

Abstract
Lewy bodies, a pathological hallmark of Parkinson's disease (PD), contain aggregated alpha-synuclein (αSyn), which is found in several modified forms and can be discovered phosphorylated, ubiquitinated and truncated. Aggregation-prone truncated species of αSyn caused by aberrant cleavage of this fibrillogenic protein are hypothesized to participate in its sequestration into inclusions subsequently leading to synaptic dysfunction and neuronal death. Here, we investigated the role of calpain cleavage of αSyn in vivo by generating two opposing mouse models. We crossed into human [A30P]αSyn transgenic (i) mice deficient for calpastatin, a calpain-specific inhibitor, thus enhancing calpain activity (SynCAST(−)) and (ii) mice overexpressing human calpastatin leading to reduced calpain activity (SynCAST(+)). As anticipated, a reduced calpain activity led to a decreased number of αSyn-positive aggregates, whereas loss of calpastatin led to increased truncation of αSyn in SynCAST(−). Furthermore, overexpression of calpastatin decreased astrogliosis and the calpain-dependent degradation of synaptic proteins, potentially ameliorating the observed neuropathology in [A30P]αSyn and SynCAST(+) mice. Overall, our data further support a crucial role of calpains, particularly of calpain 1, in the pathogenesis of PD and in disease-associated aggregation of αSyn, indicating a therapeutic potential of calpain inhibition in PD.
INTRODUCTION
Parkinson's disease (PD) is a slowly progressive disorder caused by neuronal loss of dopaminergic neurons predominantly in the substantia nigra pars compacta (1, 2). The neuropathological hallmark of PD is intracellular inclusions termed Lewy bodies (LB) that are mainly composed of alpha-Synuclein (αSyn) (2, 3).
Increasing evidence strongly suggests that αSyn inclusions accumulate in a hierarchical pattern, reflecting vulnerability of distinct neuronal populations in the PD brain and initially affecting distinct nuclei in the brain stem spreading into cerebral cortical areas in advanced PD (4–6). Intracellularly, the formation of pathological αSyn depositions occurs by a multistep process, including a conformational change and the association of the misfolded protein into protofilaments and amyloid fibrils, forming the disease specific entity (7, 8). Several studies have elucidated the role of different αSyn regions on fibrillation of αSyn. Whereas the N-terminus transforms into a helical conformation upon its binding to phospholipids (9, 10), and the NAC domain was shown to form the core region of fibrils (11), it appears that the acidic C-terminus, however, is not implicated in the observed fibrillation pathology. Instead, it preserves αSyn fibrillation by maintaining the unfolded structure of the monomeric form (12, 13). Both, neutralization of the negative charge of the C-terminus via calcium (14) and the C-terminal truncation, were associated with an enhanced aggregation and fibrillation in vitro (15, 16) and in vivo (17–19).
Several proteases are implicated in C-terminal truncation of αSyn subsequently leading to enhanced aggregation pathology. Among others, plasmin has a predominant role in cleavage of the N-terminal region (20), and MMPs, neurosin, as well as cathepsin D cleave at several sites within αSyn (21–24). Further, C-terminal fragments of αSyn derived from the lysosomal enzyme cathepsin D are a main component of αSyn assemblies (25).
Besides these proteases, calpains have been implicated in several neurodegenerative diseases, including PD (26). Calpains are calcium-dependent proteases and play an essential role in numerous physiological cellular and neuronal functions (27). Alterations in calcium homeostasis lead to pathological activation of calpains affecting negatively the function of several neuronal substrates such as αSyn, amyloid-beta and ataxin 3 (26–29). Pathological activation of calpains is also involved in synaptic dysfunction and may be critical to memory formation (27, 30, 31).
Recently, it was shown that calpain 1, one of the major isoforms of the calpain family in the brain, cleaves αSyn in vitro: soluble αSyn is cleaved at amino acid 57, whereas the cleavage sites within fibrillar αSyn are predominantly located at the C-terminus at amino acids 114 and 122, promoting the co-assembly of soluble αSyn (32, 33). Dufty et al. (3) showed that C-terminally calpain-cleaved αSyn is detectable in LB in brains of PD patients. The role of calpain in the pathogenesis of PD was supported through the inhibition of calpain in a mouse model, which prevents neuronal and behavioral deficits induced by the neurotoxin MPTP (34). Additionally, overexpression of calpastatin, the endogenous specific inhibitor of calpain, was shown to reduce calpain activity and to protect from calcium-dependent neuronal excitotoxicity in transgenic mouse brain (30). Vice versa, calpastatin depletion triggered excitotoxicity, importantly without affecting development, viability and synaptic plasticity under physiological (calcium) conditions in mice (35).
Taking advantage of mice displaying an age-dependent αSyn pathology, including its presence as proteinase K (PK)-resistant and insoluble conformation (36–38), we investigated whether calpains are implicated in the aSyn induced pathogenesis of PD. Therefore, we crossed calpastatin knockout mice (35) and calpastatin overexpressing transgenic mice (30) with mice overexpressing the human mutated [A30P]αSyn under control of the Thy-1 promoter (37). Here, we demonstrate for the first time that the overexpression of calpastatin leads to a reduction in truncated as well as aggregated αSyn and to synaptic impairment in vivo. These data indicate that calpain may be an attractive candidate target for therapeutical intervention in PD.
RESULTS
Cleavage pattern of [WT]αSyn and [A30P]αSyn in vitro
Calpain is known to recognize motifs between conformational domains, therefore resulting in a distinct cleavage pattern of soluble and fibrillized αSyn (32, 33). Since the A30P mutation of αSyn is known to increase its propensity to aggregate (39–41), we first determined whether [WT]αSyn and [A30P]αSyn display the same calpain cleavage pattern using a calpain 1 in vitro cleavage assay. Recombinant soluble as well as fibrillar human αSyn was incubated with recombinant calpain 1 and calcium (3 µm) for up to 30 min. Two different antibodies directed against the N- or C-terminus of αSyn (Fig. 1A) were used to identify the calpain 1 cleavage pattern of αSyn. Both, [WT]αSyn and [A30P]αSyn, were cleaved equally and showed a similar fragment pattern. However, the cleavage pattern between soluble and fibrillar αSyn showed clear differences. Overall, the soluble C-terminal αSyn fragments (Fig. 1B; i) were much smaller than the fibrillar C-terminal fragments (Fig. 1B; iii), whereas the N-terminal αSyn fragments (Fig. 1B; ii, iv) revealed comparable sizes. Although most of the higher-molecular fibrillized [A30P]αSyn structures were reduced after calpain digestion, a remaining strong signal at the size of ∼25–37 kDa was detected after 30 min that may indicate formation of dimeric αSyn species. These differences in higher molecular structures between the recombinant [A30P]αSyn and [WT]αSyn are most likely due to the increased propensity of the A30P mutation to form oligomers, which was observed both ex vitro (7, 42) and in PD (43). In particular, the 35–37 kDa species was previously detected as an SDS–PAGE resistant dimer (42–45). Analytical ultracentrifugation measurement confirmed the presence of dimeric αSyn representing ∼2.2% of total αSyn (Supplementary Material, Fig. S1A and B). Its strong detection level might be overestimated by an antibody preference toward accessible epitopes, as previously described (42). The incubation with the calpain inhibitor ALLN sufficiently prevented fragmentation of both [WT]αSyn and [A30P]αSyn and thus confirmed the specificity of the calpain-derived fragments of αSyn (see also Supplementary Material, Fig. S1C). Thus, our results suggest that mutant soluble and fibrillized [A30P]αSyn is readily cleaved by calpain 1, leading to accumulation of both N- and C-terminally truncated species, the latter may potentiate its conversion into dimeric structures.
Figure 1.
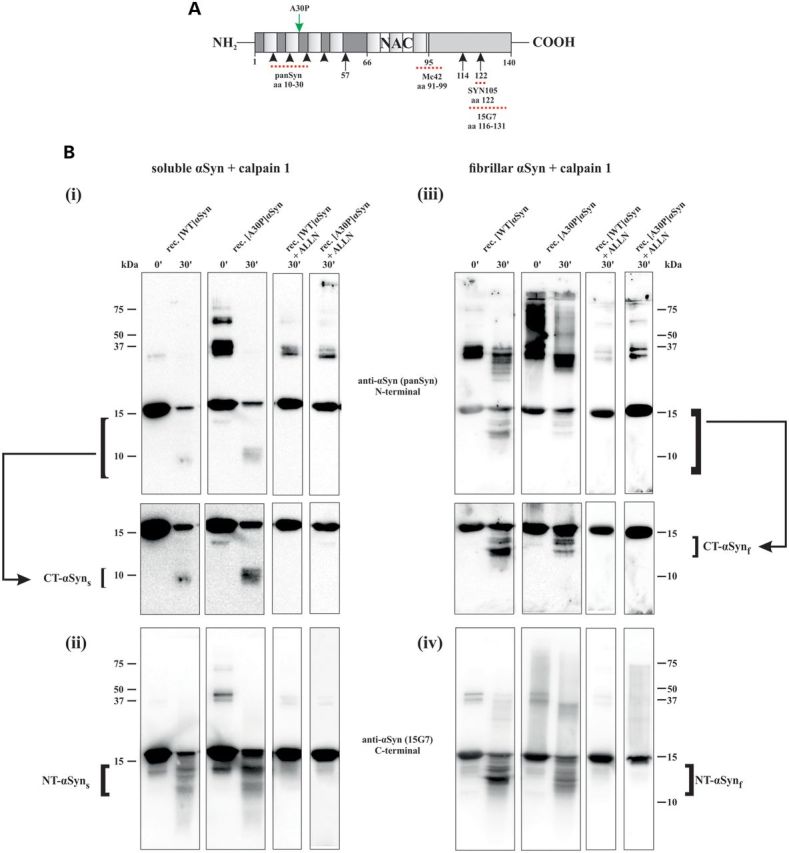
αSyn cleavage by calpain 1 in vitro. (A) Scheme of the human αSyn sequence indicating the cleavage sites of calpain 1 and the epitopes of the antibodies used in this study. The main cleavage sites (arrows) of calpain 1 are located in the N-terminus at amino acids 57 for soluble αSyn and in the C-terminus at amino acids 114 and 122 for fibrillar αSyn. (B) Human recombinant soluble (i, ii) and fibrillar (iii, iv) αSyn (WT, A30P) were incubated with recombinant calpain 1 for 30 min. Two different antibodies, directed either against the N- or the C-terminal part, were used to identify the fragment pattern of αSyn cleaved by calpain 1. The N-terminal antibody is detecting CT-αSyn, whereas the antibody targeting the C-terminus is detecting the fragments derived from cleavage in the N-terminus (NT-αSyn). Note that no gross differences between truncation of recombinant [WT]αSyn and mutant [A30P]αSyn were observed. However, there were differences in the cleavage pattern between soluble and fibrillar αSyn and residual higher oligomeric fibrillar structures of the [A30P]αSyn. To confirm the specificity of the calpain 1 derived αSyn cleavage products the inhibitor ALLN was added (Fig. 1B; + ALLN). rec, recombinant.
Generation of SynCAST(−) and SynCAST(+) mice
To investigate the role of calpain in PD-associated aggregation and neuropathology in vivo, we generated SynCAST(+) double transgenic mice overexpressing human mutated [A30P]αSyn (37) as well as human calpastatin, the only natural inhibitor of calpain (30), or crossed mice overexpressing human mutated [A30P]αSyn into a calpastatin-deficient background (38; SynCAST(−); Supplementary Material, Fig. S2). Overexpression of human mutated [A30P]αSyn and human calpastatin as well as homozygosity was proven by quantitative real-time PCR (unpublished data), western blot analyses and immunohistochemistry (Supplementary Material, Fig. S2). The knockout of calpastatin was proven by PCR (unpublished data), and overexpression of human transgenic calpastatin was validated via immunohistochemistry (Supplementary Material, Fig. S3A) and western blot analysis (Supplementary Material, Fig. S3B).
Overexpression of CAST decreases the expression of calpain 1 and results in a reduction of the calpain-mediated spectrin breakdown product
In order to analyze the expression level of one of the main calpain isoforms, calpain 1 (µ-type) and the activation of calpain in total, we performed western blot analyses with lysates of the brain stem (n = 3). The antibody against calpain 1 detects autoproteolytically cleaved (activated; 75 kDa) and full-length (latent; 80 kDa) calpain 1 (Fig. 2A). Calpain activation was additionally measured by an antibody against alpha-spectrin, a well-known substrate for calpain that yields to a specific 145 kDa breakdown product of alpha-spectrin (Fig. 2A; 44). Overexpression of human calpastatin (cast(+/+), SynCAST(+)) leads to a significant reduction in the calpain 1 expression level compared with SynCAST(−) mice (Fig. 2A and B). It also revealed a significant decreased ratio of 145 kDa calpain-cleaved and 240 kDa uncleaved alpha-spectrin when compared with [A30P]αSyn and SynCAST(−) (Fig. 2A and C). Interestingly, we found an increase in the breakdown product of calpain-cleaved spectrin, an indicator of overall calpain activity in [A30P]αSyn single transgenic mice, further elevated by its overexpression on calpastatin-deficient background (Fig. 2A and C). Thus, our data may reflect a tendency of increased calpain activity in [A30P]αSyn single transgenic mice in vivo that is further triggered by its overexpression on calpastatin-deficient background. We also performed immunohistochemical analysis for calpain 1 (Fig. 2D). Its expression pattern differed between the various mouse lines used: knockout of calpastatin in SynCAST(−) as well as overexpression of [A30P]αSyn showed a pronounced punctuated calpain1 immunoreactivity and was detected both in soma and the neuropil, whereas SynCAST(+) mice overexpressing calpastatin and wild-type mice exhibited a more weak and homogenous expression pattern (Fig. 2D).
Figure 2.

Expression of calpain 1 is changed and decreases calpain-mediated spectrin breakdown product in a mouse model of Parkinson's disease upon calpastatin overexpression. (A) Lysates of the brain stem (n = 3) were used for western blot analysis. Decreased ratio of autocleaved calpain 1 (activated; 75 kDa) to the full-length form (latent; 80 kDa) is observed upon overexpression of human calpastatin (SynCAST(+), cast(+/+)), whereas the knockout of calpastatin (SynCAST(−), cast(−/−)) leads to an increased ratio. The activity of calpain was measured using an antibody against alpha-spectrin, a well-known substrate, and its breakdown product (145 kDa). Calpain-mediated spectrin breakdown product was dramatically decreased in the brain stem of animals overexpressing calpastatin, especially when αSyn was overexpressed. (B and C) Quantification of the expression level of calpain 1 and the ratio of 145 kDa calpain-cleaved and 240 kDa uncleaved alpha-spectrin was done by densitometric measurements. The (#) sign represents the significance compared with [A30P]αSyn, and the dollar sign ($) compared with SynCAST(−). Error bars represent mean + SEM. Statistical significance was evaluated by one-way ANOVA where (**) indicates P < 0.01. (D) Staining of the brain stem with an antibody specific for both, activated and inactivated calpain 1. Immunohistochemistry revealed a punctuated staining for calpain 1 after knockout of calpastatin as well as overexpressing human [A30P]αSyn, whereas the pattern is increased after knockout. Mice overexpressing calpastatin and wild-type mice displayed a weak and homogeneous staining of calpain 1. Scale bar = 50 µm.
Overexpression of CAST leads to reduced fragmentation of αSyn
To analyze the cleavage pattern of αSyn by calpain in vivo, we performed western blot analysis of lysates of the brain stem of young (3 months; n = 3) and aged (19 months; n = 3) mice. The antibody panSyn targets the αSyn N-terminus and recognizes both, human and murine αSyn, whereas the antibody 15G7 detects only human αSyn at its C-terminus (Fig. 1 A). Thus, the N-terminal antibody recognizes C-terminally cleaved αSyn (CT-αSyn), whereas 15G7 detects the N-terminally cleaved fragments of αSyn (NT-αSyn). We did not detect a decrease of full-length αSyn (Fig. 3A, * human/murine, # human) over the time. Neither overexpression nor knockout of calpastatin revealed significant differences of the CT-αSyn1 fragment (Fig. 3B). However, the amount of the smaller soluble CT-αSyn2 fragments, which were also detected via calpain digestion of purified recombinant [A30P]αSyn (Fig. 3A, left panel), were less abundant in calpastatin overexpressing mice when compared with SynCAST(−) and [A30P]αSyn. Moreover, the amount of the CT-αSyn2 fragments strongly increased with age in [A30P]αSyn and SynCAST(−), whereas its low level remained unchanged in SynCAST(+) mice (Fig. 3B). The NT-αSyn1 fragments were almost stable over the time; however, a significant reduction was observed by calpastatin co-overexpression in SynCAST(+) (Fig. 3B). In summary, the fragmentation of αSyn is reduced by the overexpression of calpastatin, associating with activation of calpain in SynCAST(−) mice and thus provides clear evidence that calpain is involved in truncation of αSyn in vivo.
Figure 3.

Fragmentation of human αSyn is increased in the brain stem of SynCAST(−) and single transgenic [A30P]αSyn mice. (A) Brain stem of young and aged mice (n = 3) was used for western blot analysis. Asterisk is indicating murine and human full-length αSyn; number sign is highlighting human full-length αSyn. The antibody targeting the N-terminus is identifying CT-αSyn, whereas the antibody directed against the C-terminus is detecting the N-terminally cleaved αSyn (NT-αSyn). CT-αSyn cleavage products are increased with age in [A30P]αSyn and SynCAST(−), whereas the N-terminally derived fragments are almost stable over the time. Calpastatin overexpression decreases the αSyn cleavage resulting in the absence of one CT-αSyn fragment but also of NT-αSyn fragments. β-Actin was used as loading control. As an internal control recombinant soluble human [A30P]αSyn cleaved by calpain 1 was used. (B) Quantification was done by densitometric measurements and subsequent normalization of the protein level to β-actin. Error bars represent mean + SEM. Statistical significance was evaluated by one-way ANOVA where (**) indicates P < 0.01. (C) Truncation sites of αSyn derived from the brain stem of SynCAST(+), SynCAST(−) and [A30P]αSyn mice identified by nanoLC-ESI-MS/MS experiments. First amino acids of the N-terminal and last of C-terminal protein fragments are marked in the amino acid sequence.
Mass spectrometric identification of αSyn truncations in SynCAST(−) and SynCAST(+) mice
The mass spectrometric nanoLCESI-MS/MS experiments enabled unambiguous identification of 19 truncated forms of αSyn (Table 1, Fig. 3C). For the correct assignment of the αSyn truncation, we used a multistage strategy with three control instances. Hereto, we used (i) a database with all possible forms of αSyn truncations, combined with (ii) an additional search against a database containing all proteins known to be expressed in aged (19 months) mice to exclude false positive identifications for individual fragment ion spectra, and (iii) the comparison of the generated in vivo data with fragment ion analysis on recombinant [A30P]αSyn.
Table 1.
Terminal peptides of in vivo proteolyzed αSyn from mice brain stem identified by tandem mass spectrometry
| First (N)/last (C) amino acids | C- or N-terminal | Sequence | Charge (z) | m/z | Mascot score | SynCAST(+) mice* | SynCAST(−) mice* | [A30P]αSyn mice* | rec[A30P] αSyn |
|---|---|---|---|---|---|---|---|---|---|
| 12 | N | KEGVVAAAEKTK | 3 | 4109 | 43 | 8 | 5 | 22 | + |
| 14 | N | GVVAAAEKTK | 2 | 4873 | 42 | 88 | 44 | 56 | + |
| 15 | N | VVAAAEKTK | 2 | 4588 | 55 | 58 | 28 | 75 | + |
| 16 | N | VAAAEKTK | 2 | 4093 | 42 | 149 | 58 | 125 | + |
| 17 | N | AAAEKTK | 2 | 3597 | 52 | 161 | 59 | 39 | + |
| 50 | N | HGVATVAEK | 2 | 4562 | 78 | 156 | 98 | 213 | + |
| 56 | C | TKEGVVHGVATVA | 2 | 6344 | 46 | 89 | 75 | 69 | + |
| 66 | N | VGGAVVTGVTAVAQK | 2 | 6789 | 91 | 1.755 | 2.267 | 2.073 | + |
| 67 | N | GGAVVTGVTAVAQK | 2 | 6294 | 43 | 211 | 199 | 171 | + |
| 68 | N | GAVVTGVTAVAQK | 2 | 6009 | 68 | 695 | 6.660 | 2.088 | + |
| 69 | N | AVVTGVTAVAQK | 2 | 5724 | 85 | 1.595 | 1.868 | 2.629 | + |
| 72 | C | TKEQVTNVGGAVVT | 2 | 7018 | 46 | 111 | 97 | 209 | + |
| 79 | C | TKEQVTNVGGAVVTGVTAVAQ | 2 | 10151 | 80 | 39 | 32 | 69 | + |
| 80 | N | KTVEGAGSIAAATGFVK | 2 | 8040 | 132 | 24.620 | 40.500 | 14.343 | + |
| 82 | N | VEGAGSIAAATGFVKK | 2 | 7534 | 64 | 329 | 528 | 296 | + |
| 83 | N | EGAGSIAAATGFVKK | 2 | 7039 | 66 | 2.217 | 3.038 | 1.801 | + |
| 84 | N | GAGSIAAATGFVK | 2 | 5753 | 52 | 190 | 351 | 182 | + |
| 87 | N | SIAAATGFVK | 2 | 4828 | 43 | 217 | 271 | 206 | + |
| 94 | C | TVEGAGSIAAATGF | 2 | 6264 | 87 | 739 | 1.546 | 776 | + |
Amino acid (of identified peptide). *, precursor ion intensities/1000 normalized with progenesis LC-MS; +, used as positive control.
Using this strategy, we identified that the amphipathic repeat region of αSyn was affected by seven truncations. Six of them occurred within the N-terminal domain yielding protein fragments starting with amino acids 12, 14–17 and 50, and 1 was a C-terminal fragment ending at position 56. The hydrophobic NAC region was affected more often as indicated by nine N-terminal and three C-terminal fragments of the protein cleaved between amino acids 66 and 94. Presumable due to the enhanced calpain activity, we found marked elevations of protein fragmentation after amino acids 67 and 79–83 for the SynCAST(−) mice. Proteolysis of the acidic part was not detectable by this mass spectrometric setup in the investigated in vivo mouse models. It was only detectable with our quality control approach using recombinant [A30P]αSyn after tryptic digests of the previously in vitro calpain 1 cleaved protein.
Overexpression of CAST leads to a decreased number of cellular inclusions and prevents gliosis
To verify the effect of calpastatin modulation and thus calpain activation in SynCAST(−) and cast(−/−), as well as calpain inactivation in SynCAST(+) and cast(+/+) single transgenic mice on protein aggregation, we performed sequential protein extraction of the brain stem to detect insoluble αSyn by western blotting. We found a clear reduction in higher molecular weight immunosignals of aggregated αSyn in SynCAST(+) mice (Fig. 4A). We also stained brain sections with Thioflavin S (Fig. 4C; a–d), an amyloid dye that binds beta-sheet-rich amyloid structures (arrows). Since aggregated αSyn is PK resistant, we also digested slices with PK prior immunohistochemical staining with an antibody specific to human αSyn, thereby monitoring inclusions (Fig. 4C; e–l; arrowhead). Measuring the aggregate load revealed that the inactivation of calpain in SynCAST(+) leads to a reduced aggregation (Fig. 4D–G). Both, the %-area positive for Thioflavin S (Fig. 4D) and human αSyn (Fig. 4E), as well as the cellular inclusions (%) positive for Thioflavin S (Fig. 4F) and human αSyn (Fig. 4G) were significantly decreased by co-overexpression of calpastatin in SynCAST(+) mice.
Figure 4.
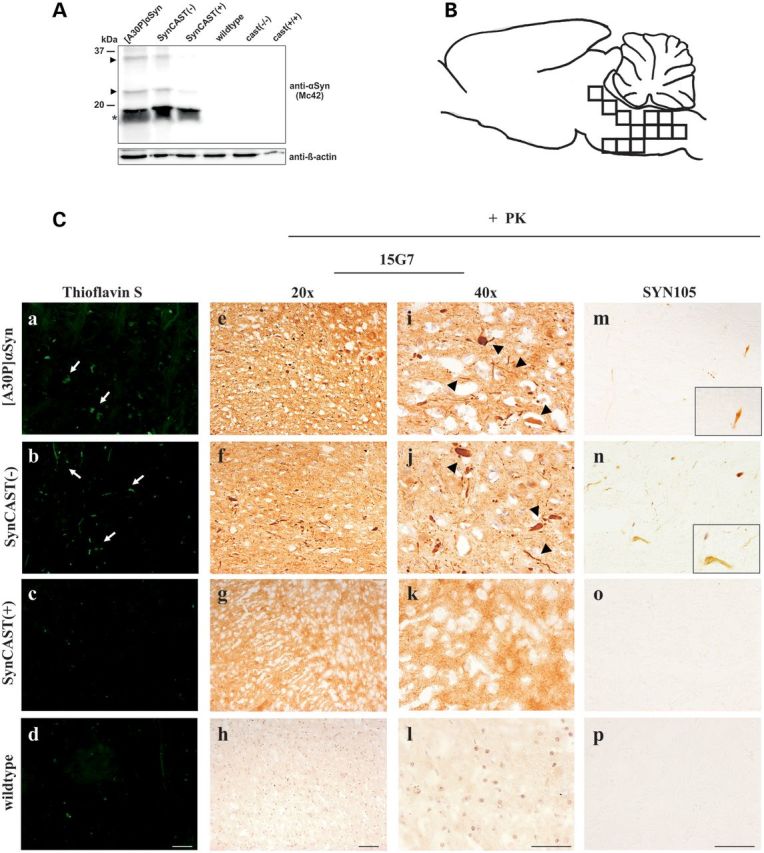

Decreased aggregate load in aged SynCAST(+) mice. (A) Brain stem proteins of 19-month-old animals were sequentially extracted as described under Materials and Methods. In the last step, the pellet was solubilized in 8 m urea plus 5% SDS. Overexpression of calpastatin results in a slight decrease in the insoluble full-length αSyn (asterisk) and a distinct decrease in high molecular weight signals of insoluble αSyn (arrowheads). (B) Schematic diagram of the mouse brain indicating the counted areas. For each slice, 12 pictures were taken across the entire brain stem. (C) Decreased numbers of aggregates were found in SynCAST(+) mice. (a–d) Brain slices were incubated with amyloid dye Thioflavin S that specifically labels β-pleated protein conformation (e–l). Sections were digested with PK and then incubated with an antibody against human αSyn (15G7). Area and cellular inclusions positive for both Thioflavin S (D and F) and PK-resistant αSyn (E and G) were significantly decreased after overexpression of human calpastatin. (m–p) Similarly, brain slices of old mice were digested with PK and were incubated with the SYN105 antibody against human αSyn cleaved by calpain. The images of the indicated genotypes revealed a positive staining for [A30P]αSyn and SynCAST(−) mice. No calpain cleaved αSyn was detected in mice overexpressing human calpastatin (SynCAST(+)). (D–G) Quantification of aggregates in the brain stem indicating a reduced aggregate load in SynCAST(+) mice. Error bars representing mean + SEM. Statistical significance was evaluated by one-way ANOVA where (***) indicates P < 0.001. Scale bar = 50 µm.
To confirm that calpain is indeed playing a role in the cleavage of αSyn in our genetically modified mouse models of PD, we used an antibody specific for human calpain-cleaved αSyn (SYN105, 42) in immunohistochemistry after PK digestion. The SYN105 antibody, kindly provided by Dr Masliah (University of California, San Diego, USA), has a strong preference to calpain-cleaved αSyn at amino acids 122 (Fig. 1A) (46, 47). SYN105 did not detect aggregates in SynCAST(+) mice at all, but showed positive aggregates in SynCAST(−) as well as in [A30P]αSyn mice (Fig. 4C; m–p). These data indicate that calpain is an important factor that promotes cleavage and aggregation of αSyn. Furthermore, we showed for the first time that calpain is already activated and cleaving αSyn in [A30P]αSyn single transgenic mice. Associated with reduced calpain activation, we also found a remarkable reduction in activated astrocytes in the brain stem of the SynCAST(+) compared with [A30P]αSyn and SynCAST(−) mice, as shown by the reduced levels of the astrocytic marker GFAP (Fig. 5). However, using TUNEL and toluidine staining, we found no indication for enhanced cell death in our models (unpublished data).
Figure 5.

Overexpression of calpastatin leads to decreased astrogliosis in SynCAST(+) mice. (A) Brain slices of 19-month-old mice were stained with the astrocytic marker GFAP and showed a decreased gliosis after calpastain overexpression. (B) Quantification of gliosis in 19-month-old mice revealed a significant decrease in GFAP reactivity in SynCAST(+) mice. Sign (#) represents the significance compared with [A30P]αSyn and the dollar sign ($) compared with SynCAST(−) mice. Error bars represent mean + SEM. Statistical significance was evaluated by one-way ANOVA where (***) indicates P < 0.001. Scale bar = 50 µm.
Overexpression of CAST ameliorates the CAST-depletion-dependent synaptic pathology
The presence of αSyn misfolding and truncations, known to interfere with SNARE complex assembly (48), prompted us to analyze synaptic proteins implicated in the vesicle cycle. In addition, calpains have not only been associated with neurodegeneration but are also involved in synaptic dysfunction (27, 30, 31, 49). To evaluate the modulatory effect of calpain on the expression level of synaptic markers, we isolated the synaptosomes with the Syn-PER Synaptic Protein Extraction Reagent (Thermo Scientific).
The total level of synaptic vesicle proteins synaptophysin, synapsin Ia/b and NSF, the latter involved in synaptic vesicle fusion, showed a significant reduction in SynCAST(−) compared with SynCAST(+) mice (Fig. 6A–D). The significant alteration of synapsin I (Fig. 6A and C) and NSF levels (Fig. 6A and D) between single-transgenic [cast (−/−)] and cast knockout mice [cast (+/+)] additionally points toward a role of calpain in vesicle cycle; and the specific reduction in synaptophysin to a synergistic effect in SynCAST(−) mice. This suggests that calpain may have a modulatory effect and contribute to a synaptic vesicle cycle defect in the present human [A30P]αSyn model of PD.
Figure 6.

Overexpression of calpastatin prevents reduction in synaptic markers. (A) Synaptosomal fraction of the brain stem of aged mice (n = 3) was used for western blot analysis and showed that loss of presynaptic proteins, like (B) synaptophysin, (C) synapsin Ia/b and (D) NSF, is reduced after calpastatin overexpression. Quantification was done by densitometric measurements and subsequent normalization of the protein level to β-actin. Sign (#) represents the significance compared with [A30P]αSyn mice, and the dollar sign ($) compared with SynCAST(−) mice. Error bars represent mean + SEM. Statistical significance was evaluated by one-way ANOVA where (**) indicates P < 0.01.
DISCUSSION
Different proteases, e.g. caspases, plasmin and MMPs, have been indicated in the cleavage of αSyn (3, 20, 21). However, increasing evidence clearly points toward a major role of calpains in several neurodegenerative diseases, including PD (26–29, 34, 50). Nonetheless, the mechanism and function of the truncation of αSyn by calpain is unclear due to conflicting results in the past. For instance, Dufty et al. (3) suggested that calpains play a role in the aggregation of αSyn, whereas Mishizen-Eberz et al. (32) concluded that calpain can degrade fibrillar αSyn and that cleavage near and within the middle region of soluble αSyn prevents its fibrillation. Certainly, there are more and more arguments that neurotoxicity and aggregation pathology in synucleinopathies is associated with the amount of CT-αSyn. Some of the indications are that CT-αSyn and CT-αSyn cleaved by calpain are detected in post mortem brain of PD patients and enriched in LBs (3). Furthermore, studies in mice showed that overexpression of the C-terminally truncated αSyn leads to pathological inclusions (3, 15, 16, 18, 51).
Here, we show that the co-overexpression of [A30P]αSyn and calpastatin, the only known endogenous inhibitor of calpains (30), leads to a reduction in the truncation of αSyn and reduced protein aggregation in SynCAST(+) mice, respectively.
In an in vitro assay, we provide evidence that [WT]αSyn and [A30P]αSyn have the same cleavage pattern and that only the fibrillation of αSyn has an influence on the cleavage pattern, which was detected by western blot analysis (Fig. 1B). These findings were similar to previously published in vitro studies (33).
As calpastatin acts as a buffering mechanism against involuntary protein cleavage during calpain activation (52) modulating constitutive calpain activation in vivo (30), we generated two contrary mouse models to investigate the long-term impact of calpain cleavage of αSyn as a model of the pathogenesis of PD. We either inactivated or activated calpain in a PD mouse model overexpressing human [A30P]αSyn (37) via overexpressing human calpastatin (30) or through the depletion of the mouse-endogenous calpastatin (35).
The studied single transgenic [A30P]αSyn mouse is a well-established model for α-synucleinopathy and was shown to form somatodendritic accumulations of insoluble αSyn through the brain and spinal cord (37). Upon aging, overexpressed human αSyn achieves a fibrillar amyloid conformation, observed by Thioflavin S and PK-resistant aggregated αSyn, that were mainly present in brain stem nuclei (36, 38). For that reason, our study focused on the impact of calpain on the pathogenesis of α-synucleinopathy in the brain stem.
We were able to modify the expression level of calpain 1 in both SynCAST(+) and SynCAST(−) mice demonstrated by immunoblots with an antibody that is targeting the autocleaved (75 kDa) as well as full-length (80 kDa) calpain 1 (Fig. 2A and B). Thus, the overexpression of calpastatin exhibits a reduced expression of activated calpain 1, whereas mice on a calpastatin-deficient background revealed increased protein levels of activated calpain 1 (Fig. 2A and B). An indicator of the activity of calpain in total is alpha-spectrin, a well-known substrate of calpain whose proteolysis results in a 145 kDa breakdown product that has been used previously as a quantitative measurement (30, 53). Indeed, the ratio of the 145 kDa calpain-cleaved and 240 kDa uncleaved spectrin substantiated a higher activity of calpain in SynCAST(−) mice, whereas the activity of calpain is significantly decreased in the SynCAST(+) animals compared with [A30P]αSyn (Fig. 2A and C). The correlation between the calpain expression level and activity is in agreement with a previous study on the impact of the calpain activation in a transgenic Alzheimer mouse model overexpressing calpastatin (29). Moreover, it was shown that calpain 1 is responsible for the C-terminally (amino acids 122) cleavage of αSyn that results in the formation of toxic fragments (3, 16, 32).
To investigate the impact of calpain on the proteolysis of αSyn in young and old animals, we performed immunoblot analysis and combined it to the use of two antibodies that permitted the detection C-terminally as well as N-terminally truncated αSyn species. The cleavage pattern was not age-dependent (Fig. 3A), but the level of the fragmented αSyn increased over the detected time period (Fig. 3A and B). In addition, SynCAST(+) mice displayed fewer fragments: N-terminal truncations were completely diminished in SynCAST(+) mice and a selective C-terminal truncated fragment (∼9 kDa) was not detectable anymore (Fig. 3). These data were confirmed by mass spectrometric analysis that allowed the identification of αSyn truncations within the N-terminus and NAC region, respectively (Table 1). However, cleavage in the C-terminal acidic region was not detected (Fig. 3C). Previous experiments on recombinant αSyn also showed that peptides cleaved after amino acids 113, 115, 119 or 122 were identifiable by mass spectrometry, but their ionization behavior is not well understood (54). These C-terminal peptides lack alkaline amino acids which act as a proton catcher for an efficient ionization process and it is most likely that this has an effect on their underrepresentation in the complex peptide mixture we used in this study.
In support of previous reports which indicated that the calpastatin–calpain system is associated with protein aggregation in neurodegenerative diseases (27–29, 55), we found a decreased aggregation load in SynCAST(+) compared with SynCAST(−) and single transgenic [A30P]αSyn mice by monitoring aggregates by the use of Thioflavin S, PK and detection of high molecular αSyn in the insoluble fraction (Fig. 4). Furthermore, we did not detect aggregates positive for calpain-cleaved αSyn in SynCAST(+) mice using an antibody targeting a major calpain cleavage site in human αSyn at amino acids 122 (32,46). All these findings are in line with previous in vitro data showing that calpain is cleaving soluble αSyn in particular in its N-terminal moiety, while fibrillar αSyn is mainly cleaved within its C-terminus (33) and is associated to a stronger aggregation phenotype (3, 15, 16, 18, 46, 51). In addition, the tendentiously increased calpain activity via overexpression of human [A30P]αSyn was further triggered by calpastatin depletion, as evidenced by calpain autocleavage, spectrin breakdown and the specific antibody SYN105 immunoreactivity. This may be explained by the fact that calpain is activated by an increased intracellular calcium concentration as a response to the overexpression of αSyn leading to oligomerization at synaptic membranes associating with an increased calcium influx (33, 56–58).
Both, the calcium-dependent protease calpain and αSyn, are localized at presynaptic terminals (33). Previous studies revealed that the overexpression of αSyn inhibits the release of neurotransmitters (59), which refer to the notion that αSyn aggregate-related synaptic dysfunctions cause neurodegeneration (60). Furthermore, N-terminal truncation and the A30P mutation are known to decrease synaptic targeting, while C-terminal truncation additionally increased the aggregation-dependent neurotoxicity in PD models (48). In addition, the level of GFAP, an indicator of astrogliosis and a classical marker for neuronal injury, associated with synaptic impairment in PD models (19, 47, 61, 62), is also increased after the activation of calpain 1 in injured spinal cord and prion-infected mice (63, 64). Recently, it was also shown that the passive immunization with a C-terminal antibody reduced the accumulation of calpain-cleaved αSyn in axons and synapses (55). Furthermore, overexpression of calpastatin as well as the passive immunization with an antibody against the C-terminus of αSyn (amino acids 118–126) prevents synapse loss and astrogliosis (29, 30, 55). Since we observed a calpain-dependent modulation of these markers, in particular the αSyn truncations (Fig. 4), reactive astrogliosis (Fig. 5) and the CAST-dependent depletion of synaptic markers (Fig. 6), were ameliorated in double transgenic SynCAST(+) mice, our data support the assumption that the calpain–calpastatin system plays an important role in aggregation and synaptic pathology in the present PD model. Alteration of synaptic marker between single transgenic cast (+/+) and cast (−/−) knockout mice may point toward a primary role of calpain in the synaptic vesicle cycle. Interestingly, cast (−/−) mice showed a significant depletion of synapsin I when compared with cast (+/+) and non-transgenic controls. Recently, Easley-Neal et al. (65) described a negative role of p25-hyperactivated cyclin-dependent kinase 5 (Cdk 5) related recruitment of synapsin I to the synapse. Therefore, the previously observed calpain-dependent aberrant activation of Cdk 5 and its known inhibition in CAST mice (66) may explain the observed alteration in synapsin I level. The additionally detected specific reduction in synaptophysin in SynCAST(−) mice, however, may relate to an adverse synergistic effect of A30P αSyn and calpain on synaptic integrity.
In summary, we show that αSyn is proteolytically cleaved by calpain in vivo and that a decreased cleavage of αSyn via calpain inhibition results not only reduced the aggregation load, but also preserved synaptic integrity and diminished astrocytosis. Therefore, the present study support a crucial role for abnormal calpain activation as well as subsequent increased truncation of αSyn in the pathogenesis of PD and offer an interesting target for prospective therapeutic approaches of synucleinopathies.
MATERIALS AND METHODS
Calpain cleavage of α-Synuclein in vitro
Recombinant wild-type ([WT]αSyn) and A30P αSyn ([A30P]αSyn) were expressed in Escherichia coli strain BL21 (DE3; Stratagene) and purified as described (67). Soluble WT or [A30P]αSyn were assembled into fibrils in assembly buffer (50 mm Tris–HCl, pH 7.5, 150 mm KCl) at 37°C, following continuous shaking at 600 rpm in an Eppendorf Thermomixer (Hamburg, Germany). Assembly was monitored using Thioflavin T binding and the fibrillar nature of the assembly was assessed after adsorption on carbon-coated 200-mesh grids, staining with 1% uranyl acetate and imaging by electron microscopy (Jeol 1400 transmission electron microscope; JEOL, Peabody, MA, USA).
The calpain cleavage assay was performed with minor changes as described previously (28, 32, 33). Briefly, 1.25 µg of recombinant soluble as well as fibrillar [WT]αSyn and [A30P]αSyn were incubated in a calpain reaction buffer containing 1000 ng calpain 1 at 37°C for 30 min. To verify the specific cleavage of αSyn by calpain, we utilized an inhibitor specific for calpain (ALLN, Sigma). The reaction was stopped by the addition of 5× Laemmli buffer and denaturation of samples at 95°C before western blot analysis.
Analytical ultracentrifugation
Sedimentation velocity measurements were carried out using an Optima XL-A ultracentrifuge (Beckman Coulter, Fullerton, CA, USA) equipped with UV-visible detection system using an AN60-Ti four-hole rotor and cells with two-channel 12 mm path length centerpieces. Soluble α-syn, 400 µl in 50 mm Tris–HCl, pH 7.5, 150 mm KCl, was spun at 50000 rpm (182000g) and 15°C. Sample displacement profiles were obtained by recording the absorbance at 280 nm every 5 min. Sedimentation coefficient continuous c(s) distribution was determined using the software Sedfit (68). The partial specific volume (0.7305 ml/g), the buffer viscosity (1.1534 cP) and density (1.00765 g/ml) were calculated with the software Sednterp. The sedimentation coefficient values were corrected to s20,w (standard solvent conditions in water at 20°C).
Generation of SynCAST(−) and SynCAST(+) mouse models
To investigate the role of calpain in the pathogenesis of PD in vivo, double transgenic mice (SynCAST(+)) were generated cross-breeding mice overexpressing human mutated [A30P]αSyn under the Thy-1 promoter (37, 38) with mice overexpressing human calpastatin (cast(+/+)) under the same promoter (Supplementary Material, Fig. S2; 30). To generate the opposite model, SynCAST(−), we crossed the same transgenic A30P mice into a calpastatin-deficient background (cast(−/−)) (Supplementary Material, Fig. S2; 35). Briefly, heterozygous [A30P]αSyn(+/−) mice were crossbred in both heterozygous cast-tg(+/−) and cast-ko(+/−) mice (P) to generate heterozygous SynCAST(+) ([A30P]αSyn(+/−)/cast-tg(+/−)), SynCAST(−) ([A30P]αSyn(+/−)/cast-ko(+/−)) (F1). Heterozygous SynCAST(+) or SynCAST(−) mice of the F1 generation were crossbred again to generate homozygous mice of all six lines: SynCAST(+), SynCAST(−), [A30P]αSyn, cast(+/+), cast(−/−) and wild-type (F2). To generate large cohorts of the same age, the homozygous F2 generation was crossbreed a last time into homozygous mice of the same generation (F3).
Calpastatin-depleted mice were identified by PCR with specific primers as described previously (35); calpastatin overexpression was validated by biochemistry and immunohistochemistry (Supplementary Material, Fig. S3). Both mice overexpressing human mutated [A30P]αSyn (forward: 5′-cagcagggacagtcagaag-3′; reverse: 5′-gaggtcgagctcaggttct-3′) and mice transgenic for human calpastatin (forward: 5′-tgtaggctccaaaaccaagg-3′; reverse: 5′- tgtcaggatccacaggcata-3′) were genotyped by qPCR with specific primers. Overexpression was confirmed by western blot analysis and immunohistochemistry (Supplementary Material, Fig. S3).
Protein extraction
Dissected mouse brain stems (n = 3) at the age of 3 and 19 months were homogenized either in TES (50 mm Tris, pH 7.5, 2 mm EDTA, 100 mm NaCl) or in TBS (pH 7.5, 10 mm Tris, 150 mm NaCl) buffer, supplemented with a cocktail of protease inhibitors (Complete; Roche Applied Science). Samples prepared with TES buffer were centrifuged at full speed for two times for 15 min. To analyze differences of the fragment pattern of soluble αSyn and the insoluble fraction between the six lines, proteins were sequentially extracted as described previously (69, 70). Briefly, brain stem was homogenized in TBS buffer, spun for 30 min at 120 000 g at 4°C and the supernatant representing the cytoplasmic fraction was collected. After three following steps, insoluble proteins were solubilized in 8 m urea plus 5% SDS. Owing to limited sample size, urea specimens of n = 3 animals were pooled to analyze insolubility of αSyn in brain stem region.
Synaptic protein extraction
To measure the expression level of different synaptic markers in the brain stem of aged mice (n = 3), we used the Syn-PER Synaptic Protein Extraction Reagent (Thermo Scientific) as described in the manual.
Immunoblot analysis
For immunoblot analysis, proteins were loaded on a SDS–PAGE and blotted afterward onto nitrocellulose membranes (Whatman). After blocking, the following primary antibodies were used: rabbit anti-αSyn panSyn (human/mouse; 1:100; Dianova) and rat anti-αSyn 15G7 (human; 1:50; Enzo Life Science), mouse anti-αSyn MC42 (1:3000; BD Transduction Laboratories), rabbit anti-calpain-1 Large Subunit (1:1000; Thermo Scientific), mouse anti-alpha-spectrin (1:500; Millipore), mouse anti-synaptophysin (1:500; Millipore), rabbit anti-synapsin 1/a/b (1:1000; Santa Cruz), rabbit anti-NSF (1:1000; Cell Signaling) and mouse anti-β-actin (1:10.000; Sigma). Secondary antibodies were either coupled to horseradish peroxidase (GE Healthcare) and diluted 1:3000 or labeled with a fluorescent dye (Li-Cor) and diluted 1:5000. Results were obtained for three animals per line each.
Mass spectrometric identification of αSyn truncations
Thirty micrograms of cytosolic whole brain stem protein lysates were electrophoresed via SDS–PAGE. After transfer, gel was silver stained, imaged (unpublished data) and sliced horizontally between 8 and 19 kDa. For silver staining, we used a method according to Jungblut and Seifert (71) with slight modifications to ensure mass spectrometric compatibility. Hereto, we used ethanol instead of methanol during fixation and incubation and we omitted glutaraldehyde in the incubation solution as well as formaldehyde in the silver nitrate solution. Gel slices were destained, rinsed and consecutively used for an overnight in-gel digest with trypsin. Peptides were extracted and after an amino acid analysis to determine the peptide concentration, 200 ng of the extracts were used for nanoLC-ESI-MS/MS protein identification experiments. Hereto, peptides were separated on an UltiMate 3000 RSLCnano HPLC system (Dionex, Sunnyvale, CA, USA). After loading the samples on a trap column (300 µm × 0.5 cm, particle size: 5 µm, pore size: 100 Å; Dionex) and washing for 5 min with 0.1% TFA, the peptides were separated on an analytical C18 column (75 µm × 25 cm, particle size: 2 µm, pore size: 100 Å; Dionex). The following solvents were used for separating the peptides with a flow rate of 400 nl/min: 0.1% formic acid (A); 84% ACN, 0.1% formic acid (B); HPLC-gradient: 4–40% B in 44 min, 40–95% B within 3 min and held at 95% B for further 5 min before the system was equilibrated for the next analysis. The HPLC system was connected to a nanoelectrospray ionization source (Thermo Fisher Scientific, USA) and ESI-MS/MS experiments were performed on a VelosPro (Thermo Fisher Scientific) ion trap mass spectrometer. MS spectra were scanned between 300 and 2000 m/z. The five most intensive ions (charge > 1) were selected for additional MS/MS fragmentation in the ion trap. Afterward, these ions were set on a dynamic exclusion list for 35 s to enable the analysis of ions with lower abundances. Fragments were generated by collision-induced dissociation on isolated ions with a collision energy of 35% and an activation time of 10 ms. The nano-LC-MS/MS data were transformed into MGF files for database searches with the Mascot® search algorithm (Matrix Science, London, UK, version 2.2.0), using a peptide mass tolerance of 0.4 Da and a fragment mass tolerance of 0.5 Da. For the protein identification, the enzyme settings were set on “trypsin” and up to two possible missed cleavage sites were considered for the database searches, as well as the following variable modifications: acetylation of protein N-terminus and oxidation of methionine. All data were searched against the Uniprot/Sprot database using a decoy strategy to exclude false positive identifications and additionally an in-house database consisting of the yeast proteome and all possible N-terminal and C-terminal truncations of αSyn was used to identify potential calpain cleavage products of this protein. Only peptide identifications with a Mascot score of >40 were considered for the analysis. Furthermore, the validity of the truncation corresponding peptide identifications was assigned by fragment ion spectra generated after tryptic digests of previously in vitro calpain cleaved recombinant [A30P]αSyn.
For the comparison of truncated αSyn in the brain of the different mouse models and to generate normalized precursor ion intensities adjusting differences in individual analysis, the nano-LC-MS/MS data were analyzed with the Progenesis LC-MS (V. 4.0) software.
Neuropathological analysis by Thioflavin S, PK and immunohistochemistry
Briefly, fixed brain tissue was incubated overnight in 30% sucrose and frozen at −20°C for 24 h. Brains were then embedded in tissue freezing medium (Leica Microsystems GmbH) and 7 µm sections were made with the cryomicrotome (JUNG CM3000, Leica). Beta-pleated protein conformations were labeled by incubating the brain sections with the amyloid dye Thioflavin S (0.01% in H2O; Sigma) for 5 min and washed three times for 1 min in 70% ethanol. PK digestion (Sigma) was carried out as described (72) and was followed by previously described immunohistochemistry (73). Briefly, after digestion with PK slices was incubated over night with the primary antibodies at following dilutions: rat anti- αSyn 15G7 (human) (1:25; 36) and SYN105 (1:200; 43). The next day, sections were preceded with the respective biotinylated secondary antibodies (1:200, Vector Laboratories) and visualized by using the Avidin-biotin (ABC) kit (Vector Laboratories) with diaminobenzidine tetrahydrochloride (DAB; Sigma) as the chromogen. For GFAP (1:100, Dako) and calpain 1 (1:250; Abcam), slices were not digested with PK and incubated with fluorescence-coupled antibody rabbit anti-Cy2 (1:250; Dianova). Cell death in the brain stem of 19 months old mice was analyzed with a terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay kit conjugated with TMR red (Roche Applied Science) as described in the manual.
Statistical analysis
Aggregates and neurodegenerative cells were quantified by acquiring 12 fields of the brain stem (Fig. 4B) with a 20× objective using an Axioplan 2 imaging microscope (Zeiss) equipped with an AxioCam HR color digital camera (Zeiss). For each genotype, three independent mice were used and two slices were analyzed each. The collected images were analyzed with Image J. Therefore, images were converted to an 8-bit format, an intensity threshold was set and the %-area positive for Thioflavin S, PK-resistant αSyn, GFAP and TUNEL was analyzed. Cellular inclusions positive for Thioflavin S and PK-resistant αSyn were counted manual by the Image J cell counter. Data were then analyzed by using GraphPad Prism by an one-way ANOVA with a significance threshold of *P < 0.05. All results are presented as mean + SEM.
SUPPLEMENTARY MATERIAL
FUNDING
The work was supported by the German National Genome Network to O.R. and K.M. (NGFNplus 01GS08134; German Ministry for Education and Research), ERA-Net NEURON - MIPROTRAN and the french “Agence Nationale de la Recherche”.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Huu Phuc Nguyen for organizing the calpastatin knockout mice and Jeannette Huebener for fruitful discussions. Katharina Stegen and Theresia Zuleger provided excellent technical support.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Takeda A., Mallory M., Sundsmo M., Honer W., Hansen L., Masliah E. Abnormal accumulation of NACP/alpha-synuclein in neurodegenerative disorders. Am. J. Pathol. 1998;152:367–372. [PMC free article] [PubMed] [Google Scholar]
- 2.Spillantini M.G., Schmidt M.L., Lee V.M., Trojanowski J.Q., Jakes R., Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 3.Dufty B.M., Warner L.R., Hou S.T., Jiang S.X., Gomez-Isla T., Leenhouts K.M., Oxford J.T., Feany M.B., Masliah E., Rohn T.T. Calpain-cleavage of alpha-synuclein: connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 2007;170:1725–1738. doi: 10.2353/ajpath.2007.061232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Braak H., Del Tredici K., Rüb U., de Vos R.A.I., Jansen Steur E.N.H., Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol. Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 5.Braak H., Ghebremedhin E., Rüb U., Bratzke H., Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. 2004;318:121–134. doi: 10.1007/s00441-004-0956-9. [DOI] [PubMed] [Google Scholar]
- 6.Irvine G.B., El-Agnaf O.M., Shankar G.M., Walsh D.M. Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson's disease: implications for pathogenesis and therapy. Proc. Natl Acad. Sci. USA. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uversky V.N., Li J., Fink a.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001;276:10737–10744. doi: 10.1074/jbc.M010907200. [DOI] [PubMed] [Google Scholar]
- 9.Zhu M., Fink A.L. Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem. 2003;278:16873–16877. doi: 10.1074/jbc.M210136200. [DOI] [PubMed] [Google Scholar]
- 10.Zhu M., Li J., Fink A.L. The association of alpha-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003;278:40186–40197. doi: 10.1074/jbc.M305326200. [DOI] [PubMed] [Google Scholar]
- 11.Iwai A., Masliah E., Yoshimoto M., Ge N., Flanagan L., de Silva H.A., Kittel A., Saitoh T. The precursor protein of non-A beta component of Alzheimer's disease amyloid is a presynaptic protein of the central nervous system. Neuron. 1995;14:467–475. doi: 10.1016/0896-6273(95)90302-x. [DOI] [PubMed] [Google Scholar]
- 12.Hong D.-P., Xiong W., Chang J.-Y., Jiang C. The role of the C-terminus of human α-synuclein: intra-disulfide bonds between the C-terminus and other regions stabilize non-fibrillar monomeric isomers. FEBS Lett. 2011;585:561–566. doi: 10.1016/j.febslet.2011.01.009. [DOI] [PubMed] [Google Scholar]
- 13.Bertoncini C.W., Jung Y.-S., Fernandez C.O., Hoyer W., Griesinger C., Jovin T.M., Zweckstetter M. Release of long-range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc. Natl Acad. Sci. USA. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lowe R., Pountney D.L., Jensen P.H., Gai W.P., Voelcker N.H. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004;13:3245–3252. doi: 10.1110/ps.04879704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li W., West N., Colla E., Pletnikova O., Troncoso J.C., Marsh L., Dawson T.M., Jäkälä P., Hartmann T., Price D.L., et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson's disease-linked mutations. Proc. Natl Acad. Sci. USA. 2005;102:2162–2167. doi: 10.1073/pnas.0406976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu C.-W., Giasson B.I., Lewis K.A., Lee V.M., Demartino G.N., Thomas P.J. A precipitating role for truncated alpha-synuclein and the proteasome in alpha-synuclein aggregation: implications for pathogenesis of Parkinson disease. J. Biol. Chem. 2005;280:22670–22678. doi: 10.1074/jbc.M501508200. [DOI] [PubMed] [Google Scholar]
- 17.Wakamatsu M., Ishii A., Iwata S., Sakagami J., Ukai Y., Ono M., Kanbe D., Muramatsu S., Kobayashi K., Iwatsubo T., et al. Selective loss of nigral dopamine neurons induced by overexpression of truncated human alpha-synuclein in mice. Neurobiol. Aging. 2008;29:574–585. doi: 10.1016/j.neurobiolaging.2006.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Ulusoy A., Febbraro F., Jensen P.H., Kirik D., Romero-Ramos M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci. 2010;32:409–422. doi: 10.1111/j.1460-9568.2010.07284.x. [DOI] [PubMed] [Google Scholar]
- 19.Tofaris G.K., Garcia Reitböck P., Humby T., Lambourne S.L., O'Connell M., Ghetti B., Gossage H., Emson P.C., Wilkinson L.S., Goedert M., et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1–120): implications for Lewy body disorders. J. Neurosci. 2006;26:3942–3950. doi: 10.1523/JNEUROSCI.4965-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim K.S., Choi Y.R., Park J.-Y., Lee J.-H., Kim D.K., Lee S.-J., Paik S.R., Jou I., Park S.M. Proteolytic cleavage of extracellular α-synuclein by plasmin: implications for Parkinson disease. J. Biol. Chem. 2012;287:24862–24872. doi: 10.1074/jbc.M112.348128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung J.Y., Park S.M., Lee C.-H., Um J.W., Lee H.J., Kim J., Oh Y.J., Lee S.-T., Paik S.R., Chung K.C. Proteolytic cleavage of extracellular secreted {alpha}-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005;280:25216–25224. doi: 10.1074/jbc.M503341200. [DOI] [PubMed] [Google Scholar]
- 22.Sevlever D., Jiang P., Yen S.-H.C. Cathepsin D is the main lysosomal enzyme involved in the degradation of alpha-synuclein and generation of its carboxy-terminally truncated species. Biochemistry. 2008;47:9678–9687. doi: 10.1021/bi800699v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ogawa K., Yamada T., Tsujioka Y., Taguchi J., Takahashi M., Tsuboi Y., Fujino Y., Nakajima M., Yamamoto T., Akatsu H., et al. Localization of a novel type trypsin-like serine protease, neurosin, in brain tissues of Alzheimer's disease and Parkinson's disease. Psychiatry Clin. Neurosci. 2000;54:419–426. doi: 10.1046/j.1440-1819.2000.00731.x. [DOI] [PubMed] [Google Scholar]
- 24.Iwata A., Maruyama M., Akagi T., Hashikawa T., Kanazawa I., Tsuji S., Nukina N. Alpha-synuclein degradation by serine protease neurosin: implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 2003;12:2625–2635. doi: 10.1093/hmg/ddg283. [DOI] [PubMed] [Google Scholar]
- 25.Oueslati A., Fournier M., Lashuel H.A. Role of post-translational modifications in modulating the structure, function and toxicity of alpha-synuclein: implications for Parkinson's disease pathogenesis and therapies. Prog. Brain Res. 2010;183:115–145. doi: 10.1016/S0079-6123(10)83007-9. [DOI] [PubMed] [Google Scholar]
- 26.Vosler P.S., Brennan C.S., Chen J. Calpain-mediated signaling mechanisms in neuronal injury and neurodegeneration. Mol. Neurobiol. 2008;38:78–100. doi: 10.1007/s12035-008-8036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J., Liu M.C., Wang K.K.W. Calpain in the CNS: from synaptic function to neurotoxicity. Sci. Signal. 2008;1:re1. doi: 10.1126/stke.114re1. [DOI] [PubMed] [Google Scholar]
- 28.Hübener J., Weber J.J., Richter C., Honold L., Weiss A., Murad F., Breuer P., Wüllner U., Bellstedt P., Paquet-Durand F., et al. Calpain-mediated ataxin-3 cleavage in the molecular pathogenesis of spinocerebellar ataxia type 3 (SCA3) Hum. Mol. Genet. 2012 doi: 10.1093/hmg/dds449. [DOI] [PubMed] [Google Scholar]
- 29.Liang B., Duan B.-Y., Zhou X.-P., Gong J.-X., Luo Z.-G. Calpain activation promotes BACE1 expression, amyloid precursor protein processing, and amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Biol. Chem. 2010;285:27737–27744. doi: 10.1074/jbc.M110.117960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rao M.V., Mohan P.S., Peterhoff C.M., Yang D.-S., Schmidt S.D., Stavrides P.H., Campbell J., Chen Y., Jiang Y., Paskevich P.a., et al. Marked calpastatin (CAST) depletion in Alzheimer's disease accelerates cytoskeleton disruption and neurodegeneration: neuroprotection by CAST overexpression. J. Neurosci. 2008;28:12241–12254. doi: 10.1523/JNEUROSCI.4119-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu H.-Y., Lynch D.R. Calpain and synaptic function. Mol. Neurobiol. 2006;33:215–236. doi: 10.1385/MN:33:3:215. [DOI] [PubMed] [Google Scholar]
- 32.Mishizen-Eberz A.J., Norris E.H., Giasson B.I., Hodara R., Ischiropoulos H., Lee V.M.-Y., Trojanowski J.Q., Lynch D.R. Cleavage of alpha-synuclein by calpain: potential role in degradation of fibrillized and nitrated species of alpha-synuclein. Biochemistry. 2005;44:7818–7829. doi: 10.1021/bi047846q. [DOI] [PubMed] [Google Scholar]
- 33.Mishizen-Eberz A.J., Guttmann R.P., Giasson B.I., Day G.A., Hodara R., Ischiropoulos H., Lee V.M.-Y., Trojanowski J.Q., Lynch D.R. Distinct cleavage patterns of normal and pathologic forms of α-synuclein by calpain I in vitro. J. Neurochem. 2003;86:836–847. doi: 10.1046/j.1471-4159.2003.01878.x. [DOI] [PubMed] [Google Scholar]
- 34.Crocker S.J., Smith P.D., Jackson-Lewis V., Lamba W.R., Hayley S.P., Grimm E., Callaghan S.M., Slack R.S., Melloni E., Przedborski S., et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson's disease. J. Neurosci. 2003;23:4081–4091. doi: 10.1523/JNEUROSCI.23-10-04081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takano J., Tomioka M., Tsubuki S., Higuchi M., Iwata N., Itohara S., Maki M., Saido T.C. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J. Biol. Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- 36.Freichel C., Neumann M., Ballard T., Müller V., Woolley M., Ozmen L., Borroni E., Kretzschmar H.A., Haass C., Spooren W., et al. Age-dependent cognitive decline and amygdala pathology in alpha-synuclein transgenic mice. Neurobiol. Aging. 2007;28:1421–1435. doi: 10.1016/j.neurobiolaging.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 37.Kahle P.J., Neumann M., Ozmen L., Muller V., Jacobsen H., Schindzielorz A., Okochi M., Leimer U., van Der Putten H., Probst A., et al. Subcellular localization of wild-type and Parkinson's disease-associated mutant alpha -synuclein in human and transgenic mouse brain. J. Neurosci. 2000;20:6365–6373. doi: 10.1523/JNEUROSCI.20-17-06365.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann M., Kahle P.J., Giasson B.I., Ozmen L., Borroni E., Spooren W., Müller V., Odoy S., Fujiwara H., Hasegawa M., et al. Misfolded proteinase K-resistant hyperphosphorylated alpha-synuclein in aged transgenic mice with locomotor deterioration and in human alpha-synucleinopathies. J. Clin. Invest. 2002;110:1429–1439. doi: 10.1172/JCI15777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conway K.a., Harper J.D., Lansbury P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998;4:1318–1320. doi: 10.1038/3311. [DOI] [PubMed] [Google Scholar]
- 40.el-Agnaf O.M.A., Irvine G.B. Aggregation and neurotoxicity of alpha-synuclein and related peptides. Biochem. Soc. Trans. 2002;30:559–565. doi: 10.1042/bst0300559. [DOI] [PubMed] [Google Scholar]
- 41.Volles M.J., Lansbury P.T. Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson's disease. Biochemistry. 2003;42:7871–7878. doi: 10.1021/bi030086j. [DOI] [PubMed] [Google Scholar]
- 42.Conway K.A., Harper J.D., Lansbury P.T. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- 43.Seidel K., Schöls L., Nuber S., Petrasch-Parwez E., Gierga K., Wszolek Z., Dickson D., Gai W.P., Bornemann A., Riess O., et al. First appraisal of brain pathology owing to A30P mutant alpha-synuclein. Ann. Neurol. 2010;67:684–689. doi: 10.1002/ana.21966. [DOI] [PubMed] [Google Scholar]
- 44.Langston J.W., Sastry S., Chan P., Forno L.S., Bolin L.M., Di Monte D.A. Novel alpha-synuclein-immunoreactive proteins in brain samples from the Contursi kindred, Parkinson's, and Alzheimer's disease. Exp. Neurol. 1998;154:684–690. doi: 10.1006/exnr.1998.6975. [DOI] [PubMed] [Google Scholar]
- 45.Jensen P.H., Nielsen M.S., Jakes R., Dotti C.G., Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J. Biol. Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- 46.Games D., Seubert P., Rockenstein E., Patrick C., Trejo M., Ubhi K., Ettle B., Ghassemiam M., Barbour R., Schenk D., et al. Axonopathy in an α-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated α-synuclein. Am. J. Pathol. 2013;182:940–953. doi: 10.1016/j.ajpath.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nuber S., Tadros D., Fields J., Overk C.R., Ettle B., Kosberg K., Mante M., Rockenstein E., Trejo M., Masliah E. Environmental neurotoxic challenge of conditional alpha-synuclein transgenic mice predicts a dopaminergic olfactory-striatal interplay in early PD. Acta Neuropathol. 2014 doi: 10.1007/s00401-014-1255-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burré J., Sharma M., Südhof T.C. Systematic mutagenesis of α-synuclein reveals distinct sequence requirements for physiological and pathological activities. J. Neurosci. 2012;32:15227–15242. doi: 10.1523/JNEUROSCI.3545-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nixon R.A., Saito K.I., Grynspan F., Griffin W.R., Katayama S., Honda T., Mohan P.S., Shea T.B., Beermann M. Calcium-activated neutral proteinase (calpain) system in aging and Alzheimer's disease. Ann. N. Y. Acad. Sci. 1994;747:77–91. doi: 10.1111/j.1749-6632.1994.tb44402.x. [DOI] [PubMed] [Google Scholar]
- 50.Gafni J., Ellerby L.M. Calpain activation in Huntington's disease. J. Neurosci. 2002;22:4842–4849. doi: 10.1523/JNEUROSCI.22-12-04842.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Daher J.P.L., Ying M., Banerjee R., McDonald R.S., Hahn M.D., Yang L., Flint Beal M., Thomas B., Dawson V.L., Dawson T.M., et al. Conditional transgenic mice expressing C-terminally truncated human alpha-synuclein (alphaSyn119) exhibit reduced striatal dopamine without loss of nigrostriatal pathway dopaminergic neurons. Mol. Neurodegener. 2009;4:34. doi: 10.1186/1750-1326-4-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Goll D.E., Thompson V.F., Li H., Wei W., Cong J. The calpain system. Physiol. Rev. 2003;83:731–801. doi: 10.1152/physrev.00029.2002. [DOI] [PubMed] [Google Scholar]
- 53.Czogalla a., Sikorski a.F. Spectrin and calpain: a “target” and a “sniper” in the pathology of neuronal cells. Cell. Mol. Life Sci. 2005;62:1913–1924. doi: 10.1007/s00018-005-5097-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmid A.W., Fauvet B., Moniatte M., Lashuel H.A. Alpha-synuclein post-translational modifications as potential biomarkers for Parkinson's disease and other synucleinopathies. Mol. Cell. Proteomics. 2013 doi: 10.1074/mcp.R113.032730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Masliah E., Rockenstein E., Mante M., Crews L., Spencer B., Adame A., Patrick C., Trejo M., Ubhi K., Rohn T.T., et al. Passive immunization reduces behavioral and neuropathological deficits in an alpha-synuclein transgenic model of Lewy body disease. PLoS ONE. 2011;6:e19338. doi: 10.1371/journal.pone.0019338. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Danzer K.M., Haasen D., Karow A.R., Moussaud S., Habeck M., Giese A., Kretzschmar H., Hengerer B., Kostka M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007;27:9220–9232. doi: 10.1523/JNEUROSCI.2617-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pieri L., Madiona K., Bousset L., Melki R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012;102:2894–2905. doi: 10.1016/j.bpj.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stefanis L. α-Synuclein in Parkinson's disease. Cold Spring Harb. Perspect. Med. 2012;2:a009399. doi: 10.1101/cshperspect.a009399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nemani V.M., Lu W., Berge V., Nakamura K., Onoa B., Lee M.K., Chaudhry F.A., Nicoll R.A., Edwards R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schulz-Schaeffer W.J. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lim Y., Kehm V.M., Lee E.B., Soper J.H., Li C., Trojanowski J.Q., Lee V.M.-Y. α-Syn suppression reverses synaptic and memory defects in a mouse model of dementia with Lewy bodies. J. Neurosci. 2011;31:10076–10087. doi: 10.1523/JNEUROSCI.0618-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nuber S., Harmuth F., Kohl Z., Adame A., Trejo M., Schönig K., Zimmermann F., Bauer C., Casadei N., Giel C., et al. A progressive dopaminergic phenotype associated with neurotoxic conversion of α-synuclein in BAC-transgenic rats. Brain. 2013;136:412–432. doi: 10.1093/brain/aws358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Du S., Rubin A., Klepper S., Barrett C., Kim Y.C., Rhim H.W., Lee E.B., Park C.W., Markelonis G.J., Oh T.H. Calcium influx and activation of calpain I mediate acute reactive gliosis in injured spinal cord. Exp. Neurol. 1999;157:96–105. doi: 10.1006/exnr.1999.7041. [DOI] [PubMed] [Google Scholar]
- 64.Gray B.C., Skipp P., O'Connor V.M., Perry V.H. Increased expression of glial fibrillary acidic protein fragments and mu-calpain activation within the hippocampus of prion-infected mice. Biochem. Soc. Trans. 2006;34:51–54. doi: 10.1042/BST0340051. [DOI] [PubMed] [Google Scholar]
- 65.Easley-Neal C., Fierro J., Buchanan J., Washbourne P. Late recruitment of synapsin to nascent synapses is regulated by Cdk5. Cell Rep. 2013;3:1199–1212. doi: 10.1016/j.celrep.2013.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sato K., Minegishi S., Takano J., Plattner F., Saito T., Asada A., Kawahara H., Iwata N., Saido T.C., Hisanaga S. Calpastatin, an endogenous calpain-inhibitor protein, regulates the cleavage of the Cdk5 activator p35 to p25. J. Neurochem. 2011;117:504–515. doi: 10.1111/j.1471-4159.2011.07222.x. [DOI] [PubMed] [Google Scholar]
- 67.Ghee M., Melki R., Michot N., Mallet J. PA700, the regulatory complex of the 26S proteasome, interferes with alpha-synuclein assembly. FEBS J. 2005;272:4023–4033. doi: 10.1111/j.1742-4658.2005.04776.x. [DOI] [PubMed] [Google Scholar]
- 68.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tofaris G.K., Razzaq A., Ghetti B., Lilley K.S., Spillantini M.G. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003;278:44405–44411. doi: 10.1074/jbc.M308041200. [DOI] [PubMed] [Google Scholar]
- 70.Casadei N., Pöhler A.-M., Tomás-Zapico C., Torres-Peraza J., Schwedhelm I., Witz A., Zamolo I., De Heer R., Spruijt B., Noldus L.P.J.J., et al. Overexpression of synphilin-1 promotes clearance of soluble and misfolded alpha-synuclein without restoring the motor phenotype in aged A30P transgenic mice. Hum. Mol. Genet. 2013 doi: 10.1093/hmg/ddt467. [DOI] [PubMed] [Google Scholar]
- 71.Jungblut P.R., Seifert R. Analysis by high-resolution two-dimensional electrophoresis of differentiation-dependent alterations in cytosolic protein pattern of HL-60 leukemic cells. J. Biochem. Biophys. Methods. 1990;21:47–58. doi: 10.1016/0165-022x(90)90044-d. [DOI] [PubMed] [Google Scholar]
- 72.Tanji K., Mori F., Mimura J., Itoh K., Kakita A., Takahashi H., Wakabayashi K. Proteinase K-resistant alpha-synuclein is deposited in presynapses in human Lewy body disease and A53T alpha-synuclein transgenic mice. Acta Neuropathol. 2010;120:145–154. doi: 10.1007/s00401-010-0676-z. [DOI] [PubMed] [Google Scholar]
- 73.Nuber S., Petrasch-Parwez E., Winner B., Winkler J., von Hörsten S., Schmidt T., Boy J., Kuhn M., Nguyen H.P., Teismann P., et al. Neurodegeneration and motor dysfunction in a conditional model of Parkinson's disease. J. Neurosci. 2008;28:2471–2484. doi: 10.1523/JNEUROSCI.3040-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.