

Abstract
AIM: To determine the alterations in rat enterocyte mitochondrial respiratory function and enzyme activities following traumatic brain injury (TBI).
METHODS: Fifty-six male SD rats were randomly divided into seven groups (8 rats in each group): a control group (rats with sham operation) and traumatic brain injury groups at 6, 12, 24 h, days 2, 3, and 7 after operation. TBI models were induced by Feendy’s free-falling method. Mitochondrial respiratory function (respiratory control ratio and ADP/O ratio) was measured with a Clark oxygen electrode. The activities of respiratory chain complex I-IV and related enzymes were determined by spectrophotometry.
RESULTS: Compared with the control group, the mitochondrial respiratory control ratio (RCR) declined at 6 h and remained at a low level until day 7 after TBI (control, 5.42 ± 0.46; 6 h, 5.20 ± 0.18; 12 h, 4.55 ± 0.35; 24 h, 3.75 ± 0.22; 2 d, 4.12 ± 0.53; 3 d, 3.45 ± 0.41; 7 d, 5.23 ± 0.24, P < 0.01). The value of phosphate-to-oxygen (P/O) significantly decreased at 12, 24 h, day 2 and day 3, respectively (12 h, 3.30 ± 0.10; 24 h, 2.61 ± 0.21; 2 d, 2.95 ± 0.18; 3 d, 2.76 ± 0.09, P < 0.01) compared with the control group (3.46 ± 0.12). Two troughs of mitochondrial respiratory function were seen at 24 h and day 3 after TBI. The activities of mitochondrial complex I (6 h: 110 ± 10, 12 h: 115 ± 12, 24 h: 85 ± 9, day 2: 80 ± 15, day 3: 65 ± 16, P < 0.01) and complex II (6 h: 105 ± 8, 12 h: 110 ± 92, 24 h: 80 ± 10, day 2: 76 ± 8, day 3: 68 ± 12, P < 0.01) were increased at 6 h and 12 h following TBI, and then significantly decreased at 24 h, day 2 and day 3, respectively. However, there were no differences in complex I and II activities between the control and TBI groups. Furthermore, pyruvate dehydrogenase (PDH) activity was significantly decreased at 6 h and continued up to 7 d after TBI compared with the control group (6 h: 90 ± 8, 12 h: 85 ± 10, 24 h: 65 ± 12, day 2: 60 ± 9, day 3: 55 ± 6, day 7: 88 ± 11, P < 0.01). The changes in α-ketoglutaric dehydrogenase (KGDH) activity were similar to PDH, except that the decrease in KGDH activity began at 12 h after TBI (12 h: 90 ± 12, 24 h: 80 ± 9, day 2: 76 ± 15, day 3: 68 ± 7, day 7: 90 ± 13, P < 0.01). No significant change in malate dehydrogenase (MDH) activity was observed.
CONCLUSION: Rat enterocyte mitochondrial respiratory function and enzyme activities are inhibited following TBI. Mitochondrial dysfunction may play an important role in TBI-induced gastrointestinal dysfunction.
Keywords: Mitochondria, Brain injury, Enterocyte, Rats, Malate dehydrogenase
Core tip: Many researchers over the years have attempted to reveal the possible mechanism involved in gastrointestinal dysfunction following traumatic brain injury (TBI). Mitochondria are thought to be the primary target of oxidative damage and play an important role in oxidative stress. However, alterations in rat enterocyte mitochondrial respiratory function and enzyme activities following TBI have not been described previously. The purpose of this study was to determine the effect of TBI on rat enterocyte mitochondrial respiratory function and enzyme activities, as well as to reveal the alterations in rat enterocyte mitochondrial function following TBI.
INTRODUCTION
Traumatic brain injury (TBI) is an increasing problem worldwide due to its high mortality and disability. There are a number of complications after TBI, one of which is gastrointestinal dysfunction[1]. Conversely, gastrointestinal dysfunction may lead to malnutrition, electrolyte imbalances, translocation of bacteria, and even systemic inflammatory response syndrome (SIRS)[2-4]. These complications delay recovery and increase mortality in patients with TBI. Many researchers over the years have attempted to reveal the possible mechanism involved in gastrointestinal dysfunction following TBI. Most previous studies have focused on the microstructure of gut mucosa, mucosal barrier disruption, mucosal blood flow, inflammatory mediators, and cytokines[5-8]. However, the exact mechanism of gastrointestinal dysfunction following TBI is still unknown.
Ischemia or hypoperfusion of intestinal mucosa often occurs after TBI in order to perfuse vital organs such as the heart and brain. When TBI occurs, there is decreased blood supply to the intestinal mucosa and intestinal mucosal epithelial cells are under high pressure due to oxidative stress[9]. Unfortunately, the energy store in the intestinal mucosa is too small to cope with ischemia or hypoperfusion, and intestinal epithelial cells are very sensitive to ischemia and hypoxia. A large number of free radicals are generated which attack the intestinal epithelial cells and destroy intestinal mucosal barrier function in the presence of ischemia. Mitochondria are an important part of oxidative phosphorylation and energy supply. In addition, they also generate reactive oxygen species (ROS) and the bulk of mitochondrial ROS are generated at the electron transport chain[10,11]. For that reason, mitochondria are thought to be the primary target of oxidative damage and play an important role in oxidative stress. However, the alterations in rat enterocyte mitochondrial respiratory function and enzyme activities following TBI have not been previously described.
The purpose of this study was to determine the effect of TBI on rat enterocyte mitochondrial respiratory function and enzyme activities, as well as to reveal the mechanisms involved in the alterations in rat enterocyte mitochondrial function following TBI.
MATERIALS AND METHODS
Rat models of TBI
Fifty-six male Sprague-Dawley rats, weighing 220-250 g, were provided by the Experimental Animal Center of Soochow University and randomly divided into seven groups (8 rats in each group): a control group (rats with sham operation) and TBI groups at 6, 12, 24 h, day 2, day 3, and day 7 after operation. The TBI models were induced as described by Feendy[12]. Briefly, rats were anesthetized and fixed in a stereotactic frame after a 12-h overnight fast. A right parietal bone window (diameter 5 mm) was drilled under aseptic conditions just behind the cranial coronal suture and beside the midline. A freefalling weight consisting of a steel rod weighing 40 g with a flat end diameter of 4 mm was allowed to fall onto a piston resting on the dura from a height of 25 cm to produce a standardized parietal contusion in the exposed intact cranial dura. The animals were sacrificed at the appropriate time points and tissue specimens were prepared. Animals in the control group were also anesthetized and fixed in the stereotactic frame with a right parietal bone window alone and no brain injury.
All rats were housed two to three per cage with ad libitum access to food and water and maintained on a 12/12-h light/dark cycle (lights on at 7:00 a.m.). All procedures were approved by the Institutional Animal Care Committee.
Preparation of enterocyte mitochondria
Enterocyte mitochondria were isolated as previously described[13]. Briefly, the experimental animals underwent laparotomy and one segment of small intestine (30-cm in length) was removed for subsequent procedures. Thirty mL of 0.01 mol/L ice-cold PBS were used to wash the intestinal contents and the gut was turned over to expose the intestinal mucosa. Intestinal epithelial cells were shaved off and placed in 50 mL of ice-cold buffer (containing 0.01 mol/L PBS and 10 mmol/L EDTA) and centrifuged at 3500 r/min for 2.5 min. The supernatant was discarded and the pellet was washed once with isolation buffer containing 25 mmol/L sucrose, 10 mmol/L Tris, 2 mmol/L EDTA, pH 7.4. The buffer was centrifuged at 3500 r/min for 2.5 min. The pellet was washed twice with isolation buffer and homogenates were prepared. The homogenates were diluted to 50 mL and centrifuged at 1700 r/min for 10 min. The supernatant was collected and centrifuged at 10500 r/min for 8 min. The mitochondrial pellet was suspended in isolation buffer. The mitochondrial protein concentration was determined using a bicinchoninic acid method with bovine serum albumin (BSA) as the standard. The above procedures were carried out at 4 °C.
Measurement of mitochondrial respiratory function
Mitochondrial respiratory function was measured using a Clark oxygen electrode. A mitochondrial suspension containing 1 mg mitochondrial protein was added to 1 mL of assay buffer at 30 °C. Oxygen consumption was measured in the absence (state 4 respiration) and presence of 0.25 mmol/L ADP (state 3 respiration) and succinate (final concentration 5 mmol/L). The respiratory control ratio (RCR) was expressed as the ratio of state 3 to state 4 activity, and ADP/O was expressed as the ratio of ADP added to that of oxygen consumed during state 3 respiration.
Measurement of mitochondrial respiratory chain complex I-IV activities
Complex I activity was assayed by monitoring the decrease in nicotinamide adenine dinucleotide (NADH) at 340 nm. The final concentration of mitochondrial protein was 30 μg/mL. The reaction was started by adding 200 μmol/L NADH and was scanned at 340 nm for 3 min. Rotenone (3 μmol/L) was added to the reaction system as a blank control. Complex II activity was assayed with mitochondria (final concentration 30 μg/mL) and the reaction was started with 10 mmol/L succinate and scanned at 600 nm for 2 min. Complex III activity was assayed in a mixture containing 250 mmol/L sucrose, 1 mmol/L EDTA, 50 mmol/L KPi, pH 6.5 (adjusted to reduce auto-oxidation of reduced CoQ1), 2 mmol/L KCN, 50 μmol/L cytochrome C, 0.1% BSA, and the reaction was started by 20 μg/mL mitochondria and 50 μmol/L reduced CoQ1, with the increase in absorption at 550 nm recorded for 2 min. Complex IV activity was assayed by monitoring the decrease in reduced cytochrome C at 550 nm.
Measurement of pyruvate dehydrogenase, α-ketoglutaric dehydrogenase, and malate dehydrogenase
The pyruvate dehydrogenase (PDH) assay was carried out according to Hinman’s method[14]. Briefly, MDA reacted mitochondrial suspension was placed in assay buffer containing 2.5 mmol/L NAD, 0.1 mmol/L coenzyme A, 0.2 mmol/L thiamin pyrophosphate, 0.3 mmol/L dithiothreitol, 1 mmol/L MgCl2, 1 mg/mL BSA, 0.05 M phosphate buffer, pH 7.8, 0.6 mmol/L INT, 0.1 mg/mL dihydrolipoic acid dehydrogenase, and the reaction was started with 5 mmol/L pyruvate and scanned at 500 nm for 5 min. The activities of α-ketoglutaric dehydrogenase (KGDH) and malate dehydrogenase (MDH) were assayed as described by Humphries[15]. Briefly, a mitochondrial suspension was diluted with water to 0.5 mg/mL. After adding different concentrations of MDA (equal volume of 0.01 mol/L HCl for control), the reaction solution was incubated at 37 °C for 5 min. An aliquot was then placed in the assay mixture containing 200 μmol/L TPP, 0.5 mmol/L NAD+, 130 μmol/L coenzyme A, 2.5 μmol/L rotenone in plate wells (final mitochondrial concentration 50 μg/mL). The reaction was started by 2 mmol/L α-ketoglutarate and scanned at 340 nm for 2 min to measure NADH generation. MDH was assayed in the same way, except the reaction was started by 5 mmol/L malate.
Statistical analysis
The software SPSS 13.0 was used for statistical analysis. The data were expressed as mean ± SD, and statistical differences were calculated by one-way analysis of variance. Statistical significance was assigned at P < 0.05.
RESULTS
Changes in mitochondrial respiratory function
As shown in Table 1, compared with the control group, RCR declined at 6, 12, 24 h, day 2, day 3 and day 7, respectively (6 h: 5.20 ± 0.18, 12 h: 4.55 ± 0.35, 24 h: 3.75 ± 0.22, day 2: 4.12 ± 0.53, day 3: 3.45 ± 0.41, day 7: 5.23 ± 0.24, P < 0.01). The phosphate-to-oxygen (P/O) level significantly decreased at 12, 24 h, day 2 and day 3, respectively (12 h: 3.30 ± 0.10, 24 h: 2.61 ± 0.21, day 2: 2.95 ± 0.18, day 3: 2.76 ± 0.09, P < 0.01).
Table 1.
Changes in mitochondrial respiratory function (mean ± SD)
| Groups | State 3 respiration | State 4 respiration | RCR | P/O |
| Control | 24.46 ± 1.71 | 4.33 ± 0.25 | 5.42 ± 0.46 | 3.46 ± 0.12 |
| TBI 6 h | 23.89 ± 1.85 | 4.54 ± 0.53 | 5.20 ± 0.18b | 3.42 ± 0.15 |
| TBI 12 h | 23.04 ± 2.30 | 4.21 ± 0.74 | 4.55 ± 0.35b | 3.30 ± 0.10b |
| TBI 24 h | 19.65 ± 1.94 | 5.42 ± 0.18 | 3.75 ± 0.22b | 2.61 ± 0.21b |
| TBI 2 d | 20.04 ± 2.01 | 5.05 ± 0.32 | 4.12 ± 0.53b | 2.95 ± 0.18b |
| TBI 3 d | 18.73 ± 1.88 | 5.27 ± 0.61 | 3.45 ± 0.41b | 2.76 ± 0.09b |
| TBI 7 d | 25.22 ± 1.76 | 4.36 ± 0.46 | 5.23 ± 0.24b | 3.43 ± 0.11 |
P < 0.01 vs control. TBI: Traumatic brain injury; RCR: Respiratory control ratio; P/O: ADP/O ratio.
Changes in mitochondrial respiratory chain complex I-IV activities
As shown in Figure 1, compared with the control group, the activities of mitochondrial complex I (6 h: 110 ± 10, 12 h: 115 ± 12, 24 h: 85 ± 9, day 2: 80 ± 15, day 3: 65 ± 16, P < 0.01) and complex II (6 h: 105 ± 8, 12 h: 110 ± 92, 24 h: 80 ± 10, day 2: 76 ± 8, day 3: 68 ± 12, P < 0.01) increased at 6 h and 12 h following TBI, and then significantly decreased at 24 h, day 2 and day 3, respectively. There were no differences in complex III and IV activities between the control and TBI groups.
Figure 1.
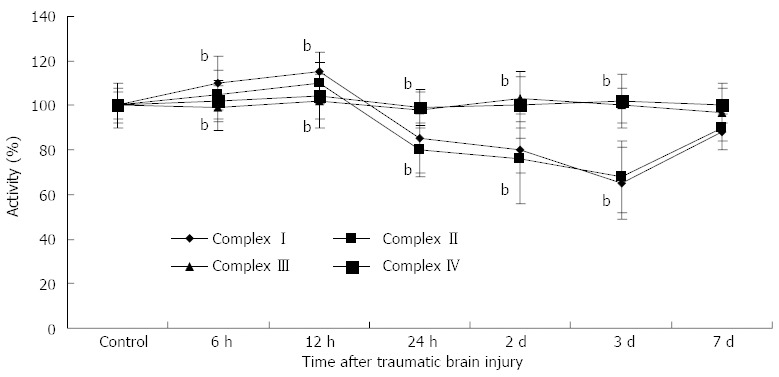
Activities of rat enterocyte mitochondrial complex I-IV after traumatic brain injury. Activities of mitochondrial complex I (6 h: 110 ± 10, 12 h: 115 ± 12, 24 h: 85 ± 9, day 2: 80 ± 15, day 3: 65 ± 16) and complex II (6 h: 105 ± 8, 12 h: 110 ± 92, 24 h: 80 ± 10, day 2: 76 ± 8, day 3: 68 ± 12) increased at 6 h and 12 h following traumatic brain injury, and then significantly decreased at 24 h, day 2 and day 3, respectively, and compared with the control group. There were no differences in complex III and IV activities between the control and TBI groups. bP < 0.01 vs control group.
Changes in PDH, KGDH, and MDH activities
As shown in Figure 2A-C, compared with the control group, PDH activity was significantly decreased at 6 h and continued up to 7 d after TBI compared with the control group (6 h: 90 ± 8, 12 h: 85 ± 10, 24 h: 65 ± 12, day 2: 60 ± 9, day 3: 55 ± 6, day 7: 88 ± 11, P < 0.01). The minimum PDH activity was seen at day 3. The changes in KGDH activity were similar to those of PDH, except that the decrease in KGDH activity began at 12 h (12 h: 90 ± 12, 24 h: 80 ± 9, day 2: 76 ± 15, day 3: 68 ± 7, day 7: 90 ± 13, P < 0.01). No significant change in MDH activity was observed.
Figure 2.
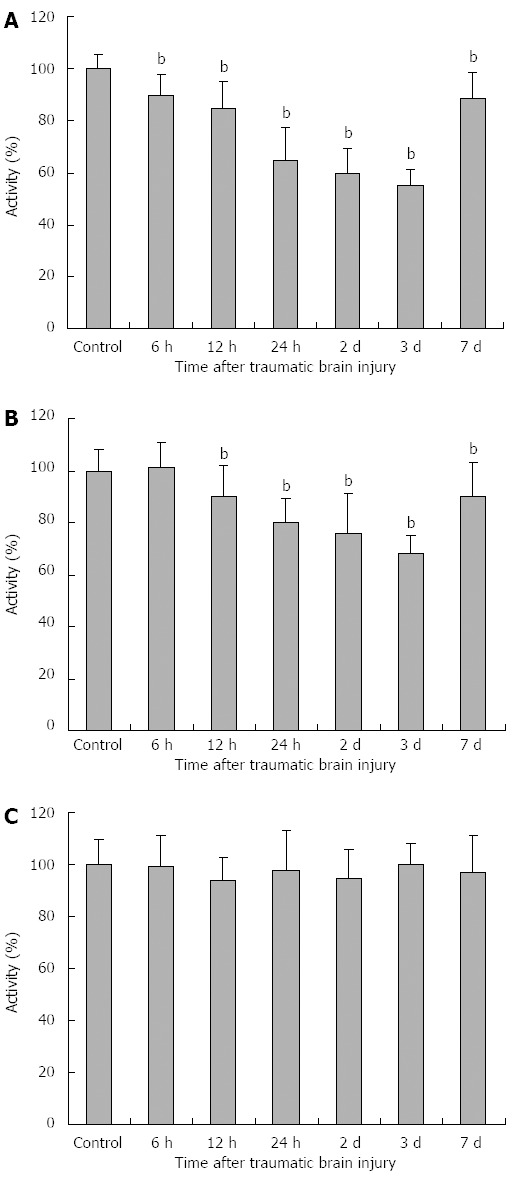
Activities of rat enterocyte mitochondrial related enzymes after traumatic brain injury. A: Pyruvate dehydrogenase (PDH) activity was significantly decreased at 6 h and continued up to 7 d after TBI compared with the control group (6 h: 90 ± 8, 12 h: 85 ± 10, 24 h: 65 ± 12, day 2: 60 ± 9, day 3: 55 ± 6, day 7: 88 ± 11). The minimum PDH activity was seen at day 3; B: The changes in α-ketoglutaric dehydrogenase (KGDH) activity were similar to those of PDH, except that the decrease in KGDH activity began at 12 h (12 h: 90 ± 12, 24 h: 80 ± 9, day 2: 76 ± 15, day 3: 68 ± 7, day 7: 90 ± 13); C: No significant change in malate dehydrogenase (MDH) activity was observed. bP < 0.01 vs control group.
DISCUSSION
Many studies have shown that the intestine may play an important role in the development and progression of SIRS, sepsis, and MODS. Thus, it is believed that the intestine is not only the target organ, but also the promoter of MODS[16-18]. Gastrointestinal dysfunction is often seen during conditions of stress, such as TBI, burn, and hemorrhagic shock, from which a sequence of complications then develop that may even be life threatening[19-21]. TBI is a major cause of death and disability throughout the world. Many patients with severe TBI often die of MODS, but not of the injury itself[22]. For these reasons, gastrointestinal dysfunction following TBI is an increasingly recognized phenomenon and is currently an important research hotspot in the bio-medical field.
Much progress has been made in the study of gastrointestinal dysfunction following TBI. Hang et al[5] confirmed that significant destruction of gut structure and impairment of barrier function could be induced by TBI 3 h after brain injury lasting for more than 7 d. An increase in intestinal mucosal permeability, decrease in intestinal motility and mucosal blood flow, and widening of intercellular tight junctions were also found in TBI rats[6,8,23]. There is evidence to suggest that increased early apoptosis of intestinal mucosal epithelial cells and attack by oxygen free radicals may contribute to stress-damage of the intestinal mucosal barrier in the early stage of TBI[24,25]. Mitochondria are involved in energy metabolism and provide energy for life activities. In addition, oxygen radicals are generated in mitochondria during oxidative phosphorylation and electron transport. In the physiological state, the production and depletion of free radicals are in equilibrium. The relative balance is destroyed when TBI occurs and vast amounts of free radicals are produced through strong lipid peroxidation. Damaged mitochondria are associated with decreased energy production and increased ROS production. In turn, increased oxidant production may aggravate mitochondrial lesions, and even activate the apoptotic signaling pathway[26]. Previous studies have revealed the occurrence of mitochondrial swelling, matrix destruction, and reduced enterocyte numbers following TBI[5,27], which are the morphological basis of mitochondrial dysfunction. Thus, we hypothesized that mitochondrial dysfunction may play an important role in TBI-induced gastrointestinal dysfunction.
However, no studies on enterocyte mitochondrial respiratory function and enzyme activities following TBI have been reported to date. In this study, we found that mitochondrial respiratory function and two related enzyme activities (PDH and KGDH) decline following TBI. The activities of mitochondrial complexes I and II are also changed after TBI. Our findings showed that TBI inhibited enterocyte mitochondrial respiratory function and related enzyme activities, suggesting that mitochondrial dysfunction may play an important role in TBI-induced gastrointestinal dysfunction.
Mitochondrial respiratory function is divided into five states. Of these, the values of states 3 and 4 are stronger for the evaluation of mitochondrial respiratory function. The ratio of state 3 to state 4 is called the mitochondrial RCR, and is the most sensitive indicator of mitochondrial respiratory function. A decreased level of RCR indicates a coupling defect in mitochondrial oxidative phosphorylation. State 4 represents permeability of the mitochondrial membrane, with an increase suggesting increased permeability[28]. The P/O ratio refers to the amount of ATP produced during the movement of two electrons through a defined electron transport chain by the reduction of an oxygen atom. The value of P/O is high in intact mitochondria, but low in damaged or dysfunctional mitochondria[29]. Mitochondrial RCR, respiratory state 4, and the value of P/O can reflect mitochondrial respiratory function to some extent. Impaired mitochondrial respiratory function in intestinal epithelial cells has been observed in animal models suffering shock or burns. Our results revealed that mitochondrial RCR decreased significantly at the early stage of TBI, and the drop in P/O ratio was later than that of RCR. These findings suggest that RCR is more susceptible to TBI than the P/O ratio. Furthermore, two troughs in mitochondrial respiratory function were found at 24 h and day 3 after TBI. We hypothesize that the formation of the first trough may be related to intestinal ischemia, while the second may be associated with intestinal reperfusion and intracranial hypertension.
The mitochondrial respiratory chain is a major source of ROS in eukaryotic cells. Dysfunction of mitochondrial respiratory chain complexes may result in an imbalance of ROS production, which has been implicated in a number of degenerative diseases, tumors, sepsis, and biological aging[30-32]. The changes in respiratory chain complex I-IV activities following TBI were discovered to be discrepant in our research. The activities of mitochondrial complexes I and II increased in the early stage of TBI and then significantly decreased until day 7. These activities were reduced to a minimum at day 3. An increase in the early course of TBI is considered to be a compensatory mechanism for energy requirements. There were no differences in complex III and IV activities between the control and TBI groups. It has been confirmed that mitochondrial complex III and IV activities decrease with age[33,34]. We speculate that complexes III and IV may correlate with chronic oxidative stress, while complexes I and II are responsible for acute mitochondrial injury.
Our findings also demonstrated that three mitochondrial dehydrogenases showed different tolerance to TBI. More specifically, PDH and KGDH activities significantly decreased in the early stage of TBI and continued up to 7 d after TBI compared with the control group. The minimum PDH and KGDH activities were seen at day 3. However, no significant change in MDH activity was observed. Long et al[35] found that the binding affinity of PDH and KGDH to malondialdehyde (MDA) was markedly higher than that of PDH. In addition, the level of mitochondrial MDA increased in the oxidative stress state. These may be the reasons for the different alterations in the three mitochondrial dehydrogenases after TBI.
The main purpose of this paper was to investigate the change in mitochondrial respiratory function and determine the mechanism of intestinal dysfunction after TBI. In fact, there may be several such reasons, including ischemia, reperfusion, and changes in intracranial pressure, which could explain intestinal dysfunction. TBI often induces the abnormal release of pituitary hormones[36-38], which can lead to intestinal dysfunction. The brain-gut axis is also an interesting possible mechanism.
However, there are several limitations in this study. Firstly, the studies were performed in rats and are still far from suitable for human experiments. Secondly, the findings were limited by the small number of studies used in the analyses. Thirdly, the studies could not be carried out in living organisms to obtain dynamic outcomes related to mitochondrial function. Finally, the results were derived only from male rats and there could be sexual dimorphism in enterocyte functions. Further study is needed to explain the mechanisms involved in the variation in the three dehydrogenases and respiratory chain complex I-IV activities. For example, mitochondrial respiratory complexes and their enzyme related gene expression could be investigated.
In conclusion, rat enterocyte mitochondrial respiratory function and the activities of PDH and KGDH declined following TBI. The activities of mitochondrial complexes I and II also changed after TBI. In turn, these alterations in mitochondrial function may aggravate existing gastrointestinal dysfunction. Mitochondrial dysfunction may play an important role in TBI-induced gastrointestinal dysfunction. Larger studies are required to explore what improvement in mitochondrial outcomes mean and how to protect mitochondrial function. Nevertheless, this study suggests a possible strategy to attenuate gastrointestinal complications after TBI by protecting mitochondrial function in intestinal epithelial cells.
ACKNOWLEDGMENTS
We would like to thank Dr. Jang-Gang Long for his technical support.
COMMENTS
Background
Traumatic brain injury (TBI) is a major cause of death and disability worldwide. Many patients with severe TBI often die of multiple organ dysfunction syndrome, but not of the injury itself. Gastrointestinal dysfunction following TBI is an increasingly recognized phenomenon, and is currently an important research hotspot in the bio-medical field.
Research frontiers
Studies have confirmed that gastrointestinal dysfunction often occurs after TBI. Increase in intestinal mucosal permeability, decrease in intestinal motility and intestinal mucosal blood flow, destruction of gut structure, impairment of barrier function, and widening of intercellular tight junctions were also found in TBI rats. There is evidence suggesting that increased early apoptosis of intestinal mucosal epithelial cells and attack by oxygen free radicals may contribute to stress-damage of the intestinal mucosal barrier in the early stage of TBI.
Applications
Mitochondrial dysfunction may play an important role in TBI-induced gastrointestinal dysfunction. It may be a new strategy to attenuate gastrointestinal complications after TBI by protecting the mitochondrial function of intestinal epithelial cells.
Terminology
Mitochondria are an important part of oxidative phosphorylation and energy supply. In addition, they also generate reactive oxygen species. Mitochondria are thought to be the primary target of oxidative damage and play an important role in oxidative stress and apoptosis. Mitochondrial respiratory function can be evaluated by respiratory control ratio, the phosphate-to-oxygen ratio, and the activities of respiratory chain complexes I-IV and related enzymes.
Peer review
This is an original and interesting paper on the study of rat enterocyte mitochondrial respiratory function and the activities of related enzymes following TBI. Their findings demonstrated that rat enterocyte mitochondrial respiratory function and activities of pyruvate dehydrogenase and α-ketoglutaric dehydrogenase decline following TBI. Activities of mitochondrial complexes I and II also changed after TBI. The study provides evidence that enterocyte mitochondrial dysfunction is induced by TBI.
Footnotes
Supported by The Scientific Research Foundation of the Chinese PLA Medical Programs, No. ms031
P- Reviewer: Chu SH, Dubost C, Gong QY, Tanriverdi F S- Editor: Gou SX L- Editor: Rutherford A E- Editor: Zhang DN
References
- 1.Reilly P. The impact of neurotrauma on society: an international perspective. Prog Brain Res. 2007;161:3–9. doi: 10.1016/S0079-6123(06)61001-7. [DOI] [PubMed] [Google Scholar]
- 2.Pilitsis JG, Rengachary SS. Complications of head injury. Neurol Res. 2001;23:227–236. doi: 10.1179/016164101101198389. [DOI] [PubMed] [Google Scholar]
- 3.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–105; discussion 1031-105;. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 4.Tan M, Zhu JC, Yin HH. Enteral nutrition in patients with severe traumatic brain injury: reasons for intolerance and medical management. Br J Neurosurg. 2011;25:2–8. doi: 10.3109/02688697.2010.522745. [DOI] [PubMed] [Google Scholar]
- 5.Hang CH, Shi JX, Li JS, Wu W, Yin HX. Alterations of intestinal mucosa structure and barrier function following traumatic brain injury in rats. World J Gastroenterol. 2003;9:2776–2781. doi: 10.3748/wjg.v9.i12.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang YB, Liu J, Yang ZX. Effects of intestinal mucosal blood flow and motility on intestinal mucosa. World J Gastroenterol. 2011;17:657–661. doi: 10.3748/wjg.v17.i5.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hang CH, Shi JX, Li JS, Li WQ, Yin HX. Up-regulation of intestinal nuclear factor kappa B and intercellular adhesion molecule-1 following traumatic brain injury in rats. World J Gastroenterol. 2005;11:1149–1154. doi: 10.3748/wjg.v11.i8.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feighery L, Smyth A, Keely S, Baird AW, O’Connor WT, Callanan JJ, Brayden DJ. Increased intestinal permeability in rats subjected to traumatic frontal lobe percussion brain injury. J Trauma. 2008;64:131–17; discussion 131-17;. doi: 10.1097/TA.0b013e3181568d9f. [DOI] [PubMed] [Google Scholar]
- 9.Wang YB, Liang PX, ZhuYQ The effects of intestinal perfusion and motivity on the intestinal permeability in rats with traumatic brain injury. Zhonghua Xiaohua Zazhi. 2007;27:103–106. [Google Scholar]
- 10.Gardeström P, Lernmark U. The contribution of mitochondria to energetic metabolism in photosynthetic cells. J Bioenerg Biomembr. 1995;27:415–421. doi: 10.1007/BF02110004. [DOI] [PubMed] [Google Scholar]
- 11.Kowald A, Kirkwood TB. Evolution of the mitochondrial fusion-fission cycle and its role in aging. Proc Natl Acad Sci USA. 2011;108:10237–10242. doi: 10.1073/pnas.1101604108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feeney DM, Boyeson MG, Linn RT, Murray HM, Dail WG. Responses to cortical injury: I. Methodology and local effects of contusions in the rat. Brain Res. 1981;211:67–77. doi: 10.1016/0006-8993(81)90067-6. [DOI] [PubMed] [Google Scholar]
- 13.Lawrence CB, Davies NT. A novel, simple and rapid method for the isolation of mitochondria which exhibit respiratory control, from rat small intestinal mucosa. Biochim Biophys Acta. 1986;848:35–40. doi: 10.1016/0005-2728(86)90157-x. [DOI] [PubMed] [Google Scholar]
- 14.Hinman LM, Blass JP. An NADH-linked spectrophotometric assay for pyruvate dehydrogenase complex in crude tissue homogenates. J Biol Chem. 1981;256:6583–6586. [PubMed] [Google Scholar]
- 15.Humphries KM, Szweda LI. Selective inactivation of alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase: reaction of lipoic acid with 4-hydroxy-2-nonenal. Biochemistry. 1998;37:15835–15841. doi: 10.1021/bi981512h. [DOI] [PubMed] [Google Scholar]
- 16.Dervenis C, Smailis D, Hatzitheoklitos E. Bacterial translocation and its prevention in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2003;10:415–418. doi: 10.1007/s00534-002-0727-5. [DOI] [PubMed] [Google Scholar]
- 17.Doig CJ, Sutherland LR, Sandham JD, Fick GH, Verhoef M, Meddings JB. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444–451. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 18.Rowlands BJ, Soong CV, Gardiner KR. The gastrointestinal tract as a barrier in sepsis. Br Med Bull. 1999;55:196–211. doi: 10.1258/0007142991902213. [DOI] [PubMed] [Google Scholar]
- 19.Xiao SC, Zhu SH, Xia ZF, Lu W, Wang GQ, Ben DF, Wang GY, Cheng DS. Prevention and treatment of gastrointestinal dysfunction following severe burns: a summary of recent 30-year clinical experience. World J Gastroenterol. 2008;14:3231–3235. doi: 10.3748/wjg.14.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bansal V, Costantini T, Kroll L, Peterson C, Loomis W, Eliceiri B, Baird A, Wolf P, Coimbra R. Traumatic brain injury and intestinal dysfunction: uncovering the neuro-enteric axis. J Neurotrauma. 2009;26:1353–1359. doi: 10.1089/neu.2008.0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Overhaus M, Toegel S, Bauer AJ. Interaction of hemorrhagic shock and subsequent polymicrobial sepsis on gastrointestinal motility. Shock. 2009;31:382–389. doi: 10.1097/SHK.0b013e3181862ea4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemp CD, Johnson JC, Riordan WP, Cotton BA. How we die: the impact of nonneurologic organ dysfunction after severe traumatic brain injury. Am Surg. 2008;74:866–872. [PubMed] [Google Scholar]
- 23.Smith K. TBI affects intestinal motility. Nat Rev Gastroenterol Hepatol. 2013;10:260. doi: 10.1038/nrgastro.2013.68. [DOI] [PubMed] [Google Scholar]
- 24.Wang YB, Yang ZX. [Effect of oxygen free radicals on bacterial translocation in rats with traumatic brain injury] Zhonghua Yi Xue Zazhi. 2010;90:1716–1718. [PubMed] [Google Scholar]
- 25.Jin W, Ni H, Dai Y, Wang H, Lu T, Wu J, Jiang J, Liang W. Effects of tert-butylhydroquinone on intestinal inflammatory response and apoptosis following traumatic brain injury in mice. Mediators Inflamm. 2010;2010:502564. doi: 10.1155/2010/502564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grivennikova VG, Vinogradov AD. Mitochondrial production of reactive oxygen species. Biochemistry (Mosc) 2013;78:1490–1511. doi: 10.1134/S0006297913130087. [DOI] [PubMed] [Google Scholar]
- 27.Mei Q, Diao L, Xu JM, Liu XC, Jin J. A protective effect of melatonin on intestinal permeability is induced by diclofenac via regulation of mitochondrial function in mice. Acta Pharmacol Sin. 2011;32:495–502. doi: 10.1038/aps.2010.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brown GC. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem J. 1992;284(Pt 1):1–13. doi: 10.1042/bj2840001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hinkle PC. P/O ratios of mitochondrial oxidative phosphorylation. Biochim Biophys Acta. 2005;1706:1–11. doi: 10.1016/j.bbabio.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 30.Dröse S, Brandt U. Molecular mechanisms of superoxide production by the mitochondrial respiratory chain. Adv Exp Med Biol. 2012;748:145–169. doi: 10.1007/978-1-4614-3573-0_6. [DOI] [PubMed] [Google Scholar]
- 31.Zhang XN, Qi M. Mitochondrion and its related disorders: making a comeback. J Zhejiang Univ Sci B. 2008;9:90–92. doi: 10.1631/jzus.B0710621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rocha M, Herance R, Rovira S, Hernández-Mijares A, Victor VM. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infect Disord Drug Targets. 2012;12:161–178. doi: 10.2174/187152612800100189. [DOI] [PubMed] [Google Scholar]
- 33.Federico A, Cardaioli E, Da Pozzo P, Formichi P, Gallus GN, Radi E. Mitochondria, oxidative stress and neurodegeneration. J Neurol Sci. 2012;322:254–262. doi: 10.1016/j.jns.2012.05.030. [DOI] [PubMed] [Google Scholar]
- 34.Morán M, Moreno-Lastres D, Marín-Buera L, Arenas J, Martín MA, Ugalde C. Mitochondrial respiratory chain dysfunction: implications in neurodegeneration. Free Radic Biol Med. 2012;53:595–609. doi: 10.1016/j.freeradbiomed.2012.05.009. [DOI] [PubMed] [Google Scholar]
- 35.Long J, Wang X, Gao H, Liu Z, Liu C, Miao M, Liu J. Malonaldehyde acts as a mitochondrial toxin: Inhibitory effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Life Sci. 2006;79:1466–1472. doi: 10.1016/j.lfs.2006.04.024. [DOI] [PubMed] [Google Scholar]
- 36.Tanriverdi F, Ulutabanca H, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F. Pituitary functions in the acute phase of traumatic brain injury: are they related to severity of the injury or mortality? Brain Inj. 2007;21:433–439. doi: 10.1080/02699050701311083. [DOI] [PubMed] [Google Scholar]
- 37.Tanriverdi F, Ulutabanca H, Unluhizarci K, Selcuklu A, Casanueva FF, Kelestimur F. Three years prospective investigation of anterior pituitary function after traumatic brain injury: a pilot study. Clin Endocrinol (Oxf) 2008;68:573–579. doi: 10.1111/j.1365-2265.2007.03070.x. [DOI] [PubMed] [Google Scholar]
- 38.Kasturi BS, Stein DG. Traumatic brain injury causes long-term reduction in serum growth hormone and persistent astrocytosis in the cortico-hypothalamo-pituitary axis of adult male rats. J Neurotrauma. 2009;26:1315–1324. doi: 10.1089/neu.2008.0751. [DOI] [PMC free article] [PubMed] [Google Scholar]