

Abstract
X-linked creatine transport (CRTR) deficiency, caused by mutations in the SLC6A8 gene, leads to intellectual disability, speech delay, epilepsy, and autistic behavior in hemizygous males. Additional diagnostic features are depleted brain creatine levels and increased creatine/creatinine ratio (cr/crn) in urine. In heterozygous females the phenotype is highly variable and diagnostic hallmarks might be inconclusive. This survey aims to explore the intrafamilial variability of clinical and brain proton Magnetic Resonance Spectroscopy (MRS) findings in males and females with CRTR deficiency. X-chromosome exome sequencing identified a novel missense mutation in the SLC6A8 gene (p.G351R) in a large family with X-linked intellectual disability. Detailed clinical investigations including neuropsychological assessment, measurement of in vivo brain creatine concentrations using quantitative MRS, and analyses of creatine metabolites in urine were performed in five clinically affected family members including three heterozygous females and one hemizygous male confirming the diagnosis of CRTR deficiency. The severe phenotype of the hemizygous male was accompanied by most distinct aberrations of brain creatine concentrations (−83% in gray and −79% in white matter of age-matched normal controls) and urinary creatine/creatinine ratio. In contrast, the heterozygous females showed varying albeit generally milder phenotypes with less severe brain creatine (−50% to −33% in gray and −45% to none in white matter) and biochemical urine abnormalities. An intrafamilial correlation between female phenotype, brain creatine depletion, and urinary creatine abnormalities was observed. The combination of powerful new technologies like exome-next-generation sequencing with thorough systematic evaluation of patients will further expand the clinical spectrum of neurometabolic diseases.
Introduction
Creatine deficiency syndromes constitute a group of neurometabolic disorders which include two autosomal recessive conditions with impaired creatine synthesis (arginine glycine amidinotransferase (AGAT) deficiency, OMIM #612718, and guanidinoacetate methyltransferase (GAMT) deficiency, OMIM #612736) as well as one X-linked defect of creatine transport (creatine transport (CRTR) deficiency, OMIM #300036). The latter is caused by mutations in the SLC6A8 gene, located on chromosome Xq28 (Longo et al. 2011). Prevalent clinical symptoms of CRTR deficiency are intellectual disability (ID), expressive speech and language delay, epilepsy, and autistic behavior (van de Kamp et al. 2013).
CRTR deficiency is a quite common cause of X-linked ID (XLID) in males with an estimated frequency of 0.3–3.5% (Arias et al. 2007; Clark et al. 2006). The majority of affected males with CRTR deficiency present with non-syndromic ID. However, in some cases mild dysmorphic features such as mid-facial hypoplasia and short stature have been described (van de Kamp et al. 2013). Diagnostic hallmarks in males with CRTR deficiency are a markedly reduced or absent brain creatine (tCr) signal measured by proton magnetic resonance spectroscopy (MRS) and an increased creatine/creatinine (cr/crn) ratio in urine. In contrast to GAMT deficiency, guanidinoacetate (GAA) levels in plasma and urine remain within normal range.
To date, numerous mutations in the SLC6A8 gene have been described (compare Leiden Open Variation Database, www.lovdnl/slc6a8). According to the X-chromosomal inheritance, females with heterozygous mutations in the SLC6A8 gene can be mildly affected or may even be asymptomatic. The variable phenotype has been ascribed to the different X-inactivation patterns (van de Kamp et al. 2011). The female phenotypes that have been reported so far comprise developmental delay as well as mild to moderate ID. In addition, some affected females show subtle cerebellar symptoms, behavioral or gastrointestinal problems, and in one case severe epilepsy (Hahn et al. 2002; Kleefstra et al. 2005; Mancardi et al. 2007; Mercimek-Mahmutoglu et al. 2010). In females, MRS has failed to demonstrate an unambiguously reduced tCr signal (Bizzi et al. 2002; Salomons et al. 2001). Moreover, elevated cr/crn ratio in urine and impaired creatine uptake in fibroblasts, the biochemical markers in affected males, seem unreliable parameters in heterozygous females (van de Kamp et al. 2011).
Here, we report a large German family with non-syndromic ID caused by a novel SLC6A8 gene mutation, which has been detected via thorough sequencing of all X-chromosome-specific exons. The survey of clinical and MRS data demonstrates the intrafamilial spectrum of clinical phenotypes in affected males and females and correlates the degree of ID with laboratory and MRS findings.
Subjects and Methods
Subjects
A large German family (D150) with intellectually disabled members in three generations was analyzed (compare family tree, Fig. 1). One affected male (II.4) with severe ID also had epilepsy. Two affected males with severe ID lived in sheltered homes (II.4 and III.4). Craniofacial dysmorphism was not present in this family. Five family members (four females: II.2, III.2, IV.1, IV.3 and one male: IV.4) were studied in detail. The healthy girl (IV.2) with normal intelligence was excluded from further investigations.
Fig. 1.
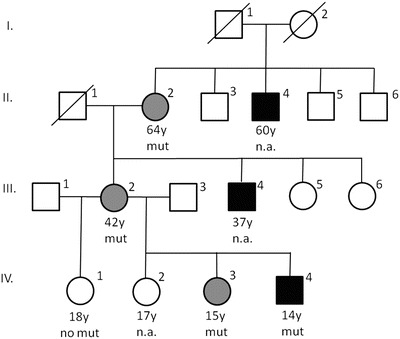
Family tree. Family members of three generations were affected. Gray symbols represent affected heterozygous females, black symbols indicate affected males. mut genetic analysis revealed the mutation p.G351R in the SLC6A8 gene, no mut the familial SLC6A8 mutation was excluded by genetic analysis, n.a. not analyzed, genetic analysis has not been performed. Subjects II.4 and III.4 are not included in Table 1. Subject II.4 (age 60 years) is known to suffer from epilepsy, severe intellectual disability, and to live in a residential home for disabled people. Subject III.4 (age 37years) is known to have a severe intellectual disability and lives in a residential home
Neuropsychological Assessment
To evaluate intellectual abilities in this family, the Wechsler Adult Intelligence Test-Third Edition was used for subjects II.2, III.2, and IV.1. To study subjects IV.3 and IV.4, the Wechsler Intelligence Scale for Children-Forth Edition was applied. Mild ID was defined as an IQ score of 50–69 and moderate ID as an IQ score of 35–49.
Sequencing of X-Chromosome-Specific Exons
Genomic DNA (3 μg) from the index patient (IV.4 in Fig. 1) was used for constructing a single-end Illumina sequencing library using the Illumina Genomic DNA Single End Sample Prep kit, according to the instructions of the manufacturer. For X-chromosome exome enrichment, we used the Agilent SureSelect Human X Chromosome Kit, which contains 47,657 RNA baits for 7,591 exons of the human X chromosome. Single-end deep sequencing was performed on the Illumina Genome Analyzer GAIIx. Read length was 76 nt. Sequences were analyzed with in-house-developed tools. Confirmation of the SLC6A8 mutation and segregation analysis in the family was done by PCR and conventional Sanger sequencing using the gene-specific primer pair 2769_10SLC6A8f TCCCGGCCTCCTACTACTTCC, 2769_10SLC6A8r CATACAGGCAATGTCGTCCA.
Proton MR-Spectroscopy
Combined MRS/MR-imaging (MRI) examinations were performed on a 3T clinical whole-body MR system (Magnetom Tim Trio, Siemens Healthcare, Erlangen, Germany) with an 8-channel receive array (Invivo, Gainesville, FL, USA). The study was approved by the institutional ethics committee.
For single voxel, MRS volumes of interest (VOI) were selected from T1- and T2-weighted images and positioned in posterior paramedian cortical gray matter (GM) (12.5 ml), in frontal (F) as well as parieto-occipital (PO) white matter (WM) (4.1 ml) (see Fig. 2). Fully relaxed spectra were obtained in each VOI by applying the short-echo time stimulated echo acquisition mode (STEAM) sequence (repetition time/echo time/mixing time: 6000/20/10 ms, 64 accumulations) (Frahm et al. 1989; Natt et al. 2005). Metabolites considered in this study include creatine and phosphocreatine (tCr) as ubiquitous compounds linked to energy metabolism, the neuroaxonal marker N-acetylaspartate and N-acetylaspartylglutamate (tNAA), choline-containing compounds (Cho) involved in membrane turnover, and the glial marker myo-inositol (Ins) (Dreha-Kulaczewski et al. 2009; Pouwels and Frahm 1998). Additionally, GAA, involved in creatine metabolism, was assessed. The absolute concentrations of these metabolites were calculated using LCModel and expressed in mmol/l (Provencher 1993).
Fig. 2.
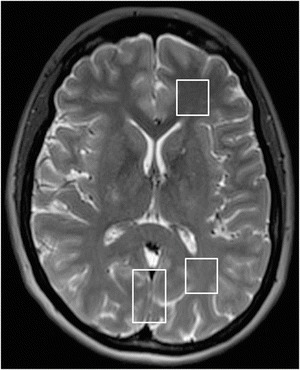
Axial T2-weighted MRI of heterozygous female subject III.2. MRI demonstrates the placement of VOI in posterior paramedian cortical gray matter and in frontal as well as parieto-occipital white matter. No signal abnormalities could be detected. Mild cerebellar atrophy is not shown
Metabolite concentrations were compared to healthy age- and region-matched control groups from our local database. Deviations of >2 standard deviations (SD) from control values were considered significant.
Biochemical Analysis
GAA and cr were measured in urine by gas chromatography– mass spectrometry with deuterated internal standards as previously described (Almeida et al. 2004; Arias et al. 2004). Values were compared to healthy age-matched controls obtained from a local database.
Results
Clinical and developmental details of six family members are summarized in Table 1. Two female subjects (IV.1, IV.3 in Fig. 1) had a history of motor and speech delay. No developmental data were available for the two females II.2 and III.2. Female IV.1 had epilepsy from her 6th to 16th year of life which was well controlled by valproic acid. Three females (III.2, IV.1, and IV.3) required special education due to ID. In addition, aggressive and hyperactive behavior was evident in female subject IV.3. All four females (II.2, III.2, IV.1, and IV.3) had IQ scores in the mild ID range (IQ 50–69). The lowest score was reached by subject III.2.
Table 1.
Clinical features, urine analysis and MRS data of individual family members
| Subject | G | Age (yrs) | Developmental and neurological details | Education | IQ |
SLC6A8 mutation p.G351R |
MRS (mmol/l) | Urine analysis | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| VOI | tCr | tNAA | Cho | Ins | cr/crn | GAA (μMol/mMol crn) | |||||||
| II.2 | f | 64 | Developmental details n.k., mild ID |
n.k. | 62e | Hetero zygous |
n.p. | 0.173 | 45.0 | ||||
| III.2 | f | 43 | Developmental details n.k., mild ID |
Special school, no profession | 50e | Hetero zygous |
GM WM F |
3.3*3.5 | 6.2 6.9 |
1.1 1.6 |
3.4 4.6 |
0.235 | 31.6 |
| IV.1 | f | 17 | MDa/SDb; Epilepsy, mild ID |
Special school, sheltered workplace | 62e | None | GM WM F |
5.2 5.0 |
7.0 7.8* |
1.1 1.7 |
3.6 3.8* |
0.047 | 37.7 |
| IV.2 | f | 16 | Normal development, normal intelligence |
High school | n.t. | n.d. | n.d. | n.d. | n.d. | ||||
| IV.3 | f | 14 | SDc, aggressive behavior, hyperactive, mild ID |
Special school | 57f | Hetero zygous |
GM WM PO |
2.6* 2.3* |
7.7 8.2 |
0.8 1.0 |
3.7 2.5 |
0.338g | 33.4 |
| IV.4 | m | 13 | MDa/SDd, aggressive behavior, moderate ID |
Special school | 40f | Hemi zygous |
GM WM PO |
0.9* 0.9* |
7.5 8.4* |
0.9 1.2 |
4.8 2.7 |
2.094g | 78.4 |
G gender, f female, m male, yrs years, n.k. not known, n.t. not tested, n.d. not done, MD motor delay, SD speech delay, IQ intelligence quotient, cr creatine, crn creatinine, GAA guanidinoacetate, ID intellectual disability, mild ID IQ score of <70, moderate ID IQ score of <50, MRS magnetic resonance spectroscopy, VOI volume of interest, tCr creatine and phosphocreatine; tNAA N-acetylaspartate and N-acetylaspartylglutamate, Cho choline-containing compounds, Ins myo-inositol, GM gray matter, WM F white matter frontal, WM PO white matter parieto-occipital
aWalking at ~2.5 years
bSingle words at ~2.5 years
cSingle words at ~2 years, short sentences at ~5 years
dSingle words at 2.7 years
eWechsler Adult Intelligence Test-Third Edition
fWechsler Intelligence Scale for Children-Forth Edition
gAbove normal range
* >2SD from control
The affected male (IV.4) presented a delay in motor and speech development as well as aggressive behavioral problems. The boy requires special education. Notably, his IQ score was the lowest of all family members and fell within the moderate ID range (IQ 35–49).
Genetic Studies
In an effort to identify the pathogenic mutations in families with XLID collected by the EUROMRX consortium and associated groups, we sequenced all X-chromosome specific exons in the index patient (IV.4 in Fig. 1) of this large family (D150). After filtering of all identified variants against publicly available data, including the 1000 Genomes project database, dbSNP135 and the Exome Variant Server, the only mutation that was predicted to be deleterious was a missense mutation in SLC6A8 (g.G>A, chrX:152959802–152959802, UCSC, hg19, p.Gly351Arg, NP_001136278; p.Gly456Arg, NP_001136277, p.Gly466Arg, NP_005620). Moreover, this change was absent in >450 X-chromosome exomes from males with XLID (data not shown). Subsequently, the presence of this SLC6A8 mutation was confirmed by PCR using a gene-specific primer set and Sanger sequencing, and it was also present in additional affected family members (Table 1).
Proton MR-Spectroscopy
Spectra from GM and WM of four subjects are shown in Fig. 3. No distinct tCr peaks could be detected in GM and WM of the hemizygous male IV.4 (Fig. 3, row A). In the heterozygous girl IV.3 (Fig. 3, row B) tCr resonances were evident but decreased when compared to controls (Fig. 3, row E). The tCr peak heights of the adult heterozygote III.2 (Fig. 3, row C) approximated those of normal controls. By contrast, the spectra obtained from the female without SLC6A8 mutation IV.1 (Fig. 3, row D) were unremarkable.
Fig. 3.
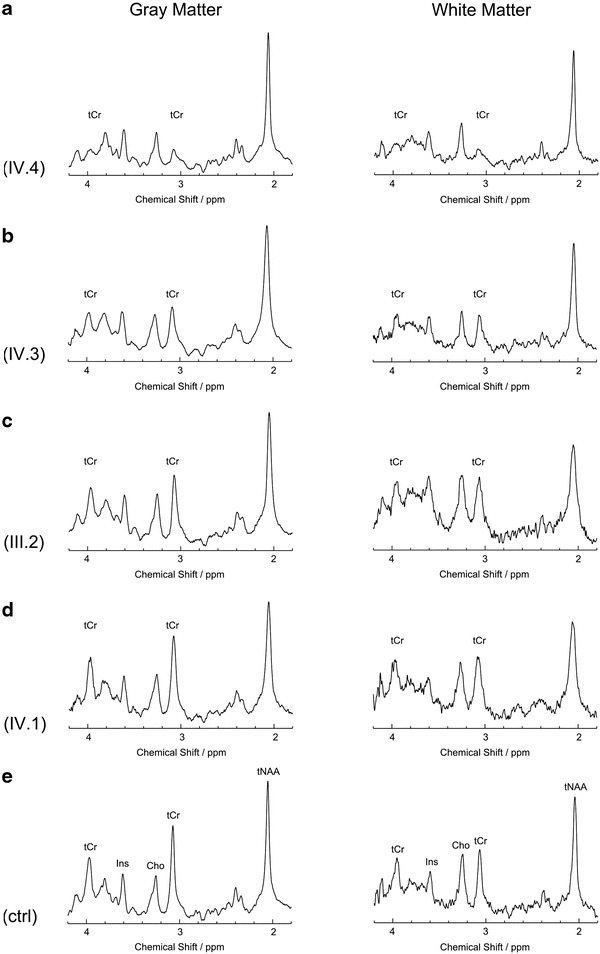
Single voxel proton MR spectra from gray and white matter of affected family members with CRTR deficiency. (Row A) male subject IV.4 hemizygous for SLC6A8 mutation. (row B) female subject IV.3 heterozygous for SLC6A8 mutation. (Row C) female subject III.2 heterozygous for SLC6A8 mutation. (Row D) female subject IV.1 without mutation. (Row E) healthy control subject (female, age 13 years). Note the variability of tCr (sum of creatine and phosphocreatine) peaks at 3.2 and 3.9 ppm between subjects. In row A no defined tCr peaks can be detected, whereas in row B they are clearly visible but reduced compared to controls (row E). In row C and D the tCr peaks are of normal heights compared to controls. Assignments of main peaks of the proton MR spectrum are added to the control spectra (row E). Note that scale of spectra varies to facilitate qualitative assessment of differences of tCr peak heights. tNAA sum of N-acetylaspartate and N-acetylaspartylglutamate, Cho choline-containing compounds, Ins myo-inositol
Quantitative analysis of tCr concentrations confirmed the most distinct reduction (GM −83%, WM −79% of normal control values in Table 2) in the affected boy (IV.4) (see Table 1). The tCr levels in the affected heterozygous sister (IV.3) were also significantly reduced compared to controls, albeit to a smaller degree (GM −50%, WM −45%). The heterozygous mother (III.2) showed a significant tCr decrease (−33%) only in GM. The tCr concentrations of the female (IV.1) without SLC6A8 mutation were within normal range. The other major metabolites showed no significant differences in GM compared to controls. In WM, high levels of tNAA in subjects IV.1 and IV.4 as well as of Ins in subject IV.1 were found.
Table 2.
Control values for MRS and urine analysis
| MRS (mmol/l) | Urine analysis | |||||
|---|---|---|---|---|---|---|
| VOI | tCr | tNAA | Cho | Ins | cr/crn | GAA (μmol/Mmol crn) |
| GM n = 22 (13.5 ± 2.6 yrs) GM n = 4 (40.6 ± 4.1 yrs) WM F n = 18 (13.6 ± 2.6 yrs) WM F n = 4 (40.6 ± 4.1 yrs) WM PO n = 21 (13.5 ± 2.3 yrs) WM PO n = 4 (40.6 ± 4.1 yrs) |
5.2 ± 0.9 4.9 ± 0.2 3.9 ± 0.6 3.9 ± 0.5 4.2 ± 0.5 3.9 ± 0.7 |
7.2 ± 1.0 7.0 ± 0.6 5.9 ± 0.9 6.1 ± 0.4 7.3 ± 0.5 7.1 ± 0.6 |
1.0 ± 0.3 1.0 ± 0.1 1.4 ± 0.3 1.7 ± 0.4 1.5 ± 0.3 1.6 ± 0.3 |
3.4 ± 0.7 3.1 ± 0.2 2.4 ± 0.6 3.4 ± 1.2 2.7 ± 0.4 2.8 ± 0.3 |
n = 169
0.011–0.25 (>12 yrs) |
n = 169
4–220 (<15 yrs) 10–160 (>15 yrs) |
n number of subjects, yrs years, cr creatine, crn creatinine, GAA guanidinoacetate, VOI volume of interest, MRS magnetic resonance spectroscopy, tCr creatine and phosphocreatine, tNAA N-acetylaspartate and N-acetylaspartylglutamate, Cho choline-containing compounds, Ins myo-inositol, GM gray matter, WM white matter, F frontal, PO parieto-occipital
No signal clearly arising from GAA (at a chemical shift of 3.8 ppm) could be detected in any of the subjects’ spectra. With GAA included into the LCModel analysis, low concentrations of GAA may be fitted in GM only, but these were comparable to normal controls and thus regarded an artifact.
The MRI of the hemizygous boy showed an unspecific small, hyperintense lesion on T2-weighted images within the left basal ganglia. Mild cerebellar vermis atrophy was detectable on the MRI of the heterozygous female subject III.2 and slightly more pronounced on the MR images of her daughter (IV.1) without SLC6A8 mutation.
Biochemical Analysis
Notably, the most pronounced increase of cr/crn urinary level was measured in the hemizygous boy (IV.4). The comparatively much lower ratio of his heterozygous sister (IV.3) (Table 1) was still elevated with respect to her age-matched control group (Table 2). Their heterozygous mother (III.2) as well as the half-sister without SLC6A8 mutation (IV.1) had normal cr/crn ratios. Urinary excretion of GAA was within normal ranges in all subjects.
Discussion
In a large family with non-syndromic XLID, X-chromosome exome screening revealed a novel mutation in the SLC6A8 gene (p.G351R, NP_001136278) leading to CRTR deficiency syndrome. The diagnosis of CRTR deficiency was subsequently confirmed by MRS of the brain as well as urinary creatine/creatinine measurement in three affected females and one affected male. The phenotypic variability within this family is in line with numerous reports about the broad range of clinical presentations in this neurometabolic disease (deGrauw et al. 2003; van de Kamp et al. 2013). Profound ID was evident in all male members. The hemizygous male index patient presented with the characteristic severe phenotype comprising ID as well as severe speech delay and behavioral disturbances. All heterozygous females showed milder symptoms. Their IQs ranged from 50–62 (Table 1). However, the familial SLC6A8 mutation was excluded in one half-sister (IV.1) of the index patient, who presented with mild ID and epilepsy. This half-sister has a different and also intellectually disabled father than subjects IV.2-IV.4 and therefore may be affected with ID of another origin. Cerebellar signs or gastrointestinal problems like chronic constipation, ileus, or megacolon, which had previously been reported in affected males and females (Kleefstra et al. 2005; van de Kamp et al. 2011), were not present in our family. Moreover, no affected family member had obvious craniofacial dysmorphism.
In the clinical setting, CRTR deficiency is usually diagnosed via a screening test in urine, which shows a strong elevation of the cr/crn ratio in affected males. In our index patient, cr/crn ratio screening in urine was problematic since he rejected urine sampling several times and wore diapers. The missense mutation in the SLC6A8 gene was detected via X-chromosome exome sequencing. Subsequently, the cr/crn ratios were evaluated in all affected heterozygous females. Interestingly, only one of the three heterozygous females had a mild increase of the urinary cr/crn ratio. Remarkably, this female patient also presented with the lowest brain tCr levels and had a more severe phenotype comprising behavioral disturbances. This observation is in line with a correlation between clinical phenotype, cerebral tCr depletion and elevation of urinary cr/crn ratio postulated by van de Kamp et al (van de Kamp et al. 2011). However, a strict correlation between brain MRS or biochemical data and IQ scores, as suggested, has not been found in the heterozygous females with CRTR deficiency described here. Yet, an inverse correlation between cerebral tCr concentrations and urinary cr/crn ratios could be seen. The small sample size precluded further statistical analysis.
CRTR deficiency syndromes are increasingly detected among patients with non-syndromic unexplained ID (Clark et al. 2006; Lion-Francois et al. 2006; Rosenberg et al. 2004). In contrast, defects in creatine synthesis seem to be much less frequent. In affected females, it is difficult to diagnose CRTR deficiency via urine analysis. In our study, a significant elevation of cr/crn ratio in urine analysis was only found in one affected female.
We performed a quantitative brain MRS study in this family on both affected genders of two generations, which revealed significant reduction of brain tCr concentration in the GM of all family members with the SLC6A8 gene mutation. In WM of the adult heterozygous female the concentration of tCr remained within normal ranges (see Table 1). The hemizygous male index patient IV.4 (Fig. 2, row A) showed almost complete absence of the tCr peaks in both GM and WM, which corresponds to previous reports (van de Kamp et al. 2013). The disparity of the tCr reduction among the heterozygous females within the same family, however, is a remarkable finding. The youngest girl with SLC6A8 mutation showed a distinct tCr decrease, which was already qualitatively appreciable in the spectra (Fig. 2, row B) and subsequently confirmed by quantification. In contrast, the slighter tCr reduction in GM of the heterozygous mother has not been evident in the spectral pattern but was disclosed only as a result of the quantification (Fig. 2, row C). The tCr concentrations in one half-sister of the index patient, in whom the familial SLC6A8 mutation was excluded, were within normal limits (Fig. 2, row D) as expected.
To date, quantitative brain MRS data of heterozygous females with CRTR deficiency have only been included in a minority of reports. Most publications are restricted to the qualitative description of a reduced peak of total creatine (tCr) in the spectrum (Cecil et al. 2003; deGrauw et al. 2003; Salomons et al. 2001). Dezertova et al. (2008) verified a decreased tCr concentration in an affected female only after quantification, thus emphasizing the importance of quantitative MRS. On the other hand, another quantitative MRS study of a cohort of eight female heterozygotes revealed a significant tCr reduction only on the group level (van de Kamp et al. 2011). Individual brain creatine concentrations were within normal ranges regardless of the region, a finding that is consistent with our results. The observation of a GAA signal as described by Sijens et al. (2005) could not be confirmed in our study.
The small unspecific hyperintense lesion in the basal ganglia seen on T2-weighted MR images of the boy is consistent with a hamartoma. The atrophy of cerebellar vermis has been detected in female family members with as well as without SLC6A8 mutation and thus is considered as unspecific.
Several therapeutic trials using creatine monohydrate, L-arginine, and glycine have so far failed to reliably improve cerebral tCr levels and/or clinical symptoms in patients with CRTR deficiency (van de Kamp et al. 2012). However, brain-specific Slc6a8 knockout mice, an animal model of human CRTR deficiency, were treated with the creatine analog cyclocreatine and did indeed show improved cognition as recently reported by Kurosawa et al. (2012). The authors were also able to demonstrate that the cyclocreatine entered the mouse brain and was successfully funneled into its metabolism. Thus, further studies using this promising therapy approach are warranted to assess its implications for humans.
Conclusion
In a large family with X-linked intellectual disability of unknown cause, the novel SLC6A8 mutation p.G351R was detected via X-chromosome exome sequencing. The diagnosis of CRTR deficiency was confirmed by MRS of the brain as well as urinary creatine/creatinine measurement, thus underlining the pathogenicity of this novel SLC6A8 mutation. Female phenotypes heterozygous for the mutation varied significantly even within one family. However, a correlation between severity of clinical symptoms, brain creatine depletion and urinary creatine abnormalities could be observed.
The combination of powerful new technologies like exome-next-generation sequencing with thorough systematic evaluation of patients as in this study will work synergistically to further expand our knowledge of the clinical spectrum of neurometabolic diseases.
Acknowledgment
This work has been supported by grants of the German Ministry of Education and Research through the German Leukonet (Grants 01GM0642 and 01GM0836; KB, SDK, JG); the MRNET (Grant 01GS08161; HHR); by the Project GENCODYS (241995), which is funded by the European Union Framework Program 7 (FP7); and the Volkswagen Stiftung (PD, GH). SDK has received funding from the Dorothea-Schloezer-Program of the Georg August University Goettingen, Germany. We thank Melanie Bienek and Ute Fischer for excellent technical assistance.
Synopsis
The combination of the powerful technology exome-next-generation sequencing with thorough systematic clinical evaluation of patients, as in this study of a family with X-linked creatine transport deficiency, will work synergistically to further expand the clinical spectrum of neurometabolic diseases.
Compliance with Ethics Guidelines
Conflict of Interest
Steffi Dreha-Kulaczewski, Vera Kalscheuer, Andreas Tzschach, HaoCougar Hu, Gunther Helms, Knut Brockmann, Almut Weddige, Peter Dechent, Gregor Schlüter, Ralf Krätzner, Hans-Hilger Ropers, Jutta Gärtner, and Birgit Zirn declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained for all patients being included in the study.
Details of the Contributions of Individual Authors
SDK: designed and conducted the MR studies, interpreted the MR data, wrote the manuscript
VMK, HHR: designed the genetic studies, interpreted the data, substantially revised the manuscript
AT, HH, GS: conducted the genetic studies, analyzed and interpreted the data
AW: conducted the neuropsychological investigations
RK: designed and conducted the biochemical studies, interpreted the data, revised the manuscript
GH, PD: designed the MR studies, interpreted the data, revised the manuscript
KB, JG, BZ: designed and conducted the clinical study, substantially revised the manuscript
Footnotes
Competing interests: None declared
Contributor Information
S. Dreha-Kulaczewski, Email: sdreha@gwdg.de
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Almeida LS, Verhoeven NM, Roos B, et al. Creatine and guanidinoacetate: diagnostic markers for inborn errors in creatine biosynthesis and transport. Mol Genet Metab. 2004;82:214–219. doi: 10.1016/j.ymgme.2004.05.001. [DOI] [PubMed] [Google Scholar]
- Arias A, Garcia-Villoria J, Ribes A. Guanidinoacetate and creatine/creatinine levels in controls and patients with urea cycle defects. Mol Genet Metab. 2004;82:220–223. doi: 10.1016/j.ymgme.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Arias A, Corbella M, Fons C, et al. Creatine transporter deficiency: prevalence among patients with mental retardation and pitfalls in metabolite screening. Clin Biochem. 2007;40:1328–1331. doi: 10.1016/j.clinbiochem.2007.07.010. [DOI] [PubMed] [Google Scholar]
- Bizzi A, Bugiani M, Salomons GS, et al. X-linked creatine deficiency syndrome: a novel mutation in creatine transporter gene SLC6A8. Ann Neurol. 2002;52:227–231. doi: 10.1002/ana.10246. [DOI] [PubMed] [Google Scholar]
- Cecil KM, DeGrauw TJ, Salomons GS, Jakobs C, Egelhoff JC, Clark JF. Magnetic resonance spectroscopy in a 9-day-old heterozygous female child with creatine transporter deficiency. J Comput Assist Tomogr. 2003;27:44–47. doi: 10.1097/00004728-200301000-00009. [DOI] [PubMed] [Google Scholar]
- Clark AJ, Rosenberg EH, Almeida LS, et al. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum Genet. 2006;119:604–610. doi: 10.1007/s00439-006-0162-9. [DOI] [PubMed] [Google Scholar]
- deGrauw TJ, Cecil KM, Byars AW, Salomons GS, Ball WS, Jakobs C. The clinical syndrome of creatine transporter deficiency. Mol Cell Biochem. 2003;244:45–48. doi: 10.1023/A:1022487218904. [DOI] [PubMed] [Google Scholar]
- Dezortova M, Jiru F, Petrasek J, et al. 1H MR spectroscopy as a diagnostic tool for cerebral creatine deficiency. Magma. 2008;21:327–332. doi: 10.1007/s10334-008-0137-z. [DOI] [PubMed] [Google Scholar]
- Dreha-Kulaczewski SF, Helms G, Dechent P, Hofer S, Gartner J, Frahm J. Serial proton MR spectroscopy and diffusion tensor imaging in infantile Balo's concentric sclerosis. Neuroradiology. 2009;51:113–121. doi: 10.1007/s00234-008-0470-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm J, Bruhn H, Gyngell ML, Merboldt KD, Hanicke W, Sauter R. Localized high-resolution proton NMR spectroscopy using stimulated echoes: initial applications to human brain in vivo. Magn Reson Med. 1989;9:79–93. doi: 10.1002/mrm.1910090110. [DOI] [PubMed] [Google Scholar]
- Hahn KA, Salomons GS, Tackels-Horne D, et al. X-linked mental retardation with seizures and carrier manifestations is caused by a mutation in the creatine-transporter gene (SLC6A8) located in Xq28. Am J Hum Genet. 2002;70:1349–1356. doi: 10.1086/340092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra T, Rosenberg EH, Salomons GS, et al. Progressive intestinal, neurological and psychiatric problems in two adult males with cerebral creatine deficiency caused by an SLC6A8 mutation. Clin Genet. 2005;68:379–381. doi: 10.1111/j.1399-0004.2005.00489.x. [DOI] [PubMed] [Google Scholar]
- Kurosawa Y, Degrauw TJ, Lindquist DM, et al. Cyclocreatine treatment improves cognition in mice with creatine transporter deficiency. J Clin Invest. 2012;122:2837–2846. doi: 10.1172/JCI59373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lion-Francois L, Cheillan D, Pitelet G, et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology. 2006;67:1713–1714. doi: 10.1212/01.wnl.0000239153.39710.81. [DOI] [PubMed] [Google Scholar]
- Longo N, Ardon O, Vanzo R, Schwartz E, Pasquali M. Disorders of creatine transport and metabolism. Am J Med Genet C Semin Med Genet. 2011;157:72–78. doi: 10.1002/ajmg.c.30292. [DOI] [PubMed] [Google Scholar]
- Mancardi MM, Caruso U, Schiaffino MC, et al. Severe epilepsy in X-linked creatine transporter defect (CRTR-D) Epilepsia. 2007;48:1211–1213. doi: 10.1111/j.1528-1167.2007.01148.x. [DOI] [PubMed] [Google Scholar]
- Mercimek-Mahmutoglu S, Connolly MB, Poskitt KJ, et al. Treatment of intractable epilepsy in a female with SLC6A8 deficiency. Mol Genet Metab. 2010;101:409–412. doi: 10.1016/j.ymgme.2010.08.016. [DOI] [PubMed] [Google Scholar]
- Natt O, Bezkorovaynyy V, Michaelis T, Frahm J. Use of phased array coils for a determination of absolute metabolite concentrations. Magn Reson Med. 2005;53:3–8. doi: 10.1002/mrm.20337. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med. 1998;39:53–60. doi: 10.1002/mrm.1910390110. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Rosenberg EH, Almeida LS, Kleefstra T, et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet. 2004;75:97–105. doi: 10.1086/422102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomons GS, van Dooren SJ, Verhoeven NM, et al. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet. 2001;68:1497–1500. doi: 10.1086/320595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sijens PE, Verbruggen KT, Oudkerk M, van Spronsen FJ, Soorani-Lunsing RJ. 1H MR spectroscopy of the brain in Cr transporter defect. Mol Genet Metab. 2005;86:421–422. doi: 10.1016/j.ymgme.2005.08.004. [DOI] [PubMed] [Google Scholar]
- van de Kamp JM, Mancini GM, Pouwels PJ, et al. Clinical features and X-inactivation in females heterozygous for creatine transporter defect. Clin Genet. 2011;79:264–272. doi: 10.1111/j.1399-0004.2010.01460.x. [DOI] [PubMed] [Google Scholar]
- van de Kamp JM, Pouwels PJ, Aarsen FK, et al. Long-term follow-up and treatment in nine boys with X-linked creatine transporter defect. J Inherit Metab Dis. 2012;35:141–149. doi: 10.1007/s10545-011-9345-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet. 2013;50:463–472. doi: 10.1136/jmedgenet-2013-101658. [DOI] [PubMed] [Google Scholar]