

Abstract
Severe hypertriglyceridemia (sHTG) (plasma triglyceride level > 10 mmol/L) due to lipoprotein lipase (LPL) deficiency is a known risk factor for acute pancreatitis. A 23-day-old male with sHTG was admitted to the Neonatal Intensive Care Unit for plasmapheresis being at high risk for acute pancreatitis. Given the potential hazard of an extracorporeal technique in a very young infant, we decided to perform an exchange transfusion (ET), a procedure widely used by neonatologists and less invasive than plasmapheresis. ET led to a dramatic reduction in plasma triglyceride level, from 93.2 to 3.8 mmol/L at the end of the procedure, without adverse events. The subsequent administration of a special formula low in fat and high in medium-chain triglycerides was effective in keeping fasting plasma triglyceride level below 5.6 mmol/L during the first 5 months of life. The sequence of LPL gene revealed that the patient was apparently homozygous for a novel nucleotide deletion (c.840delG) in exon 6 leading to a premature termination codon (p.N281Mfs*23). However, family studies revealed that while the patient’s mother was heterozygous for this mutation, the father was heterozygous for a novel deletion eliminating the whole LPL gene. The patient therefore turned out to be a compound heterozygous for two LPL gene mutations predicted to abolish LPL activity. This is the first case of sHTG treated with ET in a neonate reported in the literature. ET appears to be a safe procedure, alternative to plasmapheresis, to prevent acute pancreatitis in young infants with sHTG due to LPL deficiency.
Introduction
Hypertriglyceridemia is conventionally defined as severe (sHTG) when the level of fasting plasma triglyceride (TG) is > 10 mmol/L. This condition is often referred to as chylomicronemia because of plasma accumulation of chylomicrons in the fasting state (Brahm and Hegele 2013). Familial chylomicronemia, also known as hyperlipoproteinemia (HLP) type 1, is a rare recessive disorder which often presents during early infancy and childhood (Brahm and Hegele 2013; Feoli-Fonseca et al. 1998). Fasting serum TG level is generally > 10 mmol/L and sometimes can exceed 100 mmol/L. The clinical features include failure to thrive, eruptive xanthomas, lipemia retinalis, hepatosplenomegaly, recurrent abdominal pain, and episodes of acute pancreatitis (Brahm and Hegele 2013; Feoli-Fonseca et al. 1998; Kavazarakis et al. 2004; Avis et al. 2010; Chen et al. 2012). Familial chylomicronemia is due to a defect in the lipolytic cascade of TG-rich lipoproteins that may result from mutations in at least five different genes: LPL (encoding the lipoprotein lipase, LPL; OMIM #238600), APOC2 (encoding apolipoprotein CII, the activator of LPL; OMIM #207750), APOA5 (encoding apolipoprotein AV, also an activator of LPL; OMIM #144650), GPIHBP1 (encoding the molecular platform which, on the endothelial surface of capillaries, allows the interactions of LPL with TG-rich lipoproteins, apolipoprotein CII, and apolipoprotein AV; OMIM #612757), and LMF1 (encoding a tissue factor which allows the secretion of functional LPL and hepatic lipase, HL; OMIM #611761) (Johansen et al. 2011; Davies et al. 2012; Surendran et al. 2012). LPL deficiency due to mutations in the LPL gene is the most common disorder (95 % of cases) with an estimated prevalence of one per million individuals in most populations (Brahm and Hegele 2013).
Treatment of patients with familial chylomicronemia requires severe dietary fat restriction in order to maintain fasting TG levels below 4.5–5.6 mmol/L to reduce the risk of pancreatitis (Feoli-Fonseca et al. 1998; Leaf 2008). Plasmapheresis constitutes a therapeutic option in selected cases of sHTG for preventing this potentially life-threatening complication (Ewald and Kloer 2009; Stefanutti et al. 2009).
In this study, we report on a newborn with sHTG admitted to a Neonatal Intensive Care Unit (NICU) for therapeutic plasmapheresis by plasmafiltration, who was successfully treated with exchange transfusion (ET), a less invasive procedure than plasmapheresis. The patient was found to be a compound heterozygous for two novel LPL mutations, including the complete deletion of LPL gene.
Materials and Methods
Patient
The patient was a male neonate delivered by caesarean section after a full-term uneventful pregnancy. His birth weight was 3,305 g; he had a normal physical examination. Breastfeeding was started on day 1. He was discharged from hospital on day 6 of life in good clinical conditions with complementary feeding. His parents, apparently unrelated, were in good health as his two sisters.
On day 23 of life the capillary blood, collected for the second Guthrie screening routinely performed at the local hospital, was remarkably pink-creamy colored. Laboratory tests revealed extremely elevated plasma levels of TG (140 mmol/L) and total cholesterol (TC) (17.3 mmol/L); plasma lipase level was 209 IU/L (normal value 7–60 IU/L). Enteral feeding was suspended, an electrolyte solution was started, and the baby was transferred to our NICU after 12 h to undergo plasmafiltration.
At admission, the baby appeared in good health. Physical examination showed weight, height, and head circumference within the normal range (50th–75th percentile for his age), mild hepatomegaly, and some small yellowish papules on the face resembling eruptive xanthoma. Ophthalmoscopic examination revealed creamy-white retinal vessels, typical of lipemia retinalis. Blood chemistry confirmed the high plasma TG and TC levels (93.2 and 20.6 mmol/L, respectively); transaminase, alkaline phosphatase, total bilirubin, and serum electrolytes were within the normal range. No other biochemical data could be obtained because of the lipemic serum. Blood cell count was normal; hemoglobin level was 9.9 g/dL. Fasting was continued and the different therapeutic options were carefully evaluated. Conservative treatment only (i.e., dietary therapy) and plasmapheresis by plasmafiltration were excluded in favor of ET. A blood sample was collected from the patient and his family members for LPL gene analysis. A written informed consent was obtained from the parents before initiating the ET procedure and for DNA analysis.
Exchange Transfusion (ET) Procedure
ET is a procedure by which an infant’s blood is replaced with donor blood by repeatedly removing and replacing small aliquots of blood over a short time period. We used fresh (collected <5 days before the procedure), filtered, irradiated 0 Rh-negative packed red blood cells suspended in AB plasma in a ratio to ensure a hematocrit of 40 %. Aliquots of 10 mL of donor blood were exchanged with infant blood till a cumulative volume of exchange of 200 mL/kg (twofold and half the estimated infant blood volume) was reached. ET was performed using a central venous catheter (double-lumen 5 French) placed in the right internal jugular vein by surgical cutdown, avoiding ligation of the vessel. The “pull-push technique” through special four-way stopcock was used. The procedure was completed within 120 min. No adverse events were observed except for mild thrombocytopenia which resolved spontaneously.
Analysis of LPL Gene
The exons and the promoter region of LPL gene (NG_008855.1, Chromosome 8p22) were amplified from genomic DNA by polymerase chain reaction (PCR) and sequenced using appropriate primers (Bertolini et al. 2000). Multiplex ligation-dependent probe amplification (MLPA) (SALSA MLPA P218-B1 LPL probe mix, MRC Holland, Amsterdam, the Netherlands) was used for the detection of major rearrangements of LPL gene. The PCR products were separated on an ABI PRISM 3100 sequencer and the data analyzed by Peak Scanner™ software v1.0. The mutations were designated according to the Human Genome Variation Society, 2012 version (http://www.hgvs.org/mutnomen/recs-DNA.html). LPL protein sequence variants were designated according to http://www.hgvs.org/mutnomen/recs-prot.html.
Results
ET Procedure and Follow-Up
ET led to a dramatic reduction in plasma TG level: 3.84 mmol/L at the end of the procedure and 1.95 mmol/L 12 h later. Lipemia retinalis disappeared. Fasting and a lipid-free parenteral nutrition were continued. After 72 h from ET, laboratory tests were as follows: TG 1.03 mmol/L, TC 3.77 mmol/L, high-density lipoprotein cholesterol (HDL-C) 0.85 mmol/L, low-density lipoprotein cholesterol (LDL-C) 2.45 mmol/L; amylase, lipase, and the other biochemical parameters were within the normal range. Hemoglobin was 13.1 g/dL. After 3 days fasting, diet with a special formula low in fat and high in medium-chain triglycerides (MCTs) was started (Monogen®, 25 % of calories as fat, 90 % of fat as MCT). Plasma lipids slightly increased after starting the diet: TG 3.83 mmol/L, TC 4.13 mmol/L (HDL-C 0.54 mmol/L, LDL-C 2.25 mmol/L) after 4 days; TG 5.32 mmol/L, TC 4.13 mmol/L (HDL-C 0.62 mmol/L, LDL-C 2.09 mmol/L) after 8 days; TG 4.04 mmol/L, TC 3.20 mmol/L (HDL-C 0.57 mmol/L, LDL-C 1.47 mmol/L) after 14 days. Parenteral nutrition was discontinued and the catheter removed after 7 days from ET. The baby was discharged after 20 days in excellent clinical conditions with feeding with Monogen® milk. At the age of 5 months, the baby is still in good health and presents a normal growth (weight, height, and head circumference within the 75th percentile) and development. He is fed with the same milk and plasma lipid values are similar to those recorded at discharge: TG 4.18 mmol/L, TC 3.36 mmol/L, HDL-C 0.59 mmol/L, LDL-C 1.60 mmol/L.
Before the child was discharged, family members were evaluated for presence of dyslipidemia. In all subjects, fasting plasma lipids were found to be within the normal range for age and sex.
Analysis of LPL Gene
The resequencing of LPL gene revealed that the patient was homozygous for a single nucleotide deletion in exon 6 (c.840delG) (Fig. 1). This mutation causes a shift in the reading frame leading to a truncated protein (p.N281Mfs*23) of 302 amino acids. Therefore, the sequence of exon 6 of LPL gene was performed in all family members (parents and two sisters) available for study. This screening revealed that the mother and one sister of the patient were heterozygous carriers of the mutation (Fig. 1). The absence of the mutation in the father (Fig. 1) was inconsistent with the genotype of the patient, suggesting either a condition of non-paternity or that the father might be heterozygous carrier of a partial deletion of LPL gene eliminating exon 6. To test this hypothesis, we analyzed the DNA of all family members by MLPA and found that the patient’s father was, in fact, heterozygous for a complete deletion of LPL gene (Fig. 2) that had been transmitted to the patient. The latter therefore turned out to be a compound heterozygote with one allele harboring the c.840delG and the other harboring of deletion of the LPL gene. The LPL deletion was also found in the patient’s sister previously found to be negative for the c.840delG mutation.
Fig. 1.
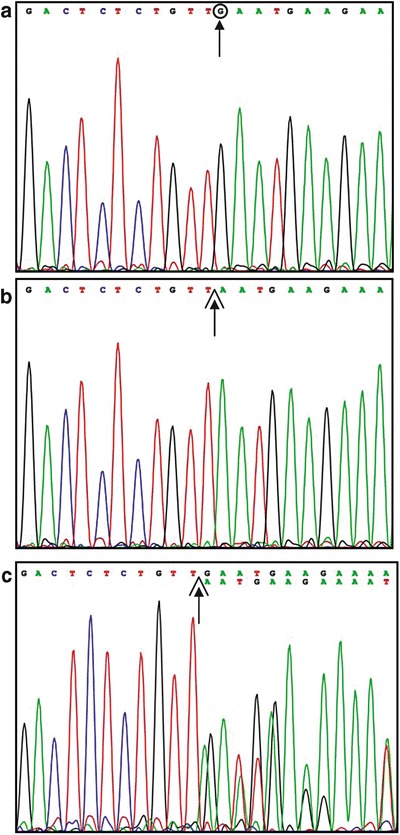
Partial nucleotide sequence of exon 6 of LPL gene. The hypertriglyceridemic patient (panel b) appears to be homozygous for a single nucleotide deletion (c.840delG). This mutation was found in patient’s mother (panel c) in heterozygous state, but not in patient’s father (panel a) whose sequence was normal
Fig. 2.
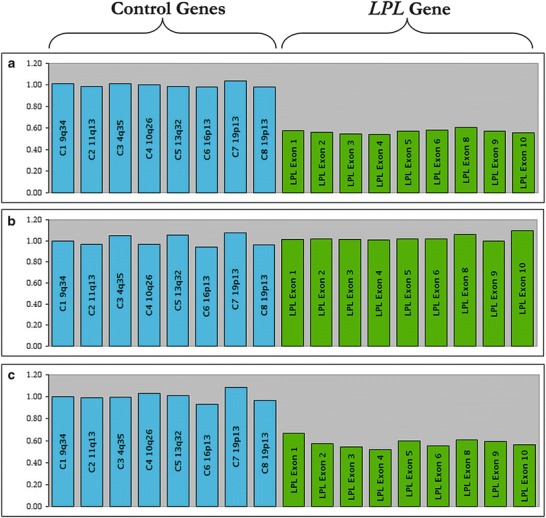
Multiplex ligation-dependent probe amplification (MLPA) of LPL gene. The height of the histograms indicates that the patient is heterozygous for a large deletion which eliminates the entire LPL gene (panel a); this deletion is present also in the patient’s father (panel c), but not in the patient’s mother (panel b)
Discussion
Clinical Aspects
In this study, we describe the clinical features and the treatment of a 23-day-old male with sHTG who was admitted to our NICU for plasmapheresis, being at high risk for acute pancreatitis. Therapeutic plasmapheresis can be used to rapidly reduce TG levels as patients with sHTG are at high risk for acute pancreatitis (Ewald and Kloer 2009; Stefanutti et al. 2009).
Since 1978, different plasmapheresis techniques (plasmafiltration, plasma exchange) have been applied to the treatment of adult patients with sHTG. The use of plasmapheresis in newborn infants raises great concern, because of the potential risks related to the extracorporeal procedure, particularly the hemodynamic effects and hemorrhagic events. Plasmafiltration would be preferred in very young infants since it requires a lower volume of extracorporeal circulation than plasma exchange, but it seems to be less effective in sHTG, as chylomicrons remain trapped in the primary plasma filter because of their large size and high molecular weight (Ewald and Kloer 2009; Stefanutti et al. 2013). Stefanutti et al. (2004, 2013) reported two cases of sHTG in very young infants (a 25-day-old male and a 3-month-old female) treated successfully with plasma exchange without adverse events. These authors introduced modifications to the standard procedure to minimize the risks. Given the little chance of success with plasmafiltration we are familiar with and our limited experience in plasma exchange plasmapheresis, we decided to perform an ET. ET was introduced in the late 1940s to decrease the mortality of hemolytic disease of the newborn and to prevent kernicterus in the surviving infants. Since then, ET has been applied to many diseases (such as high levels of unconjugated hyperbilirubinemia in the newborn due to any cause, severe anemia, disseminated intravascular coagulation, neonatal sepsis), and has become one of the most common procedures performed by neonatologists (Steiner et al. 2007). The most common ET-related adverse effects include thrombocytopenia, hypocalcemia, hyperkalemia, apnea, bradycardia, hypotension, and catheter-related complications. A retrospective study by Steiner et al. (2007) reported ET-related morbidity and mortality over a 20-year period (1986–2006) at Yale New Haven Hospital. The majority of complications were transient and, when serious adverse events occurred, they were observed in infants who were preterm and/or very sick. No deaths were related to ET during the study period.
In our patient, ET led to a significant and immediate reduction in plasma TG level without adverse events. This effect is due to the ET procedure “per se” and, possibly, to the presence in transfused blood of LPL released from blood mononuclear cells (known to produce minute amounts of LPL) (Merkel et al. 2002; Stengel et al. 1998). Dietary therapy, started after the ET procedure, was effective in maintaining fasting plasma TG level below 5.6 mmol/L over time.
To the best of our knowledge, this is the first case of sHTG treated with ET in a newborn infant reported in the literature. Given the result we achieved, we believe that ET should be taken into account by neonatologists and pediatricians to treat sHTG in very young infants in order to rapidly decrease elevated TG levels and thus avoiding the potentially fatal risk of acute pancreatitis. This observation sets the stage for a future clinical trial of exchange transfusion in newborns with sHTG due to LPL deficiency or other genetic or acquired conditions.
Molecular Aspects
The hypothesis that the patient had familial chylomicronemia was confirmed by the analysis of LPL gene, which revealed at first that he was apparently homozygous for a single nucleotide deletion (c.840delG) predicted to result in a truncated protein. This truncated protein belongs to a group of functionally inactive LPL proteins, resulting from mutations that cause the elimination of the carboxyl-terminal domain of the protein (Bertolini et al. 2000; Merkel et al. 2002). This domain is involved in the intracellular processing and secretion of LPL, the formation of functionally active LPL homodimer, the interaction of LPL with TG-rich lipoproteins, and in the binding of LPL to the LDL receptor-related proteins (LRPs) and glycosaminoglycans of the endothelial cell surface (Murthy et al. 1996; Buscà et al. 1998; Wang and Eckel 2009).
The unexpected absence of the mutation c.840delG in the patient’s father prompted us to consider the hypothesis that the father had transmitted to his son a partial deletion of LPL gene involving exon 6 (the exon harboring the c.840delG). By applying the MLPA method, we found that the patient’s father was heterozygous for a large deletion eliminating the whole LPL gene. This deletion was transmitted to the patient (who was in fact a compound heterozygote) and to one of his healthy sisters. As far as we know, this is the first report of a complete deletion of LPL gene in humans. At the moment, we do not know the size of this deletion and whether or not other nearby genes are involved. The two heterozygous carriers of this deletion (the father and the sister of the patient) appear to be in good health and to have a normal plasma lipid profile.
Acknowledgments
The authors would like to thank the patient and his family for their participation, Dr. Paolo Ernesto Villani (NICU, Department Mother and Children, Hospital “C. Poma”, Mantua, Italy) for getting the patient transferred to our NICU, Dr. Gianluigi Ardissino (Unit of Pediatric Nephrology, Fondazione IRCCS Ca’ Granda Ospedale Maggiore Policlinico, University of Milan, Milan, Italy) for helping us in evaluating different therapeutic options.
Synopsis
Exchange transfusion appears to be a safe procedure, alternative to plasmapheresis, to prevent acute pancreatitis in young infants with severe hypertriglyceridemia due to lipoprotein lipase deficiency.
Compliance with Ethics Guidelines
Conflict of Interest
Lorenza Pugni, Enrica Riva, Carlo Pietrasanta, Claudio Rabacchi, Stefano Bertolini, Cristina Pederiva, Fabio Mosca, and Sebastiano Calandra declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from patient’s parents for being included in the study. This manuscript is a case report containing no identifying patient information, and examinations and treatment of the patient fall in clinical practice.
Authors’ Contributions
Lorenza Pugni, Carlo Pietrasanta, and Fabio Mosca designed the study, performed the blood exchange transfusion and collected the data. Enrica Riva and Cristina Pederiva followed up the patient after the discharge. Sebastiano Calandra, Claudio Rabacchi, and Stefano Bertolini performed the molecular investigation and analyzed the molecular data. Lorenza Pugni and Sebastiano Calandra wrote the manuscript.
Funding Support
Genetic analysis was supported in part by a grant from Emilia-Romagna Region: RARER – Area1 (E35E09000880002) project to Sebastiano Calandra.
Footnotes
Competing interests: None declared
Contributor Information
Lorenza Pugni, Email: lorenza.pugni@mangiagalli.it.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Avis HJ, Scheffer HJ, Kastelein JJP, Dallinga-Thie GM, Wijburg FA. Pink-creamy whole blood in a 3-month-old infant with a homozygous deletion in the lipoprotein lipase gene. Clin Genet. 2010;77:430–433. doi: 10.1111/j.1399-0004.2009.01369.x. [DOI] [PubMed] [Google Scholar]
- Bertolini S, Simone ML, Pes GM, et al. Pseudodominance of lipoprotein lipase (LPL) deficiency due to a nonsense mutation (Tyr302>Term) in exon 6 of LPL gene in an Italian family from Sardinia (LPLOlbia) Clin Genet. 2000;57:140–147. doi: 10.1034/j.1399-0004.2000.570209.x. [DOI] [PubMed] [Google Scholar]
- Brahm A, Hegele RA. Hypertriglyceridemia. Nutrients. 2013;5:981–1001. doi: 10.3390/nu5030981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buscà R, Martinez M, Vilella E, et al. The carboxy-terminal region of human lipoprotein lipase is necessary for its exit from the endoplasmic reticulum. J Lipid Res. 1998;39:821–833. [PubMed] [Google Scholar]
- Chen YH, Ke ZL, Wang YX, Wang Y, Zheng YZ. Two case reports of familial chylomicronemia syndrome. Case Rep Pediatr. 2012;2012:384719. doi: 10.1155/2012/384719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BS, Beigneux AP, Fong LG, Young SG. New wrinkles in lipoprotein lipase biology. Curr Opin Lipidol. 2012;23:35–42. doi: 10.1097/MOL.0b013e32834d0b33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald N, Kloer HU. Severe hypertriglyceridemia: an indication for apheresis? Atheroscler Suppl. 2009;10:49–52. doi: 10.1016/S1567-5688(09)71810-0. [DOI] [PubMed] [Google Scholar]
- Feoli-Fonseca JC, Lévy E, Godard M, Lambert M. Familial lipoprotein lipase deficiency in infancy: clinical, biochemical and molecular study. J Pediatr. 1998;133:417–423. doi: 10.1016/S0022-3476(98)70280-X. [DOI] [PubMed] [Google Scholar]
- Johansen CT, Kathiresan S, Hegele RA. Genetic determinants of plasma triglycerides. J Lipid Res. 2011;52:189–206. doi: 10.1194/jlr.R009720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavazarakis E, Stabouli S, Gourgiotis D, et al. Severe hypertriglyceridemia in a Greek infant: a clinical, biochemical and genetic study. Eur J Pediatr. 2004;163:462–466. doi: 10.1007/s00431-004-1474-1. [DOI] [PubMed] [Google Scholar]
- Leaf DA. Chylomicronemia and the chylomicronemia syndrome: a practical approach to management. Am J Med. 2008;121:10–12. doi: 10.1016/j.amjmed.2007.10.004. [DOI] [PubMed] [Google Scholar]
- Merkel M, Eckel RH, Goldberg IJ. Lipoprotein lipase: genetics, lipid uptake, and regulation. J Lipid Res. 2002;43:1997–2006. doi: 10.1194/jlr.R200015-JLR200. [DOI] [PubMed] [Google Scholar]
- Murthy V, Julien P, Gagne C. Molecular pathobiology of human lipoprotein lipase gene. Pharmacol Ther. 1996;70:101–135. doi: 10.1016/0163-7258(96)00005-8. [DOI] [PubMed] [Google Scholar]
- Stefanutti C, Lanti A, Di Giacomo S, et al. Therapeutic apheresis in low weight patients: technical feasibility, tolerance, compliance, and risks. Transfus Apher Sci. 2004;31:3–10. doi: 10.1016/j.transci.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Stefanutti C, Di Giacomo S, Vivenzio A, et al. Therapeutic plasma exchange in patients with severe hypertriglyceridemia: a multicenter study. Artif Organs. 2009;33:1096–1102. doi: 10.1111/j.1525-1594.2009.00810.x. [DOI] [PubMed] [Google Scholar]
- Stefanutti C, Gozzer M, Pisciotta L, et al. A three month-old infant with severe hyperchylomicronemia: molecular diagnosis and extracorporeal treatment. Atheroscler Suppl. 2013;14:73–76. doi: 10.1016/j.atherosclerosissup.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Steiner LA, Bizzarro MJ, Ehrenkranz RA, Gallagher PG. A decline in the frequency of neonatal exchange transfusions and its effect on exchange-related morbidity and mortality. Pediatrics. 2007;120:27–32. doi: 10.1542/peds.2006-2910. [DOI] [PubMed] [Google Scholar]
- Stengel D, Antonucci M, Gaoua W, et al. Inhibition of LPL expression in human monocyte-derived macrophages is dependent on LDL oxidation state: a key role for lysophosphatidylcholine. Arterioscler Thromb Vasc Biol. 1998;18:1172–1180. doi: 10.1161/01.ATV.18.7.1172. [DOI] [PubMed] [Google Scholar]
- Surendran RP, Visser ME, Heemelaar S, et al. Mutations in LPL, APOC2, APOA5, GPIHBP1 and LMF1 in patients with severe hypertriglyceridaemia. J Intern Med. 2012;272:185–196. doi: 10.1111/j.1365-2796.2012.02516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]