

Abstract
Neurological dysfunction is common in humans and animals with lysosomal storage diseases. β-Mannosidosis, an autosomal recessive inherited disorder of glycoprotein catabolism caused by deficiency of the lysosomal enzyme β-mannosidase, is characterized by intracellular accumulation of small oligosaccharides in selected cell types. In ruminants, clinical manifestation is severe, and neuropathology includes extensive intracellular vacuolation and dysmyelination. In human cases of β-mannosidosis, the clinical symptoms, including intellectual disability, are variable and can be relatively mild. A β-mannosidosis knockout mouse was previously characterized and showed normal growth, appearance, and lifespan. Neuropathology between 1 and 9 months of age included selective, variable neuronal vacuolation with no hypomyelination. This study characterized distribution of brain pathology in older mutant mice, investigating the effects of two strain backgrounds. Morphological analysis indicated a severe consistent pattern of neuronal vacuolation and disintegrative degeneration in all five 129X1/SvJ mice. However, the mice with a mixed genetic background showed substantial variability in the severity of pathology. In the severely affected animals, neuronal vacuolation was prominent in specific layers of piriform area, retrosplenial area, anterior cingulate area, selected regions of isocortex, and in hippocampus CA3. Silver degeneration reaction product was prominent in regions including specific cortical layers and cerebellar molecular layer. The very consistent pattern of neuropathology suggests metabolic differences among neuronal populations that are not yet understood and will serve as a basis for future comparison with human neuropathological analysis. The variation in severity of pathology in different mouse strains implicates genetic modifiers in the variable phenotypic expression in humans.
Introduction
β-Mannosidosis (OMIM 248510) is a rare lysosomal storage disease that was first identified in Nubian goats (Jones and Dawson 1981; Jones and Laine 1981; Jones et al. 1983) and later in Salers cattle (Abbitt et al. 1991; Bryan et al. 1993; Patterson et al. 1991) and humans (Alkhayat et al. 1998; Cooper et al. 1986; Wenger et al. 1986). This autosomal recessive inherited disorder of glycoprotein catabolism is caused by a deficiency of the lysosomal enzyme β-mannosidase (MANBA; EC 3.2.1.25, 609489). MANBA acts exclusively at the last step of oligosaccharide catabolism in glycoprotein degradation and functions to cleave the β-linked mannose sugar found in N-linked oligosaccharides of glycoproteins (Neufeld 1991; Winchester et al. 2000). β-Mannosidosis is characterized by the intracellular accumulation of small oligosaccharides (mainly disaccharides and trisaccharides) in selected cell types (Jones and Laine 1981). In human cases of β-mannosidosis, the clinical symptoms, including intellectual disability, are relatively mild and are variable, even in patients with null mutations (Bedilu et al. 2002); however, little is known about the pathology of the disease. In contrast, the two ruminant animal models (goats and cows) have a severe clinical presentation at birth (Jones et al. 1983; Abbitt et al. 1991) and pathology includes extensive regionally variable myelin deficits as well as widespread cytoplasmic vacuolation (Patterson et al. 1991; Lovell and Jones 1983,1985).
A mouse model of β-mannosidosis was created by targeted disruption of the β-mannosidase gene by homologous recombination in 129X1/SvJ ES cells (Zhu et al. 2006). Homozygous mutant animals had the expected enzyme deficiency and had accumulation of disaccharide in brain tissue, but showed normal growth, appearance, and lifespan. Previous examination of mutant animals between 1 and 9 months of age showed selective, variable neuronal vacuolation with no hypomyelination, closer to what would be expected in the human β-mannosidosis phenotype given the human clinical presentation. Cell populations identified with variable vacuolation included pyramidal cells (layer V) in dorsolateral cortex, choroid plexus, specific segments of Ammon’s horn, striatum, amygdala, deep cerebellar nuclei, and spinal cord (Zhu et al. 2006).
This study was designed to define the distribution and severity of central nervous system (CNS) pathology in the isocortex and allocortex of older mice, and to determine if there are phenotypic or pathologic differences based on the clone or background of the animals. This study characterized the distribution of brain pathology in mutant mice between the ages of 15 and 20 months, investigating the effects of two different embryonic stem cell (ES) clones and two strain backgrounds: 129X1/SvJ congenic and mixed C57BL/6J x 129X1/SvJ.
Methods
Animals
A β-mannosidosis knockout mouse was produced by targeted disruption of the β-mannosidosis gene (Zhu et al. 2006). The congenic line, labeled “sv129” in Table 1, was generated by backcrossing the chimeras from the transgenic 129X1/SvJ ES cells (clone A2) to 129X1/SvJ mice. The line labeled “mixed” in Table 1 was generated by backcrossing the chimeras from the transgenic 129X1/SvJ ES cells (clones A2 and B2) to C57BL/6J mice, leading to F1 animals with mixed strain background. The resulting F1 mice of a mixed 129X1/SvJ:C57Bl/6J background were intercrossed to generate homozygotes in the F2 generation. All of the homozygous knockout mice (of both clones and both strains) lacked β-mannosidase enzyme activity and showed similar patterns of storage material (Zhu et al. 2006). This study utilized 21 homozygous mutant mice and 4 wild-type (WT) mice between the ages of 15 and 20 months. See Table 1 under Results for the age and strain for each mouse.
Table 1.
Summary of characteristics and outcomes (severity of pathology)
| ID number | Age (mo) | Sex | Status | Strain | Clone | Vacuolation | Degeneration stain |
|---|---|---|---|---|---|---|---|
| 127 | 20 | M | WT | Mixed | A2 | None | None |
| 365 | 15 | M | WT | Mixed | A2 | None | None |
| 272 | 18 | F | WT | sv129 | A2 | None | None |
| 352 | 16 | F | WT | sv129 | A2 | None | None |
| 104 | 20 | F | Mutant | Mixed | A2 | Mild/variable | None |
| 106 | 20 | F | Mutant | Mixed | A2 | Mild/variable | None |
| 131 | 20 | F | Mutant | Mixed | A2 | Mild | None |
| 149 | 19 | F | Mutant | Mixed | A2 | Mild | None |
| 150 | 19 | F | Mutant | Mixed | A2 | Mild | Mild |
| 209 | 19 | F | Mutant | Mixed | A2 | None | None |
| 154 | 19 | F | Mutant | Mixed | B2 | Mild | None |
| 155 | 19 | F | Mutant | Mixed | B2 | Mild | Mild |
| 160 | 19 | F | Mutant | Mixed | B2 | Mild/variable | Mild |
| 169 | 19 | M | Mutant | Mixed | B2 | Mild | Mild |
| 177 | 19 | M | Mutant | Mixed | B2 | Mild | Mild |
| 361 | 15 | F | Mutant | Mixed | A2 | Mild/variable | Mild/variable |
| 366 | 15 | F | Mutant | Mixed | A2 | Mild/variable | None |
| 371 | 15 | F | Mutant | Mixed | A2 | Severe | Severe |
| 376 | 15 | F | Mutant | Mixed | A2 | Mild/variable | None |
| 386 | 15 | F | Mutant | Mixed | A2 | None | None |
| 262 | 18 | F | Mutant | sv129 | A2 | Severe | Severe |
| 264 | 18 | F | Mutant | sv129 | A2 | Severe | Severe |
| 342 | 16 | F | Mutant | sv129 | A2 | Severe | Severe |
| 348 | 16 | M | Mutant | sv129 | A2 | Severe | Severe |
| 349 | 16 | F | Mutant | sv129 | A2 | Severe | Severe |
See Methods for description of mild, mild/variable, and severe classifications
All animal experiments were approved by the Institutional Animal Care and Use Committee at Michigan State University and were in accordance with the NIH Guide to the Humane Care of Laboratory Animals.
Embedding, Sectioning, and Staining
Animals were perfused transcardially with 4% paraformaldehyde. Following removal from the skull, brains were placed in 4% paraformaldehyde for 24 h, and then transferred to cacodylate storage buffer. Brains were embedded in a 5×5 array in a gelatin matrix using MultiBrain Technology (NeuroScience Associates, Knoxville, TN) as reported in previous studies (Fix et al. 1996; Ong et al. 2001). The block of embedded brains was frozen and sectioned coronally at 30 μm beginning at the olfactory bulb and proceeding to the medulla. A serial set of every sixth section was selected for staining with hematoxylin and eosin (H&E), with Weil myelin stain, and with amino cupric silver stain to reveal disintegrative degeneration (Ong et al. 2001). A few selected sections were subjected to Nissl staining, GFAP staining for astrocytes, or Iba1 staining for microglia.
Since each of the large sections cut from the block was a composite section holding individual sections from each of the brains embedded in each block, uniformity of staining was achieved across normal and mutant mice. Sections at each level were analyzed for the distribution and extent of pathology as seen in each type of stain.
Morphological Analysis
Sections from all animals at all levels were analyzed to determine the overall characteristics and severity of pathology. Charts using a 5×5 grid were developed to record observations on each section at each level without regard to status of strain or clone. The type and extent of the pathology in each structure was recorded. Severity was classified as mild, mild/variable, and severe by characterizing the extent of vacuolation or degeneration staining in specific regions. For those regions where pathology was observed in any of the animals, “severe” indicated that vacuoles or degeneration staining were prominent in all of the affected regions. “Mild” indicated less prominent pathology in those regions. “Mild/variable” indicated that mild pathology was present in some regions, but in some regions, no pathology was apparent. After the variation in severity was determined, selected sections were analyzed in greater detail to record the precise location of vacuolation and degeneration staining in each severely affected mouse.
Results
Overall Severity of Pathology Analyzed by Strain and Clone
Table 1 provides a summary of the age and strain for each mouse, indicating the overall severity of vacuolation and degeneration staining. Analysis indicated a severe consistent pattern of neuronal vacuolation and disintegrative degeneration in all five 129X1/SvJ mice (clone A2). However, the mixed background mice (A2 and B2 clones) showed substantial variability in the severity of pathology (Table 1). There was no discernible difference related to the ES clone. In the severely affected animals (all 129X1/SvJ mutant mice, one mixed background mouse), neuronal vacuolation was prominent in a consistent pattern. Thus, the background strain was an important factor in determining the phenotype of the CNS pathology.
Distribution of Vacuolation
The vacuole distribution at four selected levels is shown diagrammatically in Fig. 1. In general, vacuolation consistently occurred in specific layers of piriform cortex, retrosplenial cortex, anterior cingulate cortex, some dorsal and lateral cortical regions, and in hippocampus. The structures involved are illustrated and will be discussed as labeled in the Allen Mouse Brain Atlas (http://mouse.brain-map.org/). In the dorsal hippocampus, the only neurons showing vacuolation were pyramidal cells in the dorsal aspect of CA3 (Fig. 1c), in a narrow region at the junction of CA3 and CA2. An example of the specificity of the vacuolation is demonstrated in Fig. 2b,g in HE-stained and Weil-myelin-stained sections. In the medial isocortex, vacuolation was prominent in neurons of the anterior cingulate cortex (rostrally) and the retrosplenial cortex (caudally) (Fig. 2b,e,f). In the retrosplenial cortex, the vacuolation appeared more dense in the ventral retrosplenial cortex compared to the dorsal retrosplenial cortex and lateral agranular part, resulting in an apparent gap (Fig. 1c,d; Fig. 2b,f) between the vacuolation in medial cortex and in dorsal cortex. A similar gap is seen more rostrally (Fig. 1b), with vacuolation more prominent in the ventral anterior cingulate cortex than in the dorsal anterior cingulate cortex. In Fig. 1b,c,d and Fig. 2b,e,f, the prominent vacuolation in regions of dorsal and lateral cortex is illustrated. With respect to layers showing vacuolation, it appeared that in the medial cortex, neurons in layers 4–5 contained vacuoles, while vacuolation was prominent in layer 5 of dorsal and lateral cortex. The vacuoles were intraneuronal, as illustrated in Fig. 2d, and previously demonstrated with 1μ thick toluidine-blue-stained plastic sections and electron microscopy (Zhu et al. 2006).
Fig. 1.
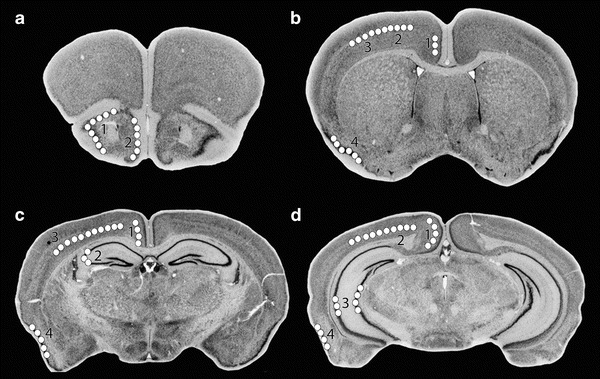
Structures showing prominent vacuolation at four levels of coronal sections. Diagrams are from The Mouse Brain Library DBA2J atlas (www.mbl.org). Regions with consistent vacuolation are shown by white circles on one side of the brain; all lesions are bilaterally symmetric. Regions involved include: 1 – molecular layer of piriform area, 2 – layer 2 of taenia tecta (a); 1 – anterior cingulate cortex, 2 – layer 5 of primary motor area, 3 – layer 5 of primary somatosensory area, 4 – molecular layer of piriform area (b); 1 – retrosplenial cortex, 2 – hippocampus CA3, 3 – layer 5 of primary motor, somatosensory, visual cortex, 4 – piriform area (c); 1 – retrosplenial cortex, 2 – layer 5 of visual cortex, 3 – parts of hippocampus including part of CA3, 4 – piriform area (d)
Fig. 2.
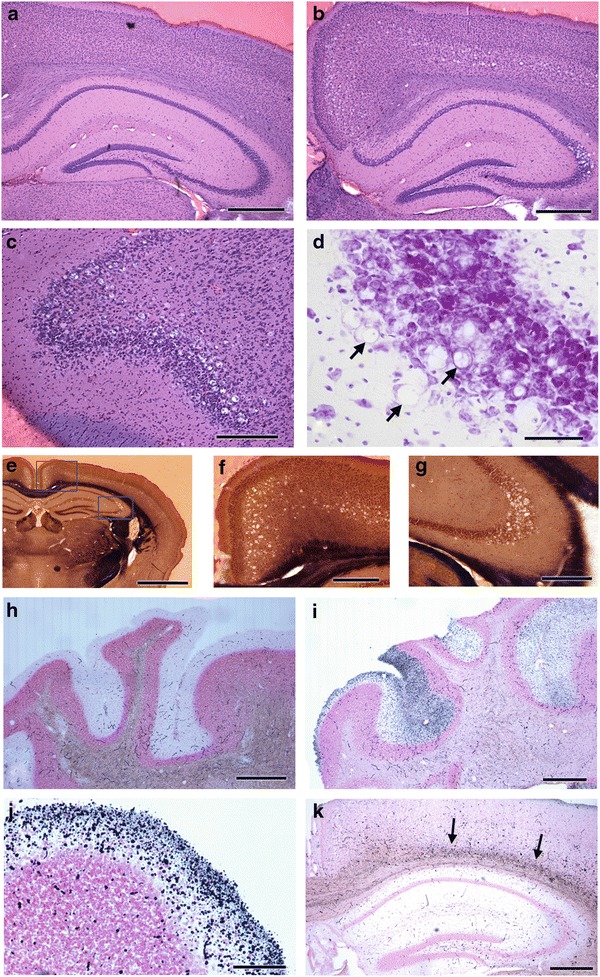
Examples of pathology in selected locations in HE-stained sections (a–d), Weil-myelin-stained sections (e–g), and amino cupric silver–stained sections (h-k). Extensive vacuolation is present in hippocampus CA3, retrosplenial cortex, and cerebral cortex layer 5 in an affected mouse (b) compared to a control mouse (a). Vacuolation is prominent in piriform area in frontal lobe (c). Enlargement of vacuoles in part of hippocampus CA3 in Nissl stain reveals intracytoplasmic vacuoles; arrows indicate cells where a flattened nucleus is visible adjacent to a vacuole (d). Low-magnification view of one hemisphere (e) demonstrates restricted locations of vacuoles in cortex and hippocampus, with higher magnification views, indicated by rectangles, showing vacuolation in retrosplenial cortex and cerebral cortex layer 5 (f) and part of hippocampus CA3 (g). The absence of cupric silver reaction product in control cerebellum molecular layer, except for red blood cells in vessels (h) contrasts with reaction product in a section of cerebellum from an affected mouse showing degeneration staining in the molecular layer, variable among folia (i). Large numbers of silver-stained puncta consistent with degenerating nerve terminals were observed in some folia (j). Localized degeneration staining was consistently observed in deep layers of cerebral cortex of affected mice (k). Scale bars and mouse ID numbers for each image: a-300 μm, #352; b-300 μm, #262; c-250 μm, #264; d-20 μm, #349; e- 900 μm, #348; f – 275 μm, #348; g-200 μm, #349; h-520 μm, #352; i-520 μm, #348; j-250 μm, #349; k-420 μm, #262
Distribution of Degeneration Staining
A consistent pattern of silver reaction product was observed only in the same β-mannosidosis mutants that showed substantial vacuolation. In the cerebellar molecular layer, large numbers of silver-stained puncta consistent with degenerating nerve terminals were observed, with variation among folia in the intensity of staining (Fig. 2i,j). No neuronal abnormalities were observed in inferior olivary nuclei (source of projection of climbing fibers to the cerebellar cortex). In the region dorsal to the hippocampus, silver-stained puncta over a linear area were observed (Fig. 2k); the most intense staining of puncta occurred in deep layers of cortex adjacent to the corpus callosum/external capsule. Mild linear wisps were present in the corpus callosum, but these were also present to some extent in the control sections.
Discussion
Lysosomal storage diseases involve dysfunction of one of the more than 50 enzymes in the lysosome, resulting in the accumulation of the associated uncatabolized substrate (Neufeld 1991; Winchester et al. 2000) or toxic intermediates (Suzuki et al. 2003). A majority of lysosomal storage diseases affect the CNS and disease presentation can range from severe and consistent to mild and variable depending on the enzyme involved, the severity of the mutation, and the species concerned. In addition, the genetic mutations may trigger complex pathogenic cascades (Walkley 2009; Lieberman et al. 2012). For example, interference with signaling events and salvage processing normally controlled by the endosomal/lysosomal system and/or effects on autophagy may represent key mechanisms accounting for the inherent complexity of lysosomal disorders.
Goats and cattle affected with β-mannosidosis have very similar severe clinical features, and affected animals die in the neonatal period if intensive care is not provided (Jones et al. 1983; Abbitt et al. 1991). Microscopic examination reveals extensive cytoplasmic vacuolation in specific cell types of the nervous system and visceral organs. The storage material does not stain with PAS and appears clear in electron microscopic examination. Regionally specific myelin deficiency is present in the CNS but not in peripheral nerves (Jones et al. 1983; Lovell and Jones 1983; Boyer et al. 1990). In contrast with ruminant β-mannosidosis, human cases have a milder and heterogeneous clinical expression, even when caused by functionally null mutations (Alkhayat et al. 1998; Bedilu et al. 2002; Cooper et al. 1990). The most severe cases are associated with intellectual disability, developmental delay, dysmorphology, frequent infections, and hearing loss. None of the human cases showed clinical signs that would indicate CNS hypomyelination. Little is known about the distribution of storage material or the severity of tissue vacuolation. Mutant β-mannosidase-deficient mice (Zhu et al. 2006) had normal growth, appearance, and lifespan, and no hypomyelination, closer to the human β-mannosidosis phenotype than the ruminant phenotype.
Reasons for the phenotypic differences among mouse, human, and ruminant β-mannosidosis are unknown, but speculation has centered on differences in the size and nature of the storage product and on the different developmental programs of the species (Cooper et al. 1990). In the lysosome, N-linked glycoproteins are degraded sequentially from the nonreducing end of the molecule. The β-mannoside linkage is unique to N-linked glycoproteins; therefore, unlike most other lysosomal enzymes, β-mannosidase has only a single substrate in the cell. In humans and rodents, the nonsequential cleavage of the terminal GlcNAc(β1-4)GlcNAc bond of the carbohydrate moiety of glycoprotein can be accomplished by chitobiase (ctbs), resulting in storage of disaccharide, Man(β1-4)GlcNAc (vanPelt et al. 1990; Winchester 2005). In ruminants, there is very low expression of chitobiase (Zhu et al. 2006; Aronson and Kuranda 1989) and storage of the trisaccharide Man(β1-4)GlcNAc(β1-4)GlcNAc predominates; novel complex oligosaccharides also accumulate (Jones and Laine 1981; Matsuura and Jones 1985). Cloning of the chitobiase gene (Balducci et al. 2008) permits the construction of a double β-mannosidase and chitobiase knockout mouse, to determine if the storage product is the trisaccharide, as in ruminants, and assess the clinical phenotype. There may also be other factors involved in the neuropathology mechanisms in different species.
In the β-mannosidase-deficient mice, there was considerable variation among two strains of mice in the severity of neuropathology as illustrated in Table 1. There was a consistent pattern of neuronal vacuolation and disintegrative degeneration in all five 129X1/SvJ mice. However, the mixed background mice showed substantial variability in the severity of pathology. The variability in severity for C57BL/6J mice is greater than in the examination of 9-month-old mice (Zhu et al. 2006); in that study, the majority of C57BL/6J mice had moderate to severe vacuolation. The influence of genetic background on knockout mouse phenotypes is well documented and can present itself as completely different phenotypes, as variations in penetrance of phenotype, or as variable expressivity of phenotype (Sanford et al. 2001). Modifier genes (Sanford et al. 2001) and/or biological robustness (Barbaric et al. 2007) may play a role in these effects, and such modifying factors may be epistatic, epigenetic, or of other origins. In β-mannosidosis, the variation in the pathology depending on genetic background indicates modifier factors are involved. One hypothesis is that protective factor(s) in the mixed strain mice, variable among animals, modifies the disease expression and determines differential susceptibility to phenotypic features such as neuronal vacuolation. Similar factors may be involved in the tremendous clinical heterogeneity among the small number of patients with β-mannosidosis, including intrafamilial variation and variation among patients with null mutations (Alkhayat et al. 1998). Thus, for future studies of this animal model, it will be important to use the congenic 129X1/SvJ strain (or characterize the phenotype in a different uniform genetic background). Identification of factors that modify the disease expression may help clarify the pathogenetic mechanisms involved in this or other lysosomal storage diseases.
In those mutant mice showing vacuolation, the locations of neuronal vacuoles were very consistent, including piriform area, taenia tecta, part of hippocampus CA3 (consistent in dorsal hippocampus but not ventral hippocampus), retrosplenial area, parts of anterior cingulate area and entorhinal cortex, and layer V in primary motor cortex, primary somatosensory cortex, primary visual cortex, and posterior parietal association cortex. The variation in vacuolation in the hippocampus may be related to multiple divisions identified by genetic and metabolic characteristics (Fanselow and Dong 2010; Thompson et al. 2008). The piriform area and taenia tecta are part of the olfactory system. The retrosplenial area is part of a network of brain regions that participates in a range of cognitive functions, including episodic memory and navigation. The entorhinal cortex mainly projects to the hippocampus, including both CA3 and CA1. The anterior cingulate area plays a role in emotional and attentive responses to internal and external stimulation. The pyramidal neurons in layer V of cerebral cortex integrate input and project to many other parts of the CNS. It is possible that the pattern of vacuolation seen in mouse β-mannosidosis would contribute to the intellectual disability and emotional changes reported in human cases. However, it is not clear why only these specific neuronal populations show vacuolation. In other lysosomal storage disorders, different, but disease-specific, patterns of neuropathology are characteristic. For example, α-mannosidosis (OMIM 248500), caused by mutations in the α-mannosidase (MAN2B1, EC 3.2.1.24, 609458) gene, has been described in cats, cattle, guinea pigs, humans, and knockout mice (Auclair and Hopwood 2007; Blanz et al. 2008; Crawley and Walkley 2007; Damme et al. 2011; Stinchi et al. 1999; Vite et al. 2001) and has somewhat different neurological manifestations and neuropathological characteristics depending on the species. Aspartylglucosaminuria (AGU, OMIM 208400) (Dunder et al. 2010; Jalanko et al. 1998; Kaartinen et al. 1996) is caused by mutations in the gene for glycosylasparaginase (AGA, EC 3.5.1.26, 613228), the enzyme necessary for hydrolysis of the protein-oligosaccharide linkage in N-linked glycoproteins. Glycosylasparaginase deficiency results in accumulation of glycoasparagines, such as aspartylglucosamine (GlcNAc-Asn), a small molecule that may be analogous to Man(β1-4)GlcNAc. In the AGU mouse model (Jalanko et al. 1998; Kaartinen et al. 1996), CNS vacuolation is prominent in neurons and astrocytes of cortical and subcortical gray matter. Further understanding of the different patterns of pathology may lead to delineation of specific metabolic characteristics of subsets of neurons that are not currently understood. Cognitive or behavioral deficits have been identified in α-mannosidosis and AGU knockout mice, and correlate with CNS pathology. Both animal models have been used successfully to explore therapeutic strategies to improve brain function (Blanz et al. 2008; Damme et al. 2011; Dunder et al. 2010).
In order to investigate a potential correlation between vacuolation and level of enzyme activity in β-mannosidosis, the distribution of lesions was compared with the distribution of mouse brain lysosomal β-mannosidase in situ hybridization and expression analysis as shown in the Allen Brain Atlas (Lein et al. 2007). There was no consistent correlation, although CA3 showed higher expression than other hippocampal regions. Some areas that were vacuolated did not show increased expression, and some regions with higher expression (e.g., Purkinje cells in cerebellum) did not show vacuolation. In general, β-mannosidase has very low expression throughout the CNS in mice (Lein et al. 2007) and in goats (Boyer et al. 1990; Lovell et al. 1994) compared to that of other glycoprotein catabolic enzymes. There is no apparent consistent correlation among gene expression, enzyme activity, and severity of vacuolation. There could be differences in levels of substrate, leading to vacuolation in cells with higher levels of substrate or other metabolic variations among brain regions. Production of antibodies to the enzyme and/or substrate would allow investigations of metabolic differences between neuronal and glial populations.
In β-mannosidosis mice, silver reaction product indicating neuronal degeneration was prominent only in the mutant mice that had severe vacuolation, suggesting that it is a consistent part of the neuropathology expression in this disease. The pattern of degeneration staining was very consistent among animals, and occurred mainly in cerebellar molecular layer and in cortical regions dorsal to the hippocampal formation (Fig. 2). Degeneration staining did not occur in areas of vacuolation or adjacent regions, suggesting that vacuolation does not lead to substantial cell body or axon loss. Further studies will be necessary to elucidate the mechanisms that lead to the degeneration staining in specific regions; results will provide new information about specific types of cellular pathophysiology. Preliminary staining of selected sections with GFAP for astrocytes and Iba1 for microglia showed some degree of reactivity in regions with vacuolation, but did not provide any additional information related to pathophysiology (data not shown).
In summary, a β-mannosidase knockout mouse model was utilized to characterize CNS pathology in mice with different strain backgrounds. The consistency of distribution of lesions in a congenic strain 129X1/SvJ reinforces the potential of this model for investigating treatment strategies important for brain function. The variation in pathology depending on genetic background indicates the complex nature of interactions that may determine the extent of cellular abnormalities and phenotypic expression. Animal models are important in the clarification of the pathophysiology/pathogenetic mechanism of disease (Suzuki et al. 2003) as well as giving insight into normal physiology and metabolism. Further investigation of the specific patterns of vacuolation and other pathology in this and related lysosomal storage diseases will lead to better understanding of specific metabolic characteristics among populations of neurons.
Acknowledgments
Processing and staining of mouse brains was generously provided by NeuroScience Associates. This work was supported by NIDDK grant DK49782 from the National Institutes of Health to KHF, and by Michigan State University Foundation grant to KHF and KLL.
Synopsis
In β-mannosidosis, the neuronal pathology observed in mice is consistent with the intellectual disability displayed in human cases, and the variation among different strains implicates genetic modifiers in variable phenotypic expression.
Author Who Serves as Guarantor
Kathryn L. Lovell, Ph.D.
Details of Funding
Processing and staining of mouse brains was generously provided by NeuroScience Associates. This work was supported by NIDDK grant DK49782 from the National Institutes of Health to KF, and by Michigan State University Foundation grant to KF and KL.
Compliance with Ethics Guidelines
Conflict of Interest
Kathryn Lovell, Mei Zhu, Meghan Drummond, Robert Switzer, and Karen Friderici declare that they have no conflict of interest. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors.
Informed Consent
Informed consent: not required. This article does not contain any studies with human subjects performed by any of the authors.
Use of Vertebrate Animals
All institutional and national guidelines for the care and use of laboratory animals were followed.
Details of the Contributions of Individual Authors
- KLL:
designed the study, participated in perfusion and brain removal, analyzed results to generate distribution of pathology, took a lead role in writing the manuscript.
- MZ:
produced the mutant mice, bred them for the current experiments, responsible for perfusion and brain removal, participated in writing the manuscript.
- MCD:
assisted with breeding mice, assisted with analysis of data, participated in writing the manuscript.
- RCS:
processed the brains for sectioning and staining, participated in photography and analysis of results, participated in writing the manuscript.
- KHF:
supervised production of the mutant mice and breeding, participated in writing the manuscript.
Footnotes
Competing interests: None declared
Contributor Information
Kathryn L. Lovell, Email: lovell@msu.edu1
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Abbitt B, Jones MZ, Kasari TR, et al. Beta-mannosidosis in twelve Salers calves. J Am Vet Med Assoc. 1991;198:109–113. [PubMed] [Google Scholar]
- Alkhayat AH, Kraemer SA, Leipprandt JR, Macek M, Kleijer WJ, Friderici KH. Human beta-mannosidase cDNA characterization and first identification of a mutation associated with human beta-mannosidosis. Hum Mol Genet. 1998;7:75–83. doi: 10.1093/hmg/7.1.75. [DOI] [PubMed] [Google Scholar]
- Aronson NN, Jr, Kuranda MJ. Lysosomal degradation of Asn-linked glycoproteins. FASEB J. 1989;3:2615–2622. doi: 10.1096/fasebj.3.14.2531691. [DOI] [PubMed] [Google Scholar]
- Auclair D, Hopwood JJ. Morphopathological features in tissues of alpha-mannosidosis guinea pigs at different gestational ages. Neuropathol Appl Neurobiol. 2007;33:572–85. doi: 10.1111/j.1365-2990.2007.00849.x. [DOI] [PubMed] [Google Scholar]
- Balducci C, Bibi L, Berg T, et al. Molecular cloning and structural organization of the gene encoding the mouse lysosomal di-N-acetylchitobiase (ctbs) Gene. 2008;416:85–91. doi: 10.1016/j.gene.2008.03.009. [DOI] [PubMed] [Google Scholar]
- Barbaric I, Miller G, Dear TN. Appearances can be deceiving: phenotypes of knockout mice. Brief Funct Genomic Proteomic. 2007;6:91–103. doi: 10.1093/bfgp/elm008. [DOI] [PubMed] [Google Scholar]
- Bedilu R, Nummy KA, Cooper A, et al. Variable clinical presentation of lysosomal beta-mannosidosis in patients with null mutations. Mol Genet Metab. 2002;77:282–290. doi: 10.1016/S1096-7192(02)00172-5. [DOI] [PubMed] [Google Scholar]
- Blanz J, Stroobants S, Lüllmann-Rauch R, et al. Reversal of peripheral and central nervous storage and ataxia after recombinant enzyme replacement therapy in alpha-mannosidosis mice. Hum Mol Genet. 2008;17:3437–3445. doi: 10.1093/hmg/ddn237. [DOI] [PubMed] [Google Scholar]
- Boyer PJ, Jones MZ, Rathke EJS, Truscott NK, Lovell KL. Regional central nervous system oligosaccharide storage in caprine beta-mannosidosis. J Neurochem. 1990;55:660–664. doi: 10.1111/j.1471-4159.1990.tb04184.x. [DOI] [PubMed] [Google Scholar]
- Bryan L, Schmutz S, Hodges SD, Snyder FF. Bovine beta-mannosidosis: pathologic and genetic findings in Salers calves. Vet Pathol. 1993;30:130–139. doi: 10.1177/030098589303000205. [DOI] [PubMed] [Google Scholar]
- Cooper A, Sardharwalla IB, Roberts MM. Human beta-mannosidase deficiency. N Engl J Med. 1986;315:1231. doi: 10.1056/NEJM198608143150722. [DOI] [PubMed] [Google Scholar]
- Cooper A, Hatton CE, Thornley M, Sardharwalla IB. Alpha- and beta-mannosidoses. J Inherit Metab Dis. 1990;13:538–548. doi: 10.1007/BF01799511. [DOI] [PubMed] [Google Scholar]
- Crawley AC, Walkley SU. Developmental analysis of CNS pathology in the lysosomal storage disease alpha-mannosidosis. J Neuropathol Exp Neurol. 2007;66:687–697. doi: 10.1097/nen.0b013e31812503b6. [DOI] [PubMed] [Google Scholar]
- Damme M, Stroobants S, Walkley SU, et al. Cerebellar alterations and gait defects as therapeutic outcome measures for enzyme replacement therapy in alpha-mannosidosis. J Neuropathol Exp Neurol. 2011;70:83–94. doi: 10.1097/NEN.0b013e31820428fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunder U, Valtonen P, Kelo E, Mononen I. Early initiation of enzyme replacement therapy improves metabolic correction in the brain tissue of aspartylglycosaminuria mice. J Inherit Metab Dis. 2010;33:611–617. doi: 10.1007/s10545-010-9158-7. [DOI] [PubMed] [Google Scholar]
- Fanselow MS, Dong H-W. Are the dorsal and ventral hippocampus functionally distinct structures? Neuron. 2010;65:7. doi: 10.1016/j.neuron.2009.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fix AS, Ross JF, Stitzel SR, Switzer RC. Integrated evaluation of central nervous system lesions: stains for neurons, astrocytes, and microglia reveal the spatial and temporal features of MK-801-induced neuronal necrosis in the rat cerebral cortex. Toxicol Pathol. 1996;24:291–304. doi: 10.1177/019262339602400305. [DOI] [PubMed] [Google Scholar]
- Jalanko A, Tenhunen K, McKinney CE, et al. Mice with an aspartylglucosaminuria mutation similar to humans replicate the pathophysiology in patients. Human Molec Genet. 1998;7:265–272. doi: 10.1093/hmg/7.2.265. [DOI] [PubMed] [Google Scholar]
- Jones MZ, Dawson G. Caprine beta-mannosidosis: inherited deficiency of beta-D-mannosidase. J Biol Chem. 1981;256:5185–5188. [PubMed] [Google Scholar]
- Jones MZ, Laine RA. Caprine oligosaccharide storage disease accumulation of beta-mannosyl (1–4) beta-N-acetylglucosaminyl (1–4) beta-N-acetylglucosamine in brain. J Biol Chem. 1981;256:5181–5184. [PubMed] [Google Scholar]
- Jones MZ, Cunningham JG, Dade AW, et al. Caprine beta-mannosidosis: clinical and pathological features. J Neuropathol Exp Neurol. 1983;42:268–285. doi: 10.1097/00005072-198305000-00005. [DOI] [PubMed] [Google Scholar]
- Kaartinen V, Mononen I, Voncken JW. A mouse model for the human lysosomal disease aspartylglycosaminuria. Nat Med. 1996;2:1375–1378. doi: 10.1038/nm1296-1375. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lieberman AP, Puertollano R, Raben N, Slaugenhaupt S, Walkley SU, Ballabio A. Autophagy in lysosomal storage disorders. Autophagy. 2012;8:719–730. doi: 10.4161/auto.19469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell KL, Jones MZ. Distribution of central nervous system lesions in beta-mannosidosis. Acta Neuropathol. 1983;62:121–126. doi: 10.1007/BF00684928. [DOI] [PubMed] [Google Scholar]
- Lovell KL, Jones MZ. Axonal and myelin lesions in beta-mannosidosis: ultrastructural characteristics. Acta Neuropathol. 1985;65:293–299. doi: 10.1007/BF00687011. [DOI] [PubMed] [Google Scholar]
- Lovell KL, Kranich RJ, Cavanagh KT. Biochemical and histochemical analysis of lysosomal enzyme activities in caprine beta-mannosidosis. Molec Chem Neuropath. 1994;21:61–74. doi: 10.1007/BF03160085. [DOI] [PubMed] [Google Scholar]
- Matsuura F, Jones MZ. Structural characterization of novel complex oligosaccharides accumulated in the caprine beta-mannosidosis kidney: Occurrence of tetra- and pentasaccharides containing a beta-linked mannose residue at the nonreducing terminus. J Biol Chem. 1985;260:15239–1524. [PubMed] [Google Scholar]
- Neufeld EF. Lysosomal storage diseases. Annu Rev Biochem. 1991;60:257–280. doi: 10.1146/annurev.bi.60.070191.001353. [DOI] [PubMed] [Google Scholar]
- Ong WY, Kumar U, Switzer RC, et al. Neurodegeneration in Niemann-Pick type C disease mice. Exp Brain Res. 2001;141:218–231. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- Patterson JS, Jones MZ, Lovell KL, Abbitt B. Neuropathology of bovine beta-mannosidosis. J Neuropathol Exp Neurol. 1991;50:538–546. doi: 10.1097/00005072-199109000-00002. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Kallapur S, Ormsby M, Doetschman T. Influence of genetic background on knockout mouse phenotypes. Gene Knockout Protocols, Methods in Molec Biol. 2001;158:217–225. doi: 10.1385/1-59259-220-1:217. [DOI] [PubMed] [Google Scholar]
- Stinchi S, Lullmann-Rauch R, Hartmann D, et al. Targeted disruption of the lysosomal alpha-mannosidase gene results in mice resembling a mild form of human alpha-mannosidosis. Hum Mol Genet. 1999;8:1365–1372. doi: 10.1093/hmg/8.8.1365. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Ezoe T, Tohyama J, Matsuda J, Vanier MT, Suzuki K. Are animal models useful for understanding the pathophysiology of lysosomal storage disease? Acta Paediatr Suppl. 2003;92:54–62. doi: 10.1111/j.1651-2227.2003.tb00223.x. [DOI] [PubMed] [Google Scholar]
- Thompson CL, Pathak SD, Jeromin A, et al. Genomic anatomy of the hippocampus. Neuron. 2008;60:1010–1021. doi: 10.1016/j.neuron.2008.12.008. [DOI] [PubMed] [Google Scholar]
- vanPelt J, Hokke CH, Dorland L, Duran M, Kamerling JP, Vliegenthart JF. Accumulation of mannosyl-beta(1–4)-N-acetylglucosamine in fibroblasts and leukocytes of patients with a deficiency of beta-mannosidase. Clin Chim Acta. 1990;187:55–60. doi: 10.1016/0009-8981(90)90261-P. [DOI] [PubMed] [Google Scholar]
- Vite CH, McGowen JC, Braund KG, et al. Histopathology, electrodiagnostic testing, and magnetic resonance imaging show significant peripheral and central nervous system myelin abnormalities in the cat model of alpha-mannosidosis. J Neuropath Exp Neurol. 2001;60:817–828. doi: 10.1093/jnen/60.8.817. [DOI] [PubMed] [Google Scholar]
- Walkley SU. Pathogenic cascades in lysosomal disease – Why so complex? J Inher Met Dis. 2009;32:181–189. doi: 10.1007/s10545-008-1040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenger DA, Sujansky E, Fennessey PV, Thompson JN. Human beta-mannosidase deficiency. N Engl J Med. 1986;315:1201–1205. doi: 10.1056/NEJM198611063151906. [DOI] [PubMed] [Google Scholar]
- Winchester B. Lysosomal metabolism of glycoproteins. Glycobiology. 2005;15:1R–15R. doi: 10.1093/glycob/cwi041. [DOI] [PubMed] [Google Scholar]
- Winchester B, Vellodi A, Young E. The molecular basis of lysosomal storage diseases and their treatment. Biochem Soc Trans. 2000;28:150–154. doi: 10.1042/bst0280150. [DOI] [PubMed] [Google Scholar]
- Zhu M, Lovell KL, Patterson JS, Saunders TL, Hughes ED, Friderici KH. Beta-mannosidosis mice: a model for the human lysosomal storage disease. Human Molec Genetics. 2006;15:493–500. doi: 10.1093/hmg/ddi465. [DOI] [PubMed] [Google Scholar]