

Abstract
Multiple acyl-CoA dehydrogenation deficiency (MADD; also known as glutaric aciduria type II) is a human autosomal recessive disease classified as one of the mitochondrial fatty-acid oxidation disorders. MADD is caused by a defect in the electron transfer flavoprotein (ETF) or ETF dehydrogenase (ETFDH) molecule, but as yet, inherited MADD has not been reported in animals. Here we present the first report of MADD in a cat. The affected animal presented with symptoms characteristic of MADD including hypoglycemia, hyperammonemia, vomiting, diagnostic organic aciduria, and accumulation of medium- and long-chain fatty acids in plasma. Treatment with riboflavin and l-carnitine ameliorated the symptoms. To detect the gene mutation responsible for MADD in this case, we determined the complete cDNA sequences of feline ETFα, ETFβ, and ETFDH. Finally, we identified the feline patient-specific mutation, c.692T>G (p.F231C) in ETFDH. The affected animal only carries mutant alleles of ETFDH. p.F231 in feline ETFDH is completely conserved in eukaryotes, and is located on the apical surface of ETFDH, receiving electrons from ETF. This study thus identified the mutation strongly suspected to have been the cause of MADD in this cat.
Introduction
Multiple acyl-CoA dehydrogenation deficiency (MADD, also known as glutaric aciduria type II, GAII, OMIM 231680) is a type of mitochondrial fatty-acid oxidation disorder (FAOD) characterized by hypoketotic hypoglycemia; metabolic acidosis; fatty infiltration of the liver, heart, and kidneys; and diagnostic organic aciduria (Frerman and Goodman 2001; Gordon 2006). In addition, MADD may be accompanied by various symptoms including vomiting, hypotonia, hyperammonemia, hepatomegaly, renal cysts, myopathy, an odor of sweaty feet, and congenital anomalies. Severe MADD appears in the neonatal period, and patients die within the first few weeks of life. However, MADD can also develop in later life. Treatment with riboflavin and l-carnitine is effective only for patients with mild symptoms (Gordon 2006; Olsen et al. 2007; Henriques et al. 2009; Law et al. 2009; Ishii et al. 2010; Lund et al. 2010).
MADD is an autosomal recessive disease, and most cases are caused by a defect of electron transfer flavoprotein (ETF) or ETF dehydrogenase (ETFDH, also known as ETF-ubiquinone oxidoreductase, EC 1.5.5.1) (Frerman and Goodman 2001; Er et al. 2011). These proteins play a role in the metabolism of fatty acids. ETF forms a heterodimer composed of α- and β-subunits, and contains flavin adenine dinucleotide (FAD) as a redox cofactor. In the mitochondrial matrix, ETF receives electrons from acyl-CoA dehydrogenase (EC 1.3.99.3) in the process of fatty acid degradation. ETFDH is a monomer, and contains FAD and a 4Fe-4S cluster as redox prosthetic groups (Frerman and Goodman 2001; Watmough and Frerman 2010). ETFDH transfer electrons from ETF to ubiquinone, which exists in the inner mitochondrial membrane and participates in electron transport system to produce ATP. In this process, electrons enter through the flavin center of ETFDH and exit via the 4Fe-4S cluster for delivery to ubiquinone. Deficiency of ETF or ETFDH leads to dysfunction of acyl-CoA dehydrogenase, resulting in accumulation of long- and medium-chain fatty acids. As metabolites of the accumulated substances, the urine and plasma levels of specific organic acids increase; these include isovaleric, isobutyric, 2-methylbutyric, glutaric, ethylmalonic, 3-hydroxyisovaleric, 2-hydroxyglutaric, 5-hydroxyhexanic, adipic, suberic, sebacic, and dodecanedioic acid, as well as isovalerylglycine, isobutyrylglycine, and 2-methylbutyrylglycine.
According to the Genetics Home Reference serviced by the U.S. National Library of Medicine (http://ghr.nlm.nih.gov/), the precise incidence of MADD is still unknown in view of its rarity. Tamaoki et al. surveyed Japanese patients with FAODs identified between 1985 and 2000, and found 14 patients with MADD out of 64 such patients (Tamaoki et al. 2002). They estimated that the frequency of FAODs in Japan is at least 1/5,000 births, implying that the frequency of MADD is about 1/20,000. Some studies have investigated the epidemiology of MADD in southern China, where c.250G>A mutation in ETFDH is carried at high frequency in patients with riboflavin-responsive MADD (Law et al. 2009; Liang et al. 2009; Er et al. 2010; Lan et al. 2010; Wen et al. 2010; Wang et al. 2011).
In animals, outbreaks of fatal MADD have been recorded in horses with atypical myopathy in particular weather conditions (Finno et al. 2006; Cassart et al. 2007; Westermann et al. 2008; van der Kolk et al. 2010; Valberg et al. 2012). In these cases, an exogenous factor was predicted to cause MADD, and a genetic mutation of ETF and ETFDH has never been recognized. In any event, MADD associated with gene mutation in animals has never been reported. Here, we report a novel case of inherited MADD that developed in a cat, which we consider to be the first of its kind to have been reported in an animal.
Materials and Methods
Subject
A 6-month-old castrated cat with a previous history of hypoglycemia and hyperammonemia was brought by its owner to the animal hospital with a chief complaint of collapse, loss of appetite, and vomiting. Blood biochemistry revealed elevated levels of ammonia and liver enzymes (NH3, 227 μg/dl; ALT, 610 U/l; and AST, more than 1,000 U/l); reference values of NH3, ALT, and AST are 23–78 μg/dl, 22–78 U/l, and 22–84 U/l, respectively, in cats. A generalized seizure also occurred during follow-up. We treated the cat for hyperammonemia with administration of lactulose and infusion of Ringer’s solution. Although we suspected hepatic encephalopathy associated with a portosystemic shunt, measurement of fasting and postprandial blood ammonia and serum bile acid (SBA) concentrations revealed no evidence of hepatic dysfunction (Table 1). In addition, computed tomographic angiography revealed no congenital vascular anomaly or any enlargement of the liver. Symptoms were improved within 10 days after the first visit. Just a year later, this cat was brought again in worse condition than that at the first visit. As a countermeasure against hepatic lipidosis accompanied by lack of energy, the cat received nutritional care via a gastrostomy tube, in addition to the treatment for hyperammonemia.
Table 1.
Diet loading test
| NH3 (μg/dl) | SBA (μmol/l) | |
|---|---|---|
| Fasting | 227 | 3.7 |
| Postprandial | 71 | 18.2 |
| Reference range | 23–78 | 0–20 |
SBA serum bile acid
Diet Loading Test
To exam hepatic function, blood ammonia and SBA concentrations were measured during fasting and postprandial periods at the first visit. Blood samples were collected after an overnight fast and at 2 hours after a meal.
GC-MS Analysis
To assay urine organic acids, GC-MS analysis was conducted in MILS INTERNATIONAL (Kanazawa, Japan) as published previously (Zhang et al. 2000). The first sample of urine was collected under the condition in loss of appetite at the second visit, when the present cat was 18 months old. At 6 days after the first sampling, we received the result suggesting MADD. The affected cat has received the oral administration of riboflavin (100–300 mg/day) and l-carnitine (100–150 mg/day) since then. At 16 days of treatment, posttreatment sample was collected after an overnight fast.
LC-MS/MS Analysis
To assay the blood levels of acylcarnitines, LC-MS/MS analysis was conducted as published previously (Chang et al. 2012). The first sample was collected under the condition in force-feeding via a gastrostomy tube at the age of 18 months, when we start the treatment with riboflavin and l-carnitine as described above. At 3 months of treatment, posttreatment sample was collected after an overnight fast. Physical value of the blood levels of acylcarnitines was obtained from 27 of healthy cats. For statistical analysis, the Smirnov-Grubbs test was used.
Cell Culture
From among cats brought to the Veterinary Teaching Hospital of Miyazaki University by their owners, small pieces of subcutaneous adipose tissues were collected by scraping from surgical specimens with the informed consent of their owners, then cultured on plastic dishes with Dulbecco’s modified Eagle medium (D6429, Sigma-Aldrich, St. Louis, MO, USA) including 10 % fetal bovine serum (Lot no. ABL0106, Biofill, Victoria, Australia), 50 μM β-mercaptoethanol (Life Technologies, Carlsbad, CA, USA), 10 units/ml penicillin, 10 μg/ml streptomycin, and 250 ng/ml amphotericin B (Antibiotic-Antimycotic, 15240-062, Life Technologies). Tissue pieces were removed from the dishes four days later, and monolayer cells were allowed to proliferate on the dishes. After the first passage, amphotericin B was not included in the medium (Penicillin-Streptomycin, Life Technologies).
RT-PCR
Total RNA was isolated from cultured cells using a RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) after the third passage. Complementary DNA was synthesized using SuperScript III Reverse Transcriptase (Life Technologies) with the oligo dT primer. For rapid amplification of cDNA ends (RACE) sequencing, a cDNA library was produced using a GeneRacer kit (Life Technologies). PCR was conducted using specific primers (Table 2) and BIOTAQ HS DNA Polymerase (Bioline, London, UK). The specificity of the reaction products was confirmed by agarose-gel electrophoresis. PCR products were also collected and sequenced. The protein conformation and cysteine bonding state were predicted using the fully automated protein structure homology-modeling server (SWISS-MODEL, http://swissmodel.expasy.org/) and the cysteine disulfide bonding state and connectivity predictor (DISULFIND, http://disulfind.dsi.unifi.it/), respectively.
Table 2.
List of primers used in this study
| Purpose | Sequence | |
|---|---|---|
| Target | Forward | Reverse |
| 5'-RACE | ||
| ETFα | * | CTTTCCAAAGGCAGATGCTC |
| ETFβ | * | GCATAGCGAGGTTCGTTGAG |
| ETFDH | * | GGGCACCATCCTTTTGTATC |
| 3'-RACE | ||
| ETFα | TACCATCACTGCAGCCAAAC | * |
| ETFβ | CCGCTGGATTTCTAGACTGG | * |
| ETFDH | CTCCCAGGTCTTCCAATGAA | * |
| Gene expression | ||
| ETFα | TACCATCACTGCAGCCAAAC | CTTTCCAAAGGCAGATGCTC |
| ETFβ | CCGCTGGATTTCTAGACTGG | GCATAGCGAGGTTCGTTGAG |
| ETFDH | CTCCCAGGTCTTCCAATGAA | GGGCACCATCCTTTTGTATC |
| GAPDH | GTTTGTGATGGGCGTGAAC | CGTATTTGGCAGCTTTCTCC |
| ORF sequence | ||
| ETFα | TGAGACTGACGGTGCTTCTAG | AAAGCATTCTGGCATCTGTGAG |
| ETFβ | ACTGACCCCGGCAGGTG | CGGGAGGGTCATAGTTTTATTGCTG |
| ETFDH | AACTTCAGCGGAGGTGTTGG | GGTAAGATAGTTTGACAACCTGTTGACTTC |
| PCR-RFLP | ||
| ETFDH | GAGGGCACCTGTTGGGAT | TCCTTGAGTCCAATTCCATAGGTT |
*Attached in GeneRacer kit
PCR-Restriction Fragment Length Polymorphism (PCR-RFLP)
Genomic DNA was isolated from cultured cells after the third passage using a QIAamp DNA Mini kit (Qiagen). PCR was conducted using specific primers (Table 2) and BIOTAQ HS DNA Polymerase (Bioline). The amplified ETFDH was reacted with the restriction enzyme PciI (New England Biolabs, Ipswich, MA, USA). The specificity of the reaction products was confirmed by agarose-gel electrophoresis.
Results
Feline MADD
We assayed urine organic acids because we suspected a metabolic disorder such as dihydropyrimidinase deficiency (OMIM 222748), which has been reported in a cat exhibiting hyperammonemia (Chang et al. 2012). Surprisingly, the results indicated a MADD-like profile, with increased levels of lactic acid, sarcosine, 3-hydroxybutyric acid, malonic acid, 3-hydroxyisovaleric acid, succinic acid, ethylmalonic acid, glutaric acid, isoverylglycine, 2-hydroxyglutaric acid, 3-hydroxyglutaric acid, hexonylglycine, suberic acid, sebacic acid, 3-hydroxysebacic acid, and suberylglycine (Fig. 1a). For definitive diagnosis, we assayed the blood levels of acylcarnitines. The results supported our diagnosis of MADD; increases of C4, C5, C6, C8, C10, C12, C14, C16, and C18 were evident, but no increase of C0, C2, and C3 (Table 3 and Fig. 2). MADD was thus confirmed, and treatment with riboflavin and l-carnitine improved the symptoms, including the elevated levels of urine organic acids and blood levels of acylcarnitines (Table 3 and Fig. 1b and 2).
Fig. 1.
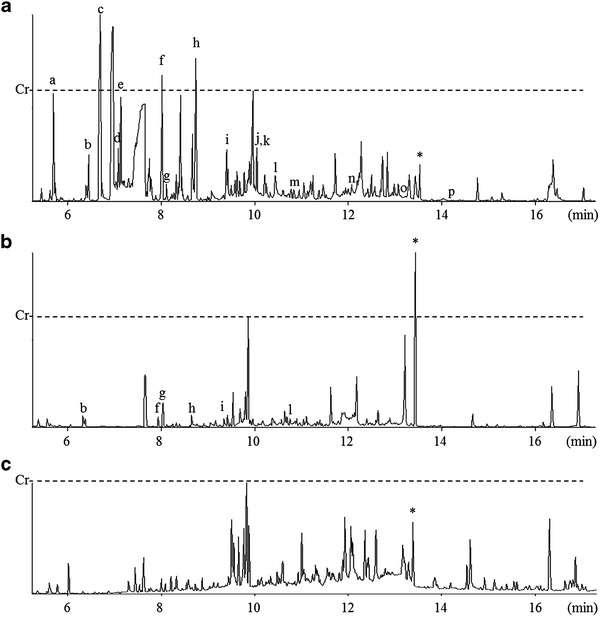
GC-MS analysis of urine organic acids. Total ion chromatograms are obtained from the cat with MADD (a) before, and (b) after treatment with riboflavin and l-carnitine. (c) Reference chromatogram is obtained from urine of a normal cat. Horizontal and vertical axes are scales of retention time and signal intensity, respectively. Scale marks represented as Cr indicates a signal intensity of creatinine in each chromatogram. Various organic acids characteristic of MADD are increased in the present cat with MADD, and treatment with riboflavin and l-carnitine suppresses the elevated levels of organic acids. a: lactic acid, b: sarcosine, c: 3-hydroxybutyric acid, d: malonic acid, e: 3-hydroxyisovaleric acid, f: succinic acid, g: ethylmalonic acid, h: glutaric acid, i: isovalerylglycine, j: 2-hydroxyglutaric acid, k: 3-hydroxyglutaric acid, l: hexonylglycine, m: suberic acid, n: sebacic acid, o: 3-hydroxysebacic acid, p: suberylglycine, and *: heptadecanoic acid added as internal standard
Table 3.
Blood levels of acylcarnitine species
| Numerical symbol | Normal feline reference value | Pretreatment | Posttreatment | |
|---|---|---|---|---|
| Mean (nmol/ml) | S.D. | Value (nmol/ml) | Value (nmol/ml) | |
| C0 | 12.7 | 6.8 | 22.1 | 29.3 |
| C2 | 7.27 | 2.38 | 7.42 | 4.98 |
| C3 | 0.55 | 0.31 | 0.38 | 0.35 |
| C4 | 0.14 | 0.06 | 9.36** | 3.49 |
| C5 | 0.15 | 0.09 | 7.5** | 2.88 |
| C5DC | 0.01 | 0.01 | 0.2** | 0.14 |
| OH-C5 | 0.16 | 0.06 | 0.13 | 0.17 |
| C5:1 | 0.02 | 0.01 | 0.02 | 0.02 |
| C6 | 0.02 | 0.02 | 1.48** | 1.12* |
| C8 | 0.02 | 0.01 | 0.72** | 0.6** |
| C10 | 0.01 | 0.01 | 0.45** | 0.57** |
| C12 | 0.02 | 0.01 | 0.46** | 0.44** |
| C14 | 0.04 | 0.02 | 0.77** | 0.44 |
| C14:1 | 0.03 | 0.02 | 1.27** | 0.93* |
| C16 | 0.26 | 0.15 | 2.27** | 0.79 |
| OH-C16 | 0.00 | 0.00 | 0.02** | 0.01 |
| C18 | 0.18 | 0.10 | 2.29** | 0.87 |
| OH-C18:1 | 0.01 | 0.01 | 0.06* | 0.04 |
| C18:1 | 0.26 | 0.23 | 1.96** | 0.67 |
**P < 0.01
*P < 0.05
Fig. 2.
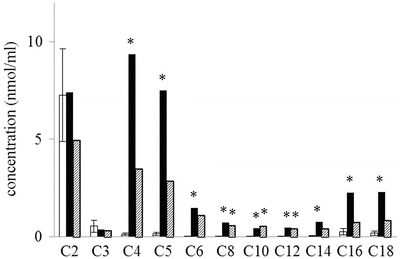
LC-MS/MS analysis of acylcarnitine species. The present cat with MADD had high blood levels of carnitines conjugated with saturated fatty acids longer than C4 (black columns). Treatment with riboflavin and l-carnitine improved the blood levels of acylcarnitines as normal (shaded columns). White columns and their error bars indicate mean and standard deviation, respectively, of the blood levels of acylcarnitines in 27 of healthy cats. *: P < 0.01
Sequencing of Feline ETFα, ETFβ, and ETFDH
To detect the gene mutation causing MADD, we tried to amplify the cDNA of feline ETFα, ETFβ, and ETFDH including the region of the open reading frame (ORF) based on the Ensembl genome browser (http://www.ensembl.org/). However, this was not successful for ETFα and ETFDH. The referenced sequence of ETFβ cDNA also seemed to lack the 5'- and 3'-untranslated regions. Therefore, we attempted to determine the complete cDNA sequences of ETFα, ETFβ, and ETFDH in normal cats. Using RACE sequencing, we identified the specific sequences (Table 4) for the feline ETFα, ETFβ, and ETFDH cDNAs, which were 1340, 841, and 2153 bp long, respectively, apparently encoding proteins of 333, 255, and 617 amino acids, respectively, and each showing 96 % similarity with their human homologs. No splicing variants were detected in any of the three genes. Based on this refined information, we succeeded in amplifying the complete ETFα, ETFβ, and ETFDH cDNAs including an ORF region from the present cat with MADD and three control cats. In cultured cells derived from adipose tissue, the cat with MADD expressed ETFα, ETFβ, and ETFDH at the same levels as those in control cats (Fig. 3).
Table 4.
List of sequenced genes
Fig. 3.
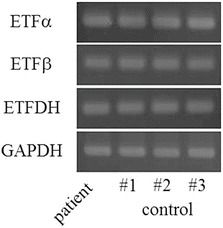
RT-PCR analysis of MADD-related genes. All of the cats examined, including the one with MADD, expressed ETFα, ETFβ, and ETFDH
Presence of a Novel Mutation in the Cat with MADD
We analyzed the sequence of amplified cDNA from all cats including that of the patient, and finally identified a specific mutation in the latter: c.692T>G in ETFDH (Fig. 4a). No other mutation was detected in ETFDH, ETFα, or ETFβ. PCR-RFLP analysis showed that the patient only carries mutant alleles of ETFDH (Fig. 4b). The c.692T>G mutation in ETFDH causes an amino acid substitution: p.F231C. To assess the importance of p.F231 for the function of ETFDH, we compared the amino acid sequences of various species using a database search, and this revealed that p.F231 in feline ETFDH is completely conserved in eukaryotes, including human (Homo sapiens), mouse (Mus musculus), amphibia (Xenopus laevis), fish (Danio rerio), insect (Drosophila melanogaster), nematode (Caenorhabditis elegans), plant (Arabidopsis thaliana), and fungus (Saccharomyces cerevisiae) (Fig. 5a). The disulfide bonding predictor, DISULFIND, showed that no abnormal disulfide bond is formed on the aberrant p.C231 in ETFDH. Analysis of the protein conformation showed that p.F231 is located on the surface of the FAD-binding domain opposite the ubiquinone domain in ETFDH (Fig. 5b).
Fig. 4.
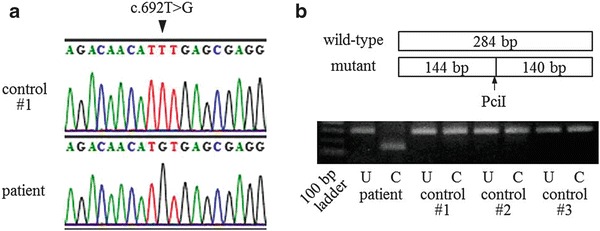
The ETFDH mutation in the present cat with MADD. (a) The mutation c.692T>G (arrow head) was identified by sequencing analysis. (b) The allelic frequency of the mutation was determined by PCR-RFLP analysis. The PciI restriction site (arrow), ACATGT, was located on the fragment amplified from the mutant ETFDH, but not on that from wild-type ETFDH. The lanes labeled U and C show the electrophoretic profiles of the ETFDH fragments without and with PciI cleavage, respectively, after PCR amplification. The fragment derived from the cat with MADD was completely cleaved by PciI
Fig. 5.
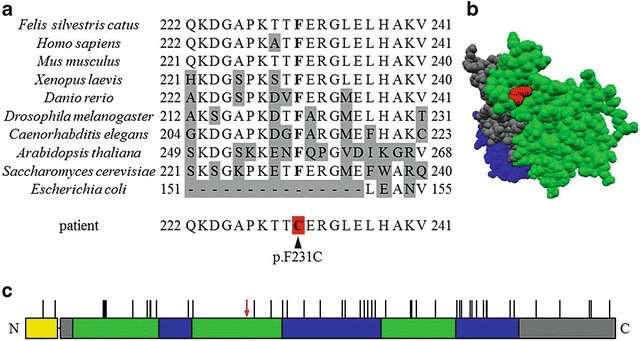
Characterization of the p.F231C mutation. (a) The peptide sequence of feline ETFDH is compared with its homologs for the amino acids covering the region p.Q222 to p.V241. Phenylalanine encoded by c.692T in feline ETFDH is conserved in ETFDH from Homo sapiens, Mus musculus, Xenopus laevis, Danio rerio, Drosophila melanogaster, Caenorhabditis elegans, Arabidopsis thaliana, and Saccharomyces cerevisiae. Escherichia coli lacks the homologous region including p.F231. The gray background indicates amino acids that differ from the feline sequence. The red background indicates the mutation site. (b) The protein conformation of feline ETFDH represented in space-fill mode predicted by SWISS-MODEL. p.F231 (red) is located on the surface of the FAD-binding domain (green) opposite the ubiquinone domain (blue). (c) The primary structure of the ETFDH precursor with the mutation locus is shown. The colors indicate the domain of the signal peptide (yellow), the 4Fe-4S cluster (gray), the FAD-binding site (green), and ubiquinone (blue). Vertical lines on each domain indicate the locus of mutations reported previously in human cases of MADD (Trakadis et al. 2012). The present novel mutation (red arrow) does not overlap with any of these mutations
Discussion
A surviving individual with MADD is rare because patients with neonatal-onset MADD die soon after birth. As an animal model of MADD has been never established, research on MADD has made little progress. The present case of inherited MADD in a cat is the first of its kind to have been reported other than in humans, indicating that inherited MADD is latent in animals.
The cat we studied presented with symptoms of hypoglycemia, hyperammonemia, vomiting, and increased levels of urine organic acids including 3-hydroxyisovaleric, ethylmalonic, glutaric, 2-hydroxyglutaric, suberic, and sebacic acids, as well as isovalerylglycine (Fig. 1a). The blood levels of medium- and long-chain acylcarnitine species were also elevated (Table 3 and Fig. 2). Therefore, the symptoms of MADD in this cat were consistent with those in humans (Frerman and Goodman 2001; Gordon 2006), suggesting that the present individual would be a valuable pathological model of human MADD.
Various mutations have been identified in ETFα, ETFβ, and ETFDH in human MADD patients. Most mutations causing riboflavin-responsive MADD have been located in ETFDH (Olsen et al. 2007; Law et al. 2009; Trakadis et al. 2012). The present cat, which responded to riboflavin treatment, also carried a mutation in ETFDH (Fig. 4), but not in ETFα or ETFβ. The mutations inherited in human patients with riboflavin-responsive MADD all cause a single amino acid substitution (Olsen et al. 2007; Law et al. 2009; Trakadis et al. 2012). The present cat expressed ETFDH mRNA at the same level as control cats (Fig. 3). The p.F231C substitution in ETFDH does not appear to lead to the formation of an abnormal disulfide bond. Therefore, cases of mild MADD responsive to riboflavin treatment may involve less severe dysfunction of ETFDH.
The mutation identified in this study has not been reported previously in human cases of MADD (Goodman et al. 2002; Olsen et al. 2003; Schiff et al. 2006; Yotsumoto et al. 2008; Er et al. 2010; Wolfe et al. 2010; Er et al. 2011; Wang et al. 2011; Trakadis et al. 2012) (Fig. 5c). p.F231 in ETFDH has been completely conserved in eukaryotes across mammal to fungus (Fig. 5a), implying that p.F231 has an important role in the function of ETFDH. Phenylalanine has a role in electron transfer in Alzheimer’s amyloid beta-peptide, methylamine dehydrogenase, sulfite oxidase, and triheme cytochrome PpcA (Davidson 2003; Pogocki 2004; Dantas et al. 2013; Davis et al. 2013). p.F231 is located on the FAD-binding domain, which is involved in the receipt of electrons from ETF, at the end opposite the ubiquinone domain, which is buried in the inner mitochondrial membrane (Fig. 5b) (Watmough and Frerman 2010). Thus, p.F231 is located on the apical surface of ETFDH, where it is exposed to the mitochondrial matrix. Therefore, the p.F231C amino acid substitution in ETFDH might hinder the receipt of electrons from ETF, followed by accumulation of reduced ETF, and leading to a deficiency of acyl-CoA dehydrogenase. There is possibility that this p.F231C substitution in ETFDH causes MADD in humans.
Our analysis of allelic frequency showed that present cat only carries mutant alleles of ETFDH (Fig. 4b). In humans, most cases of MADD are attributable to a zygotic effect on different mutations in ETF or ETFDH (Olsen et al. 2003), although the majority of patients with late-onset MADD recorded in southern China carried a homozygous mutation of c.250G>A in ETFDH (Wang et al. 2011). According to the owner, the present cat is derived from a group of dozens of stray cats, which were fed by a particular person. They were likely to breed in a small colony. The present cat might have been produced by inbreeding of ancestors carrying the mutation c.692T>G in ETFDH, although there is the possibility of a hemizygous point mutation or uniparental disomy containing ETFDH gene. Unfortunately, all three littermates and parents already died, so we could not observe genetic information about this mutation. Accordingly, we recommend avoiding unnecessary inbreeding of companion animals to prevent the production of genetic abnormalities such as the present one.
In conclusion, we have described the first case of inherited MADD in an animal, and identified a novel mutation in ETFDH suspected to be the cause. The cat we examined is the only one currently known to be diagnosed as having MADD, and the knowledge gained from it is anticipated to be valuable for the development of an effective therapy for MADD. As gene therapy can be applied for patients carrying a single gene mutation, we are currently studying the possibility of gene therapy for this cat.
Acknowledgments
The authors sincerely thank the owners of the studied cats for their participation. We are grateful to MILS INTERNATIONAL for GC-MS analysis of urine organic acids. This study was supported by a Grant-in-Aid from University of Miyazaki (3901020300: to KN) and a Grant-in-Aid from the Japan Society for the Promotion of Science (KAKENHI-B 25292187: to KN).
Synopsis
We discovered the first case of MADD in a cat.
Compliance with Ethics Guidelines
Shoichi Wakitani, Shidow Torisu, Taiki Yoshino, Kazuhisa Hattanda, Osamu Yamato, Ryuji Tasaki, Haruo Fujita, and Koichiro Nishino declare that they have no conflicts of interest. This article does not contain any studies with human subjects performed by the any of the authors. All institutional and national guidelines for the care and use of laboratory animals were followed. The work presented here was carried out in collaboration between all authors. SW and KN conceived and designed this experiment, analyzed a gene mutation, and wrote this article. ST and KH diagnosed the cat. TY conducted DNA sequencing; OY, RT, and HF conducted LC-MS/MS analysis.
Footnotes
Competing interests: None declared
Contributor Information
Koichiro Nishino, Email: aknishino@cc.miyazaki-u.ac.jpa.
Collaborators: Johannes Zschocke and K Michael Gibson
References
- Cassart D, Baise E, Cherel Y, et al. Morphological alterations in oxidative muscles and mitochondrial structure associated with equine atypical myopathy. Equine Vet J. 2007;39:26–32. doi: 10.2746/042516407X157765. [DOI] [PubMed] [Google Scholar]
- Chang HS, Shibata T, Arai S, et al. Dihydropyrimidinase deficiency: the first feline case of dihydropyrimidinuria with clinical and molecular findings. JIMD Rep. 2012;6:21–26. doi: 10.1007/8904_2012_139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantas J, Morgado L, Pokkuluri P, et al. Solution structure of a mutant of the triheme cytochrome PpcA from Geobacter sulfurreducens sheds light on the role of the conserved aromatic residue F15. Biochim Biophys Acta. 2013;1827:484–492. doi: 10.1016/j.bbabio.2012.12.008. [DOI] [PubMed] [Google Scholar]
- Davidson V. Probing mechanisms of catalysis and electron transfer by methylamine dehydrogenase by site-directed mutagenesis of alpha Phe55. Biochim Biophys Acta. 2003;1647:230–233. doi: 10.1016/S1570-9639(03)00056-6. [DOI] [PubMed] [Google Scholar]
- Davis A, Cornelison M, Meyers K, et al. Effects of mutating aromatic surface residues of the heme domain of human sulfite oxidase on its heme midpoint potential, intramolecular electron transfer, and steady-state kinetics. Dalton Trans. 2013;42:3043–3049. doi: 10.1039/c2dt31508d. [DOI] [PubMed] [Google Scholar]
- Er TK, Liang WC, Chang JG, Jong YJ. High resolution melting analysis facilitates mutation screening of ETFDH gene: applications in riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin Chim Acta. 2010;411:690–699. doi: 10.1016/j.cca.2010.01.033. [DOI] [PubMed] [Google Scholar]
- Er TK, Chen CC, Liu YY, et al. Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif. BMC Struct Biol. 2011 doi: 10.1186/1472-6807-11-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finno C, Valberg S, Wünschmann A, Murphy M. Seasonal pasture myopathy in horses in the midwestern United States: 14 cases (1998–2005) J Am Vet Med Assoc. 2006;229:1134–1141. doi: 10.2460/javma.229.7.1134. [DOI] [PubMed] [Google Scholar]
- Frerman FE, Goodman SI, et al. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric academia type II. In: Scriver CR, Beaudet AL, Sly WS, et al., editors. The metabolic and moleculer bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 2357–2365. [Google Scholar]
- Goodman S, Binard R, Woontner M, Frerman F. Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab. 2002;77:86–90. doi: 10.1016/S1096-7192(02)00138-5. [DOI] [PubMed] [Google Scholar]
- Gordon N. Glutaric aciduria types I and II. Brain Dev. 2006;28:136–140. doi: 10.1016/j.braindev.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Henriques B, Rodrigues J, Olsen R, et al. Role of flavinylation in a mild variant of multiple acyl-CoA dehydrogenation deficiency: a molecular rationale for the effects of riboflavin supplementation. J Biol Chem. 2009;284:4222–4229. doi: 10.1074/jbc.M805719200. [DOI] [PubMed] [Google Scholar]
- Ishii K, Komaki H, Ohkuma A, et al. Central nervous system and muscle involvement in an adolescent patient with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Brain Dev. 2010;32:669–672. doi: 10.1016/j.braindev.2009.08.008. [DOI] [PubMed] [Google Scholar]
- Lan MY, Fu MH, Liu YF, et al. High frequency of ETFDH c.250G > A mutation in Taiwanese patients with late-onset lipid storage myopathy. Clin Genet. 2010;78:565–569. doi: 10.1111/j.1399-0004.2010.01421.x. [DOI] [PubMed] [Google Scholar]
- Law LK, Tang N, Hui J, et al. Novel mutations in ETFDH gene in Chinese patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Clin Chim Acta. 2009;404:95–99. doi: 10.1016/j.cca.2009.02.015. [DOI] [PubMed] [Google Scholar]
- Liang WC, Ohkuma A, Hayashi Y, et al. ETFDH mutations, CoQ10 levels, and respiratory chain activities in patients with riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency. Neuromuscul Disord. 2009;19:212–216. doi: 10.1016/j.nmd.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund A, Skovby F, Vestergaard H, et al. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inherit Metab Dis. 2010;33:495–500. doi: 10.1007/s10545-009-9000-2. [DOI] [PubMed] [Google Scholar]
- Olsen R, Andresen B, Christensen E, et al. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- Olsen R, Olpin S, Andresen B, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- Pogocki D. Mutation of the Phe20 residue in Alzheimer’s amyloid beta-peptide might decrease its toxicity due to disruption of the Met35-cupric site electron transfer pathway. Chem Res Toxicol. 2004;17:325–329. doi: 10.1021/tx030044w. [DOI] [PubMed] [Google Scholar]
- Schiff M, Froissart R, Olsen R, et al. Electron transfer flavoprotein deficiency: functional and molecular aspects. Mol Genet Metab. 2006;88:153–158. doi: 10.1016/j.ymgme.2006.01.009. [DOI] [PubMed] [Google Scholar]
- Tamaoki Y, Kimura M, Hasegawa Y, et al. A survey of Japanese patients with mitochondrial fatty acid beta-oxidation and related disorders as detected from 1985 to 2000. Brain Dev. 2002;24:675–680. doi: 10.1016/S0387-7604(02)00074-8. [DOI] [PubMed] [Google Scholar]
- Trakadis Y, Kadlubowska D, Barnes R, et al. Pregnancy of a patient with multiple acyl-CoA dehydrogenation deficiency (MADD) Mol Genet Metab. 2012;106:491–494. doi: 10.1016/j.ymgme.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Valberg S, Sponseller B, Hegeman A, et al. Seasonal pasture myopathy/atypical myopathy in North America associated with ingestion of hypoglycin A within seeds of the box elder tree. Equine Vet J. 2012 doi: 10.1111/j.2042-3306.2012.00684.x. [DOI] [PubMed] [Google Scholar]
- Van der Kolk JH, Wijnberg ID, Westermann CM, et al. Equine acquired multiple acyl-CoA dehydrogenase deficiency (MADD) in 14 horses associated with ingestion of Maple leaves (Acer pseudoplatanus) covered. Mol Genet Metab. 2010;101:289–191. doi: 10.1016/j.ymgme.2010.06.019. [DOI] [PubMed] [Google Scholar]
- Wang ZQ, Chen XJ, Murong SX, et al. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD) in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G > A. J Mol Med. 2011;89:569–576. doi: 10.1007/s00109-011-0725-7. [DOI] [PubMed] [Google Scholar]
- Watmough NJ, Frerman FE. The electron transfer flavoprotein: ubiquinone oxidoreductases. Biochim Biophys Acta. 2010;1797:1910–1916. doi: 10.1016/j.bbabio.2010.10.007. [DOI] [PubMed] [Google Scholar]
- Wen B, Dai T, Li W, et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations. J Neurol Neurosurg Psychiatry. 2010;81:231–236. doi: 10.1136/jnnp.2009.176404. [DOI] [PubMed] [Google Scholar]
- Westermann C, Dorland L, Votion D, et al. Acquired multiple Acyl-CoA dehydrogenase deficiency in 10 horses with atypical myopathy. Neuromuscul Disord. 2008;18:355–364. doi: 10.1016/j.nmd.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Wolfe L, He M, Vockley J, et al. Novel ETF dehydrogenase mutations in a patient with mild glutaric aciduria type II and complex II-III deficiency in liver and muscle. J Inherit Metab Dis. 2010 doi: 10.1007/s10545-010-9246-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yotsumoto Y, Hasegawa Y, Fukuda S, et al. Clinical and molecular investigations of Japanese cases of glutaric acidemia type 2. Mol Genet Metab. 2008;94:61–67. doi: 10.1016/j.ymgme.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Zhang C, Xu K, Dave U, et al. Inborn errors of metabolism discovered in Asian department of pediatrics and mental retardation research center. J Chromatogr B Biomed Sci Appl. 2000;746:41–49. doi: 10.1016/S0378-4347(00)00087-6. [DOI] [PubMed] [Google Scholar]