

Abstract
Cardiac dysfunction is a well-known consequence of diabetes, with sustained hyperglycaemia leading to the development of a cardiomyopathy that is independent of cardiovascular disease or hypertension. Animal models of diabetes are commonly used to study the pathophysiology of diabetic cardiomyopathy, with the hope that increased knowledge will lead ultimately to better therapeutic strategies being developed. At physiological temperature, left ventricular trabeculae isolated from the streptozotocin rat model of type 1 diabetes showed decreased stress and prolonged relaxation, but with no evidence that decreased contractility was a result of altered myocardial Ca2+ handling. Although sarcoplasmic reticulum (SR) Ca2+ reuptake appeared slower in diabetic trabeculae, it was offset by an increase in action-potential duration, thereby maintaining SR Ca2+ content and favouring increased contraction force. Frequency analysis of t-tubule distribution by confocal imaging of ventricular tissue labeled with wheat germ agglutinin or ryanodine receptor antibodies showed a reduced T-power for diabetic tissue, but the differences were minor in comparison to other models of heart failure. The contractile dysfunction appeared to be the result of disrupted F-actin in conjunction with the increased type I collagen, with decreased myofilament Ca2+ sensitivity contributing to the slowed relaxation.
Keywords: Diabetic cardiomyopathy, Heart failure, Contractility, T-tubules, Excitation-contraction coupling, Calcium homeostasis
Core tip: Diabetic patients develop a cardiomyopathy that is independent of vascular disease, and is thought to develop as a direct result of the prolonged hyperglycaemia. Animal models of diabetes can help us understand the cellular mechanisms that lead ultimately to contractile dysfunction of diabetic cardiomyopathy. The streptozotocin rat model of type 1 diabetes has slowed Ca2+ transients and twitch force kinetics, with reduced myofilament Ca2+ sensitivity. Myocytes are decreased in volume in diabetic hearts, with reduced and disrupted F-actin, and type 1 collagen is increased. Together, these changes all contribute to the reduced contractility of diabetic cardiomyopathy.
INTRODUCTION
Patients with diabetes develop a cardiomyopathy that is independent of coronary artery disease and hypertension[1], and contributes to the increased mortality and morbidity of the disease[2,3]. The mechanisms that lead to development of the diabetic cardiomyopathy are poorly understood, although they appear to be a direct result of cellular damage from the hyperglycaemia. The early stages of the cardiomyopathy are associated with reduced diastolic function, with 27%-70% of asymptomatic diabetic patients showing some form of diastolic abnormality[4-6]. Later this progresses to include systolic dysfunction and heart failure[7,8]. Diabetes manifests in two forms, both of which are a result of abnormal glucose metabolism. Type I diabetes usually has its onset early in life and is characterized by insufficient insulin production, whereas type II diabetes has its origin downstream of insulin binding to its receptor, and is therefore known as insulin-resistant diabetes. Diabetic cardiomyopathy develops in both type I and type II forms of the disease[9,10].
Although the heart contains many different cell types, it is the cardiac myocytes that perform the work that enables the heart to function as a pump. With each cardiac cycle, the myocytes experience rapid changes in intracellular ion concentrations that are crucial to the hearts inotropy, lusitropy, and energy metabolism. This review will outline the ultrastructural and functional changes that contribute to the impaired contraction and relaxation characteristic of diabetic cardiomyopathy.
MECHANISMS CONTRIBUTING TO DIABETIC CARDIOMYOPATHY
Streptozotocin rat model of diabetes
Animal models have frequently been used in research into the cellular mechanisms associated with diabetes[11], with the insulin-deficient streptozotocin rat (STZ) commonly studied. Type-1 diabetes in humans is characterized by the destruction of the pancreatic β-cells, as occurs in the STZ. Streptozotocin is a naturally occurring glucose analog that is particularly toxic to the insulin-producing beta cells of the pancreatic islets. The chemical is transported into cells via the glucose transporter-2 (GLUT-2)[12]. Since the pancreatic beta cells have high levels of GLUT-2, they accumulate streptozotocin in large quantities, resulting in their destruction and the onset of a diabetic state. Rats treated with a single dose of streptozotocin (60 mg/kg) rapidly develop biochemical and functional myocardial abnormalities. They exhibit increased water consumption (180 mL/d compared to 43 mL/d for sham-injected control) and elevated plasma glucose levels (31 mmol/L compared to 4 mmol/L for control) that are sustained. Isolated cardiac muscle preparations from diabetic rats 8 wk post-injection show depressed contractility, diminished compliance and decreased inotropic drug responses[13]. Abnormalities in contraction and metabolism have been reported both in vivo and in vitro in the STZ diabetic rat model, reflecting changes at the cardiac myocyte level as a result of the sustained hyperglycaemia. The STZ rat has proved an invaluable model for investigation of the pathogenesis of type 1 diabetes and its complications, and in the development of potential new treatments for the disease[14-16].
The reduced contractility of diabetic hearts
Contraction in cardiac muscle is brought about by an increase in the myocyte intracellular Ca2+ concentration (the “Ca2+ transient”). Propagation of the action potential across the surface sarcolemma and throughout the transverse tubule system (t-tubules) opens voltage-gated L-type Ca2+ channels causing a synchronised influx of Ca2+ into the myocytes (the “Ca2+ current”). This Ca2+ current then triggers release of Ca2+ from the junctional region of the sarcoplasmic reticulum (SR) via the ryanodine receptors (RyRs) in a process termed “Ca2+-induced Ca2+-release”[17,18]. In this way the intracellular Ca2+ concentration [Ca2+]i is rapidly increased to approximately 10 times the resting level. Ca2+ then diffuses to the contractile proteins where it binds to troponin C, initiating cross-bridge cycling and force development. Excitation-contraction coupling has therefore been a major focus of those investigating the cellular mechanisms that underlie the reduced contractility of failing hearts.
Intracellular calcium transients in diabetic hearts
Measurements carried out on multicellular trabeculae isolated from the left ventricle under near physiological conditions (1.5 mmol/L [Ca2+]o, 37 °C and 5 Hz) showed trabeculae from diabetic rats had depressed contractility with prolonged contraction and relaxation in comparison to their controls, consistent with other studies[19-21].
An alteration of intracellular Ca2+ homeostasis has previously been suggested as underlying the diabetic cardiac dysfunction (for review see[22]) although, as noted, results are often contradictory. While some of these discrepancies might be attributable to the extent of disease progression (diabetic stage) and experimental conditions, very few studies have examined the [Ca2+]i control of contractility under near-physiological temperatures and rates of stimulation. Our study showed that diabetic rats had an unchanged resting [Ca2+]i level and amplitude of Ca2+ transient, despite a reduced contractility[23]. Averaged Ca2+ transients and isometric twitches at 5 Hz stimulation are shown in Figure 1 for trabeculae from control (solid line) and diabetic (dotted line) rats, superimposed for comparison. Figure 1C shows the [Ca2+]i-stress phase plot, with a right shifted relaxation phase for diabetic trabeculae which suggests diminished myofibrillar Ca2+ sensitivity.
Figure 1.
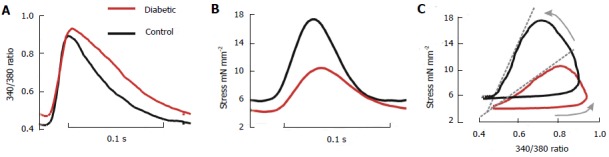
Average intracellular Ca2+ transients and isometric stress. Data were recorded from left ventricular trabeculae of diabetic (red lines) and control (black lines) hearts at 5 Hz, 37 °C, and 1.5 mmol [Ca2+]o, 7 trabeculae per group. A: Ca2+ transient (340/380 fluorescence ratio); B: Stress; C: Phase plots of the relationship between fluorescence and stress. The arrows indicate the direction of time, and the dashed grey lines accentuate the slope of the relaxation component. (Modified from Zhang et al[23]).
Figure 2 shows averaged data from trabeculae at 5 Hz stimulation and at 37 °C. Diabetic rats had prolonged time-to-peak [Ca2+]i and a prolonged time constant of Ca2+ transient decay, consistent with some other reports[20,24-26]. The slower kinetics of Ca2+ transient would contribute to the prolonged time course of cardiac contraction and relaxation in diabetic rats, but it is unclear if the reduced rate of the decay in the Ca2+ transient is sufficient to explain the slowed mechanical relaxation.
Figure 2.
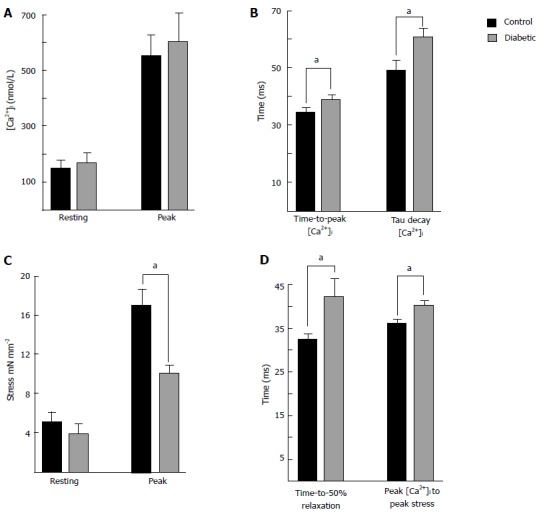
Summary of intracellular Ca2+ and isometric stress parameters. Data were recorded from left ventricular trabeculae at 37 °C, 5 Hz stimulation frequency, and 1.5 mmoL [Ca2+]o. Data are mean ± SE 8 wk post injection for control (n = 7) and diabetic (n = 8). A: Shows resting and peak [Ca2+]i. The Ca2+ transients were prolonged in diabetic trabecuae; B: Shows the time to reach peak [Ca2+]i, and the time constant of the Ca2+ transient decay; C: Shows no difference in resting stress, but peak stress was reduced in diabetic trabeculae; D: Shows the time to 50% relaxation of stress was prolonged in diabetic, as was the time from the peak of the Ca2+ transient to the peak of the twitch. aP < 0.05, diabetic vs control.
Our study showed that contractility was reduced in trabeculae from diabetic hearts, even when peak [Ca2+]i was matched between diabetic and control trabeculae by altering stimulation rate[23], suggesting that altered [Ca2+]i handling was not the primary mechanism of contractile dysfunction. The mechanical relaxation was intrinsically slower in diabetic rat hearts, which was exacerbated by the reduced rate of decrease of [Ca2+]i. In support of this idea, Figure 2D shows that the interval between the time-to-peak [Ca2+]i and the time-to-peak stress in diabetic rats was increased in comparison to control.
Analysis of electrocardiogram (ECG) in lightly anaesthetized diabetic rats prior to experimentation showed that the normalized QT interval was prolonged, implying the cardiac action potential was slower[23]. This would contribute to the prolonged Ca2+ transients observed in diabetes, but cannot explain the observed Ca2+ transient changes in full. Logarithmic plots of Ca2+ transients from control and diabetic trabeculae in Zhang et al[23] (2008) show that the linear portion of the Ca2+ fluorescence decay was delayed in trabeculae from diabetic hearts, consistent with the increase in the time-to-50% repolarization of the ventricular action potential reported in their study. Prolonged depolarization during the plateau phase of the action potential will lead also to increased L-type Ca2+ influx, although this was not shown in the Zhang et al[23] (2008) study. Frequently studies have reported changes in SERCA protein expression in explanation of observed changes to the time course of the Ca2+ transients[27,28], but decreased SERCA activity and/or expression may only contribute in part to the prolonged Ca2+ transient decay. Action potential duration is also important in determining the duration of the Ca2+ transient, and therefore the SR Ca2+ load, which in turn determines SR Ca2+ release via the RyRs[29]. ECG measurements in insulin-treated type 1 diabetic patients also show abnormal repolarization with the reports of increased QT interval and increased QT dispersion[30].
T-tubule system structure
The t-tubules are an important component of the excitation-contraction coupling system in cardiac myocytes[31]. T-tubules are an extension of the sarcolemma that project transversely into the interior of the cell adjacent to the z-line, although numerous axial connections between sarcomeres are observed[32]. This structure facilitates synchronous contraction by conducting the action potential deep within the myocyte and triggering Ca2+ release from the SR in regions located away from the cell surface. There is evidence that loss of normal transverse tubule structure is a key feature of both animal[33,34] and human heart failure[35,36]. Frequency analysis of t-tubule distribution at the z-line has been used to quantify the structural changes in t-system labelling of myocytes from rodents at different stages of heart failure[33]. This analysis exploits the periodic nature of t-tubule distribution at the z-line of sarcomere. By converting t-tubule images into the frequency domain with a fast Fourier transform, a peak associated with sarcomere spacing of 2 μm is observed in the power spectrum[37,38]. In failing myocytes the periodic pattern of t-tubule labelling is disrupted resulting in reduced sarcomere peak. This peak is termed “T-power” and provides a useful metric to quantify t-tubule structure.
Currently there is lack of comparable data for changes in t-tubules in the diabetic heart. To address this gap in knowledge we have used confocal laser scanning microscopy to examine the labelling of the t-tubules [wheat germ agglutinin (WGA)] and the ryanodine receptors (RyR), in the hearts of STZ rats with end stage heart failure as shown in Figure 3. Analysis of this labelling in the frequency domain has shown a significant but surprisingly modest decrease in T-power (or sarcomere power) in the t-tubule system of diabetic myocytes. A similar analysis of RyR labelling showed a comparable decrease in sarcomere power in diabetic myocytes. Visual inspection of the labelling in Figure 3 shows that both the structure of t-system (WGA) and the SR (RyR) are largely intact in myocytes from diabetic rat hearts, which is consistent with the comparatively normal calcium transients measured in the cardiac trabeculae of this animal model[23]. This contrasts with the dramatic loss of the t-system structure reported in non-diabetic animal heart failure. For example the t-system is dramatically remodelled in spontaneously hypertensive rat, while the labelling of RyR is largely intact[34,38]. A similar situation is seen in non-diabetic human heart failure[36]. This may turn out to be a key point of difference between diabetic and other forms of heart failure, and it remains to be seen if a similar pattern of t-system preservation is seen in the diabetic human heart. Alternatively, the lack of obvious changes in the t-tubule distribution of STZ-induced diabetic rat hearts 8 wk post injection may reflect the relatively short duration of the disease. Figure 2B shows an increase in the time-to-peak of the Ca2+ transients in longitudinal section (LV) trabeculae from diabetic hearts, which may reflect changes in excitation-contraction coupling from hyperglycaemia-induced loss of t-tubule structure.
Figure 3.
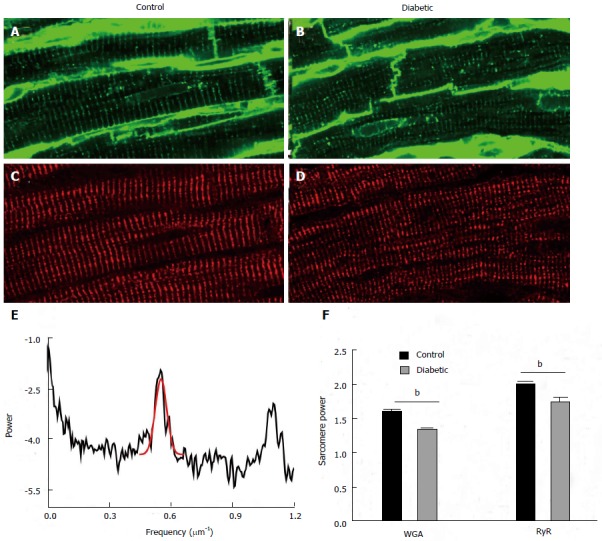
Structural changes in proteins associated with excitation-contraction coupling. Transverse tubules were visualised by labelling with wheat germ agglutinin in (A) control and (B) diabetic tissue. The same tissue sections were dual labelled with antibodies against ryanodine receptors (RyR) in (C) control and (D) diabetic tissue. The periodicity or regularity of labelling was assessed using a fast Fourier transform. An example of this analysis is shown in (E) which is the plot of the FFT in control myocyte labelled with RyR. The peak associated with sarcomeric periodicity (approximately 0.55 μm-1) is fitted with a Gaussian in red. The height of this peak is used as a metric to assess the regularity of sarcomere labelling termed “sarcomere” power. (F) This shows the mean sarcomere power for both wheat germ agglutinin and ryanodine receptor labelling from 18 cells from 3 control animals and 18 cells from 3 diabetic animals. Both wheat germ agglutinin and ryanodine receptor sarcomere power were modestly but highly significantly reduced in cells from diabetic hearts (Bonferroni corrected t test, bP < 0.01, diabetic vs control.).
Ventricular remodeling of diabetic hearts
Although intracellular Ca2+ cycling is essential to the contraction and relaxation of cardiac mycocytes, the extracellular matrix and the myofilaments within the myocytes are essential also. The contractile proteins that make up the myofilaments are the end effectors of excitation-contraction coupling, and their responsiveness to Ca2+ directly determines myocyte contractility (for reviews see[39,40]). Changes in the contractile proteins of diabetic hearts have been reported, and are likely to contribute substantially to the observed changes in contraction and relaxation. Figure 2C and D show both reduced contraction (peak stress) and slowed relaxation in LV trabeculae from diabetic rat hearts. The slower time course of contraction in trabeculae from diabetic hearts could be explained, in part, by a shift in the myosin isoenzyme distribution from the faster alpha heavy chain to the beta form as previously reported[41] (for review see[42]). Changes in other aspects of the contractile protein system have also been described in diabetic hearts. The thin filament regulatory troponin-tropomyosin complex shows decreased Ca2+ sensitivity in skinned[43,44] and intact[16] cardiac muscle preparations. The consequence of reduced Ca2+ sensitivity is increased force production for any given cytosolic Ca2+ concentration, favouring force production during systole, but decreasing relaxation which would contribute to diastolic failure.
Ultra-structural analysis by electron microscopy has revealed loss and disorganisation of actin filaments in STZ diabetic hearts[21], which was supported by confocal analysis of phalloidin labelled ventricular tissue with disorganisation and a reduction of f-actin labelling evident[16,21,23]. We have also observed that myocyte cell diameter is reduced in the STZ diabetic rat, suggesting that amount of myofilament protein per myocyte is reduced. Figure 4 shows mean ± SE data from enzymatically isolated ventricular myocytes from diabetic and control rats. Cell length was not different between groups, but both width and volume were markedly reduced in myocytes from diabetic hearts. Similar changes in f-actin content and myocyte size in the STZ diabetic rat have been reported by Kawaguchi et al[45] (1999). Pertinently these authors also identified a decrease in myocyte diameter in the diabetic human heart[45]. Changes in myocyte volume have been shown to occur as early as one week after induction of diabetes in the STZ[46]. The decreased myocyte volume is evident in the left hand side (LHS) panel of Figure 5C and D where representative tissue from the LV free wall of diabetic rat hearts shows reduced myocyte diameter. It appears then that the diabetic myocytes are atrophied. Ultrastructural changes in mitochondrial morphology have been shown by electron microscopy of diabetic rat heart, with likely consequences not only for myocyte volume but also energy metabolism[21]. Proteomic analysis of diabetic rat heart identified multitude of changes in the mitochondrial proteome[47]. The most notable changes are an increase in enzymes involved in long chain fatty acids oxidation and decrease in enzymes involved in catabolism. Metabolism of the diabetic heart is shifted from a mix of carbohydrates and fatty acids for energy supply to relying almost solely on fatty acids, with a resultant increase in the production of oxygen free radical end products[48]. Significantly the proteomic analysis also showed changes in proteins involved with oxidative stress, suggesting that impaired energy metabolism might lead to myocytes being unable to meet the energetic needs producing changes in the structure and function of the contractile machinery.
Figure 4.
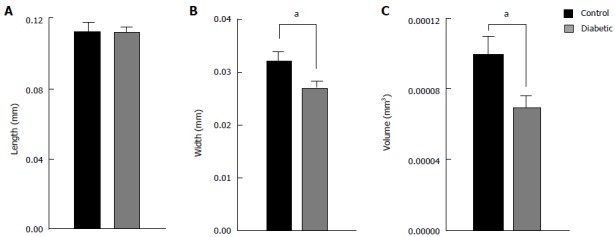
Average dimensions of isolated ventricular myocytes from diabetic and control rat hearts. Cell length (A) was not different between diabetic (n = 35) and control (n = 19) hearts, whereas cell width (B) and cell volume (C) was reduced. aP < 0.05, diabetic vs control.
Figure 5.
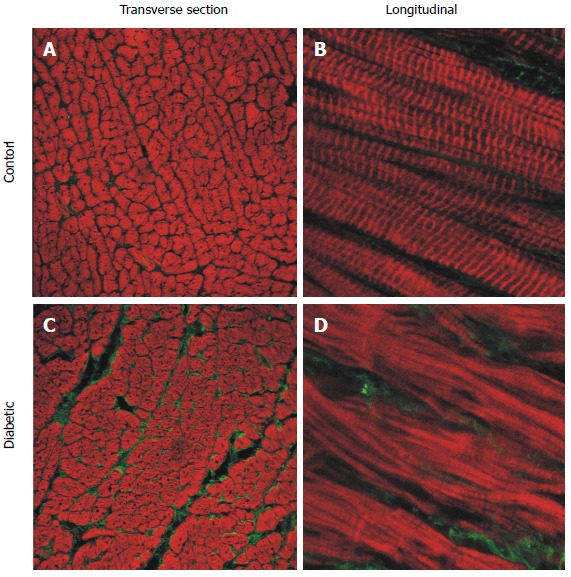
Representative confocal images of longitudinal section free wall immuno-labelled for type I collagen (green) and f-actin (red). Sections from the endocardium of control (A and B) and diabetic (C and D) rat hearts. Left hand side panels: Transverse sections from endocardium (25 × objective). Right hand side panels: Longitudinal sections (63 × objective, zoom × 3). (Modified from Zhang et al[23]).
Diabetic cardiomyopthy is also associated with increased stiffness in the left ventricle[49], and a decreased maximum rate-of-rise in developed stress[23], suggesting that cardiac compliance is reduced in diabetic rats. The extracellular matrix in healthy hearts provides a scaffolding that supports the myocytes and other tissue components, enabling the coordinated transduction of force that is necessary for the heart to function as a pump. Collagen is an important component of the extracellular matrix, with type I and type III collagens the most abundant types in ventricular tissue forming 90% of the total collagen content[50]. Figure 5 shows type I collagen is increased in diabetic rat hearts, which would contribute to the decreased ventricular compliance, with no change in type III collagen[23]. Myocardial echodensity has been reported as increased in asymptomatic diabetic patients, thought to be a result of increased collagen deposition[51]. It is proposed that increased echodensity might therefore act as an early indicator of the subsequent development of diabetic cardiomyopathy.
CONCLUSION
In conclusion, diabetic cardiomyopathy arises as a result of the sustained hyperglycaemia and the damaging effects this has on the heart. Ventricular myocytes from untreated diabetic rat hearts show contractile dysfunction after 8 wk of hyperglycaemia, with prolonged action potential duration, slower Ca2+ transient decay and reduced myofilament Ca2+ sensitivity. Gross structural changes to the myocardium are evident at this stage of the disease. Extracellular type 1 collagen is increased, t-tubules are less regular in appearance, and F-actin within myocytes is reduced in content and disrupted in appearance. We conclude that it is these structural changes that are the main contributors to the contractile dysfunction of diabetic cardiomyopathy, along with mitochondrial changes that compromise energy supply. We suggest that consideration should therefore be given in future studies to the contribution of these observed structural changes to the contractile deficit in the diabetic hearts, rather than focusing on myocyte Ca2+ handling in searching for effective treatments for diabetic cardiomyopathy.
Footnotes
P- Reviewer: Fawzy ME, Kato TS S- Editor: Ji FF L- Editor: A E- Editor: Wu HL
Supported by The Health Research Council of New Zealand
References
- 1.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 2.Sowers JR, Epstein M, Frohlich ED. Diabetes, hypertension, and cardiovascular disease: an update. Hypertension. 2001;37:1053–1059. doi: 10.1161/01.hyp.37.4.1053. [DOI] [PubMed] [Google Scholar]
- 3.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 4.Paillole C, Dahan M, Paycha F, Solal AC, Passa P, Gourgon R. Prevalence and significance of left ventricular filling abnormalities determined by Doppler echocardiography in young type I (insulin-dependent) diabetic patients. Am J Cardiol. 1989;64:1010–1016. doi: 10.1016/0002-9149(89)90799-6. [DOI] [PubMed] [Google Scholar]
- 5.Zarich SW, Arbuckle BE, Cohen LR, Roberts M, Nesto RW. Diastolic abnormalities in young asymptomatic diabetic patients assessed by pulsed Doppler echocardiography. J Am Coll Cardiol. 1988;12:114–120. doi: 10.1016/0735-1097(88)90364-6. [DOI] [PubMed] [Google Scholar]
- 6.Romano S, Di Mauro M, Fratini S, Guarracini L, Guarracini F, Poccia G, Penco M. Early diagnosis of left ventricular diastolic dysfunction in diabetic patients: a possible role for natriuretic peptides. Cardiovasc Diabetol. 2010;9:89. doi: 10.1186/1475-2840-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarich SW, Nesto RW. Diabetic cardiomyopathy. AHJ. 1989;5:1000–1012. doi: 10.1016/0002-8703(89)90236-6. [DOI] [PubMed] [Google Scholar]
- 8.Piccini JP, Klein L, Gheorghiade M, Bonow RO. New insights into diastolic heart failure: role of diabetes mellitus. Am J Med. 2004;116 Suppl 5A:64S–75S. doi: 10.1016/j.amjmed.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 9.Celentano A, Vaccaro O, Tammaro P, Galderisi M, Crivaro M, Oliviero M, Imperatore G, Palmieri V, Iovino V, Riccardi G. Early abnormalities of cardiac function in non-insulin-dependent diabetes mellitus and impaired glucose tolerance. Am J Cardiol. 1995;76:1173–1176. doi: 10.1016/s0002-9149(99)80330-0. [DOI] [PubMed] [Google Scholar]
- 10.Shimoni Y, Ewart HS, Severson D. Type I and II models of diabetes produce different modifications of K+ currents in rat heart: role of insulin. J Physiol. 1998;507(Pt 2):485–496. doi: 10.1111/j.1469-7793.1998.485bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rees DA, Alcolado JC. Animal models of diabetes mellitus. Diabet Med. 2005;22:359–370. doi: 10.1111/j.1464-5491.2005.01499.x. [DOI] [PubMed] [Google Scholar]
- 12.Schnedl WJ, Ferber S, Johnson JH, Newgard CB. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes. 1994;43:1326–1333. doi: 10.2337/diab.43.11.1326. [DOI] [PubMed] [Google Scholar]
- 13.Fein FS, Kornstein LB, Strobeck JE, Capasso JM, Sonnenblick EH. Altered myocardial mechanics in diabetic rats. Circ Res. 1980;47:922–933. doi: 10.1161/01.res.47.6.922. [DOI] [PubMed] [Google Scholar]
- 14.Dai S, McNeill JH. Ascorbic acid supplementation prevents hyperlipidemia and improves myocardial performance in streptozotocin-diabetic rats. Diabetes Res Clin Pract. 1995;27:11–18. doi: 10.1016/0168-8227(94)01013-p. [DOI] [PubMed] [Google Scholar]
- 15.Li CJ, Lv L, Li H, Yu DM. Cardiac fibrosis and dysfunction in experimental diabetic cardiomyopathy are ameliorated by alpha-lipoic acid. Cardiovasc Diabetol. 2012;11:73. doi: 10.1186/1475-2840-11-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Ward ML, Phillips AR, Zhang S, Kennedy J, Barry B, Cannell MB, Cooper GJ. Protection of the heart by treatment with a divalent-copper-selective chelator reveals a novel mechanism underlying cardiomyopathy in diabetic rats. Cardiovasc Diabetol. 2013;12:123. doi: 10.1186/1475-2840-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. J Physiol. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fabiato A, Fabiato F. Dependence of the contractile activation of skinned cardiac cells on the sarcomere length. Nature. 1975;256:54–56. doi: 10.1038/256054a0. [DOI] [PubMed] [Google Scholar]
- 19.Ren J, Davidoff AJ. Diabetes rapidly induces contractile dysfunctions in isolated ventricular myocytes. Am J Physiol. 1997;272:H148–H158. doi: 10.1152/ajpheart.1997.272.1.H148. [DOI] [PubMed] [Google Scholar]
- 20.Choi KM, Zhong Y, Hoit BD, Grupp IL, Hahn H, Dilly KW, Guatimosim S, Lederer WJ, Matlib MA. Defective intracellular Ca(2+) signaling contributes to cardiomyopathy in Type 1 diabetic rats. Am J Physiol Heart Circ Physiol. 2002;283:H1398–H1408. doi: 10.1152/ajpheart.00313.2002. [DOI] [PubMed] [Google Scholar]
- 21.Cooper GJ, Phillips AR, Choong SY, Leonard BL, Crossman DJ, Brunton DH, Saafi ‘L, Dissanayake AM, Cowan BR, Young AA, et al. Regeneration of the heart in diabetes by selective copper chelation. Diabetes. 2004;53:2501–2508. doi: 10.2337/diabetes.53.9.2501. [DOI] [PubMed] [Google Scholar]
- 22.Pierce GN, Russell JC. Regulation of intracellular Ca2+ in the heart during diabetes. Cardiovasc Res. 1997;34:41–47. doi: 10.1016/s0008-6363(97)00010-2. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Cannell MB, Phillips AR, Cooper GJ, Ward ML. Altered calcium homeostasis does not explain the contractile deficit of diabetic cardiomyopathy. Diabetes. 2008;57:2158–2166. doi: 10.2337/db08-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ishikawa T, Kajiwara H, Kurihara S. Alterations in contractile properties and Ca2+ handling in streptozotocin-induced diabetic rat myocardium. Am J Physiol. 1999;277:H2185–H2194. doi: 10.1152/ajpheart.1999.277.6.H2185. [DOI] [PubMed] [Google Scholar]
- 25.Kotsanas G, Delbridge LM, Wendt IR. Stimulus interval-dependent differences in Ca2+ transients and contractile responses of diabetic rat cardiomyocytes. Cardiovasc Res. 2000;46:450–462. doi: 10.1016/s0008-6363(00)00062-6. [DOI] [PubMed] [Google Scholar]
- 26.Norby FL, Wold LE, Duan J, Hintz KK, Ren J. IGF-I attenuates diabetes-induced cardiac contractile dysfunction in ventricular myocytes. Am J Physiol Endocrinol Metab. 2002;283:E658–E666. doi: 10.1152/ajpendo.00003.2002. [DOI] [PubMed] [Google Scholar]
- 27.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- 28.Nagai R, Zarain-Herzberg A, Brandl CJ, Fujii J, Tada M, MacLennan DH, Alpert NR, Periasamy M. Regulation of myocardial Ca2+-ATPase and phospholamban mRNA expression in response to pressure overload and thyroid hormone. Proc Natl Acad Sci USA. 1989;86:2966–2970. doi: 10.1073/pnas.86.8.2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bode EF, Briston SJ, Overend CL, O’Neill SC, Trafford AW, Eisner DA. Changes of SERCA activity have only modest effects on sarcoplasmic reticulum Ca2+ content in rat ventricular myocytes. J Physiol. 2011;589:4723–4729. doi: 10.1113/jphysiol.2011.211052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zdárská D, Pelísková P, Charvát J, Slavícek J, Mlcek M, Medová E, Kittnar O. ECG body surface mapping (BSM) in type 1 diabetic patients. Physiol Res. 2007;56:403–410. doi: 10.33549/physiolres.931021. [DOI] [PubMed] [Google Scholar]
- 31.Cheng H, Cannell MB, Lederer WJ. Propagation of excitation-contraction coupling into ventricular myocytes. Pflugers Arch. 1994;428:415–417. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- 32.Soeller C, Cannell MB. Examination of the transverse tubular system in living cardiac rat myocytes by 2-photon microscopy and digital image-processing techniques. Circ Res. 1999;84:266–275. doi: 10.1161/01.res.84.3.266. [DOI] [PubMed] [Google Scholar]
- 33.Wei S, Guo A, Chen B, Kutschke W, Xie YP, Zimmerman K, Weiss RM, Anderson ME, Cheng H, Song LS. T-tubule remodeling during transition from hypertrophy to heart failure. Circ Res. 2010;107:520–531. doi: 10.1161/CIRCRESAHA.109.212324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward ML, Crossman DJ, Cannell MB. Mechanisms of reduced contractility in an animal model of hypertensive heart failure. Clin Exp Pharmacol Physiol. 2011;38:711–716. doi: 10.1111/j.1440-1681.2011.05563.x. [DOI] [PubMed] [Google Scholar]
- 35.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci USA. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Crossman DJ, Ruygrok PN, Soeller C, Cannell MB. Changes in the organization of excitation-contraction coupling structures in failing human heart. PLoS One. 2011;6:e17901. doi: 10.1371/journal.pone.0017901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang QS, Huang XN, Yang GZ, Jiang XY, Zhou QX. Inhibitory effect of ginsenoside Rb1 on calcineurin signal pathway in cardiomyocyte hypertrophy induced by prostaglandin F2alpha. Acta Pharmacol Sin. 2007;28:1149–1154. doi: 10.1111/j.1745-7254.2007.00601.x. [DOI] [PubMed] [Google Scholar]
- 38.Song LS, Sobie EA, McCulle S, Lederer WJ, Balke CW, Cheng H. Orphaned ryanodine receptors in the failing heart. Proc Natl Acad Sci USA. 2006;103:4305–4310. doi: 10.1073/pnas.0509324103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kobayashi T, Jin L, de Tombe PP. Cardiac thin filament regulation. Pflugers Arch. 2008;457:37–46. doi: 10.1007/s00424-008-0511-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koubassova NA, Tsaturyan AK. Molecular mechanism of actin-myosin motor in muscle. Biochemistry (Mosc) 2011;76:1484–1506. doi: 10.1134/S0006297911130086. [DOI] [PubMed] [Google Scholar]
- 41.Pierce GN, Dhalla NS. Cardiac myofibrillar ATPase activity in diabetic rats. J Mol Cell Cardiol. 1981;13:1063–1069. doi: 10.1016/0022-2828(81)90296-0. [DOI] [PubMed] [Google Scholar]
- 42.Malhotra A, Sanghi V. Regulation of contractile proteins in diabetic heart. Cardiovasc Res. 1997;34:34–40. doi: 10.1016/s0008-6363(97)00059-x. [DOI] [PubMed] [Google Scholar]
- 43.Akella AB, Ding XL, Cheng R, Gulati J. Diminished Ca2+ sensitivity of skinned cardiac muscle contractility coincident with troponin T-band shifts in the diabetic rat. Circ Res. 1995;76:600–606. doi: 10.1161/01.res.76.4.600. [DOI] [PubMed] [Google Scholar]
- 44.Hofmann PA, Menon V, Gannaway KF. Effects of diabetes on isometric tension as a function of [Ca2+] and pH in rat skinned cardiac myocytes. Am J Physiol. 1995;269:H1656–H1663. doi: 10.1152/ajpheart.1995.269.5.H1656. [DOI] [PubMed] [Google Scholar]
- 45.Kawaguchi M, Asakura T, Saito F, Nemoto O, Maehara K, Miyake K, Sugai N, Maruyama Y. Changes in diameter size and F-actin expression in the myocytes of patients with diabetes and streptozotocin-induced diabetes model rats. J Cardiol. 1999;34:333–339. [PubMed] [Google Scholar]
- 46.Cagalinec M, Waczulíková I, Uličná O, Chorvat D. Morphology and contractility of cardiac myocytes in early stages of streptozotocin-induced diabetes mellitus in rats. Physiol Res. 2013;62:489–501. doi: 10.33549/physiolres.932467. [DOI] [PubMed] [Google Scholar]
- 47.Jüllig M, Hickey AJ, Middleditch MJ, Crossman DJ, Lee SC, Cooper GJ. Characterization of proteomic changes in cardiac mitochondria in streptozotocin-diabetic rats using iTRAQ™ isobaric tags. Proteomics Clin Appl. 2007;1:565–576. doi: 10.1002/prca.200600831. [DOI] [PubMed] [Google Scholar]
- 48.Lazar HL. Alterations in myocardial metabolism in the diabetic myocardium. Semin Thorac Cardiovasc Surg. 2006;18:289–292. doi: 10.1053/j.semtcvs.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 49.Candido R, Forbes JM, Thomas MC, Thallas V, Dean RG, Burns WC, Tikellis C, Ritchie RH, Twigg SM, Cooper ME, et al. A breaker of advanced glycation end products attenuates diabetes-induced myocardial structural changes. Circ Res. 2003;92:785–792. doi: 10.1161/01.RES.0000065620.39919.20. [DOI] [PubMed] [Google Scholar]
- 50.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–1652. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 51.Di Bello V, Talarico L, Picano E, Di Muro C, Landini L, Paterni M, Matteucci E, Giusti C, Giampietro O. Increased echodensity of myocardial wall in the diabetic heart: an ultrasound tissue characterization study. J Am Coll Cardiol. 1995;25:1408–1415. doi: 10.1016/0735-1097(95)00026-Z. [DOI] [PubMed] [Google Scholar]