

Abstract
Newborn screening (NBS) using tandem mass spectrometry (MS/MS) permits detection of neonates with Glutaric Aciduria-Type II (GA-II). We report follow-up of positive GA-II screens by the New England Newborn Screening Program.
Methods: 1.5 million infants were screened for GA-II (Feb 1999–Dec 2012). Specialist consult was suggested for infants with two or more acylcarnitine elevations suggestive of GA-II.
Results: 82 neonates screened positive for GA-II, 21 weighing > 1.5 kg and 61 weighing ≤ 1.5 kg. Seven (one weighing < 1.5 kg), were confirmed with GA-II. Four of these had the severe form (died < 1 week). The other three have a milder form and were identified because of newborn screening. Two (ages > 5 years) have a G-Tube in place, had multiple hospitalizations and are slightly hypotonic. The third infant remains asymptomatic (9 months old). Two GA-II carriers were also identified. The remaining positive screens were classified as false positives (FP). Six infants (> 1.5 kg) classified as FP had limited diagnostic work-up. Characteristics and outcomes of all specimens and neonates with a positive screen were reviewed, and marker profiles of the cases and FP were compared to identify characteristic profiles.
Conclusion: In addition to the severe form of GA-II, milder forms of GA-II and some GA-II carriers are identified by newborn screening. Some positive screens classified as FP may be affected with a milder form of the disorder. Characteristic GA-II profiles, quantified as GA-II indexes, may be utilized to predict probability of disorder and direct urgency of intervention for positive screens.
Introduction
Glutaric Aciduria Type II (GA-II; also known as multiple acyl-CoA dehydrogenase deficiency or ethylmalonic-adipic aciduria; OMIM 231680) is an autosomal recessive disorder of fatty acid oxidation and amino acid metabolism (Frerman and Goodman 2001). The underlying etiology is a functional deficiency of the electron transport flavoprotein (ETF, comprised of the alpha and beta subunits; ETFA, ETFB 130410) or electron transfer flavoprotein dehydrogenase (ETF-DH; EC 1.5.5.1), caused by mutations in any one of the ETFA, ETFB, or ETFDH genes (Goodman et al. 2002; Olsen et al. 2007).
GA-II can present as either a severe neonatal form with or without congenital anomalies, or as a milder late-onset disease (Al-Essa et al. 2000; Bohm et al. 1982; Curcoy et al. 2003; de Visser et al. 1986; Loehr et al. 1990; Rhead et al. 1987). Individuals with the severe neonatal form usually present within 24–48 h after birth with severe metabolic abnormalities (nonketotic hypoglycemia, metabolic acidosis, hyperammonemia, lactic acidosis), hypotonia, hepatomegaly, and cardiomegaly, and have a rapidly fatal course. Individuals with the mild form can present at any age with episodic illness (lethargy, vomiting, abdominal pain, hepatomegaly, cardiomegaly, rhabdomyolysis, ataxia) and metabolic abnormalities worsened by catabolic stress. Treatment for the severe form is ineffective. Treatment for the milder form consists of dietary modifications and supplementation with riboflavin and carnitine (Frerman and Goodman 2001)
In the severe form of the disease, a diagnosis can usually be made based on characteristic urine organic acid (UOA) and plasma acylcarnitine (PAC) profiles (Frerman and Goodman 2001). In the milder form, characteristic biochemical profiles may be exhibited only during an acute metabolic crisis. In such cases, enzymatic analysis or molecular studies may be required to make a definitive diagnosis.
Tandem mass spectrometry (MS/MS) makes it feasible to measure numerous acylcarnitines in dried blood spots enabling screening for several fatty acid oxidation defects (FAOD) and organic acidurias (OA) (Millington et al. 1990). GA-II is also detected by newborn screening (NBS). We report the experience of the New England Newborn Screening Program (NENSP) in identifying individuals at risk for GA-II, diagnostics of the positive screens, and long-term outcomes of confirmed cases born in MA, ME, RI, NH, and VT (defined “region” for this report).
Methods and Population
The NENSP has been routinely analyzing acylcarnitines and amino acid markers on NBS dried blood spot specimens since 1999. Screening specimens are obtained between 24–72 h of birth. For infants in the neonatal intensive care units (NICU) and very low birth weight infants (VLBW; Weight ≤ 1.5 kg), additional specimens are requested at 2 weeks, 1 month, and at discharge. The laboratory procedure, as previously reported (Zytkovicz et al. 2001), involves extraction from the dried blood spots into a methanol solution with stable isotope-labeled internal standards with butylation prior to analysis by MS/MS. Measurement of C2 acylcarnitine was added in 2002.
Positive screening results wherein one or more markers exceed a defined cutoff are communicated to the primary care provider with recommendations for management (which may include referral to a metabolic specialist). Diagnostic testing is performed under the guidance of the specialist. The NENSP and partner states track all positive screens from the region until resolution of diagnosis (short-term follow-up), as determined by the specialist, and tracks confirmed cases periodically to obtain long-term follow-up information. The final diagnosis and results of confirmatory studies are provided to the NENSP by the specialists. The long-term follow-up information is usually obtained from the metabolic specialist and occasionally from the primary care provider. Data elements collected include date of last specialty visit, treatment, growth parameters, episodes of metabolic crises, biochemical abnormalities, hospitalizations, and disorder-related clinical sequelae (developmental delays, hypotonia, cardiomyopathy). Information about false negatives is obtained by the NENSP by surveying metabolic specialists periodically.
Consultation with a specialist has always been recommended for all positive screens with elevations of two or more acylcarnitines suggestive of a GA-II profile, regardless of the results of a follow-up screen (if performed). However, contact algorithms have evolved during the study period. Since July 2006, the NENSP has been sorting positive screens into categories (“2006 algorithm”) based on a GA-II Index: (1) “High Risk” for GA-II are elevations of two or more acylcarnitines in conjunction with a GA-II Index [C4xC5 x C8xC14]/ [C0xC3] value of ≥ 0.005 and (2) “Risk” for GA-II are elevations of two or more acylcarnitines, but GA-II Index is < 0.005. For “High Risk” results, an urgent consultation with a specialist is recommended; for “Risk” results a non-urgent consultation is suggested. The NENSP does not have separate cutoff values for the acylcarnitines at different ages, and specimens collected beyond 30 days of life are not stratified by risk.
The cohort for this report is limited to all infants born in the defined region and screened using MS/MS by the NENSP from February 1, 1999, to December 31, 2012. A retrospective review of the NENSP data was performed and neonates with a screening profile suggestive of GA-II, in a specimen collected within the first 30 days of life, were identified. A “GA-II profile” is defined as an elevation of two or more acylcarnitines associated with at least two distinct flavin-containing acyl-CoA dehydrogenase enzymes involved in GA-II. Among the markers screened by the NENSP, these would include acylcarnitines from the following groups: (i) C4, (ii) C5, (iii) C5DC, (iv) C8, C6, C10, (v) C12, C14:1, C14. Elevations of two or more acylcarnitines consistent with another specific disorder are excluded; for example, elevation of C8, C6 with C10OH would suggest medium-chain acyl-CoA dehydrogenase deficiency (MCADD; OMIM 201450). Diagnostic and long-term follow-up data were analyzed. Short-term and long-term outcomes were analyzed for neonates with a newborn screening profile suggestive of GA-II
Results
Approximately 1.5 million neonates in the cohort received MS/MS screens. Figure 1 summarizes the characteristics and outcomes of all specimens and neonates found to have multiple acylcarnitine elevations with a “GA-II profile” in at least one specimen collected within 30 days of birth. The first out-of-range (OOR) specimen for each baby is shown, and whether this was the “initial” specimen (all collected 24–72 h of age) or “not initial” (all collected as per routine rescreening protocol). Of the 82 infants with a “GA-II profile,” 21 had birth weights > 1.5 kg. In 20, the OOR result was seen in the initial screen and only one had an initial normal followed by and an OOR result. Sixty-one specimens were from VLBW infants. In 11 VLBW infants, the OOR result was seen in the initial specimen, and in the other 50 infants on a subsequent screen. Follow-up screening specimens were received from several neonates after the OOR result, and their results are shown as either normal, abnormal (if any acylcarnitine was out-of-range), or not available. All follow-up specimens were received within a week of the OOR result. Infants are classified as True Positives (TP; Confirmed GA-II), Carriers, and False Positives (FP; Not GA-II or GA-II carriers) based on the assessment of the treating physician. The data of the FPs were reviewed to determine the extent of their diagnostic evaluations, and those with limited diagnostic evaluations are shown as incomplete work-up. All confirmed GA-II cases and carriers had an OOR GA-II profile on the initial specimen. We are aware of no false negative newborn screens for GA-II in the study cohort.
Fig. 1.
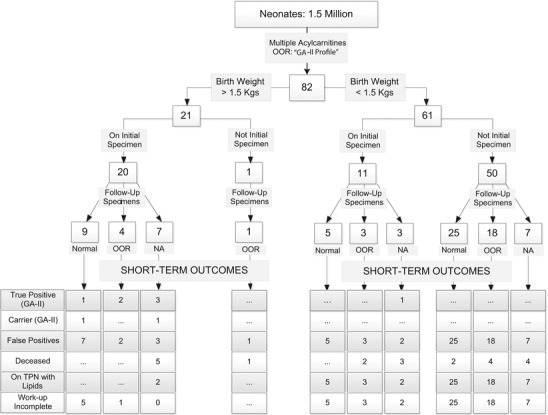
Characteristics and outcomes of all specimens and neonates found to have multiple acylcarnitine elevations with a “GA-II profile” in at least one specimen collected within 30 days of birth. Consultation with a specialist is recommended for all out-of range (OOR) screens with acylcarnitines suggestive of a GA-II profile, regardless of the results of a follow-up screen (if performed). Initial specimens were all collected at 24 to72 h of age and those shown as “Not Initial” specimens were collected at 72 h to 30 days of age. “Not Initial” specimens were collected as per routine rescreening protocols and the initial screens from these neonates were in range. False positives with limited work-up are shown as work-up incomplete. Please see text for details
The clinical presentations and confirmatory studies performed on the cases and carriers of GA-II are summarized in Table 1. Table 2 shows the concentrations of relevant acylcarnitines in all confirmed GA-II cases, carriers, and the 12 FP neonates > 1.5 kg with an OOR result on the initial screen.
Table 1.
Clinical information and results of diganotic studies of all cases and carriers of Glutaric Aciduria Type II (GA-II) identified by the New England Newborn Screening Program in the defined cohort during the period February 1999 through December 2012
| Clinical presentation | Confirmatory studies | Additional information | |||||
|---|---|---|---|---|---|---|---|
| ID | Prenatal and Birth history | Neonatal course | Urinary organic acids & acylglycines | Plasma acylcarnitines | Molecular analysis | Enzymatic studies | Autopsy findings |
| 1 | Born at 31 weeks gestation following premature rupture of membranes. Apgars 6 & 9*; Wt 1.3 kg. Agenesis of corpus callosum, complex heart defects, cleft lip & palate, enlarged echogenic kidneys and bilateral clubfeet noted at initial prenatal visit (2 weeks prior to delivery). Maternal history of a previous fetal demise at 28 weeks gestation with similar anomalies; she also had a healthy 6 year old son | Lethargic & hypotonic at birth and required endotracheal intubation. Metabolic acidosis noted that initially responded to acetate supplementation. Acute decompensation with hypotension, apnea and hypoglycemia developed on DOL 4. Investigations revealed hypoketotic hypoglycemia, hyperammonemia, metabolic acidosis, elevated CK. The hypoglycemia responded to increased IV glucose, but hyperammonemia & metabolic acidosis persisted. A severe FAOD (GA-II/CPT-II) suspected. Based on grim prognosis, ventilator support withdrawn and neonate died on DOL 7. NBS results, indicating a high risk for GA-II, were reported on DOL 7 after baby was deceased | Markedly increased lactic acid & glutaric acids. Mildly increased ethylmalonic acid, 2-hydroxyglutaric acid & its lactone, and isovalerylglycine | Elevations of acylcarnitine species of all chain lengths | … | … | Extensive lipid deposition in the heart, muscle, liver and kidneys. Bilateral cleft lip and palate, cardiomegaly with 2 small muscular ventriculoseptal defects, diffusely cystic kidneys with features of acute tubular necrosis. |
| 2 | Born at term. Apgars 9 & 9*; Wt 3.2 kg. Placental abruption during delivery suspected. No dysmorphic features noted | Pale and jittery few hours after birth with glucose of 10 mg/dl. Given dextrose bolus, and maintenance intravenous (IV) dextrose continued for 48 h. Was under observation in hospital and appeared to be doing well, when neonate turned pale and collapsed while being fed, 8 h after discontinuation of IV dextrose. NBS specimen collected a few hours prior to death indicated a high risk for GA-II | … | … | … | ETF Activity < 0.1 nmol/min/mg (Controls 0.8–2.4). Skin sample for fibroblast culture obtained post mortem | Extensive lipid deposition in heart, muscles, renal tubules and adrenal cortex. Marked thymic involution. |
| 3 | Delivered at 37 weeks of gestation due to IUGR. Apgars 7 & 8*; Wt 2.1 kg. Bilateral echogenic kidneys had been noted at 22 weeks, and IUGR & oligohydramnios noted at 32-weeks of gestation. Hypertelorism & low set ears noted at birth | Developed respiratory distress and seizures within an hour of birth and was extremely hypotonic and required endotrac heal intubation. Investigations revealed hypoketotic hypoglycemia, hyperammonemia, metabolic acidosis. A severe FAOD (GA-II/CPT-II) suspected. Head ultrasound revealed calcification in vessels around the midbrain. An echocardiogram revealed severe left ventricular dysfunction. Metabolic acidosis and hypotension refractory to treatment and baby died on DOL 2. The NBS results, indicating a high risk for GA-II, were reported after baby was deceased | Markedly increased ethylmalonic, glutaric, 2-hydroxyglutaric and adipic acids. Mildly increased methylsuccinic, suberic, sebacic, 5-hydroxyhexanoic & 7-hydroxyoctanoic acids and isovaleryl, 2-methylbutyryl & hexanoylglycines | Elevations of multiple acylcarnitine species (C4, C5, C6, C12, C14, C16, C12:1, C14:1, C14:2, C18, C18:1) suggestive of GA-II | ETFDH Gene: Two muations identified [ c.121C>T: p.R41X; c.1648_1649delCT : p.L550VfsX4] Confirmed to be in trans | … | Extensive lipid deposition in heart, muscles and adrenal cortex. Marked thymic involution. Pulmonary hypoplasia, bilobed lungs, renal dysplasia, particularly medullary with dilated tubules and cysts, focal polymicrogyria in the cerebrum and fetal osteosclerosis. |
| 4 | Born 37 weeks of gestation. Apgars 8 & 9*. Wt 3.2 kg. Hydrocephalus noted at 22 weeks of gestation. Head ultrasound at birth revealed prominent cystic areas adjacent to lateral ventricles with surrounding leukomalacia in addition to the hydrocephalus. Frontal bossing, large anterior fontanelle and hypertelorism reported | Feeding poorly on DOL-1 and had mild hypoglycemia that resolved with dextrose bolus. Lethargic and hypotonic on DOL-2, with severe hypoketotic hypoglycemia, hyperammonemia and metabolic acidosis. Hypoglycemia and metabolic acidosis resolved quickly with IV glucose, but hyperammonemia persisted. A severe FAOD (GA-II/CPT-II) suspected. Supportive measures initiated but neonate died on DOL-7. The NBS results, indicating a high risk for GA-II were reported on DOL-4 | Markedly increased lactic, ethylmalonic, methylsuccinic, glutaric & 2-hydroxyglutaric acids with mildly increased 3-hydroxybutyric, saturated & unsaturated dicarboxylic acids and isovaleryl, butyryl, hexanoyl & suberylglycines | … | … | … | |
| Follow-Up Information | |||||||
| 5 | Born 6 days post due date. Apgars 9 & 9*; Wt 3.6 kg. No dysmorphic features | No clinical issues were noted and was discharged home on DOL 3. NBS results suggesting high risk for GA-II were reported on DOL 6 and metabolic work-up initiated by specialist. The organic acid analysis was normal as was a follow-up screen collected on DOL 7. Plasma acylcarnitines revealed mild elevations of long chain acylcarnitines so a diagnosis of VLCAD suspected. Treatment (avoidance of fasting, a high carbohydrate and low fat) initiated for FAOD | During Illness at 1-1/2 years: Markedly increased ethylmalonic and adipic acids. Mildly increased methylsuccinic & 3-hydoxydicarboxylic acids and isovaleryl, butyryl, hexanoyl & suberylglycines | During Illness at 1-1/2 years: Elevations of multiple acylcarnitine species (C4, C5, C5DC, C6, C8, C10, C12, C14:2, C14:1, C14, C16) suggestive of GA-II | ETGDH Gene: Two mutations identified [c.121C>T : p.R41x; c.1448C>T : p.P483L] Confirmed to be in trans | ETF-DQ 1.4 nmol/min/mg (Controls 6.5–12.4) | Infant had 3 hospital admissions for dehydration and hypoglycemia in the first 6-months of life. Supplemental G-Tube feeding was initiated at 6 months of life. Elevations of ethylmalonic acid were noted in the urine during a hospitalization at 6 months of age, and skin samples for enzymatic studies on fibroblasts were sent out. These were supportive of SCAD and infant was diagnosed with SCAD. During another admission at 1-1/2 years of age laboratory findings suggestive of GA-II. Repeat enzymatic studies were performed and diagnosis confirmed. Riboflavin was initiated. Child continues to have episodes of biochemical abnormalities but frequency has decreased with age. Currently>10 years, child has mild hypotonia and difficulties with sustained activities but otherwise is doing well. Growth measurements are at the 50th %ile for age. |
| 6 | Born at term. Apgars 9 & 9*; Wt 3.7 kg. No dysmorphic features | Pallor and tachypnea noted at 25 h after birth attributed to an intrauterine bleed. Symptoms resolved quickly with a few hours of IV hydration and was discharged home on DOL 3. NBS results suggesting high risk for GA-II were reported on DOL 6 and metabolic work-up initiated by specialist on the same day | Markedly increased ethylmalonic & glutaric acids. Mildly increased isovaleryl, hexanoyl & suberylglycines | Elevations of multiple acylcarnitine species (C4, C5, C6, C8, C10:1, C10, C12, C14, C16, C12:1, C14:1, C14:2) suggestive of GA-II | ETFDH Gene: Two muations identified [c.1082A>G: p.Y361C; c.1648_1649delCT: p.L550VfsX4] Confirmed to be in trans | … | Treatment (avoidance of fasting, a high carbohydrate and low fat diet, carnitine and riboflavin) initiated. Supplemental G-Tube feeding initiated in 2nd year of life. Has a history of frequent episodes of biochemical abnormalities (abnormal PAC, UOA, CK and LFT’s) necessitating hospital admissions during minor illnesses but frequency has decreased with age (Current age>5 years). In addition has low energy levels and mild hypotonia but is otherwise doing well. Growth measurements are at the 50 th %ile for age. |
| 7 | Born at term. Apgars 9 & 9*; Wt 3.4 kg. No dysmorphic features | No clinical issues. NBS results suggesting high risk for GA-II were reported on DOL 4 and readmitted for diagnostic work-up. Mild hpoglycemia and abnormal LFT’s and CK noted on admission | Mildly increased ethylmalonic, glutaric, 2-hydroxyglutaric, methylsuccinic, suberic, sebacic & adipic acids and isovaleryl, 2-methylbutyryl & hexanoylglycines | Elevations of multiple acylcarnitine species (C4, C5, C6, C8, C10, C12, C14, C14:1) suggestive of GA-II | ETFDH Gene: Two mutations identified [c.250G>A:p.A84T; c.1110C>G:pG370G (Predicted to create novel splice donor site)] | … | Treatment (avoidance of fasting, a high carbohydrate and low fat diet, carnitine and riboflavin) initiated. Infant is doing well; current age 9 months. |
| 8 | Born at term. Apgars 9 & 9*; Wt 2.9 kg. No dysmorphic features | No clinical issues. NBS results suggesting risk for GA-II were reported on DOL 4 and neonate readmitted for diagnostic work-up. Mild hypoglycemia and abnormal LFT’s and CK noted on admission | No unusual organic acids detected | Mild elevations of multiple acylcarnitine species (C8, C10, C12, C14, C14:1) | ETGDH Gene: One mutation identified [858 G>A:p.W286X]. ETFA & ETFB: No mutations identified | ETF-DQ 0.70 nmol/min/mg (Controls 0.8–2.4); ETF 2.3 nmol/min/mg (Controls 0.79–2.1) | Asymptomatic. Current age 3 years. |
| 9 | Born at term. Apgars 9 & 9*; Wt 3.8 kg. No dysmorphic features | No clinical issues. NBS results suggesting risk for GA-II were reported on DOL 5 and metabolic work-up initiated by specialist the next day | No unusual organic acids detected | Normal | ETFB Gene: One variant identified [P94TfsX21]. ETFA and ETFDH: No mutations | … | Asymptomatic. Current age 1-1/2 years. |
CPT-II Carnitine palmitoyl transferase deficiency-Type II, DOL Day of life, FAOD Fatty acid oxidation defect, IV Intravenous, SCAD Short chain acyl CoA dehydrogenase deficiency, VLCAD Very long chain acyl CoA dehydrogenase deficiency
*At 1 and 5 min respectively
Table 2.
Concentrations of relevant markers and their z-scores (calculated using log normalized population). Shown here are all GA-II cases (1–7), carriers (8–9) and the false positive specimens (10–21) from defined cohort weighing > 1.5 kg who has an out-of-range screen from GA-II on initial specimen. Among the false positives, cases 16–21 are those infants which we consider as not having had complete diagnostic evaluation
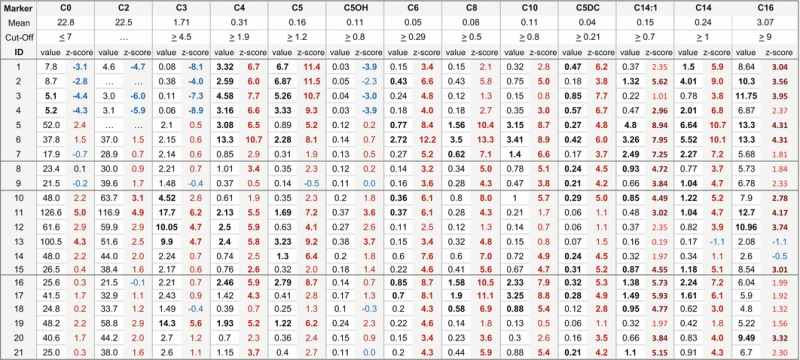
Acylcarnitine concentrations that exceed the cut-off and z-scores that exceed 3 are shown in bold
In our cohort, a total of seven cases of GA-II and 2 GA-II carriers were identified. Four of the seven cases had the severe neonatal form and are deceased. Three of these infants had malformations and a severe FAOD was clinically suspected even before an NBS screen was obtained. In the fourth neonate (Case 2) a metabolic disorder had not been considered until results of NBS became available. Three of the seven cases have a milder form of the disorder. Case 5 is notable because the characteristic biochemical profiles were not present in the diagnostic work-up in the neonatal period, and GA-II was eventually confirmed in the 2nd year of life. In this case, screening results suggesting a risk for GA-II were reported on day of life (DOL) 6. UOA and a repeat screen were normal. However, PACs revealed mild elevations of long-chain acylcarnitines and thus a diagnosis of very long chain acyl-CoA dehydrogenase deficiency (VLCAD; OMIM 201475) was entertained. Treatment in the form of avoidance of fasting and a high-carbohydrate, low-fat diet was initiated. The infant had several episodes of dehydration and hypoglycemia in the first 6 months of life, and elevations of urinary ethylmalonic acid were noted at 6 months of age. In vitro fibroblast testing for FAODs was reported as “consistent” with short-chain acyl-CoA dehydrogenase deficiency (SCAD; OMIM 201470) and the child was diagnosed with SCAD. At 1-and-1/2 years of age, UOA revealed a profile consistent with GA-II, eventually confirmed by repeat enzymatic studies. A mutation analysis was not pursued for SCAD, and it is feasible that the child harbors one or more alterations in the SCAD gene in addition to the documented mutations in the ETFDH gene. Two infants (Cases 8 and 9) were classified as GA-II carriers, based on the results of confirmatory testing, and remain asymptomatic.
Twelve of the 20 infants > 1.5 kg with an OOR result on the initial screen were classified as FP. Six were FP based on negative results of evaluations beyond initial biochemical testing (Cases 10–15). One neonate was found to have pathogenic alterations in one copy each of the MCAD and VLCAD genes. MCAD and VLCAD carriers have been detected by screening and the coincidental presence of carrier status for both could result in the multiple acylcarnitine pattern seen in this case. Whether it would lead to a clinical phenotype due to synergestic heterozygosity has not been established, but the increased C2 acylcarnitine in NBS suggests significant capacity for fasting and the individual remains asymptomatic (4 years old). The other five OOR results were attributed to non-metabolic causes such as renal or respiratory failure. Hemolysis or jaundice was reported in all five. The only neonate weighing > 1.5 kg who had a normal initial screen followed by an OOR result (mild elevations of C0, C5, C8) on a subsequent screen was on total parenteral nutrition (TPN) at time of specimen collection. Routine laboratory investigations and diagnostic UOA and PAC were normal. The infant was diagnosed with a mitochondrial complex III deficiency (OMIM 124000) due to pathogenic mutations in the BCS1L gene following extensive diagnostic studies in view of severe hypotonia and seizures since birth. Mitochondrial disease has been reported to mimic long-chain FAODs and it is possible that a GA-II profile could be seen in infants with mitochondrial disease who are receiving intravenous lipids. Six infants (Cases 16–21) had incomplete evaluation. In two of these neonates classified as FP who had an OOR screen on initial specimen, a repeat NBS was done but no further diagnostic work-up was performed. In an additional 4 of these 6 neonates, molecular studies and enzymatic analyses had not been pursued if initial biochemical testing (UOA or PAC) was not consistent with GA-II, even when no other condition/s was identified to explain the OOR screening result.
A “GA-II profile” was found in the initial specimen for 11 VLBW infants, including 1 infant confirmed to have GA-II (Case 1), and in later specimens after an initial normal screen in 50 VLBW infants. The 60 VLBW infants other than Case 1 were classified as FP based on limited evaluation, but GA-II was not on the differential of the treating physicians based on the clinical picture. These 60 infants were on TPN with lipids at the time of specimen collection. The markers most commonly responsible for the “GA-II” profile included C5 (n = 58), C8 (n = 46), C4 (n = 32), and C5DC (+C10OH; n = 27). Free carnitine (C0) was also elevated in 32 neonates, probably a result of carnitine supplementation. OOR C0, C5, and C8 were the most common combination. An elevation of C14 or the other long-chain acylcarnitines was not found in the VLBW infants.
Table 3 shows the clinical status, outcomes, and GA-II Index [C4xC5 x C8 x C14]/ [C0xC3] (and its z-score) on all babies >1.5 kg with a GA-II profile on the initial specimen. All confirmed cases (1–7) had a z-score higher than 7 while all confirmed FP (10–15) and carriers (8–9) had a z-score < 6. None of the VLBW infants on TPN with incomplete work-up had an index z-score of > 2 (data not shown). The only VLBW baby with a high GA-II index was confirmed to have GA-II (Case 1). Three of the six FP that have not had complete diagnostic work-up (16, 17, 21) had a z-score > 6. One was noted to have speech delays at 6 years. The other two are apparently asymptomatic and have no history of documented hypoglycemia at their current ages of 8 years and 9 months.
Table 3.
Clinical information and the NENSP GA-II Index shown here for all GA-II cases and Infants weighing > 1.5 kg with for GA-II initial specimen
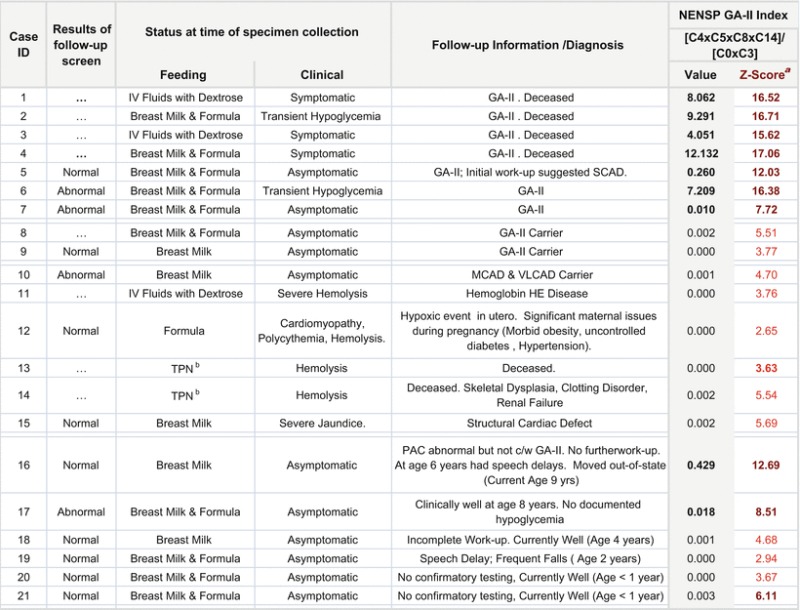
aCalculated using log normalized population
bTotal parenteral nutrition with lipids
Discussion
Seven cases of GA-II were identified in our cohort. Four had the severe form and died within a week of life. These neonates were symptomatic or deceased when NBS results were reported. Although NBS was not helpful in preventing mortality associated with the severe form, it aided clinicians in arriving at a final diagnosis, which is extremely helpful for family counseling.
For the milder forms of GA-II, a diagnosis of GA-II was pursued primarily because of NBS. Case 6 had exhibited transient hypoglycemia in the neonatal period that may have been related to GA-II, but was overlooked because it resolved quickly with a few hours of intravenous hydration. In Case 5, confirming a diagnosis of GA-II was a challenge because the characteristic biochemical abnormalities presented only in the second year of life, while previous investigations had prompted a diagnosis of SCAD. Notably all markers on the repeat NBS specimen in this infant were in range, and normal acylcarnitine levels during confirmation of abnormal NBS have been encountered in other FAODs (Browning et al. 2005). If active diagnostic work-up was not being pursued in these infants, a diagnosis of GA-II could have been easily missed or delayed. Our report documents two infants where no biochemical testing was pursued when a repeat NBS was normal, thus it is feasible that some milder cases of GA-II may be dismissed as FP.
Since riboflavin-derived cofactors are essential for the function of the flavin-containing acyl-CoA dehydrogenase enzymes, a maternal riboflavin deficiency can present with a biochemical and clinical phenotype similar to GA-II (Chiong et al. 2007). No cases of maternal riboflavin deficiency were identified in the cohort of this report; however, the maternal riboflavin status was studied in only a subset (6 of 21 of neonates > 1.5 kg) of positive screens.
A comparison of the confirmed TP with the confirmed FP (Table 2) shows that in addition to the elevations of the expected markers in varying degrees, the severe forms of GA-II have decreased levels of Free carnitine (C0), acetylcarnitine (C2), propionylcarnitine (C3), and hydroxyisovalerylcarnitine (C5OH). Low C0 concentrations are expected to be a secondary deficiency, and have been previously reported (Di Donato et al. 1986). We have observed lower C2 and C3 concentrations in other long-chain FAODs also and are not completely unexpected. The production of acetyl-CoA and thereby its carnitine conjugate (C2) is impaired in FAODs and probably accounts for the lower C2 concentrations. Similarly, it is likely that a large portion of the propionyl-CoA and its carnitine conjugate (C3) in neonates comes from beta-oxidation of odd chain fatty acids, and thus the lower C3 concentrations observed in the severe FAOD. We had observed lower concentrations of C5OH in isovaleric acidemia previously (Sahai 2011). We hypothesized that while it is known that the accumulated 3-methylcrotonyl-CoA is reversibly converted to 3-hydroxisoyvaleryl-CoA and thereby results in increased concentrations of its carnitine conjugates (C5OH) in 3-methylcrotonyl-CoA carboxylase deficiency (Van Hove et al. 1995), a reverse situation is also feasible; that is, the impaired conversion of isovaleryl-CoA to 3-methylcrotonyl-CoA due to the isovaleryl-CoA dehydrogenase deficiency in isovaleric acidemia (and GA-II) results in lower concentrations of 3-methylcrotonyl-CoA and consequently lower concentrations of 3-hydroxisoyvaleryl-CoA and its carnitine conjugates (C5OH). Based on these observations, the NENSP utilizes an index [C4xC5 x C8 x C14]/ [C0xC3] as a quantitative measure to reflect a GA-II profile; a higher value suggests a higher probability of the disorder.
The fact that three out of six of the cases highlighted in Table 3 who had not had complete diagnostic work-up had Index z-scores > 6 opens the possibility that some of these individuals are mild cases, carriers, or that the OOR screen was due to a maternal riboflavin deficiency. Sixty-one specimens with a “GA-II profile” were from VLBW infants, but one VLBW infant was confirmed to have GA-II, thus positive screens from VLBW infants cannot be ignored. Our data suggests that quantitative indexes (similar to our GA-II index) could predict risk even in this complex subset.
We propose categorizing screening results with elevations of multiple acylcarnitine species into those at “High Risk” and those at a lower “”Risk” for GA-II based on a GA-II Index in conjunction with the magnitude of the acylcarnitine concentrations. In neonates with a “High Risk” NBS, enzymatic or molecular studies need to be pursued if a diagnosis cannot be conclusively established by the initial biochemical investigations (organic acids and acylcarnitines). The algorithms available through ACMG considers elevations of C4 and C5 (+/− other acylcarnitines), a profile commonly seen in neonates on TPN, as at risk for GA-II and recommends biochemical testing, and if initial biochemical testing is normal, molecular or enzymatic testing is considered optional. The GA-II indexes and categorizations shown help refine the GA-II profile and provide better risk assessment to direct urgency of intervention and need for enzymatic molecular studies when faced with normal initial biochemical investigations in positive screens.
Acknowledgments
The authors wish to acknowledge the Massachusetts Department of Health, the Maine Department of Health and Human Services, the New Hampshire Department of Health and Human Services, the Rhode Island Department of Health, and the Vermont Department of Health for their partnership and cooperation. Further, the efforts of the treating physicians cannot be underscored. This study was supported in part by Priority Focus I and Priority Focus II funding bound to HRSA Grant U22MC03959.
Abbreviations
- C0
Free carnitine
- C10
Decanoylcarnitine
- C10OH
Hydroxydecanoylcarnitine
- C14
Tetradecanoylcarnitine
- C14:1
Tetradecenoylcarnitine
- C2
Acetylcarnitine
- C3
Propionylcarnitine
- C4
Butyrylcarnitine
- C5
Isovalerylcarnitine
- C5DC
Glutarylcarnitine
- C5OH
Hydroxyisovalerylcarnitine
- C6
Hexanoylcarnitine
- C8
Octonoylcarnitine
- DOL
Day of life
- ETF
Electron transfer flavoprotein
- ETF-DH
Electron transfer flavoprotein dehydrogenase
- FAOD
Fatty acid oxidation defects
- FP
False positive
- GA-II
Glutaric aciduria-type II
- MADD
Multiple acyl-CoA dehydrogenase deficiency
- MCAD
Medium chain acyl-CoA dehydrogenase deficiency
- MS/MS
Tandem mass spectrometry
- NBS
Newborn screening
- NENSP
New England Newborn Screening Program
- NICU
Neonatal intensive care unit
- OOR
Out of range
- SCAD
Short-chain acyl-CoA dehydrogenase deficiency
- TP
True positive
- TPN
Total parental nutrition
- VLBW
Very low birth weight
- VLCADD
Very long chain acyl-CoA dehydrogenase deficiency
Synopsis
We report the experience of the New England Newborn Screening Program in identifying individuals at risk for GA-II, the characteristics and outcomes of all specimens and neonates with a positive screen, and long-term outcomes of confirmed cases.
Conflict of Interest
All authors (I Sahai, CL Garganta, J Bailey, P James, HL Levy M Martin, E Neilan, C Phornphutkul, DA Sweetser, TH Zytkovicz and RB Eaton ) declare that they have no conflict of interest.
Informed Consent/Human or Animal Studies
This report summarizes the experience of the New England Newborn Screening Program. This chapter does not contain any studies with human or animal subjects performed by any of the authors.
Authors Attestations
The authors have not submitted similar publications previously or simultaneously.
All authors have inspected the manuscript.
All authors are in agreement for the submission.
All authors have been involved in (a) conception and design, or analysis and interpretation of data, and (b) drafting the article or revising it critically for important intellectual content.
Footnotes
Competing interests: None declared
Contributor Information
I. Sahai, Email: inderneel.sahai@umassmed.edu
Collaborators: Johannes Zschocke and K Michael Gibson
References
- al-Essa MA, Rashed MS, Bakheet SM, et al. Glutaric aciduria type II: observations in seven patients with neonatal- and late-onset disease. J Perinatol. 2000;20:120–128. doi: 10.1038/sj.jp.7200325. [DOI] [PubMed] [Google Scholar]
- Bohm N, Uy J, Kiessling M, Lehnert W. Multiple acyl-CoA dehydrogenation deficiency (glutaric aciduria type II), congenital polycystic kidneys, and symmetric warty dysplasia of the cerebral cortex in two newborn brothers. II. Morphology and pathogenesis. Eur J Pediatr. 1982;139(1):60–65. doi: 10.1007/BF00442082. [DOI] [PubMed] [Google Scholar]
- Browning MF, Larson C, Strauss A, et al. Normal acylcarnitine levels during confirmation of abnormal newborn screening in long-chain fatty acid oxidation defects. J Inherit Metab Dis. 2005;28(4):545–550. doi: 10.1007/s10545-005-0545-4. [DOI] [PubMed] [Google Scholar]
- Chiong MA, Sim KG, Carpenter K et al (2007 Sep–Oct) Transient multiple acyl-CoA dehydrogenation deficiency in a newborn female caused by maternal riboflavin deficiency. Mol Genet Metab 92(1–2):109–114 [DOI] [PubMed]
- Curcoy A, Olsen RK, Ribes A, Trenchs V, Vilaseca MA, Campistol J, Osorio JH, Andresen BS, Gregersen N. Late-onset form of beta-electron transfer flavoprotein deficiency. Mol Genet Metab. 2003;78(4):247–249. doi: 10.1016/S1096-7192(03)00024-6. [DOI] [PubMed] [Google Scholar]
- de Visser M, Scholte HR, Schutgens RB, Bolhuis PA, Luyt-Houwen IE, Vaandrager-Verduin MH, Veder HA, Oey PL. Riboflavin-responsive lipid-storage myopathy and glutaric aciduria type II of early adult onset. Neurology. 1986;36(3):367–372. doi: 10.1212/WNL.36.3.367. [DOI] [PubMed] [Google Scholar]
- Di Donato S, Frerman FE, Rimoldi M, et al. Systemic carnitine deficiency due to lack of electron transfer flavoprotein:ubiquinone oxidoreductase. Neurology. 1986;36:957–963. doi: 10.1212/WNL.36.7.957. [DOI] [PubMed] [Google Scholar]
- Frerman FE, Goodman SI. Deficiency of electron transfer flavoprotein or electron transfer flavoprotein ubiquinone oxidoreductase in glutaric acidemia type II fibroblasts. Proc Natl Acad Sci USA. 1985;82:4517–4520. doi: 10.1073/pnas.82.13.4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frerman FE, Goodman SI (2001) Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric aciduria type 2. In: Charles R Scriver, Arthur L Beaudet, William S Sly, David Valle. The metabolic and molecular bases of inherited disease. Vol. 8. McGraw-Hill, New York. pp 2357–2365
- Goodman SI, Binard RJ, Woontner MR, Frerman FE (2002 Sep–Oct). Glutaric acidemia type II: gene structure and mutations of the electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) gene. Mol Genet Metab 77(1–2):86–90 [DOI] [PubMed]
- Loehr JP, Goodman SI, Frerman FE. Glutaric acidemia type II: heterogeneity of clinical and biochemical phenotypes. Pediatr Res. 1990;27:311–315. doi: 10.1203/00006450-199003000-00024. [DOI] [PubMed] [Google Scholar]
- Millington DS, Kodo N, Norwood D, et al. Tandem mass spectrometry: a new method for acylcarnitine profiling with potential for neonatal screening for inborn errors of metabolism. J Inherit Metab Dis. 1990;13(3):321–324. doi: 10.1007/BF01799385. [DOI] [PubMed] [Google Scholar]
- Olsen RK, Andresen BS, Christensen E, et al. Clear relationship between ETF/ETFDH genotype and phenotype in patients with multiple acyl-CoA dehydrogenation deficiency. Hum Mutat. 2003;22:12–23. doi: 10.1002/humu.10226. [DOI] [PubMed] [Google Scholar]
- Olsen RK, Olpin SE, Andresen BS, et al. ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–2054. doi: 10.1093/brain/awm135. [DOI] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man (OMIM) (2012). Johns Hopkins University, Baltimore, Maryland. MIM Number: 231680
- Rhead WJ, Wolff JA, Lipson M, et al. Clinical and biochemical variation and family studies in the multiple acyl CoA dehydrogenase disorders. Pediatr Res. 1987;21:371–376. doi: 10.1203/00006450-198704000-00010. [DOI] [PubMed] [Google Scholar]
- Sahai I (2011) Multicenter validation of algorithms to improve communication of positive newborn screening results. Newborn screening and genetic testing symposium (APHL), San Diego, CA
- Van Hove JL, Rutledge SL, Nada MA, et al. 3-Hydroxyisovalerylcarnitine in 3-methylcrotonyl-CoA carboxylase deficiency. J Inherit Metab Dis. 1995;18(5):592–601. doi: 10.1007/BF02436004. [DOI] [PubMed] [Google Scholar]
- Zytkovicz TH, Fitzgerald EF, Marsden D, et al. Tandem mass spectrometric analysis for amino, organic, and fatty acid disorders in newborn dried blood spots: a two-year summary from the New England Newborn Screening Program. Clin Chem. 2001;47(11):1945–1955. [PubMed] [Google Scholar]