

Abstract
Juvenile autoimmune hepatitis (JAIH) is a progressive inflammatory liver disease, affecting mainly young girls, from infancy to late adolescence, characterized by active liver damage, as shown by high serum activity of aminotransferases, by elevated immunoglobulin G levels, high titers of serum non organ-specific and organ-specific autoantibodies, and by interface hepatitis on liver biopsy. It is a multifactorial disease of unknown etiology in which environmental factors act as a trigger in genetically predisposed individuals. Two types of JAIH are identified according to the autoantibody panel detected at diagnosis: AIH-1, characterized by the presence of anti-smooth muscle antibody and/or antinuclear antibody and AIH-2, by anti-liver-kidney microsomal antibody type 1 and/or by the presence of anti-liver cytosol type 1 antibody. Epidemiological distribution, genetic markers, clinical presentation and pattern of serum cytokines differentiate the two types of AIH suggesting possible pathogenetic mechanisms. The most effective therapy for AIH is pharmacological suppression of the immune response. Treatment should be started as soon as the diagnosis is made to avoid severe liver damage and progression of fibrosis. The aim of this review is to outline the most significant and peculiar features of JAIH, based largely on our own personal database and on a review of current literature.
Keywords: Juvenile autoimmune hepatitis, Autoimmune hepatitis, Autoantibodies, Autoimmune liver disease, Chronic hepatitis, Acute liver failure
Core tip: Juvenile autoimmune hepatitis is an inflammatory liver disease affecting mainly young girls from infancy to late adolescence, characterized by active liver damage, elevated immunoglobulin G levels, high titers of serum non organ-specific and organ-specific autoantibodies, and interface hepatitis on liver biopsy. Two types are identified according to the autoantibody panel, with differences in the epidemiological distribution, genetic markers and clinical presentation. The most effective therapy for autoimmune hepatitis is pharmacological suppression of the immune response. Treatment should be started as soon as the diagnosis is made to avoid severe liver damage and progression of fibrosis.
INTRODUCTION
Autoimmune hepatitis (AIH) results from an autoimmune attack of the liver parenchyma. The term “autoimmune hepatitis” was first employed by Mackay et al[1] in 1965 to describe “a persistent liver disease, with highly elevated levels of serum transaminase, sometimes over 1000 units, elevated serum gamma globulins, up to 6.0 g per 100 mL, piecemeal necrosis on liver biopsy with diffuse lymphoid infiltration and fibrosis, progressing to cirrhosis, various autoantibodies reactions in the serum, improving with immunosuppressive drugs”. The pattern of the serum autoantibodies that characterize patients with AIH led to a classification of this disease[2,3].
In children, after anecdotal reports of cases of chronic hepatitis and hypergammaglobulinaemia in the sixties[4], it was clearly shown that about half of the children with the histological features of chronic active hepatitis had hypergammaglobulinemia and high titers of serum autoantibodies[5] and that these patients responded in most cases to prednisone and azathioprine treatment, therefore suggesting an autoimmune mechanism[6,7]. In the same period a peculiar form of autoimmune hepatitis, now called AIH-2 was described as a distinct entity in children[8] and later confirmed in adults[9].
The aim of this review is to outline the most significant and peculiar features of juvenile AIH (JAIH), based largely from our own personal database; additionally we searched PubMed with the term of “juvenile autoimmune hepatitis”, “autoimmune hepatitis”, “epidemiology”, “pathogenesis” and “treatment”, filtered for age “birth-18 years”.
DEFINITION
AIH is a liver disease of unknown origin, pathogenetically characterized by an inflammatory liver disease, as shown by high serum activity of aminotransferases, by elevated immunoglobulin G levels, high titers of serum non organ-specific and organ-specific autoantibodies, and by interface hepatitis on liver biopsy[10]. It affects mainly young girls and spontaneously progresses to severe liver damage. Immunosuppressive therapy, which should be started as soon as diagnosis is made, induces clinical and biochemical remission in most treated patients. If untreated, cirrhosis and terminal liver failure may rapidly occur[6].
EPIDEMIOLOGY
AIH can be diagnosed at any age in both sexes. Mean annual prevalence in European adults ranges from 11.6 per 100 individuals to 17 per 100[11,12] with point prevalence in homogeneous populations, such as Alaskan natives, of 42.9 per 100 individuals[13]. Epidemiological data on JAIH are largely incomplete. There is only one recent report on the incidence and prevalence of JAIH, which was conducted in Utah, United States. In this study, the incidence and prevalence of JAIH was reported to be 0.4 and 3.0 cases per 100000 children, respectively[14]. AIH-1 is the more common type of AIH, which also affects adults and often presents at puberty, while AIH-2 is typical of pediatric age, presenting at a younger age, and even during infancy[8,15].
PATHOGENESIS
AIH is a multifactorial disease of unknown etiology. Environmental factors act as a trigger with self-perpetuating liver inflammation in predisposed individuals who carry a complex genetic background. Moreover, a defective immunoregulatory function, possibly genetically related, fails to control autoreactive clones and let the disease become clinically evident. The histological picture of interface hepatitis, in which a mononuclear and plasma cell infiltrate, which originates in the portal tracts, and disrupts the parenchymal limiting plate, morphologically illustrates this process. Among the inflammatory cells, activated T lymphocytes, positive for the CD4+ helper/inducer phenotype, predominate. These cells are believed to recognize self-antigens on the hepatocyte surface and to trigger the autoimmune liver damage[16].
Genetics
Main susceptibility HLA alleles for AIH-1 in Europe and North America were found to be DRB1*0401 and DRB1*0301. The presence of these alleles confers an increased risk of developing AIH-1 and influences some features of the disease[17]. Geographic variation of the genetic predisposition to AIH-1 exists: in some countries such as Japan, Mexico and Argentina, DR3 haplotype is poorly represented in the general population, and the principal susceptibility alleles for AIH-1 are DRB1*0404 and DRB1*0405[18-20]. European children display the typical pattern for AIH-1 of Caucasian patients with a significant prevalence of DRB1*0301 and DRB3*0101[21].
Knowledge of the genetic background of AIH-2 is limited. In Europe, DRB1*03 and DQB1*02 alleles may have an important role, whereas other studies reported an increased frequency of DRB1*07, DRB4*01 and DQB1*06. In a pediatric population from Brazil, a significant increase of DRB1*07, DRB4 and DQB1*02 was observed. Moreover, HLA-DRB1*07 allele was found significantly associated with the presence of anti-liver-kidney microsomal antibody type 1 (LKM-1) alone and HLA-DRB1*03 allele with anti-liver cytosol type 1 antibody (LC-1)[22-24].
A partial deficiency of HLA class III complement component C4, genetically determined, has been associated with JAIH[25].
Environmental factors
A number of drugs may cause unpredictable, dose-independent, immune-mediated liver damage. Autoimmune hepatitis related to halothane, tienilic acid, dihydralazine and minocycline are typically associated with LKM autoantibodies even though the molecular targets are different from AIH-2 (i.e., CYP2E1 for halothane and CYP2C9 for tienilic acid)[26].
Several viruses have been proposed as triggering factors for AIH such as HAV, measles, EBV or HSV, based on clinical or epidemiological criteria[27]. CYP2D6, the specific target of LKM-1 antibodies, shows epitopes that cross-react with homologous region of HCV, CMV and HSV[28]. Although definite evidence supporting this mechanism is lacking, it is conceivable that, infections with otherwise common viruses might lead, within a permissive genetic background, to break tolerance to self-antigens like CYP2D6, which could also be expressed, under particular conditions, on the hepatocyte surface[29].
Autoimmune reaction as a defect of regulatory function
A defect in a subpopulation of T lymphocytes regulating the immune response to liver- antigens expressed on the hepatocyte membrane has been reported in patients with AIH-1[30]. This T-cell subpopulation, bearing the interleukin 2 receptor α-chain (CD25+) and known as functional regulatory T-cells (T-regs), has been extensively studied as the putative main subset of regulatory cells for immune tolerance maintenance. In AIH patients, T-regs lymphocytes were found to be defective in number[16]. Moreover, functional studies have suggested that these cells are defective in promoting secretion of regulatory cytokines by their targets and in regulating CD4+ and CD8+ T-cell proliferation and interferon-gamma production and that they are unable to restrain monocyte activation and function[16]. However, using different markers, such as FOXP3, to identify T-regs lymphocytes, their role, in AIH, as the main immunoregulatory cells was recently challenged[31,32].
In AIH-2, the principal autoantigen (CYP2D6) is known, and the dominant epitopes target of the B and T-cell immune responses are also well characterized. On this basis, generation and expansion of HLA-restricted specific T-reg lymphocytes has been attempted and their immunomodulatory properties have been described in vitro[33]. Targeted immunotherapy with autologous infusion of ex-vivo expanded T-regs was demonstrated to induce remission of experimental AIH of mice[34].
Animal models
Advances in understanding the pathogenesis of AIH has been limited by the lack of accurate animal models. Murine models have been generated through DNA immunization with a chimeric fusion protein containing human CYP2D6 and human forminotransferase cyclodeaminase, the two self antigens of type 2 AIH, together with the extracellular region of mouse, cytotoxic T-lymphocyte antigen 4, as an immunological modulator[34,35]. Another model for AIH-2 uses CYP2D6 transgenic mice and tolerance mechanisms are overrun with the use of an adenovirus-CYP2D6 vector[36]. Immunized or infected mice developed chronic histological changes in the liver close to interface hepatitis, resembling those of AIH, with the development of a specific immune response with the production of anti-LKM1 and anti-LC-1 antibodies. A third animal model was created without the use of active immunization against xenopeptides, but using a transgenic mouse expressing chicken ovalbumin on the hepatocyte surface[37].
CLINICAL FEATURES
A specific autoantibody panel identifies two types of AIH: the presence of anti-smooth muscle antibody (SMA) and/or antinuclear antibody (ANA), in AIH-1[21,38], and LKM-1 and/or LC-1, in AIH-2[8,39]. Epidemiological distribution, genetic markers, clinical presentation and pattern of serum cytokines differentiate the two types of AIH suggesting possible pathogenetic mechanisms[40]. AIH-1 presents at any age, from infancy to the elderly, and in both sexes, while AIH-2 presents almost exclusively in childhood, with a very high incidence in females[8,39]. Patients with AIH-2 present at younger age that AIH-1, and are at higher risk to develop an acute liver failure[41]. Hypergammaglobulinemia is common in AIH-1, but it can be absent in AIH-2[8,38]. Moreover, AIH-2 is almost never associated with evidence of bile duct lesions while bile duct lesion is commonly observed in AIH-1[38]. Extra hepatic diseases of autoimmune mechanism are frequently observed in patients with both types of AIH. Autoimmune thyroid diseases (Grave’s and Hashimoto diseases) and autoimmune skin diseases such as vitiligo or alopecia are more frequently observed in AIH-2[8,38].
Three patterns of clinical onset characterize JAIH: (1) Acute onset with anorexia, nausea, vomiting and abdominal pain followed by jaundice, eventually suggesting an acute viral hepatitis, is the most frequent. In particular, patients, with AIH-2, are at higher risk than AIH-1 to develop acute liver failure with encephalopathy; (2) Insidious onset with progressive fatigue, anorexia, and intermittent jaundice lasting for several months/years before diagnosis, can be observed in about a third of patients. All these patients have clinical evidence of chronic liver disease and/or of cirrhosis at diagnosis; and (3) About 10% of patients may be asymptomatic when the liver disease is serendipitously discovered by the finding of clinical signs of chronic liver disease or by an increase of aminotransferase activity.
In a few patients, JAIH may reveal itself with symptomatic portal hypertension or with symptoms related to an extrahepatic autoimmune disease such as autoimmune thrombocytopenia, autoimmune haemolytic anemia, diabetes type 1, autoimmune thyroiditis, vitiligo, cutaneous vasculitis, uveitis, glomerulonephritis, juvenile chronic arthritis, systemic lupus erythematosus, Sjögren’s syndrome, celiac disease and inflammatory bowel disease[8,15,38].
LABORATORY FEATURES
At diagnosis, the “activity” of the liver disease can be documented by the presence of an almost constant increase of liver enzyme, in particular of serum transaminase activity that may increase up to 50 times or more the upper normal limit, while gamma glutamyltransferase (GGT) activity may be normal or only slightly elevated. An increase of GGT should suggest bile duct damage as in the case of autoimmune hepatitis/cholangitis overlap syndrome. Serum gamma globulins and immunoglobulins G are usually elevated, sometimes markedly, up to 6-8 g/L. Serum albumin may be normal in absence of liver function impairment and ascites. Serum immunoglobulin A deficiency and/or genetically determined low levels of C4 can be observed in AIH-2[8,25]. Prolonged prothrombin time suggests severe liver function impairment.
Autoantigens and autoantibodies
Recognition of pathogenetic autoantigens in AIH might be one of the key factors to develop an etiologic-based therapy. Unfortunately most of the antigens recognized by autoantibodies detected in AIH are either non organ-specific or intracellular molecules, unlikely involved in triggering autoimmune reaction. The most studied candidate autoantigens are the asialoglycoproteins receptor (ASGP-R) for type AIH-1 and the cytochrome P4502D6 (CYP2D6) for AIH-2.
The ASGP-R is an organ-specific antigen expressed in the hepatocyte membrane. Even if several experimental studies had been published, its role in pathogenesis of AIH is still controversial[42]. Both peripheral and infiltrating lymphocytes collected from adult and pediatric patients with AIH show a proliferative response to human ASGP-R[43], and a lack in T-suppressing function of CD4+ T-cells specific for ASGP-R and corrigible by immunosuppressive therapy, has been described both in patients and in their healthy relatives.
Seven isoforms of cytochrome P450 are expressed in human liver and all of these isoforms are targets of LKM reactivity in different types of autoimmune, viral or drug induced liver disease. CYP2D6 is an intracellular enzyme active in detoxification of several drugs and is the molecular target of AIH-2[44]. By effect of some cytokines, CYP2D6 can be expressed on hepatocytes surface becoming a potential target for autoreactive T-cells[29].
Detection of serum non-organ-specific autoantibodies (ANA, SMA and LKM-1) known to be associated with autoimmune liver diseases is a critical component of diagnostic criteria developed by International Autoimmune Hepatitis Group[2,3]. Their assessment should preferably be performed by indirect immunofluorescence (IIF) on frozen section of rat liver, kidney and stomach, and the presence of ANA/SMA and LKM-1 is virtually mutually exclusive. Sera screened positive for ANA/SMA should be further examined to assess the pattern of nuclear staining by the use of HEp2 cell monolayers, or to define the target of the SMA reaction[45].
The autoantibody profile does not markedly vary in the course of AIH with the exception of ANA reactivity that can be detected “de novo” in both subgroups of AIH. Autoantibodies titers varies during the course of the disease usually reducing in titer in case of remission, but also independently[46,47].
Autoantibody titers are not predictive of biochemical or histological remission. High titers at onset do not suggest a more aggressive disease and their disappearance from serum is not predictive of a better disease control during treatment or of a sustained remission in case of discontinuation of treatment.
Antinuclear and anti smooth muscle antibody, the serological hallmark of AIH-1, are usually present at high (≥ 1:100) titer, but they are not specific of AIH. ANA and SMA can, in fact, be detected in other liver diseases (viral or drug induced hepatitis, steatohepatitis and hepatocellular carcinoma) and also in non-hepatic disorders, however at lower titers.
Various patterns of ANA staining can be observed: homogeneous (60%) and speckled (15%-25%) are the most frequent, however they are not considered of clinical importance and they may vary in the same patient, during treatment. Several nuclear antigens have been identified, as a target of ANA reactivity: single and double-stranded DNA, histones, chromatin, ribonucleoprotein complexes, cyclin A and centromere, but no single AIH specific antigen has been detected so far. In AIH-1, ANA can be detected either alone or in conjunction with SMA. In children, ANA is considered positive when the titer is ≥ 1:40, however since ANA reactivity at low titer can be frequently found in children we suggest raising the positivity cut-off to at least to 1:100. Moreover, Anti dsDNA antibodies can be detected in 25% of ANA-reactive AIH-1 patients[46].
When using rat stomach as substrate for SMA: uniform IIF stain of the muscolaris mucosa, blood vessels walls (V) and parietal cell occurs. With rat kidney tissue, staining of the mesangial area of glomeruli (G) and of proximal renal tubular cells (T) also occurs. “VG” and “VGT” staining patterns are the most frequent IIF patterns encountered in AIH. SMA reactivity usually stains structural components of the cytoskeleton such as desmin and troponin. In AIH SMA reactivity is directed against filamentous (F) actin. Anti-F-actin can be detected using cultured human fibroblast or HEp2 cells. Anti-F-actin specificity is higher than SMA but anti-F-actin antibodies may be found also in viral infection, connective tissue disease and celiac disease.
Anti-LKM-1 serum reactivity defines the AIH-2, the most common type of JAIH occurring in infancy and childhood[8]. LKM-1 are present in 30%-70% of sera of patients with AIH-2 with anti-LC-1 antibody. Occasional patients with both ANA and LKM-1 have been defined as AIH-2.
LKM-1 stains hepatocytes and the proximal renal tubular cells (P3 portion) of liver and kidney sections in mice. Occasional staining of the distal renal tubules usually generates confusion with anti-mitochondrial autoantibody (AMA). AMA positivity in children is rare and Primary Biliary Cirrhosis is exceptional in pediatric age.
Anti-LC-1 is an organ-specific antibody, which homogeneously stains, in IIF, the cytoplasm of the hepatocytes, sparing the perilobular layer of central veins and without staining of the proximal renal tubules[48]. LC-1 can also be detected with both immunodiffusion and immunoblotting. LC-1 antibody reacts with forminotransferase cyclodeaminase, a 58-62 Kd liver specific antigen[49] and together with LKM-1, characterizes AIH-2. In fact LC1 reactivity can be found associated with LKM-1 in about 50% of AIH-2, but LC-1 may characterize on its own, as a sole autoantibody children with AIH-2[39].
The simultaneous presence of LKM-1 may obscure LC-1 IIF reactivity. In these cases it is necessary to use another method to detect the presence of LC-1 such as immunodiffusion, ELISA, Western blot or dot blot.
Anti-SLA is a non organ-specific antibody which target antigen is likely to be a 50 Kd protein identified as O-phosphoseryl-tRNA: selenocysteinyl-tRNA synthase. Anti-SLA is considered a specific marker for AIH-1 being present in 6% to 58% of adults and children with AIH-1, alone or in combination with SMA and/or ANA[50]. Its detection could be particularly useful in patients who are negative for conventional markers of the disease (ANA, SMA), but its diagnostic role in JAIH is not relevant.
Anti-human ASGPR is a species-specific, liver-specific autoantibody that can be detected in sera of patients with various inflammatory liver diseases, but predominantly in AIH. The absence of a commercialized assay restricts its use to few laboratories.
Anti-neutrophil cytoplasmic antibodies (ANCA) constitute a heterogeneous group of autoantibodies directed against various subcellular components of neutrophils or myeloid cells and their presence has been proven to be a reliable diagnostic tool in systemic vasculitis. They are routinely detected by IIF on ethanol fixed human neutrophils and commonly classified in cytoplasmic (cANCA), perinuclear (pANCA) and atypical (pANNA). Atypical pANCA are characterized by non-homogeneous labeling of the nuclear periphery together with multiple intranuclear fluorescent staining and have been reported in patients with autoimmune liver disease including sclerosing cholangitis associated with inflammatory colitis and in AIH-1.
LIVER HISTOLOGY
The International Autoimmune Hepatitis Group has affirmed the role of liver biopsy for the diagnosis of AIH[2,3]; liver biopsy is thus recommended in all patients suspected AIH unless there is a significant contraindication[51]. The histological hallmark of AIH is “interface hepatitis” (formerly called piecemeal necrosis) (Figure 1). A considerable amount of eosinophilic granulocytes can be observed within the portal infiltrate, especially in such cases associated to celiac disease[52].
Figure 1.
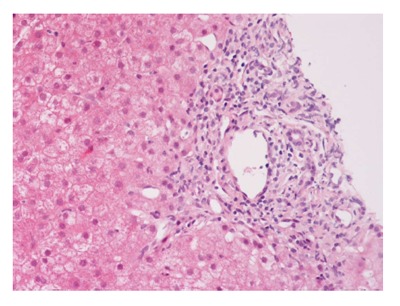
Interface hepatitis with piecemeal necrosis and lymphocyte spillover across the limiting plate.
In patients with AIH presenting as an acute liver disease, liver histology allows for differentiation between spontaneous exacerbation of a chronic liver disease (“acute-on-chronic”) and a newly developed disease. In the latter case, centrilobular zone 3 necrosis is the most typical pattern[53]. Subsequent transition to the classic features of “interface hepatitis” usually occurs. Other possible liver biopsy findings in AIH include the presence of giant multinucleated hepatocytes[54]. Moreover, diffuse giant cell transformation characterizes a distinct form of AIH in infants, associated with autoimmune hemolytic anemia[55] (Figure 2).
Figure 2.
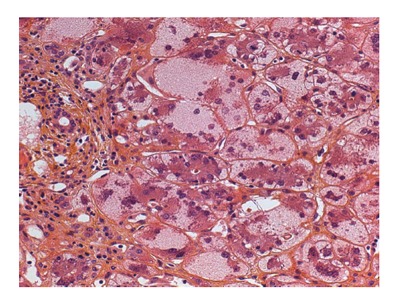
Liver biopsy of a 6 mo old infant with autoimmune hemolytic anemia showing diffuse giant cell transformation and moderate inflammatory portal infiltrate.
Massive liver cell necrosis may be present in AIH with acute severe/fulminant onset and may be associated with bridging necrosis and/or with multilobular or panlobular necrosis. These histological findings support but do not constitute firm evidence for the diagnosis of AIH.
Biliary ducts are usually not affected in AIH and the presence of lymphocytic cholangitis, or that of a mixed inflammatory infiltrate surrounding and infiltrating the bile ducts, has received a negative diagnostic rating on the IAIHG diagnostic score[3]. However, the incidental presence of bile duct inflammatory changes has been recognized in patients with AIH responding to immunosuppressive treatment[56]. In children, bile duct inflammatory changes are common in AIH-1, but are very rare or absent in AIH-2.
Liver biopsy also provides information on prognosis, identifying the presence of cirrhosis. Cirrhosis may be present at diagnosis or rapidly develop in JAIH[57]. Cirrhosis is more frequent at diagnosis in AIH-1 than in AIH-2[58], however, concerning the diagnosis of cirrhosis, “blind” percutaneous liver biopsy has been demonstrated to be of low diagnostic sensitivity, since up to 50% of patients may not be correctly diagnosed[57].
DIAGNOSIS
JAIH has variable clinical manifestations and should be considered in the diagnostic work-up of any patient with a cryptogenic liver disease. Diagnosis of JAIH basically relies on the exclusion of other possible known causes of the hepatic disease, such as chronic viral infections and Wilson’s disease and by clinical biochemical and histological “positive” criteria. Diagnosis is not challenging when all the major clinical and biochemical elements of the disease are present, such as the occurrence of an autoimmune disease in the same patient, a biochemically “active” liver disease, an elevation of serum gamma globulins, presence in serum of autoantibodies known to characterize JAIH, and compatible histopathological features on liver biopsy. However, sometimes the diagnosis may become difficult and for this reason, in 1993, an international board of physicians published a set of criteria to identify patients as having either “definite” or “probable” autoimmune hepatitis[2]. Once used primarily for scientific and research purposes, this scoring system is now widely used in clinical practice after being reviewed in 1999[3].
A simplified scoring system has since been proposed based solely on four parameters (autoantibodies, IgG levels, liver histology and exclusion of viral hepatitis), and has been validated in adults with 88% sensitivity and 97% specificity[59]. When comparing both scoring system in adults, we see that the revised original scoring system has shown greater sensitivity for the diagnosis than the simplified scoring system (100% vs 95%), while the simplified score had greater specificity (90% vs 73%) and predictability (92% vs 82%) for AIH than the revised original system[60]. The original scoring system was assessed in children using the GGT/aminotransferase ratio instead of the alkaline phosphatase/aminotransferase ratio to improve its specificity[61]. When tested in children the simplified scoring system was not proved to be effective mainly because of its low sensitivity[62]. In conclusion, no validated scoring system for the diagnosis exists for JAIH[63]. In challenging cases, once Wilson’s disease is excluded, and in absence of liver function failure, an immunosuppressive treatment should be attempted for at least 6 wk. A positive response to this treatment would suggest AIH. Moreover, relapse after immunosuppressive drug withdrawal is positively weighted in the IAIHG diagnostic criteria[2,3].
MANAGEMENT
The most effective therapy for JAIH is pharmacological suppression of the immune response. Treatment should be started as soon as the diagnosis is made to avoid severe liver damage and progression of fibrosis. Standard therapy includes a combination of prednisone and azathioprine[6] or occasionally prednisone as a monotherapy[6,7,21]. Prednisone or prednisolone is used at a higher dose that used in adults (2 mg/kg per day, up to a maximal daily dose of 60 mg/d in the adolescent) and azathioprine is administered starting from 1 mg/kg per day up to a maximum of 2.5 mg/kg per day. First line combination therapy including prednisone and azathioprine can be more effective than prednisone alone[64]. Moreover, the “steroid-sparing” effect of the azathioprine allows reducing more rapidly the steroid dose, thus tapering side effects related to the prolonged use of steroids at high dose.
The goal of the treatment is to obtain clinical and biochemical remission of the liver disease clinical signs with normalization of the “activity” of the disease (transaminase, gamma globulins) and of the liver function (prothrombin activity; INR). The definition of treatment-induced remission in JAIH should be stricter than that used in adult disease: the serum activity of aminotransferase should be maintained within the upper limit of normal, serum immunoglobulin G levels within the normal range for age, and serum autoantibodies absent or at very low titer[6,15,21]. Even clinical and biochemical remission do not always reflect histological resolution of inflammation, the proof of histological remission is not required. The rapidity and degree of response to treatment depends on the disease severity at onset. In JAIH, treatment is associated, in over 90% of cases, with a measurable clinical and laboratory response within 4 to 8 wk. Complete normalization of biochemical parameters may, however, take several months. On histopathological evaluation, the immunosuppressive treatment improves the fibrosis score, with an arrest in its progression into cirrhosis. Fibrosis control is mainly associated with regression of necroinflammatory activity[65]. Once remission is obtained, it must be maintained in the long term on the lowest possible dose of medication. Different therapeutic schedules of treatment discontinuation exist and should be tailored on individual patients. Prednisone is usually first decreased; the shift to alternate-day use of steroids may be suitable because of the lower incidence of side effect[66]. In cases of severe liver function impairment at diagnosis, liver function may further deteriorate despite an appropriate therapy. In these patients immunosuppressive therapy should be modified with the introduction of a third drug such as cyclosporine. In case of further non-response, the possibility of a liver transplant should be considered.
When complete remission is achieved, the goal of the immunosuppressive treatment is to maintain remission and to prevent relapse of the disease. Prednisone should be further reduced to the lowest dose that allows a biochemical remission. Alternate-day doses of prednisone associated to azathioprine are usually effective in maintaining remission. A relapse may occur at any time, the most frequent cause of a relapse is patient’s non-compliance. It is questionable that a histological remission has to be demonstrated through a liver biopsy in patients with clinical and biochemical remission, since the presence of histological remission has not been shown to be sufficiently indicative of an absence of possibility of relapse in the case of further reduction of the immunosuppression[6]. Liver fibrosis rarely progresses in patients who maintain a persistent biochemical remission and it can even diminish during treatment. Duration of the immunosuppressive treatment before attempting discontinuation is unknown; stopping treatment within the first two years is usually followed by a relapse[6]. We suggest that sustained remission should be maintained for at least five years, thereafter, in case of combined treatment of prednisone and azathioprine, prednisone is stopped and the patient is maintained on azathioprine monotherapy. Azathioprine monotherapy had been demonstrated to maintain remission in most patients with AIH[67]. Undetectable serum autoantibodies do not exclude the risk of relapse, but an increase of the titer of autoantibodies suggests caution in modifying the dose of immunosuppressive therapy.
Particular variant forms of AIH, as celiac disease-associated AIH on gluten free diet, might have a lower risk to relapse after treatment discontinuation[68]. In such cases a discontinuation attempt after less than 5 years of treatment could be justified.
Steroids mostly cause side effects of immunosuppressive therapy, including increase of food intake leading to moderate and reversible weight increase and a reduction of height growth. Severe side effects include obesity, growth failure, severe cosmetic changes, cutaneous striae, vertebral collapse, hyperglycemia, and cataracts, causing both visual impairment and, potentially, psychosis. Azathioprine is usually a safe drug, and cytopenia necessitating a dosage reduction is a rare event. Teratogenicity and oncogenicity issues resulting from azathioprine use in humans have not been conclusively demonstrated. Pregnancy should however be excluded in adolescent girls before starting treatment with azathioprine. However, if prolonged azathioprine treatment is needed, pregnancy has been demonstrated to be safe, in the long term, in young females with AIH[69,70]. During pregnancy, higher doses of prednisone may be an alternative option for those young women who prefer azathioprine withdrawal. However, vigilance is required at all times, and patients need careful monitoring, especially in the postpartum period, because of the possibility of relapses.
In case of non-response to conventional treatment or in the presence of severe side effects of corticosteroids the use of cyclosporine A is indicated. Cyclosporine A at a median dose of 5 mg/kg per day induces remission in children and adolescents with AIH with a initial target concentration in serum of cyclosporine of 200-250 ng/mL[71,72]. Cyclosporine treatment side effects including mild gingival hyperplasia and reversible irsutism in some patients, are usually well tolerated and disappear after reduction of the dose[73]. Normality of renal function should be verified before starting this drug. In the follow-up, once remission is obtained, the dose of cyclosporine can be reduced with a target concentration of 100 ng/mL or the patient may be shifted to conventional treatment.
In children who either did not tolerate azathioprine or did not respond to conventional treatment, mycophenolate-mofetil (MFM, 20 mg/kg per day) in addition to steroids therapy has been shown to induce and maintain remission[74]. Side effects of MFM include headache, diarrhea, dizziness, hair loss and neutropenia.
Budesonide, a steroid that is rapidly metabolized with low systemic exposure, in combination with azathioprine, has been recently shown in a trial including patients with JAIH to induce and maintain remission with fewer side effects than prednisone[75]. However, the low proportion of remission observed in this study compared to that reported in others pediatric studies using prednisone and azathioprine schedules, do not support its use as first-line treatment of JAIH[76]. Recently Rituximab, a monoclonal antibody against CD20, a B-lymphocyte surface antigen, has been successfully used in selected cases as a rescue therapy[77].
Liver transplantation should be considered as a therapeutic option for children and adolescents with JAIH and chronic end stage liver disease or in patients with acute liver failure at onset not responding to rescue immunosuppression. Five-year post-transplant survival in JAIH patients is scored at 86%[78].
LONG-TERM OUTCOME
Immunosuppressive treatment has convincingly altered the outcome of most patients with AIH[6,21,79-82]. Indeed according to previous prognostic studies on adults, 40% of patients with severe disease without treatment die within six months of diagnosis and cirrhosis eventually develops in at least 40% of untreated survivors[83,84].
On the other hand the 10-year survival rates among treated adults is 60% for those with cirrhosis on the initial liver biopsy[83,85] and more than 80% for those patients without cirrhosis at presentation[86].
The long-term outcome of JAIH still remains scarcely known, however, in case of full and prompt response to immunosuppressive therapy, the prognosis is usually satisfactory and most patients survive in the long-term with excellent quality of life and, in the majority of cases, on low dose immunosuppression.
In the five largest published series of children with AIH, overall survival rate in long-term treated patients exceeded 80% with a 5-year survival with native liver ranging between 67% and 87%; follow-up ranged from 4, 8 to 10 years[21,38,87-89].
The presence of cirrhosis on initial liver biopsy did not seem to impact long-term survival in children with AIH[87,88] while elevated total bilirubin and prolonged INR are independent risk factors of death and/or need of liver transplantation[21]. Immunosuppressive treatment requires to be prolonged in the long term in the majority of patients; however, sustained remission after treatment discontinuation has been reported in 13% to 20% of patients[21,87].
End-stage liver disease leading to liver transplantation has been reported to develop up to 14 years after diagnosis in 8% to 16% of children with JAIH compliant to immunosuppressive therapy and in absence of an evident biochemical relapse[21,87].
VARIANT FORMS OF JAIH
Giant cell hepatitis with autoimmune hemolytic anemia
Giant cell hepatitis with autoimmune hemolytic anemia is a rare entity described by Bernard et al[55] in 1981, presenting in early childhood with severe progressive liver disease in combination with Coombs positive hemolytic anemia. The clinical course is usually aggressive leading to hepatic failure and death.
The mechanism of liver disease is not known, but an autoimmune process is believed to be responsible for this component of the disease as well. A study by Whitington et al[90] has recently provided evidence that systemic B cell autoimmunity might play a pathogenetic role even if autoantibodies are usually absent, histological features characteristic of auto-immune hepatitis are missing, and the disease is highly refractory to therapy that would usually be effective in AIH. Conventional immunosuppressive treatments (steroids, azathioprine, cyclophosphamide, cyclosporine, mycophenolate) are associated with high toxicity and often produce only partial or short-lasting remission[91]. Liver transplantation is associated with high rate of disease recurrence. In more recent studies the use of anti-CD20 monoclonal antibody (Rituximab) has been reported to be effective in patients with refractory hepatitis[92]. Intravenous immunoglobulins have been also reported to be efficacious in case of severe liver function impairment at onset or during a relapse in patients treated by multiple immunosuppression although their efficacy seems to be only temporary[93].
Autoantibody-negative autoimmune hepatitis
Cryptogenic hepatitis with autoimmune features in absence of detectable serum autoantibodies is described as “autoantibody-negative AIH”[94]. It affects a small proportion of adult patients presenting with a cryptogenic liver disease with acute or chronic presentation with the clinical, biochemical and histopathological features of AIH and responsive to immunosuppressive treatment[94-96]. The comprehensive international scoring system can support, but never override the clinical diagnosis pre-treatment, and non-standard serological markers should be sought in order to enhance diagnostic confidence[3,60,97]. A 3-mo treatment trial with corticosteroids should be considered in all candidates for the diagnosis, regardless of the serological findings[98,99]. This entity has been reported in children only in small series or in single case report[100].
Celiac disease associated-AIH
Celiac disease (CD) is common in patients with AIH, especially in children, as it has been shown in a previous Italian multicenter survey[52] and in more recent small pediatric series[68,101-103]. These studies reported a prevalence of CD in up to 19% of children with JAIH. The pathogenetic role of gluten in triggering AIH is uncertain, however, both types of AIH have been described in association with CD as well as autoantibody-negative AIH[52,100]. Liver damage, as evidenced by elevated aminotransferase activity, has been reported as being present from the first observation of celiac patients in some cases while in other, CD was diagnosed by a serological screening in patients with a known AIH[101,102]. Therefore all patients with AIH should be serological screened for CD and moreover all CD patients with clinical and/or biochemical signs of liver damage should be closely followed-up to exclude an AIH, especially in the case of persistent elevation of liver enzymes on a gluten free diet.
Children with co-existent CD seem to have an apparently more favorable response to treatment, suggesting a positive effect of gluten withdrawal on AIH co-existent with CD. Gluten withdrawal might potentiate the immunosuppressive effect of the immunosuppressive drugs, maintaining remission even when the treatment has been withdrawn[68,103].
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy-associated JAIH
Autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) is a rare autosomal recessive disorder caused by mutations in autoimmune regulator gene (AIRE) inducing a loss in central immune tolerance, failure to eliminate autoreactive T cells in the thymus, and their escape to the periphery. APECED is characterized by an extremely variable pattern of destructive autoimmune reaction, mainly mediated by specific autoantibodies toward different endocrine and non-endocrine organs. Virtually, all tissues and organs may represent the target of the autoimmune attacks, thus leading to a wide spectrum of clinical features. The three main components of APECED are chronic mucocutaneous candidiasis, chronic hypoparathyroidism and Addison’s disease. Generally chronic mucocutaneous candidiasis develops first and it is often followed by chronic hypoparathyroidism, before the age of 10 years, and later on by adrenal insufficiency. In addition to the main components, the spectrum of minor manifestations may include ectodermal dystrophy, other endocrinopathies, such as hypergonadotropic hypogonadism, insulin-dependent diabetes, autoimmune thyroiditis, and pituitary dysfunction. Moreover, skin diseases (vitiligo and alopecia) and gastrointestinal disorders (chronic atrophic gastritis, pernicious anemia) and particularly AIH, may be present. AIH shows a clinical phenotype akin to AIH-2 and it is present in 15%-20% of cases with CYP1A2 and CYP2A6 as a specific target antigens[104]. Heterozygous mutations of AIRE gene have been reported in children with AIH-1 suggesting a possible predisposition role[105].
De novo autoimmune hepatitis
De novo autoimmune hepatitis, after liver transplantation, was first described in 1998 by the group of King’s College Hospital in London[106]. It is a form of late graft dysfunction characterized by abnormal liver function tests, high serum concentration of immunoglobulin, presence of autoantibodies, and histological features of interface hepatitis coupled with a rich plasma cell infiltrate[107]. This recently recognized entity affects patients transplanted for disorders other than AIH and usually of non-autoimmune nature. Since its first description several authors reported the occurrence of de novo AIH in children and adults transplanted for non-autoimmune conditions[108-115].
The pathogenesis of de novo AIH is not yet defined and there are a variety of potential mechanisms leading to autoimmune liver disease post-transplant. Possible pathogenetic mechanism include the release of autoantigens from damaged tissue, as well as molecular mimicry, whereby exposure to viruses sharing amino acid sequences with autoantigens leads to cross-reactive immunity[106]. De novo AIH responds to treatment with corticosteroids and azathioprine allowing excellent graft and patient survival. Early recognition and appropriate management are therefore essential to avoid graft loss[107,116].
CONCLUSION
Juvenile AIH is a severe liver disease of childhood and adolescence progressing rapidly toward cirrhosis and severe liver function impairment unless immunosuppressive treatment is promptly started. Its clinical spectrum is broad: from asymptomatic liver damage to acute symptomatic and even severe hepatitis. Early diagnosis is mandatory but no scoring system of sufficient sensitivity exists. Serum autoantibodies are a relevant tool, but not essential for diagnosis and liver histology has distinct but non-pathognomonic features. The vast majority of treated patients responds to the immunosuppressive treatment, but relapses are frequent and mostly related to defective compliance to treatment. Long-term outcome studies on JAIH concerning the possibility of safely stopping the immunosuppressive treatment are needed for appropriate counseling to families and patients.
Footnotes
P- Reviewer: Tannuri U S- Editor: Song XX L- Editor: A E- Editor: Liu SQ
References
- 1.Mackay IR, Weiden S, Hasker J. Autoimmune hepatitis. Ann N Y Acad Sci. 1965;124:767–780. doi: 10.1111/j.1749-6632.1965.tb19000.x. [DOI] [PubMed] [Google Scholar]
- 2.Johnson PJ, McFarlane IG. Meeting report: International Autoimmune Hepatitis Group. Hepatology. 1993;18:998–1005. doi: 10.1002/hep.1840180435. [DOI] [PubMed] [Google Scholar]
- 3.Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, Chapman RW, Cooksley WG, Czaja AJ, Desmet VJ, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol. 1999;31:929–938. doi: 10.1016/s0168-8278(99)80297-9. [DOI] [PubMed] [Google Scholar]
- 4.Grossman A, Rosenthal IM, Szanto PB. Chronic hepatitis with hypergammaglobulinemia in childhood. Report of eight cases. Pediatrics. 1962;29:933–947. [PubMed] [Google Scholar]
- 5.Odièvre M, Maggiore G, Homberg JC, Saadoun F, Couroucé AM, Yvart J, Hadchouel M, Alagille D. Seroimmunologic classification of chronic hepatitis in 57 children. Hepatology. 1983;3:407–409. doi: 10.1002/hep.1840030320. [DOI] [PubMed] [Google Scholar]
- 6.Maggiore G, Bernard O, Hadchouel M, Hadchouel P, Odievre M, Alagille D. Treatment of autoimmune chronic active hepatitis in childhood. J Pediatr. 1984;104:839–844. doi: 10.1016/s0022-3476(84)80477-1. [DOI] [PubMed] [Google Scholar]
- 7.Vegnente A, Larcher VF, Mowat AP, Portmann B, Williams R. Duration of chronic active hepatitis and the development of cirrhosis. Arch Dis Child. 1984;59:330–335. doi: 10.1136/adc.59.4.330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maggiore G, Bernard O, Homberg JC, Hadchouel M, Alvarez F, Hadchouel P, Odièvre M, Alagille D. Liver disease associated with anti-liver-kidney microsome antibody in children. J Pediatr. 1986;108:399–404. doi: 10.1016/s0022-3476(86)80880-0. [DOI] [PubMed] [Google Scholar]
- 9.Homberg JC, Abuaf N, Bernard O, Islam S, Alvarez F, Khalil SH, Poupon R, Darnis F, Lévy VG, Grippon P. Chronic active hepatitis associated with antiliver/kidney microsome antibody type 1: a second type of “autoimmune” hepatitis. Hepatology. 1987;7:1333–1339. doi: 10.1002/hep.1840070626. [DOI] [PubMed] [Google Scholar]
- 10.Maggiore G, Sciveres M. Autoimmune hepatitis: a childhood disease. Curr Pediatr Rev. 2005;1:73–90. [Google Scholar]
- 11.Boberg KM. Prevalence and epidemiology of autoimmune hepatitis. Clin Liver Dis. 2002;6:635–647. doi: 10.1016/s1089-3261(02)00021-1. [DOI] [PubMed] [Google Scholar]
- 12.Primo J, Merino C, Fernández J, Molés JR, Llorca P, Hinojosa J. [Incidence and prevalence of autoimmune hepatitis in the area of the Hospital de Sagunto (Spain)] Gastroenterol Hepatol. 2004;27:239–243. doi: 10.1016/s0210-5705(03)70452-x. [DOI] [PubMed] [Google Scholar]
- 13.Hurlburt KJ, McMahon BJ, Deubner H, Hsu-Trawinski B, Williams JL, Kowdley KV. Prevalence of autoimmune liver disease in Alaska Natives. Am J Gastroenterol. 2002;97:2402–2407. doi: 10.1111/j.1572-0241.2002.06019.x. [DOI] [PubMed] [Google Scholar]
- 14.Deneau M, Jensen MK, Holmen J, Williams MS, Book LS, Guthery SL. Primary sclerosing cholangitis, autoimmune hepatitis, and overlap in Utah children: epidemiology and natural history. Hepatology. 2013;58:1392–1400. doi: 10.1002/hep.26454. [DOI] [PubMed] [Google Scholar]
- 15.Mieli-Vergani G, Vergani D. Autoimmune liver diseases in children - what is different from adulthood? Best Pract Res Clin Gastroenterol. 2011;25:783–795. doi: 10.1016/j.bpg.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 16.Longhi MS, Ma Y, Mieli-Vergani G, Vergani D. Aetiopathogenesis of autoimmune hepatitis. J Autoimmun. 2010;34:7–14. doi: 10.1016/j.jaut.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson PT, Czaja AJ. Genetic effects on susceptibility, clinical expression, and treatment outcome of type 1 autoimmune hepatitis. Clin Liver Dis. 2002;6:707–725. doi: 10.1016/s1089-3261(02)00023-5. [DOI] [PubMed] [Google Scholar]
- 18.Yoshizawa K, Ota M, Katsuyama Y, Ichijo T, Matsumoto A, Tanaka E, Kiyosawa K. Genetic analysis of the HLA region of Japanese patients with type 1 autoimmune hepatitis. J Hepatol. 2005;42:578–584. doi: 10.1016/j.jhep.2004.12.019. [DOI] [PubMed] [Google Scholar]
- 19.Pando M, Larriba J, Fernandez GC, Fainboim H, Ciocca M, Ramonet M, Badia I, Daruich J, Findor J, Tanno H, et al. Pediatric and adult forms of type I autoimmune hepatitis in Argentina: evidence for differential genetic predisposition. Hepatology. 1999;30:1374–1380. doi: 10.1002/hep.510300611. [DOI] [PubMed] [Google Scholar]
- 20.Vázquez-García MN, Aláez C, Olivo A, Debaz H, Pérez-Luque E, Burguete A, Cano S, de la Rosa G, Bautista N, Hernández A, et al. MHC class II sequences of susceptibility and protection in Mexicans with autoimmune hepatitis. J Hepatol. 1998;28:985–990. doi: 10.1016/s0168-8278(98)80347-4. [DOI] [PubMed] [Google Scholar]
- 21.Gregorio GV, Portmann B, Reid F, Donaldson PT, Doherty DG, McCartney M, Mowat AP, Vergani D, Mieli-Vergani G. Autoimmune hepatitis in childhood: a 20-year experience. Hepatology. 1997;25:541–547. doi: 10.1002/hep.510250308. [DOI] [PubMed] [Google Scholar]
- 22.Ma Y, Bogdanos DP, Hussain MJ, Underhill J, Bansal S, Longhi MS, Cheeseman P, Mieli-Vergani G, Vergani D. Polyclonal T-cell responses to cytochrome P450IID6 are associated with disease activity in autoimmune hepatitis type 2. Gastroenterology. 2006;130:868–882. doi: 10.1053/j.gastro.2005.12.020. [DOI] [PubMed] [Google Scholar]
- 23.Djilali-Saiah I, Fakhfakh A, Louafi H, Caillat-Zucman S, Debray D, Alvarez F. HLA class II influences humoral autoimmunity in patients with type 2 autoimmune hepatitis. J Hepatol. 2006;45:844–850. doi: 10.1016/j.jhep.2006.07.034. [DOI] [PubMed] [Google Scholar]
- 24.Cassinotti A, Birindelli S, Clerici M, Trabattoni D, Lazzaroni M, Ardizzone S, Colombo R, Rossi E, Porro GB. HLA and autoimmune digestive disease: a clinically oriented review for gastroenterologists. Am J Gastroenterol. 2009;104:195–217; quiz 194, 218. doi: 10.1038/ajg.2008.10. [DOI] [PubMed] [Google Scholar]
- 25.Vergani D, Wells L, Larcher VF, Nasaruddin BA, Davies ET, Mieli-Vergani G, Mowat AP. Genetically determined low C4: a predisposing factor to autoimmune chronic active hepatitis. Lancet. 1985;2:294–298. doi: 10.1016/s0140-6736(85)90348-4. [DOI] [PubMed] [Google Scholar]
- 26.Mizutani T, Shinoda M, Tanaka Y, Kuno T, Hattori A, Usui T, Kuno N, Osaka T. Autoantibodies against CYP2D6 and other drug-metabolizing enzymes in autoimmune hepatitis type 2. Drug Metab Rev. 2005;37:235–252. doi: 10.1081/dmr-200028798. [DOI] [PubMed] [Google Scholar]
- 27.Vento S, Cainelli F. Is there a role for viruses in triggering autoimmune hepatitis? Autoimmun Rev. 2004;3:61–69. doi: 10.1016/S1568-9972(03)00053-3. [DOI] [PubMed] [Google Scholar]
- 28.Bogdanos DP, Choudhuri K, Vergani D. Molecular mimicry and autoimmune liver disease: virtuous intentions, malign consequences. Liver. 2001;21:225–232. doi: 10.1034/j.1600-0676.2001.021004225.x. [DOI] [PubMed] [Google Scholar]
- 29.Muratori L, Parola M, Ripalti A, Robino G, Muratori P, Bellomo G, Carini R, Lenzi M, Landini MP, Albano E, et al. Liver/kidney microsomal antibody type 1 targets CYP2D6 on hepatocyte plasma membrane. Gut. 2000;46:553–561. doi: 10.1136/gut.46.4.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vento S, Hegarty JE, Bottazzo G, Macchia E, Williams R, Eddleston AL. Antigen specific suppressor cell function in autoimmune chronic active hepatitis. Lancet. 1984;1:1200–1204. doi: 10.1016/s0140-6736(84)91691-x. [DOI] [PubMed] [Google Scholar]
- 31.Ferri S, Longhi MS, De Molo C, Lalanne C, Muratori P, Granito A, Hussain MJ, Ma Y, Lenzi M, Mieli-Vergani G, et al. A multifaceted imbalance of T cells with regulatory function characterizes type 1 autoimmune hepatitis. Hepatology. 2010;52:999–1007. doi: 10.1002/hep.23792. [DOI] [PubMed] [Google Scholar]
- 32.Peiseler M, Sebode M, Franke B, Wortmann F, Schwinge D, Quaas A, Baron U, Olek S, Wiegard C, Lohse AW, et al. FOXP3+ regulatory T cells in autoimmune hepatitis are fully functional and not reduced in frequency. J Hepatol. 2012;57:125–132. doi: 10.1016/j.jhep.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 33.Longhi MS, Hussain MJ, Kwok WW, Mieli-Vergani G, Ma Y, Vergani D. Autoantigen-specific regulatory T cells, a potential tool for immune-tolerance reconstitution in type-2 autoimmune hepatitis. Hepatology. 2011;53:536–547. doi: 10.1002/hep.24039. [DOI] [PubMed] [Google Scholar]
- 34.Lapierre P, Béland K, Yang R, Alvarez F. Adoptive transfer of ex vivo expanded regulatory T cells in an autoimmune hepatitis murine model restores peripheral tolerance. Hepatology. 2013;57:217–227. doi: 10.1002/hep.26023. [DOI] [PubMed] [Google Scholar]
- 35.Lapierre P, Djilali-Saiah I, Vitozzi S, Alvarez F. A murine model of type 2 autoimmune hepatitis: Xenoimmunization with human antigens. Hepatology. 2004;39:1066–1074. doi: 10.1002/hep.20109. [DOI] [PubMed] [Google Scholar]
- 36.Holdener M, Hintermann E, Bayer M, Rhode A, Rodrigo E, Hintereder G, Johnson EF, Gonzalez FJ, Pfeilschifter J, Manns MP, et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J Exp Med. 2008;205:1409–1422. doi: 10.1084/jem.20071859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buxbaum J, Qian P, Allen PM, Peters MG. Hepatitis resulting from liver-specific expression and recognition of self-antigen. J Autoimmun. 2008;31:208–215. doi: 10.1016/j.jaut.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maggiore G, Veber F, Bernard O, Hadchouel M, Homberg JC, Alvarez F, Hadchouel P, Alagille D. Autoimmune hepatitis associated with anti-actin antibodies in children and adolescents. J Pediatr Gastroenterol Nutr. 1993;17:376–381. doi: 10.1097/00005176-199311000-00007. [DOI] [PubMed] [Google Scholar]
- 39.Bridoux-Henno L, Maggiore G, Johanet C, Fabre M, Vajro P, Dommergues JP, Reinert P, Bernard O. Features and outcome of autoimmune hepatitis type 2 presenting with isolated positivity for anti-liver cytosol antibody. Clin Gastroenterol Hepatol. 2004;2:825–830. doi: 10.1016/s1542-3565(04)00354-4. [DOI] [PubMed] [Google Scholar]
- 40.Maggiore G, De Benedetti F, Massa M, Pignatti P, Martini A. Circulating levels of interleukin-6, interleukin-8, and tumor necrosis factor-alpha in children with autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 1995;20:23–27. doi: 10.1097/00005176-199501000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Maggiore G, Porta G, Bernard O, Hadchouel M, Alvarez F, Homberg JC, Alagille D. Autoimmune hepatitis with initial presentation as acute hepatic failure in young children. J Pediatr. 1990;116:280–282. doi: 10.1016/s0022-3476(05)82892-6. [DOI] [PubMed] [Google Scholar]
- 42.Rigopoulou EI, Roggenbuck D, Smyk DS, Liaskos C, Mytilinaiou MG, Feist E, Conrad K, Bogdanos DP. Asialoglycoprotein receptor (ASGPR) as target autoantigen in liver autoimmunity: lost and found. Autoimmun Rev. 2012;12:260–269. doi: 10.1016/j.autrev.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 43.Löhr H, Treichel U, Poralla T, Manns M, Meyer zum Büschenfelde KH. Liver-infiltrating T helper cells in autoimmune chronic active hepatitis stimulate the production of autoantibodies against the human asialoglycoprotein receptor in vitro. Clin Exp Immunol. 1992;88:45–49. doi: 10.1111/j.1365-2249.1992.tb03037.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gueguen M, Meunier-Rotival M, Bernard O, Alvarez F. Anti-liver kidney microsome antibody recognizes a cytochrome P450 from the IID subfamily. J Exp Med. 1988;168:801–806. doi: 10.1084/jem.168.2.801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vergani D, Alvarez F, Bianchi FB, Cançado EL, Mackay IR, Manns MP, Nishioka M, Penner E. Liver autoimmune serology: a consensus statement from the committee for autoimmune serology of the International Autoimmune Hepatitis Group. J Hepatol. 2004;41:677–683. doi: 10.1016/j.jhep.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 46.Johanet C, Beleoken E, Ballot E. Autoantibodies in autoimmune hepatitis: antinuclear antibodies (ANA) Clin Res Hepatol Gastroenterol. 2012;36:394–396. doi: 10.1016/j.clinre.2012.02.005. [DOI] [PubMed] [Google Scholar]
- 47.Couto CA, Bittencourt PL, Porta G, Abrantes-Lemos CP, Carrilho FJ, Guardia BD, Cançado EL. Antismooth muscle and antiactin antibodies are indirect markers of histological and biochemical activity of autoimmune hepatitis. Hepatology. 2014;59:592–600. doi: 10.1002/hep.26666. [DOI] [PubMed] [Google Scholar]
- 48.Martini E, Abuaf N, Cavalli F, Durand V, Johanet C, Homberg JC. Antibody to liver cytosol (anti-LC1) in patients with autoimmune chronic active hepatitis type 2. Hepatology. 1988;8:1662–1666. doi: 10.1002/hep.1840080632. [DOI] [PubMed] [Google Scholar]
- 49.Lapierre P, Hajoui O, Homberg JC, Alvarez F. Formiminotransferase cyclodeaminase is an organ-specific autoantigen recognized by sera of patients with autoimmune hepatitis. Gastroenterology. 1999;116:643–649. doi: 10.1016/s0016-5085(99)70186-1. [DOI] [PubMed] [Google Scholar]
- 50.Johanet C, Ballot E. Auto-antibodies in autoimmune hepatitis: anti-soluble liver antigen (SLA) Clin Res Hepatol Gastroenterol. 2012;36:244–246. doi: 10.1016/j.clinre.2011.10.013. [DOI] [PubMed] [Google Scholar]
- 51.Gleeson D, Heneghan MA. British Society of Gastroenterology (BSG) guidelines for management of autoimmune hepatitis. Gut. 2011;60:1611–1629. doi: 10.1136/gut.2010.235259. [DOI] [PubMed] [Google Scholar]
- 52.Caprai S, Vajro P, Ventura A, Sciveres M, Maggiore G. Autoimmune liver disease associated with celiac disease in childhood: a multicenter study. Clin Gastroenterol Hepatol. 2008;6:803–806. doi: 10.1016/j.cgh.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 53.Hofer H, Oesterreicher C, Wrba F, Ferenci P, Penner E. Centrilobular necrosis in autoimmune hepatitis: a histological feature associated with acute clinical presentation. J Clin Pathol. 2006;59:246–249. doi: 10.1136/jcp.2005.029348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Devaney K, Goodman ZD, Ishak KG. Postinfantile giant-cell transformation in hepatitis. Hepatology. 1992;16:327–333. doi: 10.1002/hep.1840160208. [DOI] [PubMed] [Google Scholar]
- 55.Bernard O, Hadchouel M, Scotto J, Odièvre M, Alagille D. Severe giant cell hepatitis with autoimmune hemolytic anemia in early childhood. J Pediatr. 1981;99:704–711. doi: 10.1016/s0022-3476(81)80388-5. [DOI] [PubMed] [Google Scholar]
- 56.Czaja AJ, Carpenter HA. Autoimmune hepatitis with incidental histologic features of bile duct injury. Hepatology. 2001;34:659–665. doi: 10.1053/jhep.2001.27562. [DOI] [PubMed] [Google Scholar]
- 57.Vajro P, Hadchouel P, Hadchouel M, Bernard O, Alagille D. Incidence of cirrhosis in children with chronic hepatitis. J Pediatr. 1990;117:392–396. doi: 10.1016/s0022-3476(05)81078-9. [DOI] [PubMed] [Google Scholar]
- 58.Floreani A, Liberal R, Vergani D, Mieli-Vergani G. Autoimmune hepatitis: Contrasts and comparisons in children and adults - a comprehensive review. J Autoimmun. 2013;46:7–16. doi: 10.1016/j.jaut.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 59.Hennes EM, Zeniya M, Czaja AJ, Parés A, Dalekos GN, Krawitt EL, Bittencourt PL, Porta G, Boberg KM, Hofer H, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology. 2008;48:169–176. doi: 10.1002/hep.22322. [DOI] [PubMed] [Google Scholar]
- 60.Czaja AJ. Performance parameters of the diagnostic scoring systems for autoimmune hepatitis. Hepatology. 2008;48:1540–1548. doi: 10.1002/hep.22513. [DOI] [PubMed] [Google Scholar]
- 61.Ebbeson RL, Schreiber RA. Diagnosing autoimmune hepatitis in children: is the International Autoimmune Hepatitis Group scoring system useful? Clin Gastroenterol Hepatol. 2004;2:935–940. doi: 10.1016/s1542-3565(04)00396-9. [DOI] [PubMed] [Google Scholar]
- 62.Hiejima E, Komatsu H, Sogo T, Inui A, Fujisawa T. Utility of simplified criteria for the diagnosis of autoimmune hepatitis in children. J Pediatr Gastroenterol Nutr. 2011;52:470–473. doi: 10.1097/MPG.0b013e3181fc1e0b. [DOI] [PubMed] [Google Scholar]
- 63.Ferri PM, Ferreira AR, Miranda DM, Simões E Silva AC. Diagnostic criteria for autoimmune hepatitis in children: a challenge for pediatric hepatologists. World J Gastroenterol. 2012;18:4470–4473. doi: 10.3748/wjg.v18.i33.4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Vitfell-Pedersen J, Jørgensen MH, Müller K, Heilmann C. Autoimmune hepatitis in children in Eastern Denmark. J Pediatr Gastroenterol Nutr. 2012;55:376–379. doi: 10.1097/MPG.0b013e3182602b20. [DOI] [PubMed] [Google Scholar]
- 65.Ferreira AR, Roquete ML, Toppa NH, de Castro LP, Fagundes ED, Penna FJ. Effect of treatment of hepatic histopathology in children and adolescents with autoimmune hepatitis. J Pediatr Gastroenterol Nutr. 2008;46:65–70. doi: 10.1097/01.mpg.0000304456.84552.13. [DOI] [PubMed] [Google Scholar]
- 66.Clark JH, Fitzgerald JF. Effect of exogenous corticosteroid therapy on growth in children with HBsAg-negative chronic aggressive hepatitis. J Pediatr Gastroenterol Nutr. 1984;3:72–76. doi: 10.1097/00005176-198401000-00016. [DOI] [PubMed] [Google Scholar]
- 67.Johnson PJ, McFarlane IG, Williams R. Azathioprine for long-term maintenance of remission in autoimmune hepatitis. N Engl J Med. 1995;333:958–963. doi: 10.1056/NEJM199510123331502. [DOI] [PubMed] [Google Scholar]
- 68.Nastasio S, Sciveres M, Riva S, Filippeschi IP, Vajro P, Maggiore G. Celiac disease-associated autoimmune hepatitis in childhood: long-term response to treatment. J Pediatr Gastroenterol Nutr. 2013;56:671–674. doi: 10.1097/MPG.0b013e31828b1dfa. [DOI] [PubMed] [Google Scholar]
- 69.Terrabuio DR, Abrantes-Lemos CP, Carrilho FJ, Cançado EL. Follow-up of pregnant women with autoimmune hepatitis: the disease behavior along with maternal and fetal outcomes. J Clin Gastroenterol. 2009;43:350–356. doi: 10.1097/MCG.0b013e318176b8c5. [DOI] [PubMed] [Google Scholar]
- 70.Heneghan MA, Norris SM, O’Grady JG, Harrison PM, McFarlane IG. Management and outcome of pregnancy in autoimmune hepatitis. Gut. 2001;48:97–102. doi: 10.1136/gut.48.1.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Alvarez F, Ciocca M, Cañero-Velasco C, Ramonet M, de Davila MT, Cuarterolo M, Gonzalez T, Jara-Vega P, Camarena C, Brochu P, et al. Short-term cyclosporine induces a remission of autoimmune hepatitis in children. J Hepatol. 1999;30:222–227. doi: 10.1016/s0168-8278(99)80065-8. [DOI] [PubMed] [Google Scholar]
- 72.Debray D, Maggiore G, Girardet JP, Mallet E, Bernard O. Efficacy of cyclosporin A in children with type 2 autoimmune hepatitis. J Pediatr. 1999;135:111–114. doi: 10.1016/s0022-3476(99)70339-2. [DOI] [PubMed] [Google Scholar]
- 73.Sciveres M, Caprai S, Palla G, Ughi C, Maggiore G. Effectiveness and safety of ciclosporin as therapy for autoimmune diseases of the liver in children and adolescents. Aliment Pharmacol Ther. 2004;19:209–217. doi: 10.1046/j.1365-2036.2003.01754.x. [DOI] [PubMed] [Google Scholar]
- 74.Aw MM, Dhawan A, Samyn M, Bargiota A, Mieli-Vergani G. Mycophenolate mofetil as rescue treatment for autoimmune liver disease in children: a 5-year follow-up. J Hepatol. 2009;51:156–160. doi: 10.1016/j.jhep.2009.02.024. [DOI] [PubMed] [Google Scholar]
- 75.Woynarowski M, Nemeth A, Baruch Y, Koletzko S, Melter M, Rodeck B, Strassburg CP, Pröls M, Woźniak M, Manns MP. Budesonide versus prednisone with azathioprine for the treatment of autoimmune hepatitis in children and adolescents. J Pediatr. 2013;163:1347–1353.e1. doi: 10.1016/j.jpeds.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 76.Mieli-Vergani G, Vergani D. Budesonide for juvenile autoimmune hepatitis? Not yet. J Pediatr. 2013;163:1246–1248. doi: 10.1016/j.jpeds.2013.06.064. [DOI] [PubMed] [Google Scholar]
- 77.D’Agostino D, Costaguta A, Álvarez F. Successful treatment of refractory autoimmune hepatitis with rituximab. Pediatrics. 2013;132:e526–e530. doi: 10.1542/peds.2011-1900. [DOI] [PubMed] [Google Scholar]
- 78.Martin SR, Alvarez F, Anand R, Song C, Yin W. Outcomes in children who underwent transplantation for autoimmune hepatitis. Liver Transpl. 2011;17:393–401. doi: 10.1002/lt.22244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murray-Lyon IM, Stern RB, Williams R. Controlled trial of prednisone and azathioprine in active chronic hepatitis. Lancet. 1973;1:735–737. doi: 10.1016/s0140-6736(73)92125-9. [DOI] [PubMed] [Google Scholar]
- 80.Stellon AJ, Hegarty JE, Portmann B, Williams R. Randomised controlled trial of azathioprine withdrawal in autoimmune chronic active hepatitis. Lancet. 1985;1:668–670. doi: 10.1016/s0140-6736(85)91329-7. [DOI] [PubMed] [Google Scholar]
- 81.Cook GC, Mulligan R, Sherlock S. Controlled prospective trial of corticosteroid therapy in active chronic hepatitis. Q J Med. 1971;40:159–185. doi: 10.1093/oxfordjournals.qjmed.a067264. [DOI] [PubMed] [Google Scholar]
- 82.Maggiore G, Bernard O, Hadchouel M, Alagille D. Life-saving immunosuppressive treatment in severe autoimmune chronic active hepatitis. J Pediatr Gastroenterol Nutr. 1985;4:655–658. [PubMed] [Google Scholar]
- 83.Soloway RD, Summerskill WH, Baggenstoss AH, Geall MG, Gitnićk GL, Elveback IR, Schoenfield LJ. Clinical, biochemical, and histological remission of severe chronic active liver disease: a controlled study of treatments and early prognosis. Gastroenterology. 1972;63:820–833. [PubMed] [Google Scholar]
- 84.Mistilis SP, Skyring AP, Blackburn CR. Natural history of active chronic hepatitis. I. Clinical features, course, diagnostic criteria, morbidity, mortality and survival. Australas Ann Med. 1968;17:214–223. doi: 10.1111/imj.1968.17.3.214. [DOI] [PubMed] [Google Scholar]
- 85.Krawitt EL. Autoimmune hepatitis. N Engl J Med. 2006;354:54–66. doi: 10.1056/NEJMra050408. [DOI] [PubMed] [Google Scholar]
- 86.Feld JJ, Dinh H, Arenovich T, Marcus VA, Wanless IR, Heathcote EJ. Autoimmune hepatitis: effect of symptoms and cirrhosis on natural history and outcome. Hepatology. 2005;42:53–62. doi: 10.1002/hep.20732. [DOI] [PubMed] [Google Scholar]
- 87.Saadah OI, Smith AL, Hardikar W. Long-term outcome of autoimmune hepatitis in children. J Gastroenterol Hepatol. 2001;16:1297–1302. doi: 10.1046/j.1440-1746.2001.02615.x. [DOI] [PubMed] [Google Scholar]
- 88.Radhakrishnan KR, Alkhouri N, Worley S, Arrigain S, Hupertz V, Kay M, Yerian L, Wyllie R, Feldstein AE. Autoimmune hepatitis in children--impact of cirrhosis at presentation on natural history and long-term outcome. Dig Liver Dis. 2010;42:724–728. doi: 10.1016/j.dld.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 89.Deneau M, Book LS, Guthery SL, Jensen MK. Outcome after Discontinuation of Immunosuppression in Children with Autoimmune Hepatitis: A Population-Based Study. J Pediatr. 2014;164:714–719.e2. doi: 10.1016/j.jpeds.2013.12.008. [DOI] [PubMed] [Google Scholar]
- 90.Whitington PF, Vos MB, Bass LM, Melin-Aldana H, Romero R, Roy CC, Alvarez F. Humoral immune mechanism of liver injury in giant cell hepatitis with autoimmune hemolytic anemia. J Pediatr Gastroenterol Nutr. 2014;58:74–80. doi: 10.1097/MPG.0b013e3182a98dbe. [DOI] [PubMed] [Google Scholar]
- 91.Maggiore G, Sciveres M, Fabre M, Gori L, Pacifico L, Resti M, Choulot JJ, Jacquemin E, Bernard O. Giant cell hepatitis with autoimmune hemolytic anemia in early childhood: long-term outcome in 16 children. J Pediatr. 2011;159:127–132.e1. doi: 10.1016/j.jpeds.2010.12.050. [DOI] [PubMed] [Google Scholar]
- 92.Shores D, Kobak G, Pegram LD, Whitington PF, Shneider BL. Giant cell hepatitis and immune thrombocytopenic purpura: reversal of liver failure with rituximab therapy. J Pediatr Gastroenterol Nutr. 2012;55:e128–e130. doi: 10.1097/MPG.0b013e3182359002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lega S, Maschio M, Taddio A, Maggiore G, Ventura A. Giant cell hepatitis with Coombs-positive haemolytic anaemia: steroid sparing with high-dose intravenous immunoglobulin and cyclosporine. Acta Paediatr. 2013;102:e137–e139. doi: 10.1111/apa.12114. [DOI] [PubMed] [Google Scholar]
- 94.Czaja AJ. Autoantibody-negative autoimmune hepatitis. Dig Dis Sci. 2012;57:610–624. doi: 10.1007/s10620-011-2017-z. [DOI] [PubMed] [Google Scholar]
- 95.Gassert DJ, Garcia H, Tanaka K, Reinus JF. Corticosteroid-responsive cryptogenic chronic hepatitis: evidence for seronegative autoimmune hepatitis. Dig Dis Sci. 2007;52:2433–2437. doi: 10.1007/s10620-006-9665-4. [DOI] [PubMed] [Google Scholar]
- 96.Czaja AJ, Carpenter HA, Santrach PJ, Moore SB, Homburger HA. The nature and prognosis of severe cryptogenic chronic active hepatitis. Gastroenterology. 1993;104:1755–1761. doi: 10.1016/0016-5085(93)90656-w. [DOI] [PubMed] [Google Scholar]
- 97.Czaja AJ. Comparability of probable and definite autoimmune hepatitis by international diagnostic scoring criteria. Gastroenterology. 2011;140:1472–1480. doi: 10.1053/j.gastro.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 98.Czaja AJ, Rakela J, Ludwig J. Features reflective of early prognosis in corticosteroid-treated severe autoimmune chronic active hepatitis. Gastroenterology. 1988;95:448–453. doi: 10.1016/0016-5085(88)90503-3. [DOI] [PubMed] [Google Scholar]
- 99.Czaja AJ. Corticosteroids or not in severe acute or fulminant autoimmune hepatitis: therapeutic brinksmanship and the point beyond salvation. Liver Transpl. 2007;13:953–955. doi: 10.1002/lt.21088. [DOI] [PubMed] [Google Scholar]
- 100.Quail MA, Russell RK, Bellamy C, Mieli-Vergani G, Gillett PM. Seronegative autoimmune hepatitis presenting after diagnosis of coeliac disease: a case report. Eur J Gastroenterol Hepatol. 2009;21:576–579. doi: 10.1097/MEG.0b013e3282fa1400. [DOI] [PubMed] [Google Scholar]
- 101.Tosun MS, Ertekin V, Selimoğlu MA. Autoimmune hepatitis associated with celiac disease in childhood. Eur J Gastroenterol Hepatol. 2010;22:898–899. doi: 10.1097/MEG.0b013e32832faf09. [DOI] [PubMed] [Google Scholar]
- 102.El-Shabrawi M, El-Karaksy H, Mohsen N, Isa M, Al-Biltagi M, El-Ansari M. Celiac disease in children and adolescents with autoimmune hepatitis: a single-centre experience. J Trop Pediatr. 2011;57:104–108. doi: 10.1093/tropej/fmq057. [DOI] [PubMed] [Google Scholar]
- 103.Di Biase AR, Colecchia A, Scaioli E, Berri R, Viola L, Vestito A, Balli F, Festi D. Autoimmune liver diseases in a paediatric population with coeliac disease - a 10-year single-centre experience. Aliment Pharmacol Ther. 2010;31:253–260. doi: 10.1111/j.1365-2036.2009.04186.x. [DOI] [PubMed] [Google Scholar]
- 104.Clemente MG, Meloni A, Obermayer-Straub P, Frau F, Manns MP, De Virgiliis S. Two cytochromes P450 are major hepatocellular autoantigens in autoimmune polyglandular syndrome type 1. Gastroenterology. 1998;114:324–328. doi: 10.1016/s0016-5085(98)70484-6. [DOI] [PubMed] [Google Scholar]
- 105.Lankisch TO, Mourier O, Sokal EM, Habes D, Lacaille F, Bridoux-Henno L, Hermeziu B, Lenaerts C, Strassburg CP, Jacquemin E. AIRE gene analysis in children with autoimmune hepatitis type I or II. J Pediatr Gastroenterol Nutr. 2009;48:498–500. doi: 10.1097/MPG.0b013e31818550de. [DOI] [PubMed] [Google Scholar]
- 106.Kerkar N, Hadzić N, Davies ET, Portmann B, Donaldson PT, Rela M, Heaton ND, Vergani D, Mieli-Vergani G. De-novo autoimmune hepatitis after liver transplantation. Lancet. 1998;351:409–413. doi: 10.1016/S0140-6736(97)06478-7. [DOI] [PubMed] [Google Scholar]
- 107.Mieli-Vergani G, Vergani D. De novo autoimmune hepatitis after liver transplantation. J Hepatol. 2004;40:3–7. doi: 10.1016/j.jhep.2003.10.022. [DOI] [PubMed] [Google Scholar]
- 108.Andries S, Casamayou L, Sempoux C, Burlet M, Reding R, Bernard Otte J, Buts JP, Sokal E. Posttransplant immune hepatitis in pediatric liver transplant recipients: incidence and maintenance therapy with azathioprine. Transplantation. 2001;72:267–272. doi: 10.1097/00007890-200107270-00018. [DOI] [PubMed] [Google Scholar]
- 109.Hernandez HM, Kovarik P, Whitington PF, Alonso EM. Autoimmune hepatitis as a late complication of liver transplantation. J Pediatr Gastroenterol Nutr. 2001;32:131–136. doi: 10.1097/00005176-200102000-00007. [DOI] [PubMed] [Google Scholar]
- 110.Heneghan MA, Portmann BC, Norris SM, Williams R, Muiesan P, Rela M, Heaton ND, O’Grady JG. Graft dysfunction mimicking autoimmune hepatitis following liver transplantation in adults. Hepatology. 2001;34:464–470. doi: 10.1053/jhep.2001.26756. [DOI] [PubMed] [Google Scholar]
- 111.Gupta P, Hart J, Millis JM, Cronin D, Brady L. De novo hepatitis with autoimmune antibodies and atypical histology: a rare cause of late graft dysfunction after pediatric liver transplantation. Transplantation. 2001;71:664–668. doi: 10.1097/00007890-200103150-00016. [DOI] [PubMed] [Google Scholar]
- 112.Spada M, Bertani A, Sonzogni A, Petz W, Riva S, Torre G, Melzi ML, Alberti D, Colledan M, Segalin A, et al. A cause of late graft dysfunction after liver transplantation in children: de-novo autoimmune hepatitis. Transplant Proc. 2001;33:1747–1748. doi: 10.1016/s0041-1345(00)02826-8. [DOI] [PubMed] [Google Scholar]
- 113.Salcedo M, Vaquero J, Bañares R, Rodríguez-Mahou M, Alvarez E, Vicario JL, Hernández-Albújar A, Tíscar JL, Rincón D, Alonso S, et al. Response to steroids in de novo autoimmune hepatitis after liver transplantation. Hepatology. 2002;35:349–356. doi: 10.1053/jhep.2002.31167. [DOI] [PubMed] [Google Scholar]
- 114.Clemente MG, Vajro P, Musu MP, Cicotto L, Ciccimarra E, Mandato C, De Virgiliis S. Autoimmune manifestations in children transplanted for non- autoimmune liver diseases. J Hepatol. 2001;34:45 (abstract). [Google Scholar]
- 115.Petz W, Sonzogni A, Bertani A, Spada M, Lucianetti A, Colledan M, Gridelli B. A cause of late graft dysfunction after pediatric liver transplantation: de novo autoimmune hepatitis. Transplant Proc. 2002;34:1958–1959. doi: 10.1016/s0041-1345(02)03137-8. [DOI] [PubMed] [Google Scholar]
- 116.Gibelli NE, Tannuri U, Mello ES, Cançado ER, Santos MM, Ayoub AA, Maksoud-Filho JG, Velhote MC, Silva MM, Pinho-Apezzato ML, et al. Successful treatment of de novo autoimmune hepatitis and cirrhosis after pediatric liver transplantation. Pediatr Transplant. 2006;10:371–376. doi: 10.1111/j.1399-3046.2005.00470.x. [DOI] [PubMed] [Google Scholar]