

Abstract
It was estimated that from 2002 to 2008 the risk of developing cancer increased a quarter-fold in men and two-fold in women due to excessive BMI. Obesity, metabolic syndrome and type 2 diabetes mellitus are strictly related and are key pathogenetic factors of non-alcoholic fatty liver disease (NAFLD), the most frequent liver disease worldwide. The most important consequence of the “metabolic epidemics” is the probable rise in the incidence of hepatocarcinoma (HCC), and NAFLD is the major causative factor. Adipose tissue is not merely a storage organ where lipids are preserved as an energy source. It is an active organ with important endocrine, paracrine, and autocrine actions in addition to immune functions. Adipocytes produce a wide range of hormones, cytokines, and growth factors that can act locally in the adipose tissue microenvironment and systemically. In this article, the main roles of insulin growth factor (IGF)-1 and IGF-2 are discussed. The role of IGF-2 is not only confined to HCC, but it may also act in early hepato-carcinogenesis, as pre-neoplastic lesions express IGF-2 mRNA. IGF-1 and IGF-2 interact with specific receptors (IGF-1R and IGF-2R). IGF-1R is over-expressed in in vitro and in animal models of HCC and it was demonstrated that IGF ligands exerted their effects on HCC cells through IGF-1R and that it was involved in the degeneration of pre-neoplastic lesions via an increase in their mitotic activity. Both IGF-2R and TGF β, a growth inhibitor, levels are reduced in human HCC compared with adjacent normal liver tissues. Another key mechanism involves peroxisome proliferator-activated receptor (PPAR)γ. In in vitro studies, PPARγ inhibited various carcinomas including HCC, most probably by regulating apoptosis via the p21, p53 and p27 pathways. Finally, as a clinical consequence, to improve survival, efforts to achieve a “healthier diet” should be promoted by physicians and politicians.
Keywords: Hepatocarcinoma, Non-alcoholic fatty liver disease, Insulin growth factor, Peroxisome proliferator-activated receptor γ, Adipokines
Core tip: Obesity, metabolic syndrome and type 2 diabetes mellitus are strictly related and are key pathogenetic factors in non-alcoholic fatty liver disease, one of the most frequent liver diseases worldwide. It is necessary to stress that one of the most important consequences of the “metabolic epidemics” is the probable rise in the incidence of cancers, particularly hepatocarcinoma. Thus, to improve survival, efforts to achieve a “healthier diet” should be promoted by physicians and politicians, even though no changes in genes have been observed in the postprandial state induced after the acute effect of specific diets in patients exhibiting non-alcoholic fatty liver disease and insulin resistance.
COMMENTARY ON HOT TOPICS
Major risk factors for the development of hepatocellular carcinoma (HCC) are infection with hepatitis B virus (HBV) and hepatitis C virus (HCV), at least in developed countries. Moreover, the etiology of HCC is multifactorial and, in addition to infections, other factors such as alcohol consumption and intake of aflotoxin-contaminated food were found to contribute to HCC development[1]. Recently, it was determined that non-alcoholic fatty liver disease (NAFLD) is related to obesity, diabetes and metabolic syndrome (MS)[2]. NAFLD, the abnormal accumulation of fat in the liver, affects approximately 20%-30% of the total population in Western countries and the strict clinical and patho-physiological links with obesity, insulin resistance (IR) and type 2 diabetes mellitus (T2DM) have led to the recent suggestion that NAFLD can be considered a new criterion of MS[3]. HCC is the most common primary liver malignancy and is one of the most lethal cancers worldwide[4]. Epidemiologically, the incidence and mortality of HCC has increased over the past several decades in the United States, Japan and several European countries[4-7]. A correlation was noted between the raised incidence of HCC and obesity: overweight and obese individuals had an increased risk of developing HCC of 17% and 90%, respectively, compared to those with normal weight[8,9]. In 2013 an interesting study evaluated the performance status in HCC subjects and found that patients with worse performance status had not only higher Child-Turcotte-Pugh and Model for End-Stage Liver Disease scores, but also larger tumor volume, more frequent vascular invasion and T2DM (P < 0.001). T2DM was a prognostic predictor of increased risk of mortality. These data support the role of T2DM, not only in the genesis of HCC, but also in its progression and therefore its treatment[10]. An increased risk of developing HCC and having a worse prognosis after this diagnosis was demonstrated not only in T2DM patients, but also in obese subjects and those with the criteria of MS[11]. The purpose of our review is to summarize the principal findings on this issue focusing on the principal pathophysiologic aspects and epidemiologic evidence.
HCC DEVELOPMENT
In recent decades many authors have focused their attention on the prevalence and/or incidence of HCC in subjects affected by MS, T2DM and obesity. Despite very recent data showing an association between liver cancer and MS, at least in HBV-and HCV-endemic areas such as Southern Taiwan[12], in 2012 this association was questioned in five meta-analyses, which all concluded that both increased body mass index (BMI) and altered glucose metabolism influence the risk of developing HCC. A systematic literature search in 2012 identified five studies evaluating the relationship between HCC and MS, T2DM and obesity. The results of this meta-analysis are shown in Table 1[13-17].
Table 1.
Results of a meta-analysis of hepatocellular carcinoma and metabolic syndrome, type 2 diabetes mellitus and obesity in 2012
| Ref. | Database | Results |
| Esposito et al[13] | 43 articles, including 38940 cases of cancer | Metabolic syndrome was associated with liver cancer (relative risk 1.43, P < 0.0001), especially in Asian populations |
| Chen et al[14] (2012) | 26 prospective studies, including 25337 primary liver cancer cases | Overweight subjects have an increased risk of primary liver cancer of 1.48 (95%CI: 1.31-1.67), and obese subjects of 1.83 (95%CI: 1.59-2.11) |
| Tanaka et al[15] (2012) | 9 cohort studies on Japanese populations | Overweight/obese individuals had a relative risk of 1.74 (95%CI: 1.33-2.28) for developing liver cancer |
| Wang et al[16] (2012) | 17 case-control studies and 32 cohort studies | Statistically significant increased risk of HCC prevalence among diabetic individuals (RR = 2.31, 95%CI: 1.87-2.84). The pooled risk estimate of 17 case-control studies (OR = 2.40, 95%CI: 1.85-3.11) was slightly higher than that of 25 cohort studies (RR = 2.23, 95%CI: 1.68-2.96). Metformin treatment was potentially protective. Long duration of diabetes and sulfonylureas or insulin treatment possibly increased HCC risk. Increased risk of HCC mortality (RR = 2.43, 95%CI: 1.66-3.55) for individuals with (vs without) diabetes |
| Wang et al[17] (2012) | 25 cohort studies | Among these, 18 studies showed that DM was associated with an increased incidence of HCC (SRRs = 2.01, 95%CI: 1.61-2.51), compared with individuals without DM |
| There was a statistically significant heterogeneity among these studies (Q = 136.68, P < 0.001, I(2) = 87.6%). Analyses sub-grouped by controlling confounders revealed that the increased incidence of HCC was independent of geographic location, alcohol consumption, history of cirrhosis, or infections with hepatitis B (HBV) or hepatitis C virus (HCV). In addition, DM was also positively associated with HCC mortality (SRR = 1.56; 95%CI: 1.30-1.87) |
When examining the possible impact of lipid and glucose metabolism in the induction and progression of HCC, it should be remembered that there are many known risk factors associated with HCC, however, HCV and HBV infections are the most studied. Other known risk factors are alcohol abuse and toxin exposure (such as aflatoxin)[18]. In this scenario it is fundamental to emphasize that, even though the possibility of developing HCC in HBV or HCV subjects is higher than in patients affected by NAFLD or non-alcoholic steatohepatitis (NASH), it is also true that MS and its hepatic manifestation (NAFLD and then NASH) affects 20%-30% of the general population in Western countries, and its incidence is increasing (Table 2[19-30]). Based on these data, Siegel et al[21] estimated that 200000-500000 individuals are potentially at risk of developing HCC.
Table 2.
Principal risk factors for developing hepatocellular carcinoma, epidemiological evidence
| Risk factors for HCC | Incidence of HCC | Epidemiology |
| NAFLD | 1%-2% overall[19] | The prevalence of NAFLD in the general population of Western countries is 20%-30%[20] |
| NASH | 4%-27% overall[21] | About 2%-3% of the general population is estimated to have NASH[20] |
| HBV | 3%-8% overall[22] | Two billion people worldwide have been exposed to HBV, and 400 million people have chronic HBV infection[24] |
| 0%-5% per year[23] | ||
| HCV | 1%-7% overall[25] | About 150 million people worldwide are infected with HCV[28] |
| 2%-8% per year[26,27] | ||
| Alcohol | 1%-2% per year in cirrhotics[29] | The WHO reports about two billion alcohol consumers worldwide and 76.3 million people with diagnosable alcohol use disorders[30] |
HCC: Hepatocellular carcinoma; NAFLD: Non-alcoholic fatty liver disease; HCV: Hepatitis C virus; HBV: Hepatitis B virus; WHO: World Health Organization.
Epidemiological evidence concerning impaired glucose metabolism, insulin resistance and diabetes
In 2012, Campbell et al[31] conducted a large prospective cohort study which included over one million subjects and found that diabetic women and men had an increased risk of 1.40 (95%CI: 1.05-1.86) and 2.26 (95%CI: 1.89-2.70) for developing HCC, respectively. The increased incidence of HCC in obese and diabetic subjects has led to the necessity to characterize the role of MS in liver carcinogenesis. Glucose intolerance, hyperglycemia, T2DM, obesity, hypertension and dyslipidemia are the key components in MS[32]. Turati et al[33] carried out a case-control study in Southern Italy, and found not only an increased incidence of HCC in subjects affected by MS, but also that the risk increases with the number of MS criteria. Although the only two MS components associated with HCC were T2DM and obesity, the risk was increased four-fold in subjects with ≥ 2 MS components, and to over six-fold in subjects without chronic infection with HBV and/or HCV, compared with non-MS subjects.
Even if the role of impaired glucose metabolism is important in hepato-carcinogenesis, its impact on HCC natural history is still controversial. Recently, Howell et al[34] compared diabetic and-non diabetic subjects treated for HCC and found that there was no difference in survival between the two groups. In contrast, a meta-analysis by Wang et al[35] found that patients with coexisting T2DM had a shorter survival time and a higher risk for tumor recurrence after curative treatments. This evidence was subsequently confirmed and associated with macro-vascular invasion[36,37]. Moreover, Abe et al[38] demonstrated that intensive care of T2DM and abstinence from alcohol consumption improved prognosis in HCC patients. In some reports, authors deduced that in HIV/HCV positive subjects the presence of T2DM favors the progression of chronic liver disease to HCC[39-41]. These are only two examples in many studies to show that T2DM can worsen the prognosis of subjects with other known risk factors for the development of HCC.
Epidemiological evidence concerning obesity
The World Health Organization (WHO) estimates that more than 1.4 billion people are overweight (BMI > 25 kg/m2), and more than 500 million are obese (BMI ≥ 30 kg/m2) worldwide[42]. It was estimated that obesity in the United States is responsible for up to 20% of all cancer deaths in women and 14% in men[43]. A recent meta-analysis focused on the incidence of various malignancies across 30 European countries in relation to increased BMI (BMI ≥ 25 kg/m2) and found that in 2002 over 70000 new cancer cases were attributable to excess body fat[44]. This meta-analysis did not investigate the risk of developing HCC in obese subjects, but many other reports showed that, not only MS and T2DM, but also obesity, related or unrelated to other signs or symptoms of MS, is a risk factor for the development of HCC[45]. In a 14-year prospective cohort study based on more 1200000 Koreans, cancer of the liver was found to be the second most common cancer in obese men (after gallbladder cancer) and the third in obese women (after breast and pancreatic cancer), indicating a positive association between BMI and the frequency of other cancers[46]. When the principal results of cohort studies from 1966 to 2007 are summarized, the relative risks of HCC were 1.17 (95%CI: 1.02-1.34) for overweight and 1.89 (95%CI: 1.51-2.36) for obese subjects[47], similar to the data shown in a meta-analysis by Tanaka et al[15] in 2012.
An Italian study found that subjects with a BMI > 25 had an approximately two-fold increase in HCC risk (OR = 1.9, 95%CI: 0.9-3.9) compared to normal weight subjects and the risk rose when HbsAg-negative and HCV-negative subjects were investigated (OR = 3.5)[48].
In 2002, Nair et al[49] focused their attention on the possibility that obesity might have a negative impact on the prognosis of HCC patients treated with curative surgical therapy such as orthotopic liver transplantation (OLT). Obesity was an independent predictor of mortality after OLT, especially long-term survival which was significantly lower in obese subjects. The authors deduced that the increased incidence of cardiovascular events was the main reason for these results. In a subsequent study by Mathur et al[50], it was confirmed that obesity predicts a poorer outcome after OLT for HCC, and that recurrence of HCC was doubled in overweight and obese patients.
LINKS BETWEEN NAFLD AND HCC
The relationship between T2DM, MS and HCC seems to be closely related to NAFLD development. However, it is still controversial as to whether these diseases should be considered as risk factors for the development of HCC independently of the presence of NAFLD. The relationships between T2DM, MS and HCC seem to be closely related to NAFLD development. Recent studies showed that the majority of obese patients or those with MS develop NAFLD. In a single topic conference held by the AASLD, it was shown that up to 70% of T2DM and obese subjects exhibited various degrees of NAFLD[51]. In accordance with this statement, the occurrence of NAFLD in the absence of MS seems to be relatively uncommon. In a study by Marchesini et al[52], 18% of normal weight subjects developed NAFLD, however, this percentage rose dramatically in obese and MS subjects, reaching 67%. Logistic regression analysis demonstrated that the presence of MS carried a high risk of a more severe form of NAFLD, i.e., NASH, with an OR of 3.2 after correction for sex, age, and BMI.
Fujii et al[53] in 2013 published an interesting study on a new NASH-HCC animal model that demonstrated the relationship between NAFLD-NASH, T2DM, MS and HCC development. A group of healthy male mice were treated with streptozocin (STZ) and a high-fat diet (HFD) or with STZ only or no treatment, and a group of healthy female mice were treated with STZ and a HFD. The infusion of STZ early after birth led to pancreatic islet injury and to diabetes, in addition, hepato-cellular injury with pathological fat accumulation, increased lipogenesis and fatty acid oxidation was induced by the HFD. At 6 wk after the beginning of the study, SFD-HFD mice developed fatty liver and NASH after 8 wk. Pericellular fibrosis around central veins was noted at 8 and 12 wk. HCC developed in all STZ-HFD male mice after 16 wk, but not in STZ-HFD female mice or in STZ male mice. Comparing these results the authors concluded that T2DM predisposes to HCC, but liver inflammation, NASH, and fibrosis were interrelated processes and were essential for HCC evolution.
HCC develops in two thirds of cases against the background of chronic liver disease caused by HCV and/or HBV infections or by alcohol abuse or hemochromatosis[54,55]. A third of HCC patients are classified as having cryptogenic cirrhosis due to the absence of these underlying diseases. To date, MS seems to be the principal noxious stimuli causing the majority of cryptogenic cirrhosis cases[56]. Moreover, recent evidence has shown that obesity and T2DM have a negative prognostic impact on the natural history of HCC. As we previously mentioned, Turati et al[33] in a recent report found a high risk of developing HCC in subjects with at least one of the MS components, increasing to up to four-fold if two or more factors were present. Moreover, obesity increased the risk of developing HCC and HCC mortality by 2-5 times, and T2DM doubled the HCC risk independently of the presence of alcoholic liver disease, viral hepatitis, or other demographic variables. A subsequent report also found that, in patients with viral hepatitis (HCV or HBV), the simultaneous presence of obesity and T2DM multiplied this risk by 100-fold[57].
Even in the non-fibrotic liver, MS seems to be a risk factor for the development of HCC, similar to that in the cirrhotic liver. Kawada et al[58] enrolled a total of 1168 patients with HCC and found that HCC developed in 75% of cases with non-cirrhotic liver affected by NASH (6 of 8 NASH subjects). This result was of poor significance due to the small number of examined cases. However, in 2012 Baffy et al[59] in an interesting review reported that from 2004 to 2011 at least 116 cases of HCC had been demonstrated in histologically-confirmed NAFLD without cirrhosis. It seems that simple MS and NAFLD, in the absence of NASH, can promote the development of HCC, as shown by Guzman et al[60].
MECHANISMS IN DETERMINING HCC
Major role of the IGF pathway
Hyperglycemia exerts deleterious metabolic effects on the liver, disrupting glucose, lipid, bile acid and triglyceride metabolism and leading to the impairment of several cellular processes[61-63]. It should be stressed that IR leads to an inflammatory systemic state, with hyper-production of pro-inflammatory cytokines such as TNFα and IL-6, and some adipokines such as leptin, whereas other molecules with anti-inflammatory properties are decreased, such as adiponectin[64]. These findings are not surprising as IR is almost always associated with obesity, and both are fundamental components of MS. In this sense, it is obvious that IR and obesity, through the development of a systemic chronic inflammatory state, leads to the promotion of inflammation and fibrosis in the liver, which are prodromal signs of hepato-carcinogenesis[65]. Decreased adiponectin level is almost always found in subjects with IR and in subjects with increased BMI in the setting of NASH and is linked to the development of hepatic fibrosis[66].
Moreover, IR and hyperinsulinemia can up-regulate the production of insulin-like growth factor-1 (IGF-1) and generally deregulate the insulin-like growth factor (IGF) pathway[67].
The aberrant activation of growth factor signaling pathways is an important mechanism in the development and progression of HCC. The most studied growth factor signaling pathways are the transforming growth factor α (TGF-α)/EGF-R, transforming growth factor β (TGF β)/TβR, hepatocyte growth factor/MET and wingless (Wnt/frizzled/β-catenin)-signaling pathways[68]. Coupled with these, dysregulation of the IGF pathway, involved in the fine regulation of proliferation and anti-apoptosis of HCC cells, is of critical importance[69,70] (Figure 1[70]).
Figure 1.
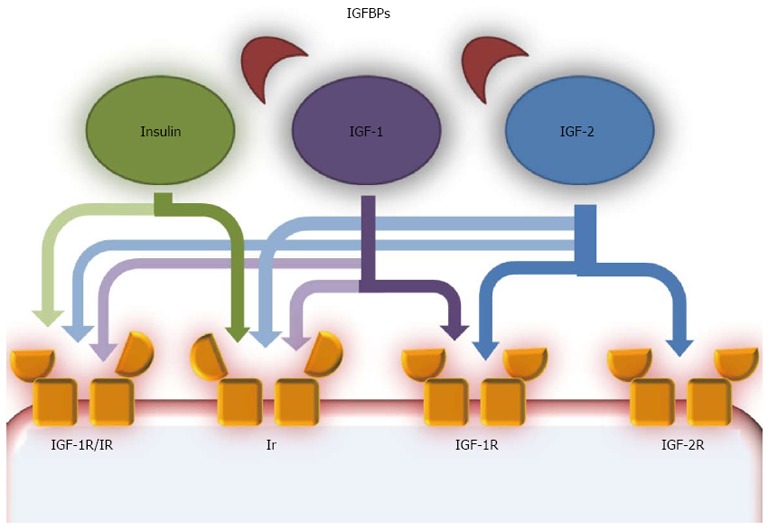
Insulin growth factor pathway scheme. The insulin-like growth factor (IGF) pathway is composed of several components. IGF ligands are IGF-1 and IGF-2, two peptides that share high similarities with insulin. A family of carrier proteins, called insulin-like growth factor binding proteins (IGFBPs), bind IGF-1 and IGF-2 in the blood. IGF-1 and IGF-2 act as autocrine, paracrine and endocrine growth factors and are mostly produced in the liver, especially IGF-1 in the postnatal era and IGF-2 during fetal development. They can act on various receptors, but have higher affinity with IGF-1R, a tyrosine kinase receptor structurally similar to the insulin receptor (Ir). IGF-1 can also bind Ir, but with lower affinity than IGF-1R, while IGF-2 binds Ir only during fetal development. IGF-2R is structurally similar to IGF-1R, but binds only IGF-2 and most probably acts on this growth hormone with an inhibitory effect, as a clearance site. Another receptor is a hybrid receptor consisting of insulin and IGF-1R hemireceptors which preferentially binds IGF-1, while insulin does not have optimal binding[70].
Many studies have demonstrated that IGF-1 mRNA was under-expressed in HCC tissues[71], while in animal models and in humans with HCC, IGF-2 was overexpressed[70]. These molecules are mostly produced in the liver and it was proposed that the reduction in IGF-1 production may be correlated with reduced hepatic function, due to the simultaneous presence of chronic liver disease or cirrhosis, or with the reduced expression of growth hormone receptors in tumoral tissues[71,72].
A separate discussion should be included for IGF-2. Physiologically the expression of IGF-2 is restricted during fetal development. A mono-allelic, maternally imprinted, IGF-2 is produced and in adulthood it is replaced by a bi-allelic form[72]. During hepato-carcinogenesis, reactivation of the fetal promoter leads to the overexpression of fetal IGF-2[73]. IGF-2 has a stimulatory effect on cell proliferation[74] and angiogenesis[75], in addition, it has an anti-apoptotic effect and its concentrations are positively associated with the expression of vascular endothelial growth factor (VEGF) in in vitro studies[76]. The role of IGF-2 is not only confined to HCC, but it may also act in early hepato-carcinogenesis, as pre-neoplastic lesions express IGF-2 mRNA[77,78].
To determine their physiological and pathological functions, IGF-1 and IGF-2 need to interact with specific receptors (IGF-1R and IGF-2R). IGF-1R is overexpressed in in vitro and in animal models of HCC[72,74] and it was demonstrated that IGF ligands exerted their effects on HCC cells through IGF-1R, and that it was involved in the degeneration of pre-neoplastic lesions via an increase in their mitotic activity[79]. IGF-2R is a clearance site for IGF-2, so it may exert inhibitory effects on the IGF pathway[80]. IGF-2R was under-expressed in in vitro studies, animal models and human HCC[70]. The levels of IGF-2R and TGFβ, another growth inhibitor, are reduced in human HCC compared with adjacent normal liver tissues[81]. From all of these findings, it is easy to imagine that the IGF substrates, IRS-1 and IRS-2, are both able to promote HCC formation and progression. Conversely, IGF-binding proteins (IGFBPs), due to the reduced bio-availability of free IGF-1 and IGF-2 in the bloodstream, seem to be able to inhibit the growth of HCC. Indeed IGFBP-3 and IGFBP-7 levels are decreased in HCC tissues and the addition of these molecules in HCC models reduces the growth and volume of hepatic tumor formation[70].
On the basis of the evidence linking IR and T2DM to HCC, in the last decade many studies have tried to analyze the possible role of anti-diabetic drugs in HCC therapy. Recently Singh et al[82] conducted a meta-analysis and concluded that some agents, such as metformin, can reduce the risk of HCC, while others, such as insulin, can increase the risk of HCC. In the same report, thiazolidinediones (TZD) did not seem to modify the risk, but several in vitro[83] and animal[84] studies have found that these molecules might act as regulators of the cell cycle, inhibiting HCC growth.
Role of transcriptional factors
TZD acts on peroxisome proliferator-activated receptors (PPAR). PPARα, γ, and δ are members of the nuclear receptor superfamily of ligand-activated transcription factors that have central roles in the storage and catabolism of fatty acids. Each of the three PPAR subtypes is expressed in a distinct, tissue-specific pattern. PPARα is highly expressed in the liver, heart, kidney, skeletal muscle, and brown adipose tissues which are metabolically very active. PPARγ is most highly expressed in white and brown adipose tissue, large intestine, and spleen. In contrast to PPARα and PPARy, which are abundantly expressed in just a few tissues, PPARδ is virtually expressed in all tissues at comparable levels. Another important function of PPARs is to regulate the cell cycle, and due to this property, they are involved in carcinogenesis[85].
PPARα seems to inhibit carcinogenesis, having anti-angiogenetic activity, via the production of thrombospondin, and anti-inflammatory properties (it suppresses interleukin 1β, TNF and ICAM1 expression)[86-88]. On the other hand, the stimulation of PPARα by agonists induces hepatomegaly and HCC following long-term therapy in animal models[89]. Physiologic stimulation of PPARα may suppress cancer through the modulation of microenvironment and microcirculation, however, its continuous abnormal stimulation may promote hepato-carcinogenesis[90]. PPARγ is overexpressed in fatty liver and is constitutionally expressed in adipose tissue and macrophages[91]. There are contrasting data on its role in hepato-carcinogenesis as it is over-, normally or under-expressed in various studies[83,92,93]. In in vitro studies, PPARγ inhibits various carcinomas including HCC, most probably by regulating apoptosis via the p21, p53 and p27 pathways[90]. A recent study by Pang et al[94] confirmed the inhibitory action of PPARγ on hepato-carcinogenesis, via the up-regulation of plasminogen activator factor 1 (PAI 1).
Role of pro-inflammatory and anti-inflammatory molecules
Adipose tissue is not merely a storage organ where lipids are preserved as an energy source. It is an active organ with important endocrine, paracrine, and autocrine actions as well as immune functions. Adipocytes produce a wide range of hormones, cytokines, and growth factors that can act locally in the adipose tissue microenvironment and systemically[95]. Pathologic hypertrophy and hyperplasia of adipocytes that can be found in obese subjects lead to a dysregulation of many endocrine functions with important consequences[96]. The maturation of adipocytes is impaired in obesity. This leads to a relative increase in pre-adipocyte populations and consequently a modification in the local microenvironment. Pre-adipocytes produce cytokines with pro-inflammatory and angiogenetic properties, ideal for their own proliferation. Cytokines overproduced in adipocytes during obesity include interleukin (IL)-6, Il-8, Il-1β, tumor necrosis factor α (TNFα), VEGF, and chemokine ligand 2 and 5 (CCL2 and CCL5), which can promote the recruitment of immune cells and vasculogenesis[97]. Moreover, the production of proinflammatory and chemoattractant molecules by adipose tissue leads to macrophage recruitment and activation, which are able to increase the chronic low-grade inflammatory response[98]. The pro-inflammatory state is also able to self-renew as many of the overexpressed cytokines, such as TNFα, IL-1β, TGFβ and interferon γ, are able to block the maturation of pre-adipocytes to adipocytes. The low-grade chronic inflammatory state was linked to IR in obese mice[99], and in particular TNFα seems to play a fundamental role by inhibiting the tyrosine phosphorylation of Ir[100] and enhancing the production of another important pro-inflammatory cytokine, IL-6[101]. Shimizu et al[102] recently reviewed the principal evidence linking obesity, inflammation and HCC development.
Two important adiponectins are involved in this process. Adiponectin has anti-angiogenetic and anti-proliferative properties, but is also under-expressed in obese subjects in favor of an increased expression of leptin[103,104]. Levels of adiponectin are inversely proportional to BMI[105], thus, hypo-adiponectinemia is associated with the development of most consequences of obesity, such as cerebrovascular diseases and dyslipidemia[106]. Animal models showed that hypo-adiponectinemia could have a role in hepato-carcinogenesis, especially when inflammation and necrosis are present (NASH)[107,108]. Studies on humans have shown that adiponectin might be useful as a prognostic factor in HCC as its serum concentration seems to be positively correlated with a poorer prognosis[109,110], even if in early HCC it seems to have less relevance[111].
Complex role of leptin, visfatin and PAI-1
Leptin is a protein encoded by the ob gene[112] involved in the regulation of body weight and energy balance and it is produced mainly by adipocytes[113]. Obese patients have increased levels of circulating leptin, and are also resistant to its activity[103]. In the liver, leptin prevents lipid accumulation and lipotoxicity[114]. On the other hand, animal models have shown that it has pro-fibrogenic properties, increasing the expression of procollagen I, TGF-β1, smooth muscle actin and increasing the production of tissue inhibitor of metalloprotease 1 in activated hepatic stellate cells (HSCs)[115,116], the principal players in liver fibrosis. Moreover, leptin promotes HSC proliferation inhibiting their apoptosis[117], and activated HSCs are themselves able to produce leptin[118]. This adipokine production, and consequently its action, is impaired not only in obese, but also in NAFLD subjects with or without NASH, even if the relationships between its serum levels are, to date, not correlated to the severity of liver fibrosis, as expected due to its fibrogenic properties[114]. The possible role of leptin in hepato-carcinogenesis is likely due to its pro-inflammatory[119,120] and pro-fibrogenic[121] actions, however, recent studies have also found that increased leptin levels are associated with the induction of VEGF in HSCs[122] and with venous invasion in renal cancer[123,124], suggesting a possible similar role in HCC. However, there are many contrasting data on this issue. The immune-modulator effect of leptin may lead to natural killer cell proliferation and activation and consequently to a reduction in tumor size[125]. Two subsequent studies by Wang et al[126,127] support the affirmation that leptin may have positive prognostic significance in HCC, improving overall survival.
Although leptin and adiponectin are the most studied adipokines, adipose tissue produces a wide range of molecules classified in this group. Some cytokines (TNFα, TGFβ, IL-1β, IL-6, IL-8, IL-10), chemokines and acute phase proteins (haptoglobin, serum amyloid-A, and PAI-1) are classified in this group[128]. As we previously described, they are involved in the initiation and progression of the systemic, chronic inflammatory status found in obese subjects, which in turn is related to HCC pathogenesis. In 1999, a study found that PAI-1 could inhibit the invasion and proliferation of HCC cells in vitro[129]. This evidence contrasts with the concept that PAI-1 is overexpressed in obese subjects and that higher concentrations of this adipokine worsen IR and impair lipid metabolism[130]. Recently, the first theory was confirmed in a report that correlated inhibition of HCC progression with PPARγ stimulation via PAI-1 activation[94]. Conversely, visfatin, another adipokine, seems to have a negative impact on hepato-carcinogenesis, indicating that adipokines have different roles in HCC progression[131].
CONTRASTING EVIDENCE
Chiang et al[132] in an up-to-date prospective cohort study based on nationwide health screening units, found that T2DM (adjusted HR = 3.38) was positively associated with deaths due to HCC. Surprisingly, hypertriglyceridemia (HR = 0.38) and hypercholesterolemia (HR = 0.50) were inversely associated with HCC mortality. MS, as defined by the American Heart Association/National Heart Lung Blood Institute criteria (HR = 0.63) or by the International Diabetes Federation criteria (HR = 0.62), was inversely associated with deaths due to HCC, especially in men[132]. This finding has not been proved by another contemporary systemic review and meta-analysis analyzing four studies (3 cohorts and 1 case control) with a total of 829651 participants. The age range of the participants was between 30 and 84 years. The combined analysis showed an overall 81% increased risk of HCC in cases with MS (relative risk, 1.81)[133].
CONCLUSION
The global cancer burden doubled in the last thirty years of the twentieth century, and it is estimated that it will similarly increase between 2000 and 2020 and nearly triple by 2030[134]. In 2005, the WHO produced a document which emphasized that more than 50% of total cancers developed in countries where obesity was a prominent risk factor[135]. Subsequently, it was estimated that from 2002 to 2008 the risk of developing cancer increased a quarter-fold in men and two-fold in women due to excessive BMI. Obesity, MS and T2DM are strictly related and are key pathogenetic factors of NAFLD, one of the most frequent liver diseases worldwide.
It is of crucial relevance to remember that one of the most important consequences of the “metabolic epidemics” is the probable rise in the incidence of cancers, such as HCC[70,80], even though other causes inducing HCC should not be neglected (Figure 2). Therefore, to improve survival[136], efforts to achieve a “healthier diet” should be promoted by physicians and politicians, even though no changes in genes were observed in the postprandial state induced after the acute effect of specific diets in patients exhibiting NAFLD and IR, who showed peripheral adipose tissue dysfunction and exhibited inappropriately low leptin biosynthesis[137]. Finally, new treatments for T2DM are expected to reduce the future burden of T2DM-related HCC[138].
Figure 2.
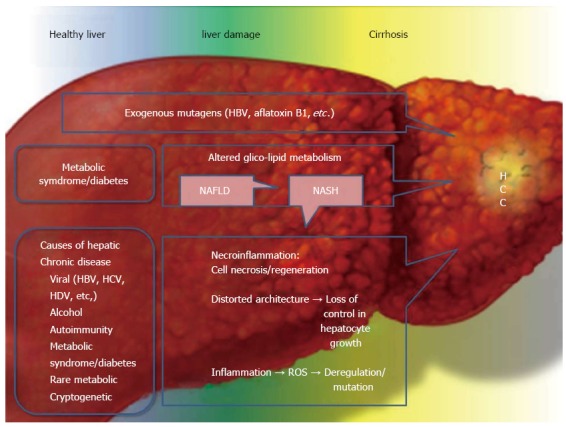
Main causes and concomitant causes of hepatocellular carcinoma, focusing on the metabolic syndrome and type 2 diabetes mellitus. Hepatocellular carcinoma (HCC) development is often the final step in liver damage during chronic liver disease. Classically the step between a “normal” liver and HCC is liver cirrhosis. Liver cirrhosis causes disruption of the delicate and complex hepatic architecture. The formation of cirrhotic nodules has, per se, an important role in HCC development as the hepatic architecture is chronically replaced by fibrous tissue. This process is on the one hand, an attempt to repair the liver damage, and on the other hand, it leads to an increased hepatocellular turn-over with impaired vascularization, increased necrosis, apoptosis and inflammation. The final step is loss of control and the formation of malignant hepatic cells. It is easily understood that almost all causes of chronic liver damage, resulting in cirrhosis, can develop HCC. Recently type 2 diabetes (T2DM), MS and obesity have been identified as risk factors for HCC in the presence or absence of cirrhosis. In summary, these diseases are able to increase and accelerate the chronic necroinflammatory process present in cirrhotic liver, but their action is also important in the first steps of chronic liver damage. The altered metabolism of gluco-lipids leads to fat accumulation in the liver, causing non-alcoholic fatty liver disease (NAFLD). Fat is an important inducer of inflammation and can alter the normal cellular turn-over acting both locally in the liver and systemically via paracrine, autocrine and endocrine actions. Nonalcoholic steatohepatitis (NASH) is an eventual evolution of NAFLD and it is possible that not only NASH, with necroinflammation leading to cirrhosis, but also “simple” fatty liver can induce cancer development, as a first step, in the liver. This point merits further investigation. A separate discussion needs to be included for the so called “mutagens”. HBV and aflatoxin β1 are the two most important mutagens involved in HCC development and in the non-cirrhotic liver. HBV: Hepatitis B virus; HCV: Hepatitis C virus; HDV: Hepatitis D virus.
Footnotes
P- Reviewer: Nseir WB, Puy RV S- Editor: Qi Y L- Editor: Webster JR E- Editor: Zhang DN
References
- 1.McGlynn KA, London WT. The global epidemiology of hepatocellular carcinoma: present and future. Clin Liver Dis. 2011;15:223–243, vii-x. doi: 10.1016/j.cld.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao J, Xie L, Yang WS, Zhang W, Gao S, Wang J, Xiang YB. Risk factors of hepatocellular carcinoma--current status and perspectives. Asian Pac J Cancer Prev. 2012;13:743–752. doi: 10.7314/apjcp.2012.13.3.743. [DOI] [PubMed] [Google Scholar]
- 3.Tarantino G, Finelli C. What about non-alcoholic fatty liver disease as a new criterion to define metabolic syndrome? World J Gastroenterol. 2013;19:3375–3384. doi: 10.3748/wjg.v19.i22.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Llovet JM, Burroughs A, Bruix J. Hepatocellular carcinoma. Lancet. 2003;362:1907–1917. doi: 10.1016/S0140-6736(03)14964-1. [DOI] [PubMed] [Google Scholar]
- 5.Eheman C, Henley SJ, Ballard-Barbash R, Jacobs EJ, Schymura MJ, Noone AM, Pan L, Anderson RN, Fulton JE, Kohler BA, et al. Annual Report to the Nation on the status of cancer, 1975-2008, featuring cancers associated with excess weight and lack of sufficient physical activity. Cancer. 2012;118:2338–2366. doi: 10.1002/cncr.27514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.La Vecchia C, Lucchini F, Franceschi S, Negri E, Levi F. Trends in mortality from primary liver cancer in Europe. Eur J Cancer. 2000;36:909–915. doi: 10.1016/s0959-8049(00)00052-6. [DOI] [PubMed] [Google Scholar]
- 7.Shibuya K, Yano E. Regression analysis of trends in mortality from hepatocellular carcinoma in Japan, 1972-2001. Int J Epidemiol. 2005;34:397–402. doi: 10.1093/ije/dyh358. [DOI] [PubMed] [Google Scholar]
- 8.Blonski W, Kotlyar DS, Forde KA. Non-viral causes of hepatocellular carcinoma. World J Gastroenterol. 2010;16:3603–3615. doi: 10.3748/wjg.v16.i29.3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clark JM. The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006;40 Suppl 1:S5–10. doi: 10.1097/01.mcg.0000168638.84840.ff. [DOI] [PubMed] [Google Scholar]
- 10.Hsu CY, Lee YH, Hsia CY, Huang YH, Su CW, Lin HC, Lee RC, Chiou YY, Lee FY, Huo TI. Performance status in patients with hepatocellular carcinoma: determinants, prognostic impact, and ability to improve the Barcelona Clinic Liver Cancer system. Hepatology. 2013;57:112–119. doi: 10.1002/hep.25950. [DOI] [PubMed] [Google Scholar]
- 11.Duan XF, Tang P, Li Q, Yu ZT. Obesity, adipokines and hepatocellular carcinoma. Int J Cancer. 2013;133:1776–1783. doi: 10.1002/ijc.28105. [DOI] [PubMed] [Google Scholar]
- 12.Chen CT, Chen JY, Wang JH, Chang KC, Tseng PL, Kee KM, Chen PF, Tsai LS, Chen SC, Lin SC, et al. Diabetes mellitus, metabolic syndrome and obesity are not significant risk factors for hepatocellular carcinoma in an HBV- and HCV-endemic area of Southern Taiwan. Kaohsiung J Med Sci. 2013;29:451–459. doi: 10.1016/j.kjms.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 13.Esposito K, Chiodini P, Colao A, Lenzi A, Giugliano D. Metabolic syndrome and risk of cancer: a systematic review and meta-analysis. Diabetes Care. 2012;35:2402–2411. doi: 10.2337/dc12-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Y, Wang X, Wang J, Yan Z, Luo J. Excess body weight and the risk of primary liver cancer: an updated meta-analysis of prospective studies. Eur J Cancer. 2012;48:2137–2145. doi: 10.1016/j.ejca.2012.02.063. [DOI] [PubMed] [Google Scholar]
- 15.Tanaka K, Tsuji I, Tamakoshi A, Matsuo K, Ito H, Wakai K, Nagata C, Mizoue T, Sasazuki S, Inoue M, et al. Obesity and liver cancer risk: an evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jpn J Clin Oncol. 2012;42:212–221. doi: 10.1093/jjco/hyr198. [DOI] [PubMed] [Google Scholar]
- 16.Wang P, Kang D, Cao W, Wang Y, Liu Z. Diabetes mellitus and risk of hepatocellular carcinoma: a systematic review and meta-analysis. Diabetes Metab Res Rev. 2012;28:109–122. doi: 10.1002/dmrr.1291. [DOI] [PubMed] [Google Scholar]
- 17.Wang C, Wang X, Gong G, Ben Q, Qiu W, Chen Y, Li G, Wang L. Increased risk of hepatocellular carcinoma in patients with diabetes mellitus: a systematic review and meta-analysis of cohort studies. Int J Cancer. 2012;130:1639–1648. doi: 10.1002/ijc.26165. [DOI] [PubMed] [Google Scholar]
- 18.Mittal S, El-Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol. 2013;47 Suppl:S2–S6. doi: 10.1097/MCG.0b013e3182872f29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matteoni CA, Younossi ZM, Gramlich T, Boparai N, Liu YC, McCullough AJ. Nonalcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology. 1999;116:1413–1419. doi: 10.1016/s0016-5085(99)70506-8. [DOI] [PubMed] [Google Scholar]
- 20.Bellentani S, Scaglioni F, Marino M, Bedogni G. Epidemiology of non-alcoholic fatty liver disease. Dig Dis. 2010;28:155–161. doi: 10.1159/000282080. [DOI] [PubMed] [Google Scholar]
- 21.Siegel AB, Zhu AX. Metabolic syndrome and hepatocellular carcinoma: two growing epidemics with a potential link. Cancer. 2009;115:5651–5661. doi: 10.1002/cncr.24687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fattovich G. Natural history and prognosis of hepatitis B. Semin Liver Dis. 2003;23:47–58. doi: 10.1055/s-2003-37590. [DOI] [PubMed] [Google Scholar]
- 23.Beasley RP, Hwang LY. Epidemiology of hepatocellular carcinoma in viral hepatitis and liver disease. Vyas AN, Dienstag JL, Hoofuagle JH, editors. New York: Grune and Stratton; 1984. pp. 209–224. [Google Scholar]
- 24. Available from: http://www.who.int/mediacentre/factsheets/fs204/en/
- 25.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132:2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 26.Niederau C, Lange S, Heintges T, Erhardt A, Buschkamp M, Hürter D, Nawrocki M, Kruska L, Hensel F, Petry W, et al. Prognosis of chronic hepatitis C: results of a large, prospective cohort study. Hepatology. 1998;28:1687–1695. doi: 10.1002/hep.510280632. [DOI] [PubMed] [Google Scholar]
- 27.Degos F, Christidis C, Ganne-Carrie N, Farmachidi JP, Degott C, Guettier C, Trinchet JC, Beaugrand M, Chevret S. Hepatitis C virus related cirrhosis: time to occurrence of hepatocellular carcinoma and death. Gut. 2000;47:131–136. doi: 10.1136/gut.47.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Available from: http://www.who.int/mediacentre/factsheets/fs164/en/
- 29.Morgan TR, Mandayam S, Jamal MM. Alcohol and hepatocellular carcinoma. Gastroenterology. 2004;127:S87–S96. doi: 10.1053/j.gastro.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 30.World Health Organization. Global Status Report on Alcohol 2004. Geneva, Switzerland: Dept. of Mental Health and Substance Abuse; 2004. p. 88. [Google Scholar]
- 31.Campbell PT, Newton CC, Patel AV, Jacobs EJ, Gapstur SM. Diabetes and cause-specific mortality in a prospective cohort of one million U.S. adults. Diabetes Care. 2012;35:1835–1844. doi: 10.2337/dc12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuzawa Y, Funahashi T, Nakamura T. The concept of metabolic syndrome: contribution of visceral fat accumulation and its molecular mechanism. J Atheroscler Thromb. 2011;18:629–639. doi: 10.5551/jat.7922. [DOI] [PubMed] [Google Scholar]
- 33.Turati F, Talamini R, Pelucchi C, Polesel J, Franceschi S, Crispo A, Izzo F, La Vecchia C, Boffetta P, Montella M. Metabolic syndrome and hepatocellular carcinoma risk. Br J Cancer. 2013;108:222–228. doi: 10.1038/bjc.2012.492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Howell J, Yiu M, Gibson R, Thomson B, Stella D, Gorelik A, Prichard PJ, Nicoll AJ. Type 2 diabetes does not worsen prognosis in hepatocellular carcinoma. Clin Res Hepatol Gastroenterol. 2011;35:214–220. doi: 10.1016/j.clinre.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 35.Wang WM, Xu Y, Yang XR, Wang YH, Sun HX, Fan J. Prognostic role of diabetes mellitus in hepatocellular carcinoma patients after curative treatments: a meta-analysis. Hepatobiliary Pancreat Dis Int. 2011;10:346–355. doi: 10.1016/s1499-3872(11)60059-3. [DOI] [PubMed] [Google Scholar]
- 36.Connolly GC, Safadjou S, Kashyap R, Chen R, Orloff MS, Hezel AF. Diabetes mellitus impacts risk of macrovascular invasion in patients undergoing transplantation for hepatocellular carcinoma. BMC Gastroenterol. 2013;13:9. doi: 10.1186/1471-230X-13-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ting CT, Chen RC, Chen CC, Liu MH, Chu D, Kuo NW. Diabetes worsens the surgical outcomes in cirrhotic patients with hepatocellular carcinoma. Tohoku J Exp Med. 2012;227:73–81. doi: 10.1620/tjem.227.73. [DOI] [PubMed] [Google Scholar]
- 38.Abe H, Aida Y, Ishiguro H, Yoshizawa K, Miyazaki T, Itagaki M, Sutoh S, Aizawa Y. Alcohol, postprandial plasma glucose, and prognosis of hepatocellular carcinoma. World J Gastroenterol. 2013;19:78–85. doi: 10.3748/wjg.v19.i1.78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kuske L, Mensen A, Müllhaupt B, Negro F, Semela D, Moradpour D, Malé PJ, Heim MH, Malinverni R, Cerny A, et al. Characteristics of patients with chronic hepatitis C who develop hepatocellular carcinoma. Swiss Med Wkly. 2012;142:w13651. doi: 10.4414/smw.2012.13651. [DOI] [PubMed] [Google Scholar]
- 40.Arase Y, Kobayashi M, Suzuki F, Suzuki Y, Kawamura Y, Akuta N, Kobayashi M, Sezaki H, Saito S, Hosaka T, et al. Effect of type 2 diabetes on risk for malignancies includes hepatocellular carcinoma in chronic hepatitis C. Hepatology. 2013;57:964–973. doi: 10.1002/hep.26087. [DOI] [PubMed] [Google Scholar]
- 41.Salmon D, Bani-Sadr F, Loko MA, Stitou H, Gervais A, Durant J, Rosenthal E, Quertainmont Y, Barange K, Vittecoq D, et al. Insulin resistance is associated with a higher risk of hepatocellular carcinoma in cirrhotic HIV/HCV-co-infected patients: results from ANRS CO13 HEPAVIH. J Hepatol. 2012;56:862–868. doi: 10.1016/j.jhep.2011.11.009. [DOI] [PubMed] [Google Scholar]
- 42. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/
- 43.Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625–1638. doi: 10.1056/NEJMoa021423. [DOI] [PubMed] [Google Scholar]
- 44.Renehan AG, Soerjomataram I, Tyson M, Egger M, Zwahlen M, Coebergh JW, Buchan I. Incident cancer burden attributable to excess body mass index in 30 European countries. Int J Cancer. 2010;126:692–702. doi: 10.1002/ijc.24803. [DOI] [PubMed] [Google Scholar]
- 45.Schlesinger S, Aleksandrova K, Pischon T, Fedirko V, Jenab M, Trepo E, Boffetta P, Dahm CC, Overvad K, Tjønneland A, et al. Abdominal obesity, weight gain during adulthood and risk of liver and biliary tract cancer in a European cohort. Int J Cancer. 2013;132:645–657. doi: 10.1002/ijc.27645. [DOI] [PubMed] [Google Scholar]
- 46.Jee SH, Yun JE, Park EJ, Cho ER, Park IS, Sull JW, Ohrr H, Samet JM. Body mass index and cancer risk in Korean men and women. Int J Cancer. 2008;123:1892–1896. doi: 10.1002/ijc.23719. [DOI] [PubMed] [Google Scholar]
- 47.Larsson SC, Wolk A. Overweight, obesity and risk of liver cancer: a meta-analysis of cohort studies. Br J Cancer. 2007;97:1005–1008. doi: 10.1038/sj.bjc.6603932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Polesel J, Zucchetto A, Montella M, Dal Maso L, Crispo A, La Vecchia C, Serraino D, Franceschi S, Talamini R. The impact of obesity and diabetes mellitus on the risk of hepatocellular carcinoma. Ann Oncol. 2009;20:353–357. doi: 10.1093/annonc/mdn565. [DOI] [PubMed] [Google Scholar]
- 49.Nair S, Verma S, Thuluvath PJ. Obesity and its effect on survival in patients undergoing orthotopic liver transplantation in the United States. Hepatology. 2002;35:105–109. doi: 10.1053/jhep.2002.30318. [DOI] [PubMed] [Google Scholar]
- 50.Mathur A, Franco ES, Leone JP, Osman-Mohamed H, Rojas H, Kemmer N, Neff GW, Rosemurgy AS, Alsina AE. Obesity portends increased morbidity and earlier recurrence following liver transplantation for hepatocellular carcinoma. HPB (Oxford) 2013;15:504–510. doi: 10.1111/j.1477-2574.2012.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic steatohepatitis: summary of an AASLD Single Topic Conference. Hepatology. 2003;37:1202–1219. doi: 10.1053/jhep.2003.50193. [DOI] [PubMed] [Google Scholar]
- 52.Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- 53.Fujii M, Shibazaki Y, Wakamatsu K, Honda Y, Kawauchi Y, Suzuki K, Arumugam S, Watanabe K, Ichida T, Asakura H, et al. A murine model for non-alcoholic steatohepatitis showing evidence of association between diabetes and hepatocellular carcinoma. Med Mol Morphol. 2013;46:141–152. doi: 10.1007/s00795-013-0016-1. [DOI] [PubMed] [Google Scholar]
- 54.Davila JA, Morgan RO, Shaib Y, McGlynn KA, El-Serag HB. Hepatitis C infection and the increasing incidence of hepatocellular carcinoma: a population-based study. Gastroenterology. 2004;127:1372–1380. doi: 10.1053/j.gastro.2004.07.020. [DOI] [PubMed] [Google Scholar]
- 55.Franceschi S, Raza SA. Epidemiology and prevention of hepatocellular carcinoma. Cancer Lett. 2009;286:5–8. doi: 10.1016/j.canlet.2008.10.046. [DOI] [PubMed] [Google Scholar]
- 56.Davis GL, Dempster J, Meler JD, Orr DW, Walberg MW, Brown B, Berger BD, O’Connor JK, Goldstein RM. Hepatocellular carcinoma: management of an increasingly common problem. Proc (Bayl Univ Med Cent) 2008;21:266–280. doi: 10.1080/08998280.2008.11928410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen CL, Yang HI, Yang WS, Liu CJ, Chen PJ, You SL, Wang LY, Sun CA, Lu SN, Chen DS, et al. Metabolic factors and risk of hepatocellular carcinoma by chronic hepatitis B/C infection: a follow-up study in Taiwan. Gastroenterology. 2008;135:111–121. doi: 10.1053/j.gastro.2008.03.073. [DOI] [PubMed] [Google Scholar]
- 58.Kawada N, Imanaka K, Kawaguchi T, Tamai C, Ishihara R, Matsunaga T, Gotoh K, Yamada T, Tomita Y. Hepatocellular carcinoma arising from non-cirrhotic nonalcoholic steatohepatitis. J Gastroenterol. 2009;44:1190–1194. doi: 10.1007/s00535-009-0112-0. [DOI] [PubMed] [Google Scholar]
- 59.Baffy G, Brunt EM, Caldwell SH. Hepatocellular carcinoma in non-alcoholic fatty liver disease: an emerging menace. J Hepatol. 2012;56:1384–1391. doi: 10.1016/j.jhep.2011.10.027. [DOI] [PubMed] [Google Scholar]
- 60.Guzman G, Brunt EM, Petrovic LM, Chejfec G, Layden TJ, Cotler SJ. Does nonalcoholic fatty liver disease predispose patients to hepatocellular carcinoma in the absence of cirrhosis? Arch Pathol Lab Med. 2008;132:1761–1766. doi: 10.5858/132.11.1761. [DOI] [PubMed] [Google Scholar]
- 61.Evans JL, Goldfine ID, Maddux BA, Grodsky GM. Oxidative stress and stress-activated signaling pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev. 2002;23:599–622. doi: 10.1210/er.2001-0039. [DOI] [PubMed] [Google Scholar]
- 62.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, Fruchart JC, Kuipers F, Staels B. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–898. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 63.Chandrasekaran K, Swaminathan K, Chatterjee S, Dey A. Apoptosis in HepG2 cells exposed to high glucose. Toxicol In Vitro. 2010;24:387–396. doi: 10.1016/j.tiv.2009.10.020. [DOI] [PubMed] [Google Scholar]
- 64.Hashimoto E, Tokushige K. Hepatocellular carcinoma in non-alcoholic steatohepatitis: Growing evidence of an epidemic? Hepatol Res. 2012;42:1–14. doi: 10.1111/j.1872-034X.2011.00872.x. [DOI] [PubMed] [Google Scholar]
- 65.Fierbinteanu-Braticevici C, Negreanu L, Tarantino G. Is fatty liver always benign and should not consequently be treated? J Physiol Pharmacol. 2013;64:3–9. [PubMed] [Google Scholar]
- 66.Gannagé-Yared MH, Khalife S, Semaan M, Fares F, Jambart S, Halaby G. Serum adiponectin and leptin levels in relation to the metabolic syndrome, androgenic profile and somatotropic axis in healthy non-diabetic elderly men. Eur J Endocrinol. 2006;155:167–176. doi: 10.1530/eje.1.02175. [DOI] [PubMed] [Google Scholar]
- 67.Ish-Shalom D, Christoffersen CT, Vorwerk P, Sacerdoti-Sierra N, Shymko RM, Naor D, De Meyts P. Mitogenic properties of insulin and insulin analogues mediated by the insulin receptor. Diabetologia. 1997;40 Suppl 2:S25–S31. doi: 10.1007/s001250051393. [DOI] [PubMed] [Google Scholar]
- 68.Villanueva A, Newell P, Chiang DY, Friedman SL, Llovet JM. Genomics and signaling pathways in hepatocellular carcinoma. Semin Liver Dis. 2007;27:55–76. doi: 10.1055/s-2006-960171. [DOI] [PubMed] [Google Scholar]
- 69.Breuhahn K, Schirmacher P. Reactivation of the insulin-like growth factor-II signaling pathway in human hepatocellular carcinoma. World J Gastroenterol. 2008;14:1690–1698. doi: 10.3748/wjg.14.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu J, Zhu AX. Targeting insulin-like growth factor axis in hepatocellular carcinoma. J Hematol Oncol. 2011;4:30. doi: 10.1186/1756-8722-4-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su TS, Liu WY, Han SH, Jansen M, Yang-Fen TL, P’eng FK, Chou CK. Transcripts of the insulin-like growth factors I and II in human hepatoma. Cancer Res. 1989;49:1773–1777. [PubMed] [Google Scholar]
- 72.Scharf JG, Dombrowski F, Ramadori G. The IGF axis and hepatocarcinogenesis. Mol Pathol. 2001;54:138–144. doi: 10.1136/mp.54.3.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tovar V, Alsinet C, Villanueva A, Hoshida Y, Chiang DY, Solé M, Thung S, Moyano S, Toffanin S, Mínguez B, et al. IGF activation in a molecular subclass of hepatocellular carcinoma and pre-clinical efficacy of IGF-1R blockage. J Hepatol. 2010;52:550–559. doi: 10.1016/j.jhep.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Scharf JG, Schmidt-Sandte W, Pahernik SA, Ramadori G, Braulke T, Hartmann H. Characterization of the insulin-like growth factor axis in a human hepatoma cell line (PLC) Carcinogenesis. 1998;19:2121–2128. doi: 10.1093/carcin/19.12.2121. [DOI] [PubMed] [Google Scholar]
- 75.Bae MH, Lee MJ, Bae SK, Lee OH, Lee YM, Park BC, Kim KW. Insulin-like growth factor II (IGF-II) secreted from HepG2 human hepatocellular carcinoma cells shows angiogenic activity. Cancer Lett. 1998;128:41–46. doi: 10.1016/s0304-3835(98)00044-5. [DOI] [PubMed] [Google Scholar]
- 76.Yao N, Yao D, Wang L, Dong Z, Wu W, Qiu L, Yan X, Yu D, Chen J, Sai W, et al. Inhibition of autocrine IGF-II on effect of human HepG2 cell proliferation and angiogenesis factor expression. Tumour Biol. 2012;33:1767–1776. doi: 10.1007/s13277-012-0436-x. [DOI] [PubMed] [Google Scholar]
- 77.Breuhahn K, Vreden S, Haddad R, Beckebaum S, Stippel D, Flemming P, Nussbaum T, Caselmann WH, Haab BB, Schirmacher P. Molecular profiling of human hepatocellular carcinoma defines mutually exclusive interferon regulation and insulin-like growth factor II overexpression. Cancer Res. 2004;64:6058–6064. doi: 10.1158/0008-5472.CAN-04-0292. [DOI] [PubMed] [Google Scholar]
- 78.Lahm H, Gittner K, Krebs O, Sprague L, Deml E, Oesterle D, Hoeflich A, Wanke R, Wolf E. Diethylnitrosamine induces long-lasting re-expression of insulin-like growth factor II during early stages of liver carcinogenesis in mice. Growth Horm IGF Res. 2002;12:69–79. doi: 10.1054/ghir.2002.0261. [DOI] [PubMed] [Google Scholar]
- 79.Yao WF, Liu JW, Sheng GL, Huang DS. Blockade of IGF-IR exerts anticancer effects in hepatocellular carcinoma. Mol Med Rep. 2011;4:719–722. doi: 10.3892/mmr.2011.486. [DOI] [PubMed] [Google Scholar]
- 80.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8:915–928. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 81.Uchida K, Kondo M, Takeda S, Osada H, Takahashi T, Nakao A, Takahashi T. Altered transcriptional regulation of the insulin-like growth factor 2 gene in human hepatocellular carcinoma. Mol Carcinog. 1997;18:193–198. [PubMed] [Google Scholar]
- 82.Singh S, Singh PP, Singh AG, Murad MH, Sanchez W. Anti-diabetic medications and the risk of hepatocellular cancer: a systematic review and meta-analysis. Am J Gastroenterol. 2013;108:881–91; quiz 892. doi: 10.1038/ajg.2013.5. [DOI] [PubMed] [Google Scholar]
- 83.Yu J, Qiao L, Zimmermann L, Ebert MP, Zhang H, Lin W, Röcken C, Malfertheiner P, Farrell GC. Troglitazone inhibits tumor growth in hepatocellular carcinoma in vitro and in vivo. Hepatology. 2006;43:134–143. doi: 10.1002/hep.20994. [DOI] [PubMed] [Google Scholar]
- 84.Borbath I, Leclercq I, Moulin P, Sempoux C, Horsmans Y. The PPARgamma agonist pioglitazone inhibits early neoplastic occurrence in the rat liver. Eur J Cancer. 2007;43:1755–1763. doi: 10.1016/j.ejca.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 85.Kliewer SA, Xu HE, Lambert MH, Willson TM. Peroxisome proliferator-activated receptors: from genes to physiology. Recent Prog Horm Res. 2001;56:239–263. doi: 10.1210/rp.56.1.239. [DOI] [PubMed] [Google Scholar]
- 86.Grau R, Punzón C, Fresno M, Iñiguez MA. Peroxisome-proliferator-activated receptor alpha agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem J. 2006;395:81–88. doi: 10.1042/BJ20050964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Panigrahy D, Kaipainen A, Huang S, Butterfield CE, Barnés CM, Fannon M, Laforme AM, Chaponis DM, Folkman J, Kieran MW. PPARalpha agonist fenofibrate suppresses tumor growth through direct and indirect angiogenesis inhibition. Proc Natl Acad Sci USA. 2008;105:985–990. doi: 10.1073/pnas.0711281105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Pineda Torra I, Gervois P, Staels B. Peroxisome proliferator-activated receptor alpha in metabolic disease, inflammation, atherosclerosis and aging. Curr Opin Lipidol. 1999;10:151–159. doi: 10.1097/00041433-199904000-00009. [DOI] [PubMed] [Google Scholar]
- 89.Reddy JK, Rao S, Moody DE. Hepatocellular carcinomas in acatalasemic mice treated with nafenopin, a hypolipidemic peroxisome proliferator. Cancer Res. 1976;36:1211–1217. [PubMed] [Google Scholar]
- 90.Kimura O, Kondo Y, Shimosegawa T. PPAR Could Contribute to the Pathogenesis of Hepatocellular Carcinoma. PPAR Res. 2012;2012:574180. doi: 10.1155/2012/574180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Willson TM, Lambert MH, Kliewer SA. Peroxisome proliferator-activated receptor gamma and metabolic disease. Annu Rev Biochem. 2001;70:341–367. doi: 10.1146/annurev.biochem.70.1.341. [DOI] [PubMed] [Google Scholar]
- 92.Schaefer KL, Wada K, Takahashi H, Matsuhashi N, Ohnishi S, Wolfe MM, Turner JR, Nakajima A, Borkan SC, Saubermann LJ. Peroxisome proliferator-activated receptor gamma inhibition prevents adhesion to the extracellular matrix and induces anoikis in hepatocellular carcinoma cells. Cancer Res. 2005;65:2251–2259. doi: 10.1158/0008-5472.CAN-04-3037. [DOI] [PubMed] [Google Scholar]
- 93.Koga H, Sakisaka S, Harada M, Takagi T, Hanada S, Taniguchi E, Kawaguchi T, Sasatomi K, Kimura R, Hashimoto O, et al. Involvement of p21(WAF1/Cip1), p27(Kip1), and p18(INK4c) in troglitazone-induced cell-cycle arrest in human hepatoma cell lines. Hepatology. 2001;33:1087–1097. doi: 10.1053/jhep.2001.24024. [DOI] [PubMed] [Google Scholar]
- 94.Pang X, Wei Y, Zhang Y, Zhang M, Lu Y, Shen P. Peroxisome proliferator-activated receptor-γ activation inhibits hepatocellular carcinoma cell invasion by upregulating plasminogen activator inhibitor-1. Cancer Sci. 2013;104:672–680. doi: 10.1111/cas.12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rajala MW, Scherer PE. Minireview: The adipocyte--at the crossroads of energy homeostasis, inflammation, and atherosclerosis. Endocrinology. 2003;144:3765–3773. doi: 10.1210/en.2003-0580. [DOI] [PubMed] [Google Scholar]
- 96.Gesta S, Tseng YH, Kahn CR. Developmental origin of fat: tracking obesity to its source. Cell. 2007;131:242–256. doi: 10.1016/j.cell.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 97.Gilbert CA, Slingerland JM. Cytokines, obesity, and cancer: new insights on mechanisms linking obesity to cancer risk and progression. Annu Rev Med. 2013;64:45–57. doi: 10.1146/annurev-med-121211-091527. [DOI] [PubMed] [Google Scholar]
- 98.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112:1796–1808. doi: 10.1172/JCI19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87–91. doi: 10.1126/science.7678183. [DOI] [PubMed] [Google Scholar]
- 100.Hotamisligil GS, Budavari A, Murray D, Spiegelman BM. Reduced tyrosine kinase activity of the insulin receptor in obesity-diabetes. Central role of tumor necrosis factor-alpha. J Clin Invest. 1994;94:1543–1549. doi: 10.1172/JCI117495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Osterreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010;140:197–208. doi: 10.1016/j.cell.2009.12.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Shimizu M, Tanaka T, Moriwaki H. Obesity and hepatocellular carcinoma: targeting obesity-related inflammation for chemoprevention of liver carcinogenesis. Semin Immunopathol. 2013;35:191–202. doi: 10.1007/s00281-012-0336-6. [DOI] [PubMed] [Google Scholar]
- 103.Kamada Y, Takehara T, Hayashi N. Adipocytokines and liver disease. J Gastroenterol. 2008;43:811–822. doi: 10.1007/s00535-008-2213-6. [DOI] [PubMed] [Google Scholar]
- 104.Marra F, Bertolani C. Adipokines in liver diseases. Hepatology. 2009;50:957–969. doi: 10.1002/hep.23046. [DOI] [PubMed] [Google Scholar]
- 105.Kadowaki T, Yamauchi T, Kubota N, Hara K, Ueki K, Tobe K. Adiponectin and adiponectin receptors in insulin resistance, diabetes, and the metabolic syndrome. J Clin Invest. 2006;116:1784–1792. doi: 10.1172/JCI29126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen MP, Tsai JC, Chung FM, Yang SS, Hsing LL, Shin SJ, Lee YJ. Hypoadiponectinemia is associated with ischemic cerebrovascular disease. Arterioscler Thromb Vasc Biol. 2005;25:821–826. doi: 10.1161/01.ATV.0000157784.25920.a7. [DOI] [PubMed] [Google Scholar]
- 107.Fukushima J, Kamada Y, Matsumoto H, Yoshida Y, Ezaki H, Takemura T, Saji Y, Igura T, Tsutsui S, Kihara S, et al. Adiponectin prevents progression of steatohepatitis in mice by regulating oxidative stress and Kupffer cell phenotype polarization. Hepatol Res. 2009;39:724–738. doi: 10.1111/j.1872-034X.2009.00509.x. [DOI] [PubMed] [Google Scholar]
- 108.Asano T, Watanabe K, Kubota N, Gunji T, Omata M, Kadowaki T, Ohnishi S. Adiponectin knockout mice on high fat diet develop fibrosing steatohepatitis. J Gastroenterol Hepatol. 2009;24:1669–1676. doi: 10.1111/j.1440-1746.2009.06039.x. [DOI] [PubMed] [Google Scholar]
- 109.Sadik NA, Ahmed A, Ahmed S. The significance of serum levels of adiponectin, leptin, and hyaluronic acid in hepatocellular carcinoma of cirrhotic and noncirrhotic patients. Hum Exp Toxicol. 2012;31:311–321. doi: 10.1177/0960327111431091. [DOI] [PubMed] [Google Scholar]
- 110.Wang SN, Yang SF, Tsai HH, Lee KT, Yeh YT. Increased adiponectin associated with poor survival in hepatocellular carcinoma. J Gastroenterol. 2013:Epub ahead of print. doi: 10.1007/s00535-013-0898-7. [DOI] [PubMed] [Google Scholar]
- 111.Miuma S, Ichikawa T, Taura N, Shibata H, Takeshita S, Akiyama M, Motoyoshi Y, Ozawa E, Fujimoto M, Kawashimo H, et al. The level of fasting serum insulin, but not adiponectin, is associated with the prognosis of early stage hepatocellular carcinoma. Oncol Rep. 2009;22:1415–1424. doi: 10.3892/or_00000583. [DOI] [PubMed] [Google Scholar]
- 112.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425–432. doi: 10.1038/372425a0. [DOI] [PubMed] [Google Scholar]
- 113.Jéquier E. Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci. 2002;967:379–388. doi: 10.1111/j.1749-6632.2002.tb04293.x. [DOI] [PubMed] [Google Scholar]
- 114.Chitturi S, Farrell G, Frost L, Kriketos A, Lin R, Fung C, Liddle C, Samarasinghe D, George J. Serum leptin in NASH correlates with hepatic steatosis but not fibrosis: a manifestation of lipotoxicity? Hepatology. 2002;36:403–409. doi: 10.1053/jhep.2002.34738. [DOI] [PubMed] [Google Scholar]
- 115.Cao Q, Mak KM, Ren C, Lieber CS. Leptin stimulates tissue inhibitor of metalloproteinase-1 in human hepatic stellate cells: respective roles of the JAK/STAT and JAK-mediated H2O2-dependant MAPK pathways. J Biol Chem. 2004;279:4292–4304. doi: 10.1074/jbc.M308351200. [DOI] [PubMed] [Google Scholar]
- 116.Cao Q, Mak KM, Lieber CS. Leptin enhances alpha1(I) collagen gene expression in LX-2 human hepatic stellate cells through JAK-mediated H2O2-dependent MAPK pathways. J Cell Biochem. 2006;97:188–197. doi: 10.1002/jcb.20622. [DOI] [PubMed] [Google Scholar]
- 117.Saxena NK, Titus MA, Ding X, Floyd J, Srinivasan S, Sitaraman SV, Anania FA. Leptin as a novel profibrogenic cytokine in hepatic stellate cells: mitogenesis and inhibition of apoptosis mediated by extracellular regulated kinase (Erk) and Akt phosphorylation. FASEB J. 2004;18:1612–1614. doi: 10.1096/fj.04-1847fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ikejima K, Takei Y, Honda H, Hirose M, Yoshikawa M, Zhang YJ, Lang T, Fukuda T, Yamashina S, Kitamura T, et al. Leptin receptor-mediated signaling regulates hepatic fibrogenesis and remodeling of extracellular matrix in the rat. Gastroenterology. 2002;122:1399–1410. doi: 10.1053/gast.2002.32995. [DOI] [PubMed] [Google Scholar]
- 119.Faggioni R, Feingold KR, Grunfeld C. Leptin regulation of the immune response and the immunodeficiency of malnutrition. FASEB J. 2001;15:2565–2571. doi: 10.1096/fj.01-0431rev. [DOI] [PubMed] [Google Scholar]
- 120.La Cava A, Matarese G. The weight of leptin in immunity. Nat Rev Immunol. 2004;4:371–379. doi: 10.1038/nri1350. [DOI] [PubMed] [Google Scholar]
- 121.Kitade M, Yoshiji H, Kojima H, Ikenaka Y, Noguchi R, Kaji K, Yoshii J, Yanase K, Namisaki T, Asada K, et al. Leptin-mediated neovascularization is a prerequisite for progression of nonalcoholic steatohepatitis in rats. Hepatology. 2006;44:983–991. doi: 10.1002/hep.21338. [DOI] [PubMed] [Google Scholar]
- 122.Aleffi S, Petrai I, Bertolani C, Parola M, Colombatto S, Novo E, Vizzutti F, Anania FA, Milani S, Rombouts K, et al. Upregulation of proinflammatory and proangiogenic cytokines by leptin in human hepatic stellate cells. Hepatology. 2005;42:1339–1348. doi: 10.1002/hep.20965. [DOI] [PubMed] [Google Scholar]
- 123.Liao LM, Schwartz K, Pollak M, Graubard BI, Li Z, Ruterbusch J, Rothman N, Davis F, Wacholder S, Colt J, et al. Serum leptin and adiponectin levels and risk of renal cell carcinoma. Obesity (Silver Spring) 2013;21:1478–1485. doi: 10.1002/oby.20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li L, Gao Y, Zhang LL, He DL. Concomitant activation of the JAK/STAT3 and ERK1/2 signaling is involved in leptin-mediated proliferation of renal cell carcinoma Caki-2 cells. Cancer Biol Ther. 2008;7:1787–1792. doi: 10.4161/cbt.7.11.6837. [DOI] [PubMed] [Google Scholar]
- 125.Elinav E, Abd-Elnabi A, Pappo O, Bernstein I, Klein A, Engelhardt D, Rabbani E, Ilan Y. Suppression of hepatocellular carcinoma growth in mice via leptin, is associated with inhibition of tumor cell growth and natural killer cell activation. J Hepatol. 2006;44:529–536. doi: 10.1016/j.jhep.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 126.Wang SN, Yeh YT, Yang SF, Chai CY, Lee KT. Potential role of leptin expression in hepatocellular carcinoma. J Clin Pathol. 2006;59:930–934. doi: 10.1136/jcp.2005.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wang SN, Chuang SC, Yeh YT, Yang SF, Chai CY, Chen WT, Kuo KK, Chen JS, Lee KT. Potential prognostic value of leptin receptor in hepatocellular carcinoma. J Clin Pathol. 2006;59:1267–1271. doi: 10.1136/jcp.2005.033464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Trayhurn P, Bing C, Wood IS. Adipose tissue and adipokines--energy regulation from the human perspective. J Nutr. 2006;136:1935S–1939S. doi: 10.1093/jn/136.7.1935S. [DOI] [PubMed] [Google Scholar]
- 129.Morita Y, Hayashi Y, Kanamaru T, Itoh T, Suzuki S, Yamamoto M, Kuroda Y, Itoh H. Inhibitory role of plasminogen activator inhibitor-1 in invasion and proliferation of HLE hepatocellular carcinoma cells. Jpn J Cancer Res. 1999;90:747–752. doi: 10.1111/j.1349-7006.1999.tb00810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Liang X, Kanjanabuch T, Mao SL, Hao CM, Tang YW, Declerck PJ, Hasty AH, Wasserman DH, Fogo AB, Ma LJ. Plasminogen activator inhibitor-1 modulates adipocyte differentiation. Am J Physiol Endocrinol Metab. 2006;290:E103–E113. doi: 10.1152/ajpendo.00605.2004. [DOI] [PubMed] [Google Scholar]
- 131.Ninomiya S, Shimizu M, Imai K, Takai K, Shiraki M, Hara T, Tsurumi H, Ishizaki S, Moriwaki H. Possible role of visfatin in hepatoma progression and the effects of branched-chain amino acids on visfatin-induced proliferation in human hepatoma cells. Cancer Prev Res (Phila) 2011;4:2092–2100. doi: 10.1158/1940-6207.CAPR-11-0340. [DOI] [PubMed] [Google Scholar]
- 132.Chiang CH, Lee LT, Hung SH, Lin WY, Hung HF, Yang WS, Sung PK, Huang KC. Opposite association between diabetes, dyslipidemia, and hepatocellular carcinoma mortality in the middle-aged and elderly. Hepatology. 2014;59:2207–2215. doi: 10.1002/hep.27014. [DOI] [PubMed] [Google Scholar]
- 133.Jinjuvadia R, Patel S, Liangpunsakul S. The association between metabolic syndrome and hepatocellular carcinoma: systemic review and meta-analysis. J Clin Gastroenterol. 2014;48:172–177. doi: 10.1097/MCG.0b013e3182a030c4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Boyle P, Levin B. World Cancer Report 2008. In: Press W, editor. Geneva: IARC Nonserial Publication; 2008. [Google Scholar]
- 135.World Health Organization. Global action against cancer. Geneva: World Health Organization Press; 2005. [Google Scholar]
- 136.Finelli C, Sommella L, Gioia S, La Sala N, Tarantino G. Should visceral fat be reduced to increase longevity? Ageing Res Rev. 2013;12:996–1004. doi: 10.1016/j.arr.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 137.Paniagua JA, Escandell-Morales JM, Gil-Contreras D, Berral de la Rosa FJ, Romero-Jimenez M, Gómez-Urbano A, Sanchez-Lopez A, Bellido E, Poyato A, Calatayud B, et al. Central obesity and altered peripheral adipose tissue gene expression characterize the NAFLD patient with insulin resistance: Role of nutrition and insulin challenge. Nutrition. 2014;30:177–185. doi: 10.1016/j.nut.2013.07.017. [DOI] [PubMed] [Google Scholar]
- 138.Marchesini G, Forlani G. Diabetes and hepatocellular cancer risk: not only a matter of hyperglycemia. Hepatology. 2012;55:1298–1300. doi: 10.1002/hep.25646. [DOI] [PubMed] [Google Scholar]