to be a milder allele (El-Hattab et al. 2010). This same mutation in the homozygous state was then reported in a man with onset of symptoms at age 14 who was still living at age 21 (Blakely et al. 2012). A second man was reported with compound heterozygosity for the mutations KM88-89ML and L143X with initial symptoms at age 34 (Garone et al. 2012). These cases highlight that MPV17-mediated disorders – and likely mitochondrial DNA depletion syndromes in general – can present in adulthood with multisystem involvement, and the scarcity of such reports in the literature suggests that the range of adult phenotypes is yet to be fully appreciated. Here, we report the first known case of adult-onset MPV17-mediated neurohepatopathy in a female, who experienced secondary amenorrhea as the putative first symptom of the disorder.
Case Report
We report a 25-year-old woman of second-cousin Pakistani parents who, after normal cognitive and motor development and normal menarche, presented at age 18 years with secondary amenorrhea. Her only other health concern was a short stature of 4’11”, while her adult siblings were 5’4” and 5’6”.
Up to this time, she had been physically active without difficulties. Shortly after her periods ceased, she developed a steppage gait and weakness in her ankles that required orthotics. She also noticed mild numbness in her lower extremities. The numbness and weakness progressed slowly and eventually involved the upper extremities. Electromyography and nerve conduction studies showed a severe axonal peripheral neuropathy (Table 1). Nerve biopsy confirmed these findings (Fig. 1). A concomitantly performed muscle biopsy did not contain sufficient viable muscle tissue to quantify mitochondrial DNA. Difficulty with memory and word-finding developed, and a brain MRI at age 24 showed a leukodystrophy with diffuse and symmetrical white matter involvement (Fig. 2) concerning for a genetic syndrome.
Table 1.
Motor and sensory nerve conduction studies
| Normal | Patient | |||
|---|---|---|---|---|
| SNAP (μV) | NCV (m/s) | SNAP (μV) | NCV (m/s) | |
| Sensory nerves | ||||
| Right sural | >5 | >41 | NR | NR |
| Left sural | >5 | >41 | NR | NR |
| Left median | >14 | >44 | 12 | 57 |
| CMAP (mV) | NCV (m/s) | CMAP (mV) | NCV (m/s) | |
| Motor nerves | ||||
| Left peroneal (TA) | >2 | >41 | 0.6 | 30 |
| Left tibial | >2 | >41 | NR | NR |
| Right tibial | >3 | >41 | NR | NR |
| Left median | >4 | >49 | 7.6 | 51 |
SNAP sensory nerve action potential, CMAP compound muscle action potential, NR no response obtained
Fig. 1.
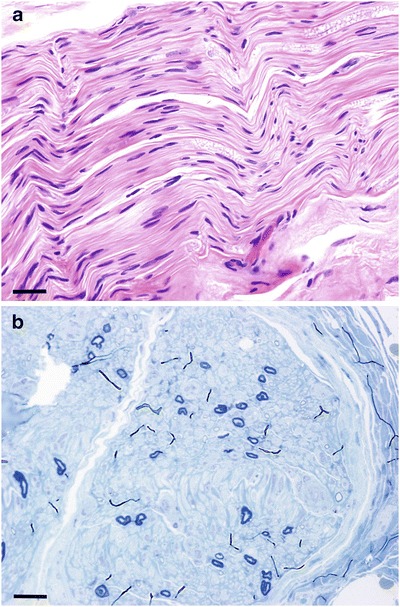
(a) Hematoxylin and eosin stain, 400x magnification, calibration bar is 0.1 mm. Significantly decreased amount of myelinated fibers without evidence of digestion chambers or inflammation. (b) Toluidine blue stain, 400x magnification, calibration bar is 0.1 mm. Severe (>95 %) loss of myelinated axons without onion bulbs or regenerating clusters
Fig. 2.
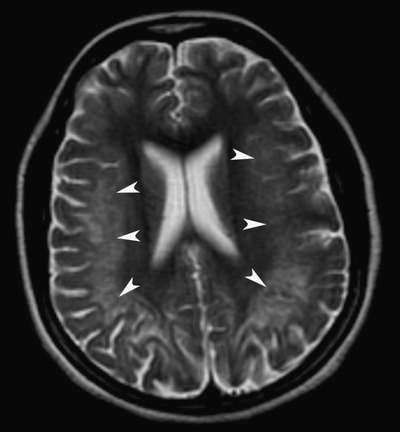
T2-weighted axial MRI image shows confluent and symmetrical hyperintensities within the deep and subcortical white matter of the posterior frontal and parietal cerebral hemispheres (arrowheads) involving the subcortical U-fibers, with sparing of the prefrontal cortices. There is associated cortical thinning. These hyperintensities did not show enhancement after the administration of IV gadolinium
Between the time of the onset of symptoms and her diagnosis, she was repeatedly hospitalized for worsening weakness, and during these admissions additional involved organ systems were noted. She developed a mild, permanent lactic acidosis, and the level of ammonia was intermittently elevated. Chronic thrombocytopenia and a macrocytic anemia also developed, with hemoglobin 9.5 g/dL and MCV 105 fL (upper limit of normal <100 fL), and normal serum levels of vitamin B12. Elevated blood glucose values developed and were treated with insulin and later with sitagliptin (Januvia). Hepatomegaly was noted and abdominal CT showed a nodular appearing liver, splenomegaly, and gastroesophageal varices consistent with cirrhosis. Portal hypertension was confirmed by both direct portal pressure measurement and upper endoscopy showing moderate esophageal varices. She also had evidence of synthetic liver dysfunction with an INR of 1.8 despite administration of vitamin K. A liver biopsy showed cirrhosis as well as steatosis and hepatocyte swelling suggestive of steatohepatitis despite her thin body habitus. Electron microscopy showed most hepatocytes to contain abundant mitochondria of varying sizes and shapes, some dilated and others with a nonuniform distribution of cristae. Quantification of mitochondrial DNA on the liver biopsy sample was not performed.
At age 25, her exam was notable for appropriate mental status, normal cranial nerve and fundal exam, striking atrophy of the muscles of the hands and lower legs with diminished strength in the finger extensors, abductor digiti minimi, and marked weakness of the tibialis anterior, gastrocnemius, and toe extensors and flexors. Deep tendon reflexes were absent in the triceps, patellae, and ankles. Pain sensation was intact in the fingertips but impaired in a stocking distribution to the mid-shin level bilaterally. Vibration sensation was intact at the fingertips. Vibration sensation was absent distal to the knees bilaterally. Joint position sensation was moderately impaired at the toes. She had a steppage gait and was too unsteady to attempt Romberg testing. The liver edge was palpable 3cm below the costal margin. The spleen could not be palpated.
Based on the clinical findings of brain and liver involvement, a hepatocerebral mitochondrial DNA sequencing panel was sent to Baylor College of Medicine, which revealed an apparently homozygous P98L mutation in MPV17. Given the known consanguinity of the parents it is most probable that this patient was indeed homozygous for this mutation, but because sequencing of the parents was not performed, we cannot formally exclude a deletion or sequencing failure affecting one allele. Additionally, a heterozygous mutation Q139X in OPA3 was detected. Homozygous mutations in OPA3 cause 3-methylglutaconic aciduria type 3, characterized by childhood onset of optic atrophy, a choreoathetoid movement disorder, and elevated urine 3-methylglutaconate. Our patient lacked these findings, indicating that the heterozygous OPA3 mutation was an incidental finding. The patient succumbed to a respiratory infection shortly after diagnostic results were received.
This is the second report of a patient homozygous for the MPV17 mutation P98L (Blakely et al. 2012) and the first in a female. Features such as amenorrhea, probably due to ovarian failure, and macrocytic anemia have not been previously reported for syndromes associated with MPV17.
Discussion
The connection between MPV17 and a hepatocerebral mitochondrial DNA depletion syndrome has been known since 2006 (Spinazzola et al. 2006), when an integrative genomics strategy suggested a mitochondrial gene resided in a chromosomal region linked with hepatocerebral disease in infants. Previously, MPV17 had been thought to be a peroxisomal protein, but studies indicated that it was in fact localized to the mitochondrial inner membrane (Spinazzola et al. 2006). Since this initial breakthrough, little additional insight has been gained into how mutations in this gene cause mitochondrial DNA depletion, or why the liver and nervous system are disproportionately affected. Other genes associated with hepatocerebral mitochondrial DNA depletion such as POLG and DGUOK have clear roles in mitochondrial DNA synthesis and maintenance. Deletion of the yeast ortholog of MPV17 recapitulates the DNA depletion phenotype, and experiments in yeast have suggested that MPV17 may be important for anaplerosis of TCA cycle intermediates and for maintaining mitochondrial integrity during stress conditions (Dallabona et al. 2010). It is important to note that P98, the residue mutated in this patient, is conserved across evolution in organisms including worms and yeast (Blakely et al. 2012). Further insight into the molecular function of MPV17 may provide better diagnostic or therapeutic approaches to patients with DNA depletion syndromes.
Adult-onset disease caused by mutations in MPV17 was only recently recognized. As in other reported cases, our patient did not have any upper motor neuron findings or disabling cognitive deficits despite the abnormal imaging that showed a leukoencephalopathy (Blakely et al. 2012). She did report some memory and word-finding difficulties as discussed above. The polyneuropathy in these syndromes is axonal, with a dropout of both large- and small-diameter nerve fibers, consistent with the patient’s weakness, gait abnormality, and loss of sensation.
Liver involvement in patients with MPV17 mutations is quite common but typically manifests in neonates and in children under the age of 5 (Nogueira et al. 2012). Manifestations can include steatohepatitis, cholestasis, cirrhosis, and liver failure (Holve et al. 1999). The mechanism is thought to be from impaired mitochondrial oxidative phosphorylation and fatty acid oxidation, resulting in impaired bile flow and steatosis, cell death, and fibrogenesis (Lee and Sokol 2007). Interestingly, our patient’s presentation included steatohepatitis and cirrhosis with portal hypertension that was not symptomatic until adulthood, but then progressed rapidly within a year as would be consistent with cases in much younger patients (Holve et al. 1999). We speculate that this course reflects a threshold of mitochondrial DNA depletion that is reached later in life in adult cases, but is then followed by rapid hepatic deterioration. Unfortunately, medical therapies for mitochondrial hepatopathies are largely ineffective (Lee and Sokol 2007). Liver transplantation was considered in this patient but not pursued due to concurrent neuromuscular disease, poor functional status, and her severe respiratory infection, which proved to be her ultimate cause of death.
To our knowledge, this is the third report of an adult, and the first female, with an MPV17-mediated mitochondrial neurohepatopathy. Even though a mitochondrial DNA depletion syndrome satisfactorily accounts for the majority of her symptoms, it must be noted, however, given the known consanguinity of the patient’s parents, that the coexistence of another recessive disorder cannot be fully excluded. This patient’s initial presentation was with amenorrhea after normal menarche. Because no other adult females with MPV17 mutations have been reported, it is possible that MPV17 has a critical but unappreciated function in the postpubertal ovary. The role of mitochondrial DNA depletion in ovarian function has not been extensively studied, but there are reports of reduced mitochondrial DNA content in the blood of women with premature ovarian failure (Kim et al. 2004), and mutations in POLG were associated with premature ovarian failure and mitochondrial DNA depletion (Luoma et al. 2004; Pagnamenta et al. 2006). Women with premature ovarian failure may represent milder forms of mitochondrial depletion syndromes that have not yet caused typical heptocerebral symptoms, and should be considered for evaluation for mitochondrial disorders if nervous system or hepatic disease are present or develop. Finally, mitochondrial DNA depletion syndromes ought to be considered when severe axonal polyneuropathies occur in young people with coexisting cryptogenic hepatic failure or leukoencephalopathy.
Acknowledgment
The authors would like to thank Robert B. Layzer, MD, from the University of California, San Francisco, for his help in evaluating this patient.
Summary
Mitochondrial DNA depletion syndromes can present in adulthood with multisystem involvement, including amenorrhea and megaloblastic anemia as newly described in this report.
Compliance with Ethics Guidelines
Bryce Mendelsohn has received consulting fees from Counsyl.
Neil Mehta, Bilal Hameed, Melike Pekmezci, Seymour Packman, and Jeffrey Ralph declare that they have no conflicts of interest.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). The studies described in this report are of a retrospective nature regarding clinical care and do not require informed consent. No identifying information is provided. No animals were used in this study.
Drs. Mendelsohn, Mehta, Hameed, Packman, and Ralph all clinically evaluated the patient and provided critical insight into medical and diagnostic management. Dr. Pekmezci reviewed and reported the pathology samples described herein. All authors contributed to and critically reviewed the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Bryce A. Mendelsohn, Email: mendelsohnb@peds.ucsf.edu
Collaborators: Johannes Zschocke and K Michael Gibson
References
- AlSaman A, Tomoum H, Invernizzi F, Zeviani M. Hepatocerebral form of mitochondrial DNA depletion syndrome due to mutation in MPV17 gene. Saudi J Gastroenterol. 2012;18(4):285–289. doi: 10.4103/1319-3767.98439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely EL, Butterworth A, Hadden RD, et al. MPV17 mutation causes neuropathy and leukoencephalopathy with multiple mtDNA deletions in muscle. Neuromuscul Disord. 2012;22(7):587–591. doi: 10.1016/j.nmd.2012.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallabona C, Marsano RM, Arzuffi P, et al. Sym1, the yeast ortholog of the MPV17 human disease protein, is a stress-induced bioenergetic and morphogenetic mitochondrial modulator. Hum Mol Genet. 2010;19(6):1098–1107. doi: 10.1093/hmg/ddp581. [DOI] [PubMed] [Google Scholar]
- El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. 2013;10(2):186–198. doi: 10.1007/s13311-013-0177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Li FY, Schmitt E, Zhang S, Craigen WJ, Wong LJ. MPV17-associated hepatocerebral mitochondrial DNA depletion syndrome: new patients and novel mutations. Mol Genet Metab. 2010;99(3):300–308. doi: 10.1016/j.ymgme.2009.10.003. [DOI] [PubMed] [Google Scholar]
- Garone C, Rubio JC, Calvo SE, et al. MPV17 mutations causing adult-onset multisystemic disorder with multiple mitochondrial DNA deletions. Arch Neurol. 2012;69(12):1648–1651. doi: 10.1001/archneurol.2012.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holve S, Hu D, Shub M, Tyson RW, Sokol RJ. Liver disease in Navajo neuropathy. J Pediatr. 1999;135(4):482–493. doi: 10.1016/S0022-3476(99)70172-1. [DOI] [PubMed] [Google Scholar]
- Karadimas CL, Vu TH, Holve SA, et al. Navajo neurohepatopathy is caused by a mutation in the MPV17 gene. Am J Hum Genet. 2006;79(3):544–548. doi: 10.1086/506913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, Lee SH, Cho SW, et al. The quantitative analysis of mitochondrial DNA copy number in premature ovarian failure patients using the real-time polymerase chain reaction. Korean J Obstet Gynecol. 2004;47(1):16–24. [Google Scholar]
- Lee WS, Sokol RJ. Liver disease in mitochondrial disorders. Semin Liver Dis. 2007;27(3):259–273. doi: 10.1055/s-2007-985071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luoma P, Melberg A, Rinne JO, et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004;364(9437):875–882. doi: 10.1016/S0140-6736(04)16983-3. [DOI] [PubMed] [Google Scholar]
- Nogueira C, de Souza CF, Husny A, Derks TG, Santorelli FM, Vilarinho L. MPV17: fatal hepatocerebral presentation in a Brazilian infant. Mol Genet Metab. 2012;107(4):764. doi: 10.1016/j.ymgme.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Pagnamenta AT, Taanman JW, Wilson CJ, et al. Dominant inheritance of premature ovarian failure associated with mutant mitochondrial DNA polymerase gamma. Hum Reprod. 2006;21(10):2467–2473. doi: 10.1093/humrep/del076. [DOI] [PubMed] [Google Scholar]
- Spinazzola A, Viscomi C, Fernandez-Vizarra E, et al. MPV17 encodes an inner mitochondrial membrane protein and is mutated in infantile hepatic mitochondrial DNA depletion. Nat Genet. 2006;38(5):570–575. doi: 10.1038/ng1765. [DOI] [PubMed] [Google Scholar]