

Abstract
Objective: Identifying phenylalanine hydroxylase (PAH) mutations associated with sapropterin response in phenylketonuria (PKU) would be an advantageous means to determine clinical benefit to sapropterin therapy.
Methods: Sapropterin response, defined as a ≥30 % reduction in phenylalanine (Phe) levels after a dose of 10 mg/kg/day sapropterin for week one and 20 mg/kg/day for week two in 112 PKU patients aged 4–45 years, was assessed in an outpatient setting. PAH was sequenced in all patients. Mutations were correlated with sapropterin response. Dietary Phe intake was increased over a 6-week period in responsive patients.
Results: Forty-six of 112 patients were sapropterin responsive. Genotypes p.[L48S];[L48S] and p.[Y414C];[Y414C] were always associated with response at a low dose. The mutation Y414C (present on 16 alleles) was most frequently associated with response. Patients with presence of the mutation L48S on at least one allele (12 alleles in 7 patients) always showed response to sapropterin. Responsive patients had a mean Phe tolerance increase of 189 % (range 11–742 %). In the 66 nonresponders, mutations R408W (38 alleles) and IVS12+1G>A (18 alleles) were detected most frequently. Genotypes [IVS12+1G>A];[IVS12+1G>A], p.[L348V];[R408W], p.[P281L];[P281L], p.[R158Q];[R408W], and p.[R261Q];[R408W] were always associated with nonresponse.
Conclusion: Data from the study contributes to growing evidence of the relationship between PAH genotype and PKU phenotype. In most cases, response to sapropterin therapy cannot be predicted based on the presence of a single mutation on one allele alone, although the complete PAH genotype may help to predict sapropterin responsiveness in PKU patients.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2013_263) contains supplementary material, which is available to authorized users.
Background
Phenylketonuria (PKU) is an autosomal recessive metabolic disorder resulting from mutations in the gene encoding phenylalanine hydroxylase (PAH) (Scriver and Kaufman 2001). More than 800 mutations have been identified (BIOPKU database, http://www.biopku.org/home/pah.asp, accessed Sep 2013), which lead to PAH deficiency and a wide range of clinical phenotypes. The deficient PAH activity results in a decreased ability to convert phenylalanine (Phe) to tyrosine (Tyr) and leads to toxic accumulation of Phe in the brain (Scriver et al. 2008). In developed countries, patients are identified at birth through newborn screening programs and classified by clinical phenotype. Minimum pretreatment Phe levels, requiring initiation of a Phe-restricted diet, vary between different countries. In Germany, patients with Phe levels >600 μmol/L (Weglage et al. 1996; Burgard et al. 1999) are treated with such a diet.
Although the Phe-restricted diet prevents the severe intellectual dysfunction associated with untreated PKU (National Institutes of Health Consensus Development Panel 2001), subtle neurocognitive deficits are still reported in individuals with PKU treated early and continuously with the Phe-restricted diet (Enns et al. 2010). As well, severely restricted diets, such as the Phe-restricted diet, are associated with a risk of nutritional deficiencies and present a substantial psychosocial burden and challenges with adherence, especially for adolescents who often fail to comply with the strict recommendations (Walter et al. 2002).
In 1999, Kure et al. published results of a study that identified a subgroup of PKU patients, who responded to exogenous BH4 with reduced blood Phe levels independent of the Phe-restricted diet (Kure et al. 1999). Initially thought to be primarily due to higher levels of available BH4, it was later determined to be caused in part by stabilization of PAH protein conformation and by preventing inactivation of the enzyme (Thöny et al. 2004; Gersting et al. 2010). Sapropterin dihydrochloride (sapropterin, Kuvan®) received marketing approval in Germany in April 2009 for individuals with PKU who are sapropterin responsive to reduce Phe levels and allows responders to modify their strict dietary regimen (Levy et al. 2007; Lee et al. 2008; Trefz et al. 2009a).
Most patients in the European Union (EU) must undergo a sapropterin response test as described in the EU summary of products characteristics (SmPC). Many protocols exist to determine sapropterin responsiveness in patients with PKU, varying significantly between continents and even between countries and clinics within a country (Blau et al. 2009). EU recommendations may include a 48-h sapropterin response test (Blau et al. 2009) under hospital supervision. An oral Phe loading of 100 mg/kg of body weight prior to the first dose of sapropterin (Muntau et al. 2002), which allows a more significant response to sapropterin for patients whose Phe levels are within recommended range by the Phe-restricted diet, was suggested as well (Staudigl et al. 2011). However, not all sapropterin response test protocols require a Phe loading test (Blau et al. 2009). For example, the United States (US) Food and Drug Administration (FDA) does not recommend the Phe loading component for sapropterin response testing, and does not require the patient to be hospitalized at any point of the response testing period (Kuvan® US Prescribing Information).
Both protocols first determine the patient’s baseline blood Phe level. In the United States, the FDA-approved initial sapropterin dose is 10 mg/kg for 1 week and if a reduction in Phe level is not achieved, the dose is increased to 20 mg/kg and monitored for a maximum of 1 month (Kuvan® US Prescribing Information). In the EU, if the Phe level is well controlled, sapropterin treatment is initiated at 20 mg/kg/day. The Phe level is assessed at the 24-h mark. If it has not been reduced by ≥30 %, a second dose of sapropterin 20 mg/kg/day is given. The blood Phe level is reassessed at 24 h. In case of a decrease in Phe level <30 % after the 48 h of testing, the patient is classified as nonresponsive and sapropterin treatment is discontinued (Blau et al. 2009).
Existing protocols for sapropterin response testing pose challenges and have limitations. The strict dietary control and increased frequency of blood Phe monitoring required during the response test period may impact patients’ compliance.
The use of PAH mutational analysis rather than sapropterin response testing protocols could potentially eliminate these challenges and reduce false-negative responses due to noncompliance and false-positive results due to natural fluctuations in Phe levels. The ability to predict long-term response to sapropterin therapy in individual patients through alternative means could positively impact patient outcomes. The PAH genotype could be an important factor for predicting response to sapropterin with mutations associated with BH4 response dispersed throughout the gene. However, although the nature and position of the mutations determines their effect on the activity of the PAH enzyme and the clinical phenotype of the patient, to date, no conclusive data exists to allow prediction of sapropterin responsiveness solely based on genotype.
Our primary objective in this Phase IV open-label study was to evaluate alternative methods for identifying individuals with PKU as responsive to sapropterin therapy including impact of genotype, individual mutation, and location of mutation in the PAH gene. We aimed to identify the influence of individual PAH mutations on response based on Phe levels and dietary Phe intake.
Methods
Patient Enrollment
This study was approved by the local ethical committee Ethikkommission der Ärztekammer Westfalen-Lippe. All PKU patients >4 years of age with plasma Phe levels ≥600 μmol/L at diagnosis currently being followed at our metabolic center at Münster University Children’s Hospital were contacted by mail to request participation in the trial. Exclusion criteria were age of <4 years, proven BH4 deficiency, and pregnancy. All patients gave informed consent to participate.
Sapropterin Response Testing
Before receiving treatment, patients had to have a minimum blood Phe level of 360 μmol/L and maintain stable blood Phe levels with <20 % fluctuation from baseline at three consecutive blood Phe draws (Fig. 1). To achieve this, plasma Phe levels were evaluated weekly by capillary blood draws. Patients’ dietary regimens were assessed regularly by dieticians. If Phe levels were <360 μmol/L, patients were asked to increase dietary Phe intake slightly, but unlike in the protocol from Blau et al. (2009), a 100 mg/kg Phe challenge was not systematically performed. After having achieved three stable Phe levels, the patients were seen in the outpatient clinic for blood sampling for PAH mutation analysis and to obtain an additional blood Phe level prior to the first dose of sapropterin.
Fig. 1.
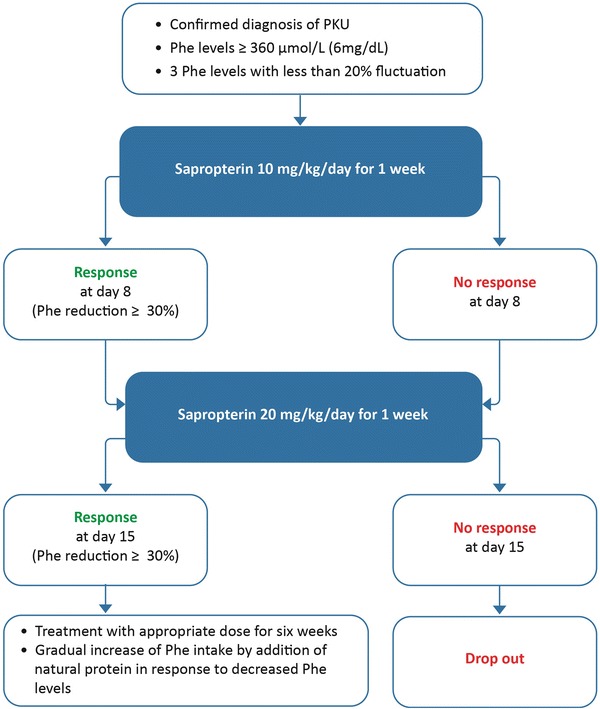
Study design
Sapropterin was dissolved in water or apple juice and was applied orally together with some food (Kuvan® US Prescribing Information). Sapropterin was initially administered at a dose of 10 mg/kg/day at the time of the outpatient visit. This dose was repeated at home every 24 h for another 6 days. At day 8 and regardless of the patient’s response to sapropterin, the sapropterin dose was increased to 20 mg/kg per day for another 7 days to test the individual response to the higher dose to determine, if the higher dose has an additional effect on lowering Phe levels. The dietary regimen of the patients remained unchanged throughout the 2 weeks of the response test. Plasma Phe levels were measured at 24 h and 7 days after the first dose of sapropterin and 24 h and 7 days after the increased dose. All blood samples were taken in fasted state prior to the daily sapropterin intake.
Extension Period After Sapropterin Response Resting
Sapropterin was discontinued for patients who did not show a ≥30 % decrease in Phe levels after 14 days. Those with ≥30 % decrease in Phe levels continued to be administered with sapropterin at a dose of 20 mg/kg/day regardless of whether response occurred at 10 mg/kg/day or 20 mg/kg/day. To determine the patients’ individual increase in Phe tolerance due to sapropterin treatment in responsive patients, dietary Phe content was gradually increased over the subsequent 6 weeks through the addition of natural protein in the diet, potentially in form of natural bread or pasta products. The amount of Phe-free amino acid supplements required was reduced as appropriate. Phe levels were monitored weekly and were maintained within the age-appropriate target levels. It should be noted that these levels are currently under debate. We applied the target levels recommended for Germany: 0–9 years: 40–240 μmol/L; >9–15 years: 40–900 μmol/L; >15 years: 40–1200 μmol/L) (Schweitzer-Krantz and Burgard 2000). After the 8 weeks of the study period, the patients were seen on a regular basis in our outpatient metabolic clinic for dietary assessment and physical examination. Phe levels were monitored every 2 weeks.
Determination of Phenylalanine Plasma Levels
Phe levels were determined in plasma samples from capillary EDTA blood draws (100 μl EDTA blood) performed by the patients or their caregivers at home. Plasma samples were analyzed by ion exchange chromatography in an Eppendorf-Biotronik LC 3000 Amino Acid Analyzer® (Eppendorf GmbH, Wesseling-Berzdorf, Germany).
Mutation Analysis
Genomic DNA from each individual patient was isolated from EDTA blood samples according to a standard protocol using the EZ1DNA Blood Kit® (Qiagen GmbH, Hilden, Germany). PCR products were directly sequenced bidirectionally using the BigDye Terminator v3.1 Cycle Sequencing Kit on an ABI 3730 Genetic Analyzer according to the manufacturer’s protocol (Applied Biosystems, Foster City, California, USA). All 13 exons of the PAH gene plus their exon–intron boundaries were analyzed. Primer sequences are available on request.
Reference accession number for normal alleles was ENSG00000171759.2 for PAH. Mutation designation was according to the official mutation nomenclature (http://www.hgvs.org/mutnomen/). To validate nomenclature of mutations, we used the program Mutalyzer 2.0 β-2 (http://www.mutalyzer.nl/2.0/). Mutations were confirmed by carrier analysis of parents.
Results
Patient Enrollment and Response Testing
The first patient was enrolled in the study in January 2010 and the last patient in October 2011. Three hundred and fifty-three patients were contacted by mail. Of these, we did not receive a response from 153 patients (43.3 %). Seventy patients (19.8 %) refused to take part in the study for a variety of reasons, including time constraints, effort required to comply with the strict regimen, safety concerns, satisfaction with current dietary regimen, aversion to tablets, and doubt about being sapropterin responsive.
One hundred and thirty (36.8 %) patients contacted by mail consented and underwent sapropterin response testing. However, 16 patients were excluded, because they were not able to maintain stable Phe levels with less than 20 % fluctuation from baseline. One patient refused to have mutation analysis performed, and one was excluded due to noncompliance to the response test protocol. The remaining 112 (31.7 %) patients completed the response test by December 2011 (Table 1): 46 (41.1 %) patients were identified as responders (Table 2), while 66 (58.9 %) of patients who completed the response test did not show a reduction of ≥30 % in their serum Phe levels and were classified as nonresponsive to sapropterin therapy (Table 3). The median Phe levels in responders versus nonresponders during the test period are shown in Fig. 2.
Table 1.
Demographic and baseline data for patients who completed the trial by December 2011
| Demographic | |
|---|---|
| Completed trial N (% of those contacted) | 112 (31.7 %) |
| Nationality N (%) | |
| German | 84 (75 %) |
| Turkish | 14 (13 %) |
| Russian | 9 (8 %) |
| Other | 5 (4 %) |
| Gender – male N (%) | 59 (53 %) |
| Age mean (median; min, max) years | 18 (15.7; 4, 45) |
| Baseline Phe levels mean (median; min, max) μmol/L | 795.3 (754.5; 340.8, 1884) |
| Baseline Phe tolerance mean (median; min, max) mg/kg | 14.88 (12.4; 2.84, 51.19) |
| Sapropterin responsive patients N (%) | 46 (41.1 %) |
| Responsive at 10 mg/kg/day | 22 (19.6 %) |
| Responsive only at 20 mg/kg/day | 24 (21.4 %) |
Table 2.
Baseline Phe levels and baseline Phe tolerance in all responders and corresponding changes in Phe levels and dietary Phe intake in all patients with continued treatment
| Dosage for response | |
|---|---|
| 10 mg/kg/day N | 22 |
| Patients with continuing treatment and increase in Phe tolerance N (%) | 15 (68.2 %)a |
| Baseline Phe levels μmol/L mean (median; min, max) | 607.3 (562.5; 403.8, 942) |
| Percentage decrease in Phe levels (median; min, max) | 49.8 (44.9; 30, 84) |
| Baseline Phe tolerance mean (median; min, max) mg/kg | 15.6 (15.3; 6.4, 36.9) |
| Mean percentage increase in Phe tolerance (median; min, max) | 220.7 (150; 25, 742) |
| 20 mg/kg/day excluding patients who responded at 10 mg/kg/day N | 24 |
| Patients with continuing treatment and increase in Phe tolerance N (%) | 15 (62.5 %)b |
| Baseline Phe levels μmol/L mean (median; min, max) | 673.1 (516.8;. 360, 1366.8) |
| Percentage decrease in Phe levels (median; min, max) | 47.5 (42; 30, 77) |
| Baseline Phe tolerance mean (median; min, max) mg/kg | 13.42 (11.9; 4.3, 42.4) |
| Mean percentage increase in Phe tolerance (median; min, max) | 157.7 (122.2; 11, 450) |
a5 patients withdrew from follow-up due to noncompliance, 1 patient withdrew consent and 1 showed an unexplained increase of Phe levels after testing period despite constant Phe intake
b5 patients did not show increased Phe tolerance despite response, 1 responsive patient showed an unexplained increase in Phe levels after testing period despite constant Phe intake, 3 patients withdrew consent
Table 3.
Baseline Phe levels and baseline Phe tolerance in all non-responders
| Non-responders | |
|---|---|
| N | 66 |
| Baseline Phe levels μmol/L mean (median; min, max) | 916.8 (900.6; 340.8, 1884) |
| Baseline Phe tolerance mean (median; min, max) mg/kg | 15.16 (11.64; 2.84, 51.19) |
Fig. 2.
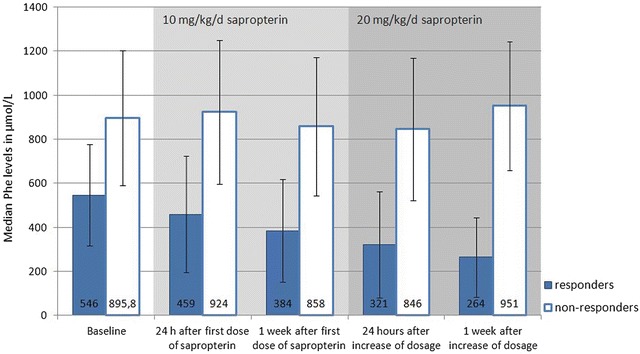
Progress of median Phe levels during test period. Median Phe levels of 46 responders and 66 nonresponders are shown
Modification of Dietary Phe Intake Within the Six-Week Extension Period
Sapropterin treatment was stopped in 5 of the 46 patients identified as sapropterin responsive after the response test period due to noncompliance to the protocol and in 4 patients due to patient withdrawal of consent. Two patients showed an unexplained increase in Phe levels despite constant Phe intake and treatment was subsequently stopped in these patients as well. In the remaining 35 responders, dietary Phe was gradually increased with natural proteins in the daily diet. Sapropterin treatment was stopped in 5 further patients, who were initially classified as responders, because these patients showed elevated Phe levels while increasing the dietary Phe intake in the 6-week period.
In the 30 sapropterin-responsive patients, dietary Phe intake could be increased by a mean value of 189 % (median 128 %, range 11–742 %) compared to baseline while maintaining blood Phe levels in the age-appropriate range according to the German national guidelines (Schweitzer-Krantz and Burgard 2000) with a dose of 20 mg/kg/day. In these patients, average Phe tolerance increased from 13.8 mg/kg/day before sapropterin treatment to 35.2 mg/kg/day after 6 weeks of sapropterin therapy.
Conversely, the amount of amino acid supplements could be reduced on average of 41.5 % from 0.76 g/kg/day before sapropterin therapy to 0.46 g/kg/day for these patients after 6 weeks of sapropterin therapy (Table 2).
PAH Mutations in PKU Patients Associated with Response and with Nonresponse to Sapropterin Treatment
Mutation analysis of PAH was performed in all the 112 patients, who completed the observational trial for sapropterin response. The bi-allelic underlying PAH mutations were detected in 108 (96.4 %) patients. In 4 (3.6 %) patients, only one pathogenic mutation on one allele could be detected. The failure to detect the mutation on the other allele was likely based on our sequencing approach, which did not cover intronic mutations or large deletions within the PAH gene.
Several genotypes were consistently associated with nonresponse and low dose response to sapropterin treatment (Supplementary Table 1). Genotypes always associated with nonresponse to sapropterin at any dose were [IVS12+1G>A];[IVS12+1G>A], p.[L348V];[R408W], p.[P281L];[P281L], p.[R158Q];[R408W], and p.[R261Q];[R408W]. Genotypes always associated with low dose response were p.[L48S];[L48S] and p.[Y414C];[Y414C]. Thus, there were more genotypes that consistently predicted nonresponse than response to sapropterin therapy, and no genotype consistently predicted high dose response (Supplementary Table 1).
Mutations associated with response were dispersed throughout the PAH gene. In our study, we observed a high frequency of discriminative N-terminal mutations (regulatory domain, amino acid position 1-142) in the group of responders. Several mutations in the regulatory domain of PAH, for example I65T or R68S, were only present in responders and not detected in any of the 66 nonresponders. Mutations in the catalytic domain of the PAH protein (amino acid position 143-410) did not consistently predict response or nonresponse to sapropterin therapy.
Frequency of mutations found in our cohort varied, with the most common being the R408W mutation, present on 48 alleles of 42 patients. Several mutations were predictive of response or nonresponse. Y414C, L48S, I65T, F331S and R68S were always associated with response, while R158Q was consistently associated with nonresponse.
Discussion
In our cohort of 112 patients who participated in the study, 46 patients (41.1 %) showed a significant response on sapropterin therapy, consistent with the estimated response rate among PKU patients reported by Fiege and Blau (2007). Patients who responded at 10 mg/kg had higher baseline Phe tolerance and seemed to also have higher increase in Phe tolerance, suggesting mild/moderate PKU (Table 2). Of those “low dose responders,” two probands were homozygous carriers of Y414C and five probands homozygous for L48S (Supplementary Table 1). Most of all responders show a clear increase in Phe tolerance (Table 2), with 7 of the 30 responders, treated continuously with sapropterin, being able to stop their Phe-restricted diet altogether while continuing a vegetarian diet.
Based on our cohort, PAH genotypes are helpful for predicting low dose response and nonresponse to sapropterin (Supplementary Table 1). However, no genotypes in our study were found to consistently predict high dose response. The genotype p.[P281L];[R408W] was found in six patients in our cohort (Supplementary Table 1). Of these, 5 were nonresponsive to sapropterin, yet one patient had a 31 % decrease in Phe levels at 20 mg/kg/day of sapropterin and was deemed responsive. While one might suspect change in dietary intake resulted in the decrease in Phe concentrations, this individual also had an 11 % increase in Phe tolerance, proving clinical benefit of treatment and thereby suggesting that this particular genotype cannot reliably discriminate between nonresponders and high dose response to treatment, consistent with the results of other studies (Trefz et al. 2009b). There are several other genotypes (Supplementary Table 1), which were found both, in nonresponders and in responders, underlying the difficulties in predicting sapropterin response only based on patients’ genotype.
Interestingly, one individual of six found with homozygous R408W, the mutation most frequently found in our cohort and most commonly found throughout Europe associated with classical PKU (Eisensmith et al. 1995; Zschocke 2003; Zurflüh et al. 2008; Karacic et al. 2009; Dobrowolski et al. 2009) and generally nonresponse to sapropterin treatment (Dobrowolski et al. 2009), responded to sapropterin already at a low dose while the remaining five individuals were classified as nonresponsive. However, the responsive individual had increasing Phe levels with treatment despite consistent Phe intake, resulting in cessation of sapropterin treatment and the individual being classified as a false-positive responder. As a result, all individuals with homozygous R408W may be considered nonresponsive to sapropterin, consistent with findings in other studies (Dobrowolski et al. 2009). Also consistent with other findings, IVS12+1G>A was always associated with nonresponse when on both alleles (Polak et al. 2013; Utz et al. 2012) or when associated with the mutation R408W (Utz et al. 2012).
Genotypes with homozygous mutations are more likely to predict sapropterin response than compound heterozygous mutations (Supplementary Table 1). Homozygous Y414C, located in the tetramerization domain, is associated with response even at the low dose of sapropterin. In other studies, Y414C has been associated with response even when combined with other mutations strongly associated with nonresponse, such as R408W (Trefz et al. 2009b; Utz et al. 2012). Interestingly, in our study when Y414C was present on only one allele, it was found in patients with both an increased Phe tolerance of <100 % and in those with an increase >300 % after sapropterin therapy, suggesting that the mutation on the allele in trans of this particular mutation is important for predicting the full clinical benefit.
Homozygous L48S, also frequently found in Southern Europe (Zurflüh et al. 2008), was always associated with sapropterin response even at the 10 mg/kg/day dose and with a high increase of Phe tolerance on BH4 therapy in our cohort (Supplementary Table 1), consistent with other studies (Zurflüh et al. 2008). One patient carrying this mutation was able to tolerate >300 % more natural protein than before sapropterin therapy. L48S, previously detected on 24 alleles of sapropterin responders (BIOPKU database, http://www.biopku.org/BioPKU_DatabasesBIOPKU.asp), was associated with response even when present on only one allele.
In our study, a high frequency of mutations in the regulatory domain was found in the group of responders. However, in the literature mutations in the regulatory domain have been inconsistent in predicting response between studies, with some revealing sapropterin responsiveness even in classic PKU patients (Wang et al. 2007) and other studies have shown inconsistent responses with identical mutations having significantly differing response rates (Trefz et al. 2009b). Mutations in the predicted catalytic domain were associated with both response and nonresponse to sapropterin treatment. The F331S mutation was always associated with sapropterin response when present on both alleles in two patients of our cohort, while R158Q on at least one allele was always associated with nonresponse to sapropterin. While p.[R261Q];[R408W], found in five individuals, was always associated with nonresponse, individually the mutations R261Q (Staudigl et al. 2011) and R408W (Utz et al. 2012) failed to discriminate between responders and nonresponders in our study, consistent with what is reported in the literature.
Although it may be expected that patients receive increased support in our clinical trial in comparison to that available in a regular outpatient setting, almost 20 % of patients still refused to take part, some citing time constraints and effort required to comply with the strict regimen. Of those who participated, 18 withdrew before completion due to noncompliance or difficulties in reaching the required stability of pretest blood Phe values. These results reinforce the need for alternative, less rigorous means to determine clinical benefit to sapropterin treatment.
The most significant risk identified in our study and relevant to all response test protocols was illustrated by seven patients who were initially classified as sapropterin responsive whose blood Phe levels rose significantly either despite unchanged Phe intake or when challenged with an increase in dietary Phe intake. In these patients, we stopped sapropterin during the 6-week diet modification period. We hypothesize that the response test was not correctly performed in these patients and that the observed decrease of Phe levels within the 2-week response test period merely reflected random fluctuations of their serum Phe levels or potential changes in diet during the response test protocol. As well, this limited control of the patient’s diet may be the main argument against outpatient sapropterin response testing. However, combining the response test with a second phase of gradual increase of dietary Phe intake as done with our protocol allows efficient detection of false-positive responders, as seen with our patients. As well, the extended test time and the risk of false-positive results are outweighed by the increased comfort for patients being tested at home and a more cost-effective process than hospitalization.
Conclusions
Individual genotype influences response to sapropterin therapy in PKU patients. Based on our study, we can conclude that PAH genotypes can help predicting low dose and nonresponse to sapropterin. However, it still needs to be shown to what extent genotype information can be used to predict in vivo BH4 responsiveness, and as stated by Staudigl et al. the evaluation of sapropterin treatment should involve “personalized procedures to safely identify and treat patients with BH4-responsive PAH deficiency” (Staudigl et al. 2011).
Based on the limited number of patients included in our study and inconsistent results in the literature, it does not seem appropriate to preclude a particular mutation as not associated with sapropterin responsiveness due to the absence of this particular mutation in our group of responders or conclude that a group of patients will be responsive based on being classified as responder in this group. Still, the results of our study contribute to the growing body of literature on PAH genotypes associated with sapropterin response, and support performing sapropterin response testing in an outpatient setting as a feasible and reliable option to assess sapropterin responsiveness.
Electronic Supplementary Material
Supplementary Table 1 Genotype and responsiveness to sapropterin treatment at high and low doses
Acknowledgements
This study and medical writing support was supported by an unrestricted grant from Merck Serono S.A. Medical writing support was provided by Judy Wiles of Facet Communications Incorporated.
Footnotes
Sarah Leuders and Eva Wolfgart contributed equally to this study
Competing interests: None declared
Contributor Information
Frank Rutsch, Email: rutschf@ukmuenster.de.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Blau N, Bélanger-Quintana A, Demirkol M, et al. Optimizing the use of sapropterin (BH4) in the management of phenylketonuria. Mol Genet Metab. 2009;96:158–163. doi: 10.1016/j.ymgme.2009.01.002. [DOI] [PubMed] [Google Scholar]
- Burgard P, Bremer HJ, Bührdel P, et al. Rationale for the German recommendations for phenylalanine level control in phenylketonuria 1997. Eur J Pediatr. 1999;158:46–54. doi: 10.1007/s004310051008. [DOI] [PubMed] [Google Scholar]
- Dobrowolski SF, Borski K, Ellingson CC, Koch R, Levy HL, Naylor EW. A limited spectrum of phenylalanine hydroxylase mutations is observed in phenylketonuria patients in western Poland and implications for treatment with 6R tetrahydrobiopterin. J Hum Genet. 2009;54:335–339. doi: 10.1038/jhg.2009.37. [DOI] [PubMed] [Google Scholar]
- Eisensmith RC, Goltsov AA, O’Neill C, et al. Recurrence of the R408W mutation in the phenylalanine hydroxylase locus in Europeans. Am J Hum Genet. 1995;56:278–286. [PMC free article] [PubMed] [Google Scholar]
- Enns GM, Koch R, Brumm V, et al. Suboptimal outcomes in patients with PKU treated early with diet alone: revisiting the evidence. Mol Genet Metab. 2010;101:99–109. doi: 10.1016/j.ymgme.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Fiege B, Blau N (2007) Assessment of tetrahydrobiopterin (BH4) responsiveness in phenylketonuria. J Pediatr 150(6):627–630 [DOI] [PubMed]
- Gersting SW, Lagler FB, Eichinger A, et al. Pahenu1 is a mouse model for tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency and promotes analysis of the pharmacological chaperone mechanism in vivo. Hum Mol Genet. 2010;19:2039–2049. doi: 10.1093/hmg/ddq085. [DOI] [PubMed] [Google Scholar]
- Karacic I, Meili D, Saravka V, et al. Genotype-predicted tetrahydrobiopterin (BH4)-responsiveness and molecular genetics in Croatian patients with phenylalanine hydroxylase (PAH) deficiency. Mol Genet Metab. 2009;97:165–171. doi: 10.1016/j.ymgme.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Kure S, Hou DC, Ohura T, et al. Tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency. J Pediatr. 1999;135:375–378. doi: 10.1016/S0022-3476(99)70138-1. [DOI] [PubMed] [Google Scholar]
- Kuvan® US Prescribing Information. http://www.kuvan.com/hcp/kuvan-full-prescribing-information.html, Accessed Sep 2013
- Lee P, Treacy EP, Crombez E, et al. Safety and efficacy of 22 weeks of treatment with sapropterin dihydrochloride in patients with phenylketonuria. Am J Med Genet A. 2008;146A:2851–2859. doi: 10.1002/ajmg.a.32562. [DOI] [PubMed] [Google Scholar]
- Levy H, Milanowski A, Chakrapami A, et al. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase II randomised placebo-controlled study. Lancet. 2007;370:504–510. doi: 10.1016/S0140-6736(07)61234-3. [DOI] [PubMed] [Google Scholar]
- Muntau AC, Röschinger W, Habich M, et al. Tetrahydrobiopterin as an alternative treatment for mild phenalketonuria. N Engl J Med. 2002;347:2122–2132. doi: 10.1056/NEJMoa021654. [DOI] [PubMed] [Google Scholar]
- National Institutes of Health Consensus Development Panel National Institutes of Health Consensus Development Conference Statement: Phenylketonuria: screening and management, October 16-18, 2000. Pediatrics. 2001;108:972–982. doi: 10.1542/peds.108.4.972. [DOI] [PubMed] [Google Scholar]
- Polak E, Ficek A, Radvanszky J et al (2013) Phenylalanine hydroxylase deficiency in the Slovak population: genotype-phenotype correlations and genptype-based predictions of BH4-responsiveness. Gene 526(2):347–355 [DOI] [PubMed]
- Schweitzer-Krantz S, Burgard P. Survey of national guidelines for the treatment of phenylketonuria. Eur J Pediatr. 2000;159(Suppl 2):70–73. doi: 10.1007/PL00014385. [DOI] [PubMed] [Google Scholar]
- Scriver CR, Kaufman S. Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. Metabolic and molecular basis of inherited disease. New York: McGraw-Hill; 2001. pp. 1667–1724. [Google Scholar]
- Scriver CR, Levy H, Donlon J (2008) Hyperphenylalaninemia: phenylalanine hydroxylase deficiency. In: Valle D, Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A (eds) The online metabolic and molecular bases of inherited disease. http://www.ommbid.com. chapter 77
- Staudigl M, Gersting SW, Danecka MK. The interplay between genotype, metabolic state and cofactor treatment governs phenylalanine hydroxylase function and drug response. Hum Mol Genet. 2011;20:2628–2641. doi: 10.1093/hmg/ddr165. [DOI] [PubMed] [Google Scholar]
- Thöny B, Ding Z, Martinez A. Tetrahydrobioterin protects phenylalanine hydroxylase activity in vivo: implications for tetrahydrobiopterin-responsive hyperphenylalaninemia. FEBS Lett. 2004;577:507–511. doi: 10.1016/j.febslet.2004.10.056. [DOI] [PubMed] [Google Scholar]
- Trefz FK, Burton BK, Longo N, et al. Efficacy of sapropterin dihydrochloride in increasing phenylalanine tolerance in children with phenylketonuria: a Phase III, randomized, double-blind, placebo controlled study. Pediatrics. 2009;154:700–707. doi: 10.1016/j.jpeds.2008.11.040. [DOI] [PubMed] [Google Scholar]
- Trefz FK, Scheible D, Götz H, Frauendienst-Egger G. Significance of genotype in tetrahydrobiopterin-responsive phenylketonuria. J Inherit Metab Dis. 2009;32:22–26. doi: 10.1007/s10545-008-0940-8. [DOI] [PubMed] [Google Scholar]
- Utz JRJ, Lorentz CP, Markowitz D, et al. START, a double blind, placebo-controlled pharmacogenetic test of responsiveness to sapropterin dihydrochloride in phenylketonuria patients. Mol Genet Metab. 2012;105:193–197. doi: 10.1016/j.ymgme.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Walter JH, White FJ, Hall SK, et al. How practical are recommendations for dietary control in phenylketonuria? Lancet. 2002;360:55–57. doi: 10.1016/S0140-6736(02)09334-0. [DOI] [PubMed] [Google Scholar]
- Wang L, Surendran S, Michals-Matalon K, et al. Mutations in the regulatory domain of phenylalanine hydroxylase and response to tetrahydrobiopterin. Genet Test. 2007;11:174–178. doi: 10.1089/gte.2006.0520. [DOI] [PubMed] [Google Scholar]
- Weglage J, Ullrich K, Pietsch M et al (1996) Untreated non-phenylketonuric-hyperphenylalaninaemia: intellectual and neurological outcome. Eur J Pediatr 155(Suppl 1):S26–S28 [DOI] [PubMed]
- Zschocke J. Phenylketonuria mutations in Europe. Hum Mutat. 2003;21:345–356. doi: 10.1002/humu.10192. [DOI] [PubMed] [Google Scholar]
- Zurflüh MR, Zschocke J, Lindner M, et al. Molecular genetics of tetrahydrobiopterin responsive phenylalanine hydroxylase deficiency. Hum Mutat. 2008;29:167–175. doi: 10.1002/humu.20637. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1 Genotype and responsiveness to sapropterin treatment at high and low doses