

Abstract
Background
Triple negative breast cancer (TNBC) is characterized by lack of expression of both estrogen and progesterone receptor as well as lack of overexpression or amplification of HER2. Despite an increased probability of response to chemotherapy, many patients resistant to current chemotherapy regimens suffer from a worse prognosis compared to other breast cancer subtypes. However, molecular determinants of response to chemotherapy specific to TNBC remain largely unknown. Thus, there is a high demand for biomarkers potentially stratifying triple negative breast cancer patients for neoadjuvant chemotherapies or alternative therapies.
Methods
In order to identify genes correlating with both the triple negative breast cancer subtype as well as response to neoadjuvant chemotherapy we employed publicly available gene expression profiles of patients, which had received neoadjuvant chemotherapy. Analysis of tissue microarrays as well as breast cancer cell lines revealed correlation to the triple negative breast cancer subtype. Subsequently, effects of siRNA-mediated knockdown on response to standard chemotherapeutic agents as well as radiation therapy were analyzed. Additionally, we evaluated the molecular mechanisms by which SFRP1 alters the carcinogenic properties of breast cancer cells.
Results
SFRP1 was identified as being significantly overexpressed in TNBC compared to other breast cancer subtypes. Additionally, SFRP1 expression is significantly correlated with an increased probability of positive response to neoadjuvant chemotherapy. Knockdown of SFRP1 in triple negative breast cancer cells renders the cells more resistant to standard chemotherapy. Moreover, tumorigenic properties of the cells are modified by knockdown, as shown by both migration or invasion capacity as well reduced apoptotic events. Surprisingly, we found that these effects do not rely on Wnt signaling. Furthermore, we show that pro-apoptotic as well as migratory pathways are differentially regulated after SFRP1 knockdown.
Conclusion
We could firstly show that SFRP1 strongly correlates with the triple negative breast cancer subtype and secondly, that SFRP1 might be used as a marker stratifying patients to positively respond to neoadjuvant chemotherapy. The mechanisms by which tumor suppressor SFRP1 influences carcinogenic properties of cancer cells do not rely on Wnt signaling, thereby demonstrating the complexity of tumor associated signaling pathways.
Keywords: Triple negative breast cancer, SFRP1, Chemotherapy sensitivity, Prognostic marker
Background
Triple negative breast cancer (TNBC) is defined by the lack of both estrogen receptor (ER) and progesterone receptor (PR) expression as well as overexpression or amplification of the human epidermal growth factor receptor HER2 [1-3]. Patients suffering from TNBC are not eligible for endocrine or HER2 targeted therapies, thus rendering chemotherapy the only therapeutic option, which may be accompanied by antiangiogenic approaches such as bevacizumab in the palliative setting [2,4,5]. Up to 15% of all breast cancer patients are diagnosed with TNBC [3]. Due to high recurrence rates and an increased risk of visceral and cerebral metastases these patients have a poorer prognosis in comparison to other breast cancer subtypes [6-8]. However, patients suffering from TNBC do have an increased probability of positive response to anthracycline/taxane- containing neoadjuvant chemotherapy. Thus, by achieving a pathologic complete response after neodajuvant chemotherapy the prognosis is as good as in other breast cancer subtypes [9]. Consequently, as chemotherapy sensitivity is one of the most important prognostic factors, it is inevitable to identify biomarkers and potential mediators of chemotherapy sensitivity in patients with TNBC.
The scientific goal of this study was to identify biomarkers, which may serve as mediators of chemotherapy sensitivity in TNBC. By using global gene expression profiles of patients receiving neoadjuvant chemotherapy we could identify secreted frizzled receptor protein 1 (SFRP1) as being correlated with the triple negative breast cancer subtype. Furthermore, we found a positive correlation of SFRP1 expression and response to neoadjuvant chemotherapy.
SFRP1 has been described to antagonize canonical Wnt signaling by binding to Wnt proteins or Wnt receptors, thereby inhibiting their downstream signaling activity [10]. In a plethora of solid tumors, including colorectal cancer, ovarian cancer, prostate cancer and lung cancer, it has been shown that SFRP1 is inactivated by promoter hypermethylation [11-15]. In breast cancer, hypermethylation of the SFRP1 promoter has been correlated to poor prognosis, presumably due to elevated levels of Wnt signaling [16,17].
By analyzing the molecular role of SFRP1 in triple negative breast cancer cells via siRNA mediated knockdown we found changes in carcinogenic properties of breast cancer cells, e.g. increased migration and invasion potential as well as reduced apoptotic events. Furthermore, we observed an increased resistance to standard cytostatic agents. Surprisingly, although SFRP1 is known to act via canonical Wnt signaling, our data suggests that its influence on triple negative breast cancer cells is apparently not mediated via this pathway.
In summary, we could show that tumor suppressor and Wnt signaling antagonist SFRP1 is correlated with the most aggressive subtype of breast cancer, i.e. triple negative breast cancer; but also with positive response to neoadjuvant chemotherapy. This makes SFRP1 a potential biomarker for future stratification of triple negative breast cancer patients. Additionally, SFRP1 seems to be involved in regulatory processes necessary for tumorigenic cancer cells, e.g. regulation of apoptosis as well as migration and adhesion processes. Surprisingly, however, these mechanisms are not mediated by canonical Wnt signaling.
Results
SFRP1 expression correlates with the TNBC subtype and response to neoadjuvant chemotherapy
In order to identify genes involved in chemotherapy response in patients suffering from TNBC we made use of a published dataset analyzing global gene expression profiles of breast cancer patients receiving neoadjuvant chemotherapy [18]. This dataset combines pretreatment gene expression profiles with response to neoadjuvant chemotherapy, e.g. showing either residual disease (n = 99) or pathologic complete response (n = 34). By analyzing both gene expression profiles as well as response to neoadjuvant chemotherapy we could firstly, show that SFRP1 expression correlates with the triple negative breast cancer subtype and secondly, demonstrate association between expression of SFRP1 and positive response to neoadjuvant chemotherapy, i.e. achievement of a pathologic complete response (Figure 1A, Tables 1, 2).We next sought to analyze the expression of SFRP1 in breast cancer tissue specimen. Therefore, we performed immunohistochemical analyses of tissue microarrays of breast cancer patients previously categorized as being either TNBC or non-TNBC. We detected different levels of SFRP1 expression mostly located in the tumor tissue (Figure 1B). When analyzing the distinct scores of expression of SFRP1 we found strong correlation of SFRP1 expression and the triple negative breast cancer subtype (Figure 1C).In addition, by using quantitative real time PCR and western blot analyses we found expression of SFRP1 in 5 out of 6 triple negative breast cancer cell lines (HCC-1937, MDA-MB-468, BT-20, MDA-MB-453 and HCC-1806), whereas no expression was detected in the TNBC cell line MDA-MB-231 as well as the non-TNBC cell lines SKBR-3 or MCF-7 (Figure 1D). These results clearly demonstrate a correlation of expression of SFRP1 and the triple negative breast cancer subtype. Additionally, there seems to be a link between the response to neoadjuvant chemotherapy and expression of SFRP1.
Figure 1.
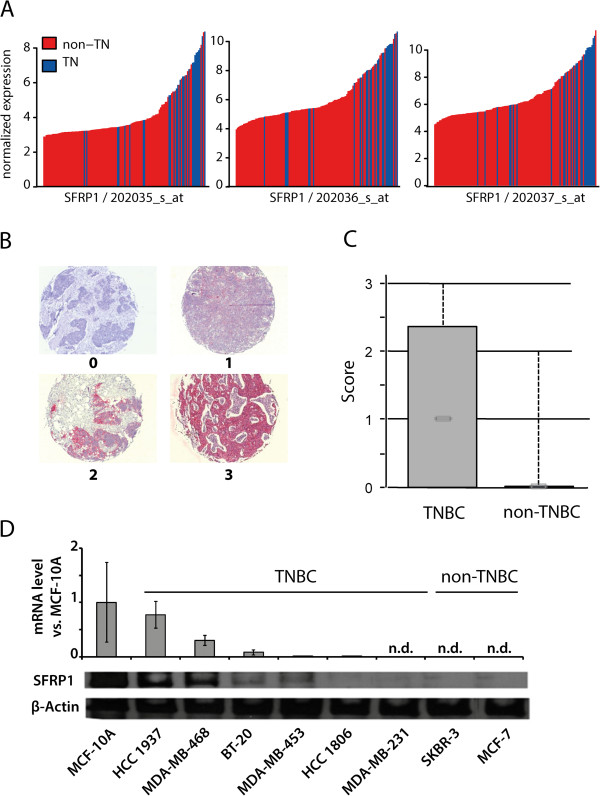
Expression of SFRP1 correlates with the triple negative breast cancer subtype. A) SFRP1 was found to be upregulated in TNBC compared to non-TNBC; shown here are all three probesets 202035_s_at, 202036_s_at and 202037_s_at (TN = triple negative). B) Expression of SFRP1 in breast cancer tissue specimens of 362 patients (TNBC = 37patients); scores 0 (negative), 1 (weak positive), 2 (positive), 3 (strong positive). C) Boxplot analysis of immunohistochemical staining. D) Protein and mRNA levels of SFRP1 in different breast (cancer) cell lines (non-tumorigenic epithelial cell line: MCF-10A, TNBC cell lines: HCC1937, MDA-MB-468, BT-20, MDA-MB-453, HCC 1806 and MDA-MB-231; non-TNBC cell lines SKBR-3 and MCF-7) (n.d. = not detected).
Table 1.
Expression of SFRP1 correlates with the triple negative breast cancer subtype
| Probeset | Gene ID | Adjusted p-value | Upregulated | Mean non-TN | Mean TN |
|---|---|---|---|---|---|
| 202035_s_at |
SFRP1 |
1.4 10E - 5 |
TNBC |
3.844 |
5.879 |
| 202036_s_at |
SFRP1 |
5.5 10E - 6 |
TNBC |
5.734 |
7.966 |
| 202037_s_at | SFRP1 | 3.0 10E - 6 | TNBC | 6.187 | 8.424 |
Table 2.
Expression of SFRP1 correlates with positive response to neoadjuvant chemotherapy
| Probeset | Gene ID | Adjusted p-value | Upregulated | Mean RD | Mean pCR |
|---|---|---|---|---|---|
| 202035_s_at |
SFRP1 |
0.017 |
pCR |
4.993 |
6.833 |
| 202036_s_at |
SFRP1 |
0.035 |
pCR |
7.116 |
8.881 |
| 202037_s_at | SFRP1 | 0.021 | pCR | 7.553 | 9.364 |
Knockdown of SFRP1 increases resistance against both chemotherapeutic agents as well as radiotherapy in triple negative breast cancer cells
In order to analyze the role of SFRP1 in triple negative breast cancer cells, we performed siRNA-mediated knockdown of SFRP1 in the triple negative breast cancer cell lines MDA-MB-468 and HCC-1806. Efficiency of knockdown was proven via quantitative real-time PCR and Western Blot analysis (Figure 2A).We next sought to evaluate the influence of SFRP1 knockdown to standard triple negative breast cancer chemotherapy. Therefore, we analyzed the response to chemotherapeutic agents after SFRP1 knockdown by cell viability assay. Interestingly, downregulation of SFRP1 expression rendered triple negative breast cancer cell line MDA-MB-468 more resistant to the chemotherapeutic agents paclitaxel, cisplatinum and doxorubicin (Figure 2B, left column). Another triple negative breast cancer cell line, HCC-1806, showed only increased resistance to paclitaxel (Figure 2B, right column). Thus, the knockdown of SFRP1 renders cancer cells more resistant to standard chemotherapeutic treatment.
Figure 2.
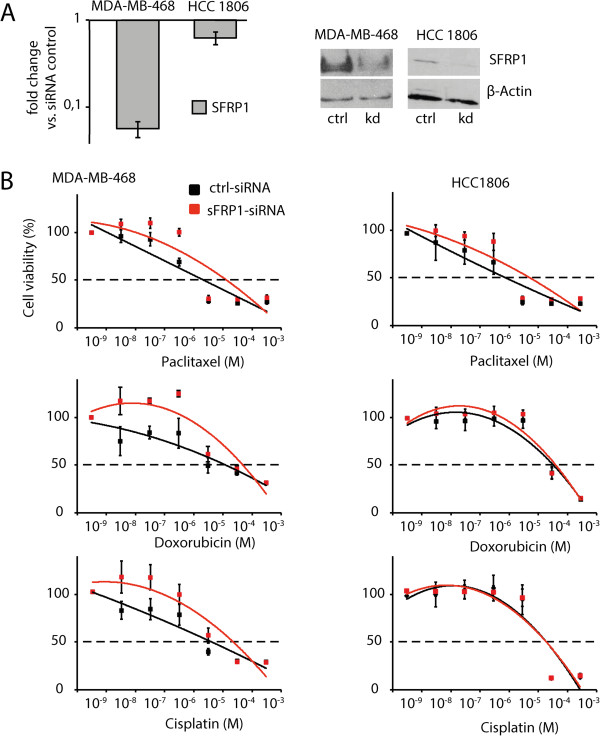
Downregulation of SFRP1 renders triple negative breast cancer cells more resistant to standard chemotherapy. A) mRNA (left) and protein (right) levels of SFRP1 after siRNA mediated knockdown, as determined by qPCR and Western-Blotting, respectively. B) Chemotherapy sensitivity of MDA-MB-468 and HCC-1806 after SFRP1 knockdown was determined using MTT cell viability assay of control vs. SFRP1 knockdown cells after treatment with cytotoxic agents (significance in MDA-MB-468 for paclitaxel ***p < 0.001 for 10 pM-1 nM, for doxorubicin ***p < 0.001 for 500 pM-50 nM and for cisplatin *p < 0.05 for 50 nM-50 μM; no significant changes were found in HCC-1806), error bars = SD, n = 3.
Knockdown of SFRP1 enhances the carcinogenic properties of triple negative breast cancer cells
Next, we sought to explore the impact of SFRP1 on tumorigenic properties of breast cancer cells. Hence, we analyzed both migration as well as invasion potential of breast cancer cell line MDA-MB-468 after SFRP1 knockdown. The migratory potential was not significantly increased after knockdown. However, invasion through matrigel-coated membranes was significantly increased by about 30% after SFRP1 knockdown (Figure 3A, B (left, center)). Additionally, we thought whether the rate of apoptosis is influences upon SFRP1 knockdown. Therefore, we analyzed the amount of apoptotic and necrotic cells via flow cytometry. We found a slight decrease of apoptotic as well as necrotic cells after SFRP1 knockdown (Figure 3C, D). These results demonstrate a link between expression of SFRP1 and carcinogenic properties of breast cancer cells.
Figure 3.
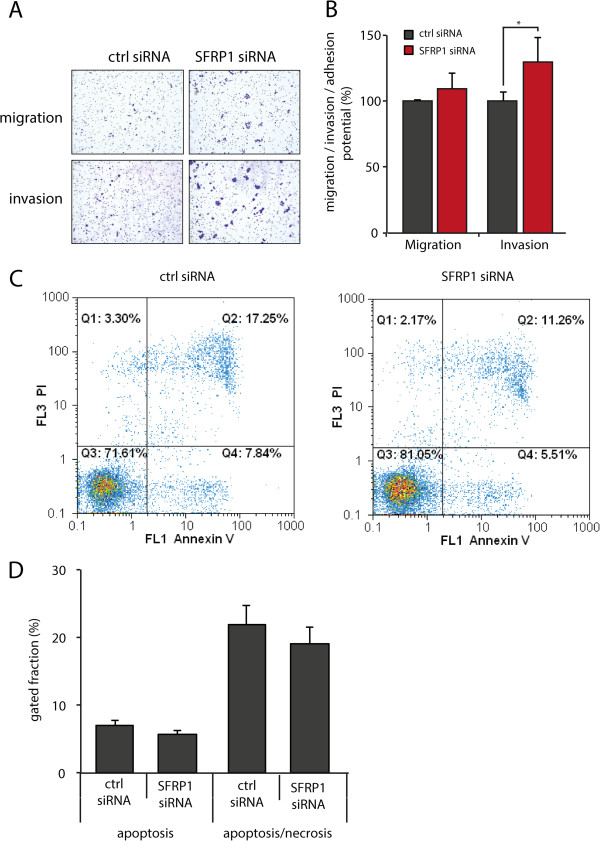
Downregulation of SFRP1 changes carcinogenic properties of triple negative breast cancer cells. A) Representative pictures of migration and invasion filters after SFRP1 knockdown in MDA-MB-468 cells. B) Quantitative analysis of migration and invasion assays after SFRP1 knockdown revealed increase in migration of about 15% and increase in invasion potential of about 30% ((*p < 0.05), error bars = SD, n = 3) C/D) Flow cytometry analysis using Annexin V staining showed a slight decrease of cells undergoing apoptotic (C: Q4) as well as late apoptotic or necrotic events (C: Q1 and Q2).
The changes of tumorigenic potential of cells after SFRP1 knockdown do not rely on Wnt signaling
SFRP1 is known to antagonize Wnt signaling activity via binding to either Wnt proteins or frizzled receptors, thereby blocking the intracellular signaling cascade [17]. Thus, we hypothesized that loss of SFRP1 might activate canonical Wnt signaling activity. Therefore, we performed a TOP-Flash/FOP-Flash luciferase assay in order to analyze the effects of SFRP1 knockdown on the triple negative breast cancer cell line MDA-MB-468. Surprisingly, we were not able to detect any changes in Wnt signaling activity of breast cancer cells after SFRP1 knockdown (Figure 4A (left)). As Wnt signaling activity is known to be very low in breast cancer cells, we made use of a known Wnt activator, LiCl, in order to stimulate Wnt signaling activity [19-21]. Treatment of cells with LiCl leads to a significant increase of Wnt signaling activity of about 30-fold. However, no significant changes were found in SFRP1 knockdown cells compared to control cells (Figure 4A (right)). Additionally, when analyzing the cellular localization of β-Catenin by immunofluorescence staining we were not able to detect any enhanced nuclear localization of β-Catenin after SFRP1 knockdown, typically a hallmark of activated Wnt signaling (Figure 4B).
Figure 4.
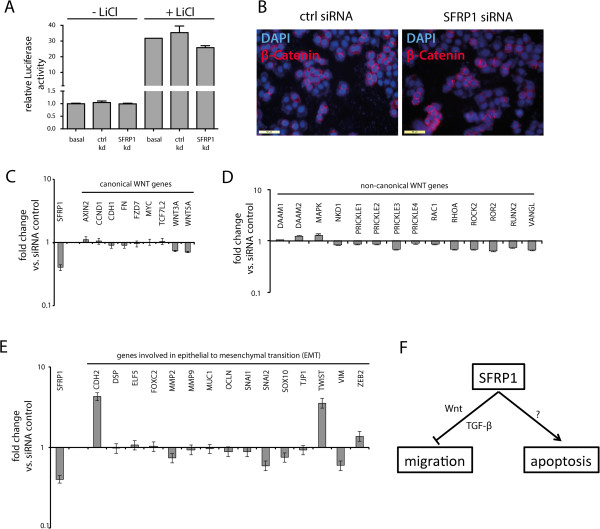
Influence of SFRP1 on carcinogenic properties does not rely on canonical Wnt signaling. A) Luciferase assays showed neither changes in Wnt signaling activity upon SFRP1 knockdown in breast cancer cells grown in standard medium (left 3 rows), nor in breast cancer cells grown in starvation medium followed by Wnt stimulation using LiCl treatment (right 3 rows). B) Immunofluorescence analysis showed no changes in cellular localization of β-Catenin after SFRP1 knockdown. C/D) mRNA levels of known canonical Wnt target genes (C) as well as members of the non-canonical Wnt signaling pathway (D) revealed no significant changes upon SFRP1 knockdown. E) mRNA levels of EMT related genes after SFRP1 knockdown. F) Model of action of SFRP1 in breast cancer.
We also performed global gene expression profile analysis via microarray analysis after SFRP1 knockdown in MDA-MB-468 cells. When analyzing effects on known canonical Wnt target genes, we were not able to detect any significant changes in gene expression (Table 3). In order to validate the microarray gene expression analysis data, we also performed quantitative real time PCR analysis. Although we found a decrease of approximately 60% in SFRP1 expression upon siRNA mediated knockdown, no significant changes of known Wnt target genes were observed (Figure 4C). Furthermore, we analyzed the expression levels of known members of the non-canonical Wnt signaling pathway [22-24]. However, we could not detect any significant changes in expression levels of these genes (Figure 4D). Since the Wnt signaling pathway is also correlated with the phenomenon of epithelial to mesenchymal transition, a process in which epithelial cells gain characteristics of mesenchymal cells, e.g. increased migratory or invasive potential, we also analyzed the expression levels of a plethora of known EMT related genes [25]. Although we found an increase in known EMT related genes N-Cadherin (CDH2) as well as TWIST, the majority of EMT genes remained unaffected after SFRP1 knockdown (Figure 4E). Thus, the effects of SFRP1 knockdown on carcinogenic properties on triple negative breast cancer cells do not appear to be mediated by elevated levels of canonical nor non-canonical Wnt signalling.
Table 3.
Expression of known Wnt target genes in SFRP1-depleted MDA-MB-468 cells
| Name | SEQ_ID | Ratio |
|---|---|---|
| TCF7L2 |
NM_030756 |
2.37 |
| MMP7 |
NM_002423 |
2.28 |
| FN |
NM_054034 |
1.57 |
| CDH1 |
AB025105 |
1.37 |
| CDH1 |
AB025106 |
1.31 |
| FZD7 |
BC015915 |
1.26 |
| VEGF |
AY263145 |
1.2 |
| AXIN2 |
BC101533 |
1.19 |
| VEGF |
M27281 |
1.16 |
| CLDN1 |
BC012471 |
1.16 |
| VEGF |
S85192 |
1.16 |
| CDH1 |
NM_004360 |
1.16 |
| AXIN2 |
NM_004655 |
1.13 |
| CLDN1 |
NM_021101 |
1.08 |
| JUN |
BC006175 |
1.07 |
| MYC |
NM_002467 |
1.01 |
| BGLAP |
NM_199173 |
0.97 |
| MYCBP |
NM_012333 |
0.95 |
| ID2 |
NM_002166 |
0.94 |
| CCND1 |
NM_053056 |
0.94 |
| CCND1 |
BC001501 |
0.94 |
| PPARD |
NM_001039694 |
0.94 |
| MYCBP |
BC008686 |
0.9 |
| SFRP1 | NM_003012 | 0.28 |
Only 2 of several known Wnt target genes show increased expression after SFRP1 knockdown (List of genes available from: The Wnt homepage; http://www.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes).
SFRP1 influences effects of cell adhesion and apoptosis
To gain insights into the mechanism underlying effects of SFRP1 knockdown on carcinogenic properties of triple negative breast cancer cells, we performed global gene expression profiling after SFRP1 knockdown followed by gene ontology analysis. When analyzing the list of genes showing upregulation of more than 30% after knockdown by using DAVID gene ontology database (http://david.abcc.ncifcrf.gov, [26]) we found several genes involved in cell adhesion, cell motion as well as signaling activity (Table 4). On the other hand, when analyzing genes showing downregulation of more than 25% after SFRP1 knockdown, we primarily found genes involved in positive regulation of apoptosis (Table 5), which is consistent with our observed decrease in apoptotic events in these cells (Figure 3C, D). Thus, SFRP1 knockdown positively regulates mechanisms of cell adhesion as well as the survival of cells presumably by inhibiting signals usually associated with apoptosis.
Table 4.
Gene ontology analysis of genes showing upregulation of > 130% after SFRP1 knockdown in MDA-MB-468 cells
| Term | Count | p-Value |
|---|---|---|
| Intracellular signaling cascade |
68 |
7.40E-04 |
| Positive regulation of transferase activity |
20 |
1.20E-03 |
| Cell motion |
31 |
2.20E-03 |
| Regulation of transferase activity |
26 |
2.20E-03 |
| Biological adhesion |
41 |
2.80E-03 |
| Cell adhesion |
41 |
2.90E-03 |
| Positive regulation of kinase activity |
18 |
4.40E-03 |
| Monosaccharide transport |
6 |
4.70E-03 |
| Regulation of kinase activity |
24 |
5.40E-03 |
| Cell migration | 20 | 5.60E-03 |
Table 5.
Gene ontology analysis of genes showing downregulation of < 75% after SFRP1 knockdown in MDA-MB-468 cells
| Term | Count | p-Value |
|---|---|---|
| Negative regulation of cell proliferation |
19 |
1.60E-05 |
| Positive regulation of apoptosis |
20 |
5.00E-05 |
| Positive regulation of programmed cell death |
20 |
5.50E-05 |
| Positive regulation of cell death |
20 |
5.90E-05 |
| Regulation of cellular localization |
14 |
1.50E-04 |
| Regulation of apoptosis |
28 |
1.60E-04 |
| Regulation of programmed cell death |
28 |
1.90E-04 |
| Induction of apoptosis by intracellular signals |
7 |
2.00E-04 |
| Regulation of cell death | 28 | 2.00E-04 |
Discussion
Triple negative breast cancer is the most aggressive breast cancer subtype associated with poor prognosis as well as high recurrence rates. Since patients suffering from TNBC do have an unfavorable prognosis mostly due to limited therapeutic options, this cancer subtype has gained much attention regarding the development of novel targeted therapies. However, a high number of TNBC patients do positively respond to neoadjuvant chemotherapy. As a result, achieving a pathologic complete response (pCR) is correlated with good prognosis similar to other breast cancer subtypes [9]. Thus, the identification of patients responding to neoadjuvant chemotherapy would greatly improve the therapeutic options for TNBC. Other TNBC patients, however, would need to be treated differently, e.g. by anti-angiogenic treatment.
By using a published dataset analyzing differential gene expression profiles as well as response to neoadjuvant chemotherapeutic treatment of breast cancer patients we could show a correlation of SFRP1 expression and the triple negative breast cancer subtype.
We could demonstrate that breast cancer cell lines showing increased SFRP1 expression are associated with the triple negative phenotype, similar to published results showing higher expression in basal like cancer cell lines compared to luminal cell lines [14]. Interestingly, we mainly found increased expression of SFRP1 in triple negative breast cancer cell lines, which molecularly belong to basal A subtype described by Neve et al. (HCC1937, MDA-MB-468 and BT20) [27]. Cell lines of basal A subtype display more epithelial characteristics, whereas basal B cell lines are shown to be more invasive presumably due to spindle-like morphology displaying mesenchymal as well as stem/progenitor-like characteristics.
Furthermore, the basal A subtype has been correlated with increased response to neoadjuvant chemotherapy when compared to the basal B subtype [28,29]. In line with these observations we could show that expression of SFRP1 is also correlated with the achievement of a pathologic complete response after neoadjuvant chemotherapy. Thus, SFRP1 might become a useful biomarker to stratify triple negative breast cancer patients, which might benefit from neoadjuvant treatment. However, for clinical applications using SFRP1 expression as a prognostic biomarker, a proper platform is needed e.g. immunohistochemistry (IHC) or fluorescent in situ hybridization (FISH) followed by conversion into dichotomous status [30].
SFRP1 belongs to the family of 5 secreted frizzled receptor proteins, which show homology to the frizzled proteins, surface receptors for Wnt proteins [31]. SFRP1 has been described to antagonize canonical Wnt signaling by binding to either Wnt ligand proteins or frizzled receptors, thereby inhibiting the downstream signaling cascade [17]. SFRP1 has been linked to a number of solid tumors, e.g. colon cancer, ovarian cancer, prostate cancer or breast cancer [11-17]. It has been shown that the SFRP1 promoter is hypermethylated in these entities, thereby inactivating SFRP1 expression and its protein translation.
When analyzing the influence of loss of SFRP1 in triple negative breast cancer cells we found an increase of tumor-associated characteristics, e.g. increase in migration and invasion capacity, reduced apoptotic events as well as resistance to cytotoxic chemotherapy. Increased Wnt signaling is known to regulate tumor progression mechanisms as well as resistance to chemotherapy or radiation [32-34]. Thus, we initially hypothesized that knockdown of SFRP1 might result in increased Wnt signaling activity, thereby promoting tumor-associated properties of cells. This would be in line with previously published data showing reduced xenograft growth after SFRP1 overexpression in breast cancer cells presumably due to blockade of canonical Wnt signaling activity [35].
Surprisingly, however, we were neither able to detect Wnt activation by TOPFlash luciferase assays, changes in cellular localization of β-Catenin, nor detect any significant upregulation of known Wnt target genes. Therefore, we propose a different mechanism at which SFRP1 influences tumorigenic properties like invasion potential or resistance to chemotherapeutic agents. However, a Wnt dependent effect may also occur in triple negative cancers as in vivo Wnt signals may be supplied from the tumor stroma. Thus, Wnt dependent effects may also be contributing to the in vivo responsiveness of triple negative breast cancer patients to chemotherapy.
In addition to its known role in Wnt signaling, recent reports also demonstrate novel roles for SFRP1 signaling. One report showed binding of SFRP1 to thrombospondin-1, thereby inhibiting cancer cell adhesion and migration. This binding was conducted via the netrin related motif of SFRP1 [36]. These data are in accordance with our observation of increased invasiveness of SFRP1-depleted cells. Another study demonstrated an increased sensitivity of cells to TGF-β signaling upon SFRP1 reduction [37]. The TGF-β pathway is involved in epithelial to mesenchymal transition (EMT) as well as cellular migration in later stage mammary tumors, despite its known function as a tumor suppressor in early stage malignancies [38-40]. However, as expression of a majority of known EMT related genes are not substantially altered upon SFRP1 knockdown (Figure 4E), EMT may be of minor relevance in our experimental system. Recently, another study demonstrated a link between reduction of SFRP1 and reduction of apoptosis in vitro [41]. By using global gene expression profiles after SFRP1 knockdown, gene ontology analyses revealed upregulation of genes involved in migration processes, whereas genes involved in the positive regulation of apoptosis were downregulated (Tables 4, 5). Thus, chemotherapy might be reinforced by inhibition of apoptosis after SFRP1 knockdown. This view is supported by our observation of a slight decrease of apoptotic events upon SFRP1 depletion (Figure 3C, D). Apparently, pathways different from Wnt signaling presumably regulate processes that lead to increase of tumorigenic properties of cancer cells.
Conclusions
Our study sheds light on the complex regulatory network of mammary tumorigenesis and tumor progression, proposing a model in which SFRP1 regulates either invasive processes via canonical Wnt signaling but also via different pathways, e.g. TGF-β signaling as well as apoptotic processes via so far unknown mechanisms (Figure 4F).
Furthermore, we conclude that SFRP1 might be clinically used to stratify patients, which suffer from triple negative breast cancer for responding to neoadjuvant chemotherapy. As the reduction of SFRP1 is in line with increased aggressiveness of cancer cells, its overexpression might be an approach to explore novel therapeutic projections [35]. Thus, an increased level of SFRP1 might sensitize triple negative breast cancer patients towards chemotherapy, thereby improving prognosis of this aggressive breast cancer subtype. Nevertheless, future analyses have to be undertaken to explore the role of SFRP1 in regulating mammary tumor progression, particularly progression of triple negative breast cancer.
Methods
Cell culture, chemicals
The human mammary epithelial cell line MCF10a and the cancerous cell lines HCC 1937, MDA-MB 468, BT-20, MDA-MB 453, HCC 1806, MDA-MB 231, SKBR-3 and MCF-7 were supplied from ATCC and cultured under recommended conditions. Medium, trypsin-EDTA, PBS, fetal calf serum and horse serum were received from PAA Laboratories.
In order to chemically stimulate Wnt signaling, cells were starved in medium without serum for 24 h followed by incubation with 10 mM LiCl (SIGMA Aldrich), which inhibits GSK3β, thereby activating Wnt signaling [19,21].
Microarray gene expression analyses
A published microarray dataset was used for differential gene expression analysis [18]. For gene expression analysis in patients, triple negative breast cancer was defined using clinical measurements for ER, PR and HER2 as described previously and compared to the remaining cases merged as non-TNBC. Response to neoadjuvant chemotherapy was defined as absence of invasive breast cancer cells at the time of definitive surgery [42] and dichotomized as either pCR (n = 34) or residual invasive disease (RD; n = 99). Among cases with TNBC, 13 cases had pCR and 14 cases had RD. Gene expression data was processed and normalized using the robust multiarray average normalization algorithm as implemented in R-package affy version 1.32.0 [43].
Differential gene expression between patient subgroups was assessed using Welch's t statistic. The resulting p values were adjusted for control of the false discovery rate (FDR) according to Benjamini and Hochberg's method [44]. Analyses were performed in R using the Bioconductor multitest package version 2.10.0 [45].
For microarray experiments after SFRP1 knockdown in MDA-MB-468 cells, mRNA was converted into cDNA by using Superscript Double-Stranded cDNA Synthesis Kit (Invitrogen) according to manufactures protocol. Fluorescence labeling was performed using NimbleGen One-Color DNA Labeling Kit followed by hybridization onto arrays (NimbleGen human gene expression 12 × 135k arrays) according to protocol. By using DEVA software raw data was extracted. Further normalization was performed using GeneSpring Software. Normalized values were imported into gene ontology database DAVID (http://david.abcc.ncifcrf.gov; [26].
For expression analysis Wnt target genes were identified using the Wnt Homepage (http://www.stanford.edu/group/nusselab/cgi-bin/wnt/target_genes).
Western Blot analysis
Cells were incubated with RIPA buffer (10 mM NaF, 1 mM Na3VO4, 10 mM β-Glycerophosphate, 7.6 mM Tris pH 7.4, 52 mM NaCl, 0.4% Triton X-100, 0.8 mM EDTA, proteinase inhibitor (SIGMA Aldrich). Protein quantification was performed via BCA assay (Pierce) according to the manufacturer’s protocol. SDS page electrophoresis and blotting were performed using standard protocols. Detection was performed using SFRP1 antibody (SIGMA Aldrich, SAB2900383) and β-Actin antibody (BioLegend, clone # 2 F1-1) and SuperSignal West Pico Chemiluminescent Substrate (Pierce). Bands were visualized with AGFA developer and fixer (AGFA).
Quantitative real-time PCR
RNA isolation was performed using NucleoSpin RNA Kits (Macherey-Nagel) with on-column DNase digestion. Reverse transcription for real-time quantitative polymerase chain reaction (RT-qPCR) was performed using MMLV reverse transcriptase (USB (Affymetrix)) and Oligo-dT15 priming at 42°C for 1 hour and at 60°C for 10 minutes. A cDNA equivalent of 50 ng total RNA was used as template in a total reaction volume of 20 μl with Power SYBR Green PCR mix (Invitrogen) on an Step One Plus cycler (ABI). Primers were added at 0.375 μM each. Calculations were based on the ΔΔCt method using two housekeeping genes for normalization. Real time primer sequences can be found in supplemental Table 1 (Additional file 1: Table S1).
siRNA mediated mRNA knockdown
siRNA mediated knockdown assays were implemented using SFRP1 siRNA (part no 4392422) and negative control siRNA (part no 4390844) (Applied Biosystems) in combination with DharmaFECT (ThermoScientific) transfection reagent according to the manufacturer’s protocol. Efficacy of knockdown was analyzed by qPCR and Western blotting 48 h – 72 h after transfection.
Cell migration / invasion assay
For migration assays, cell culture inserts equipped with 8 μm membranes were used (Falcon). For invasion assays, BioCoat Matrigel invasion chambers (BD Biosciences) were used according to the manufacturer’s protocols. Briefly, 24 – 48 h after transfection, 2 – 5 ×104 cells were seeded into cell culture inserts in medium without serum. The lower chamber was filled with medium containing serum as chemoattractant. 48 – 96 h after seeding cells, which passed the membranes, were fixed and stained using Diff-Quik staining set according to manufacturer’s protocol (Medion Diagnostics). Stained filters were mounted on microscope slides with VitroClud (Langenbrinck). Quantitative analysis was done by cell counting using Image J software.
Luciferase assay
Luciferase assays were performed using TOPFlash or FOPFlash plasmids (addgene plasmid numbers 1256 and 12457, respectively) along with renilla normalization construct (pRL-TK, Promega) using Lipofectamine 2000 (Invitrogen). Luciferase constructs were transfected 48 h after siRNA transfection. Starvation medium as well as normal medium was changed the next day. After additional 24 h, cells were harvested and processed according to the DualGlo Luciferase protocol (Promega). Relative Luciferase activity was normalized to the activity of the FOPFlash mutant vector control.
Immunohistochemistry/Immunocytochemistry
The study was approved by the local ethical review committee (Research ethics committee of the Medical Association Westfalen-Lippe and Westphalian Wilhelms University; ethical vote: 2013-156-f-S). We used tissue microarrays of 362 patients. Of these, 37 were diagnosed as being TNBC by missing expression of ER, PR and HER2. Immunohistochemistry of formalin-fixed, paraffin-embedded tissue microarrays was performed using primary antibody (SFRP1, Epitomics, clone# EPR7003) and biotinylated secondary antibodies (DAKO). Detection was performed using Chromogen Red (DAKO) and H&E (Merck). Slides were embedded with Scientific Cytoseal (Thermo Scientific Fisher).
For immunocytochemistry, cells were fixed with phosphate buffered formalin. Cells were blocked with 10% Aurion (DAKO) in PBS for 1 h. Cells were washed and incubated with primary antibody (β-Catenin, Cell Signaling, # 9587) diluted with Dako REALTM Antibody Diluent (overnight at 4°C). Fluorescent visualization was carried out using suitable Alexa Fluor-conjugated secondary antibody (1:600) together with 4′,6-diamidino-2-phenylindole (1:400) in in Dako REALTM Antibody Diluent) for 1 h at RT.
Chemotherapy sensitivity assay
For analysis of chemotherapy sensitivity, cells were incubated with cytotoxic agents using decreasing concentrations: paclitaxel (10 pM - 1 μM), doxorubicin hydrochloride (500 pM - 50 μM), cis-diamineplatinum II dichloride (50 nM - 5 mM). After 96 hours, cell viability was determined via MTT (Thiazolyl Blue Tetrazolium Bromide) (all substances were received from SIGMA Aldrich) according to the manufacturer’s protocol. Measurements were performed at least in triplicates. Significance was calculated via one-side Welch’s t-test.
Flow cytometry
Following transfection cells were stained for apoptosis as well as apoptosis/necrosis using the annexin V test kit from Becton Dickinson (San José, USA). Flow cytometric cell analysis and quantification of cell death took place on a flow cytometer (CyFlow Space, Partec, Germany) as described previously [46,47].
Consent
Written informed consent was obtained from the patients for the publication of this report and any accompanying images.
Abbreviations
SFRP1: Secreted frizzled receptor protein 1; HER2: Human epidermal growth factor receptor 2; TNBC: Triple negative breast cancer; siRNA: Small interfering RNA; ER: Estrogen receptor; PR: Progesterone receptor; PCR: Polymerase chain reaction; LiCl: Lithium chloride; pCR: Pathologic complete response; IHC: Immunohistochemistry; FISH: Fluorescence in situ hybridization; RD: Residual disease; EMT: Epithelial to mesenchymal transition.
Competing interests
The authors declare no conflict of interests.
Authors’ contribution
C.B. conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, final approval of manuscript, C.H. conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing; C.R. data analysis and interpretation; S.S. and L.B. provision of study materials; G.H. and M.G. data analysis and interpretation; B.G. and P.J.B. collection and/or assembly of data, data analysis and interpretation; L.K. conception and design, financial support; C.L.: conception and design, data analysis and interpretation. All authors read and approved the final manuscript.
Supplementary Material
Sequences of qPCR primers used in this study.
Contributor Information
Christof Bernemann, Email: Christof.Bernemann@ukmuenster.de.
Carolin Hülsewig, Email: carolin.huelsewig@gmail.com.
Christian Ruckert, Email: Christian.Ruckert@uni-muenster.de.
Sarah Schäfer, Email: sarah.a.schafer@googlemail.com.
Lena Blümel, Email: lena.bluemel@ukmuenster.de.
Georg Hempel, Email: georg.hempel@uni-muenster.de.
Martin Götte, Email: goettema@ukmuenster.de.
Burkhard Greve, Email: greveb@uni-muenster.de.
Peter J Barth, Email: Ludwig.Kiesel@ukmuenster.de.
Ludwig Kiesel, Email: Ludwig.Kiesel@ukmuenster.de.
Cornelia Liedtke, Email: Cornelia.Liedtke@uksh.de.
Acknowledgements
We thank Dorothea Godulla, Annette van Dülmen and Birgit Pers for technical assistance. We also acknowledge support by Deutsche Forschungsgemeinschaft and Open Access Publication Fund of University of Muenster. Parts of this project were funded by the internal funds of the medical faculty of Muenster University IMF (Innovative Medizinische Forschung) – BE 1 1 12 02.
References
- Carey L, Winer E, Viale G, Cameron D, Gianni L. Triple-negative breast cancer: disease entity or title of convenience? Nat Rev Clin Oncol. 2010;7:683–692. doi: 10.1038/nrclinonc.2010.154. [DOI] [PubMed] [Google Scholar]
- Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N. Triple-negative breast cancer–current status and future directions. Ann Oncol. 2009;20:1913–1927. doi: 10.1093/annonc/mdp492. [DOI] [PubMed] [Google Scholar]
- Rakha EA, El-Sayed ME, Green AR, Lee AHS, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. 2007;109:25–32. doi: 10.1002/cncr.22381. [DOI] [PubMed] [Google Scholar]
- Miller K, Wang M, Gralow J, Dickler M, Cobleigh M, Perez EA, Shenkier T, Cella D, Davidson NE. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N Engl J Med. 2007;357:2666–2676. doi: 10.1056/NEJMoa072113. [DOI] [PubMed] [Google Scholar]
- Santana-Davila R, Perez EA. Treatment options for patients with triple-negative breast cancer. J Hematol Oncol. 2010;3:42. doi: 10.1186/1756-8722-3-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheang MCU, Voduc D, Bajdik C, Leung S, McKinney S, Chia SK, Perou CM, Nielsen TO. Basal-like breast cancer defined by five biomarkers has superior prognostic value than triple-negative phenotype. Clin Cancer Res. 2008;14:1368–1376. doi: 10.1158/1078-0432.CCR-07-1658. [DOI] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA. Triple-Negative Breast Cancer: Clinical Features and Patterns of Recurrence. Clin Cancer Res. 2007;13:4429–4434. doi: 10.1158/1078-0432.CCR-06-3045. [DOI] [PubMed] [Google Scholar]
- Ruijter TC, Veeck J, Hoon JPJ, Engeland M, Tjan-Heijnen VC. Characteristics of triple-negative breast cancer. J Cancer Res Clin Oncol. 2010;137:183–192. doi: 10.1007/s00432-010-0957-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, Symmans WF, Gonzalez-Angulo AM, Hennessy B, Green M, Cristofanilli M, Hortobagyi GN, Pusztai L. Response to Neoadjuvant Therapy and Long-Term Survival in Patients With Triple-Negative Breast Cancer. J Clin Oncol. 2008;26:1275–1281. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–2634. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- Caldwell GM, Jones C, Gensberg K, Jan S, Hardy RG, Byrd P, Chughtai S, Wallis Y, Matthews GM, Morton DG. The Wnt antagonist sFRP1 in colorectal tumorigenesis. Cancer Res. 2004;64:883–888. doi: 10.1158/0008-5472.can-03-1346. [DOI] [PubMed] [Google Scholar]
- Fukui T, Kondo M, Ito G, Maeda O, Sato N, Yoshioka H, Yokoi K, Ueda Y, Shimokata K, Sekido Y. Transcriptional silencing of secreted frizzled related protein 1 (SFRP 1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene. 2005;24:6323–6327. doi: 10.1038/sj.onc.1208777. [DOI] [PubMed] [Google Scholar]
- Lodygin D, Epanchintsev A, Menssen A, Diebold J, Hermeking H. Functional epigenomics identifies genes frequently silenced in prostate cancer. Cancer Res. 2005;65:4218–4227. doi: 10.1158/0008-5472.CAN-04-4407. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Watkins DN, Jair K-W, Schuebel KE, Markowitz SD, Dong Chen W, Pretlow TP, Yang B, Akiyama Y, van Engeland M, Toyota M, Tokino T, Hinoda Y, Imai K, Herman JG, Baylin SB. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36:417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- Takada T, Yagi Y, Maekita T, Imura M, Nakagawa S, Tsao S-W, Miyamoto K, Yoshino O, Yasugi T, Taketani Y, Ushijima T. Methylation-associated silencing of the Wnt antagonist SFRP1 gene in human ovarian cancers. Cancer Sci. 2004;95:741–744. doi: 10.1111/j.1349-7006.2004.tb03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeck J, Niederacher D, An H, Klopocki E, Wiesmann F, Betz B, Galm O, Camara O, Dürst M, Kristiansen G, Huszka C, Knüchel R, Dahl E. Aberrant methylation of the Wnt antagonist SFRP1 in breast cancer is associated with unfavourable prognosis. Oncogene. 2006;25:3479–3488. doi: 10.1038/sj.onc.1209386. [DOI] [PubMed] [Google Scholar]
- Yang Z-Q, Liu G, Bollig-Fischer A, Haddad R, Tarca AL, Ethier SP. Methylation-associated silencing of SFRP1 with an 8p11-12 amplification inhibits canonical and non-canonical WNT pathways in breast cancers. Int J Cancer. 2009;125:1613–1621. doi: 10.1002/ijc.24518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, Rouzier R, Sneige N, Ross JS, Vidaurre T, Gómez HL, Hortobagyi GN, Pusztai L. Pharmacogenomic Predictor of Sensitivity to Preoperative Chemotherapy With Paclitaxel and Fluorouracil, Doxorubicin, and Cyclophosphamide in Breast Cancer. J Clin Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci U S A. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, Schutte M, Clevers H. Phosphatidylinositol 3-Kinase Signaling Does Not Activate the Wnt Cascade. J Biol Chem. 2009;284:35308–35313. doi: 10.1074/jbc.M109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, Klein PS. Glycogen synthase kinase-3beta haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnsdorf EJ, Tummala P, Jacobs CR. Non-Canonical Wnt Signaling and N-Cadherin Related β-Catenin Signaling Play a Role in Mechanically Induced Osteogenic Cell Fate. PLoS One. 2009;4:e5388. doi: 10.1371/journal.pone.0005388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Rubin B, Bodine PVN, Billiard J. Wnt5a induces homodimerization and activation of Ror2 receptor tyrosine kinase. J Cell Biochem. 2008;105:497–502. doi: 10.1002/jcb.21848. [DOI] [PubMed] [Google Scholar]
- Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA, Inanlou MR, Chiu E, Buchanan M, Hosein AN, Basik M, Wrana JL. Exosomes Mediate Stromal Mobilization of Autocrine Wnt-PCP Signalingin Breast Cancer Cell Migration. Cell. 2012;151:1542–1556. doi: 10.1016/j.cell.2012.11.024. [DOI] [PubMed] [Google Scholar]
- Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial– mesenchymal transition. Nat Rev Mol Cell Biol. 2014;15:178–196. doi: 10.1038/nrm3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe J-P, Tong F, Speed T, Spellman PT, DeVries S, Lapuk A, Wang NJ, Kuo W-L, Stilwell JL, Pinkel D, Albertson DG, Waldman FM, McCormick F, Dickson RB, Johnson MD, Lippman M, Ethier S, Gazdar A, Gray JW. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer JA, Chakravarthy AB, Rosenbluth JM, Mi D, Seeley EH, De Matos Granja-Ingram N, Olivares MG, Kelley MC, Mayer IA, Meszoely IM, Means-Powell JA, Johnson KN, Tsai CJ, Ayers GD, Sanders ME, Schneider RJ, Formenti SC, Caprioli RM, Pietenpol JA. Identification of markers of taxane sensitivity using proteomic and genomic analyses of breast tumors from patients receiving neoadjuvant paclitaxel and radiation. Clin Cancer Res. 2010;16:681–690. doi: 10.1158/1078-0432.CCR-09-1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y, Pietenpol JA. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest. 2011;121:2750–2767. doi: 10.1172/JCI45014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Freidlin B, Simon R. Biomarker-adaptive threshold design: a procedure for evaluating treatment with possible biomarker-defined subset effect. JNCI J of the National Cancer Institute. 2007;99:1036–1043. doi: 10.1093/jnci/djm022. [DOI] [PubMed] [Google Scholar]
- Bovolenta P, Esteve P, Ruiz JM, Cisneros E, Lopez-Rios J. Beyond Wnt inhibition: new functions of secreted Frizzled-related proteins in development and disease. J Cell Sci. 2008;121:737–746. doi: 10.1242/jcs.026096. [DOI] [PubMed] [Google Scholar]
- Clevers H, Nusse R. Wnt/b-Catenin Signaling and Disease. Cell. 2012;149:1192–1205. doi: 10.1016/j.cell.2012.05.012. [DOI] [PubMed] [Google Scholar]
- Holland JD, Klaus A, Garratt AN, Birchmeier W. Wnt signaling in stem and cancer stem cells. Curr Opin Cell Biol. 2013;25(2):254–264. doi: 10.1016/j.ceb.2013.01.004. [DOI] [PubMed] [Google Scholar]
- Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- Matsuda Y, Schlange T, Oakeley EJ, Boulay A, Hynes NE. WNT signaling enhances breast cancer cell motility and blockade of the WNT pathway by sFRP1 suppresses MDA-MB-231 xenograft growth. Breast Cancer Res. 2009;11:R32. doi: 10.1186/bcr2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Manso G, Calzada MJ, Chuman Y, Sipes JM, Xavier CP, Wolf V, Kuznetsova SA, Rubin JS, Roberts DD. sFRP-1 binds via its netrin-related motif to the N-module of thrombospondin-1 and blocks thrombospondin-1 stimulation of MDA-MB-231 breast carcinoma cell adhesion and migration. Arch Biochem Biophys. 2011;509:147–156. doi: 10.1016/j.abb.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger KJ, Chenausky KL, Murray ME, Schneider SS. SFRP1 reduction results in an increased sensitivity to TGF-β signaling. BMC Cancer. 2011;11:59. doi: 10.1186/1471-2407-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka RS, Dumont N, Ritter CA, Dugger TC, Brantley DM, Chen J, Easterly E, Roebuck LR, Ryan S, Gotwals PJ, Koteliansky V, Arteaga CL. Blockade of TGF-beta inhibits mammary tumor cell viability, migration, and metastases. J Clin Invest. 2002;109:1551–1559. doi: 10.1172/JCI15234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka-Cook RS, Shin I, Yi JY, Easterly E, Barcellos-Hoff MH, Yingling JM, Zent R, Arteaga CL. Activated type I TGFbeta receptor kinase enhances the survival of mammary epithelial cells and accelerates tumor progression. Oncogene. 2006;25:3408–3423. doi: 10.1038/sj.onc.1208964. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003;100:8621–8623. doi: 10.1073/pnas.1633291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauger KJ, Schneider SS. Tumour supressor secreted frizzled related protein 1 regulates p53-mediated apoptosis. Cell Biol Int. 2013;38:124–130. doi: 10.1002/cbin.10176. [DOI] [PubMed] [Google Scholar]
- Mazouni C, Peintinger F, Wan-Kau S, Andre F, Gonzalez-Angulo AM, Symmans WF, Meric-Bernstam F, Valero V, Hortobagyi GN, Pusztai L. Residual ductal carcinoma in situ in patients with complete eradication of invasive breast cancer after neoadjuvant chemotherapy does not adversely affect patient outcome. J Clin Oncol. 2007;25:2650–2655. doi: 10.1200/JCO.2006.08.2271. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001;125:279–284. doi: 10.1016/s0166-4328(01)00297-2. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechlers M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JYH, Zhang J. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greve B, Dreffke K, Rickinger A, Könemann S, Fritz E, Eckardt-Schupp F, Amler S, Sauerland C, Braselmann H, Sauter W, Illig T, Schmezer P, Gomolka M, Willich N, Bölling T. Multicentric investigation of ionising radiation-induced cell death as a predictive parameter of individual radiosensitivity. Apoptosis. 2009;14:226–235. doi: 10.1007/s10495-008-0294-6. [DOI] [PubMed] [Google Scholar]
- Greve B, Kelsch R, Spaniol K, Eich HT, Götte M. Flow cytometry in cancer stem cell analysis and separation. Cytometry A. 2012;81:284–293. doi: 10.1002/cyto.a.22022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Sequences of qPCR primers used in this study.